

Abstract
In diabetic patients and animal models of diabetes mellitus (DM), circulating endothelial progenitor cell (EPC) number is lower than in normoglycaemic conditions and EPC angiogenic properties are inhibited. Stromal cell derived factor-1 (SDF-1) plays a key role in bone marrow (BM) c-kit+ stem cell mobilization into peripheral blood (PB), recruitment from PB into ischemic tissues and differentiation into endothelial cells. The aim of the present study was to examine the effect of DM in vivo and in vitro, on murine BM-derived c-kit+ cells and on their response to SDF-1. Acute hindlimb ischemia was induced in streptozotocin-treated DM and control mice; circulating c-kit+ cells exhibited a rapid increase followed by a return to control levels which was significantly faster in DM than in control mice. CXCR4 expression by BM c-kit+ cells as well as SDF-1 protein levels in the plasma and in the skeletal muscle, both before and after the induction of ischemia, were similar between normoglycaemic and DM mice. However, BM-derived c-kit+ cells from DM mice exhibited an impaired differentiation towards the endothelial phenotype in response to SDF-1; this effect was associated with diminished protein kinase phosphorylation. Interestingly, SDF-1 ability to induce differentiation of c-kit+ cells from DM mice was restored when cells were cultured under normoglycaemic conditions whereas c-kit+ cells from normoglycaemic mice failed to differentiate in response to SDF-1 when they were cultured in hyperglycaemic conditions. These results show that DM diminishes circulating c-kit+ cell number following hindlimb ischemia and inhibits SDF-1-mediated AKT phosphorylation and differentiation towards the endothelial phenotype of BM-derived c-kit+ cells.
Keywords: diabetes mellitus, stem cell, SDF-1, chemokine, PI3K/AKT
Introduction
Several studies, in human beings and in animal models, have shown that circulating endothelial progenitor cells (EPCs) are recruited into ischemic tissues [1, 2], where they contribute to recovery of perfusion by differentiating into endothelial cells [3] and producing angiogenic factors, thereby enhancing collateral vessel formation [4]. Stromal cell derived factor-1 (SDF-1), via phosphoinositide 3-kinase (PI3K)/AKT activation appears to play an important role in EPC function [5, 6].
SDF-1 exerts a chemoattractive function on haematopoietic stem cells [7] and modulates integrin receptors affinity to extracellular matrix components [8–10]. Further, a prior work from our laboratory has shown that SDF-1 induces adhesion-dependent differentiation of bone marrow (BM)-derived c-kit+ progenitors into endothelial cells onto extracellular matrix components as well as their recruitment from the BM in response to acute hindlimb ischemia [11].
Patients with type 1 and 2 diabetes mellitus (DM) exhibit impaired new blood vessel development in response to ischemia [12, 13], including cardiac and limb ischemia and skin ulcers [14, 15], and it has been suggested that EPCs defects may contribute to diabetic vascular complications. In these patients, mobilization and proliferation of circulating EPCs [12, 16–18] is impaired. Further, in animal models of streptozotocin (STZ)-induced, type 1-like diabetes [19], and type 2-like diabetes due to obesity [20] EPC transplantation following acute hindlimb ischemia fails to induce neovascularization. The role of SDF-1 in diabetic vascular disease is still poorly characterized. It has been shown that CD34+ cells from diabetic patients exhibit a marked decrease in SDF-1-induced migration [21]. In addition, in normoglycaemic rats SDF-1 plasma levels increase following acute hindlimb ischemia/reperfusion and this response is abolished in diabetic rats [22], whereas in diabetic mice SDF-1 expression in skin ulcers is lower than in normoglycaemic controls [23]. In contrast, it has been found a marked increase in SDF-1 mRNA in the mesentery of STZ-treated diabetic rats [24].
In the present study, we used a mouse model of STZ-induced DM to analyse the time course of c-kit+ cell mobilization after hindlimb ischemia as well as SDF-1 expression in the plasma and in the skeletal muscle. Further, we studied the effect of DM and hyperglycaemia on SDF-1-ability to induce BM-derived c-kit+ cell differentiation into endothelial cells and of DM to induce AKT phosphorylation in vitro.
Materials and methods
Animal models
Diabetes mellitus (DM) was induced in 2-month-old Swiss CD1 male mice injected intraperitoneally with 40 mg/kg STZ (Sigma-Aldrich, St. Luis, MO, USA) in 0.05 M Na citrate (pH 4.5) daily for 5 days, as previously described [25]. Control mice were exposed to an identical protocol, in the absence of STZ treatment. Detection of glycaemic levels was performed as described in online Supporting Information. At 1 month following STZ treatment, hyperglycaemic mice (>200 mg/dl) were used for in vitro and in vivo experiments. In some experiments, mice underwent femoral artery dissection under general anaesthesia to induce hindlimb ischemia [25]. Sham operated animals underwent the same treatment of ischemic mice without femoral artery dissection and were used as controls. Limb perfusion index was determined by laser Doppler perfusion imaging before and at different time-points (1, 3, 7, 14, 21, 28 days) before and after femoral artery dissection [26]. For this analysis the limbs were shaved and the perfusion index was defined as the ratio between the perfusion of ischemic and controlateral paw.
Cell isolation and culture methods
BM c-kit+ cells were isolated from control and DM mice by magnetic cell sorting (MINI-MACS; Miltenyi Biotech, Bergisch, Gladbach, Germany), as previously described [11].
Differentiation assays were performed in glass chamber slides (Nalgene, Rochester, NY, USA) coated with 20 μg/ml fibronectin (FN) in RPMI medium (Invitrogen, Eugene, OR, USA) containing 5 mM glucose, supplemented with 5% foetal calf serum (FCS; Sigma-Aldrich) either in the presence or the absence of 100 ng/ml SDF-1 (R&D System, Minneapolis, MN, USA) or SDF-1 inactivated by boiling (SDF-1 B), at the same concentration. After 1 week cells were identified by Ac-LDL-DiI uptake and counted as described [11]. For immunostaining, cells were fixed with 4% paraformaldehyde in PBS. In some differentiation assays of BM-derived c-kit+ cells from normoglycaemic mice, 10 μM LY294022 (LY) (Sigma-Aldrich), a selective inhibitor of PI3K activity, was added to the medium, for 1 week, either in the presence or in the absence of 100 ng/ml SDF-1.
In some experiments, the effect of high glucose on SDF-1-induced c-kit+ cell differentiation towards the endothelial lineage was examined. In these studies, c-kit+ cells were expanded for 1 week in Stem Span serum free medium (Stem Cell Technologies, Vancouver, Canada) containing the following recombinant human cytokines: 100 ng/ml SCF, 20 ng/ml IL-3, 20 ng/ml IL-6, 100 ng/ml Flt-3 ligand (R&D Systems) [27]. Since stem span medium contains 25 mM glucose, hyperglycaemia was achieved by adding glucose to achieve a final concentration of 50 mM whereas the control medium was supplemented with 25 mM mannitol to achieve a similar osmolality and the final glucose concentration was 25 mM. After 1 week, cell expansion the differentiation assay was performed for one additional week in RPMI medium as described above. It is noteworthy that the RPMI medium contains 5 mM glucose; therefore, hyperglycaemia was achieved by supplementing this medium with glucose to achieve a final glucose concentration of 30 mM whereas the control medium was supplemented with 25 mM mannitol to achieve the same osmolality and keep the glucose concentration at 5 mM. SDF-1 (100 ng/ml) was either present or absent throughout the 2 weeks duration of this experiment.
Immunofluorescence, clonogenic and chemotaxis assays are described in Supporting Information material.
Flow cytometry
C-kit, CXCR4, Sca-1, CD34, KDR and α4 integrin receptor VLA-4 expression were evaluated by flow cytometry. Freshly isolated c-kit+ cells from normoglycaemic and DM mice were incubated in PBS containing 0.5% FCS for 20 min. on ice with fluorochrome-conjugated monoclonal antibodies recognizing murine c-kit (clone 2B8), CD34 (clone RAM34), (BD Biosciences Pharmingen, San Diego, CA, USA), Flk-1/KDR(VEGFR2) (clone 89106, R&D System), Sca-1 (clone E13–161.7), CXCR4 (clone 2B11), α4 integrin (clone R1–2) (BD Biosciences Pharmingen) at 0.8–2 mg/ml and antigen-presenting cell conjugated lineage antibody cocktail (BD Biosciences Pharmingen). BM-mononuclear cells (BM-MNCs), BM-derived c-kit+ cells and peripheral blood (PB)-mononuclear cells (PB-MNCs) were analysed by FACScalibur Fluorescence-Activated Cell Sorter (BD Biosciences Pharmingen); 1 × 104 and 5 × 104 gated events were acquired, respectively. FACS analysis of AKT phosphorylation (pAKT) was performed as follows: total BM cells were incubated overnight (37°C; 5% CO2 atmosphere) in starvation medium (IMDM: Sigma-Aldrich). Subsequently, cells were washed in PBS containing 0.5% bovine serum albumin (BSA) and incubated with FITC-coniugated anti-murine c-kit antibody for 20 min. at 21°C. SDF-1 (100 ng/ml) was added to induce AKT phosphorylation. Cells were incubated for additional 10 min. at 37°C, thereafter cells were fixed with PBS containing 2% paraformaldehyde for 10 min. at 21°C. Cells were then permeabilized with 100 μl PBS containing 0.5% BSA and 0.5% saponin and incubated for 5 min. at 21°C. Finally, 20 μl of PE-conjugated anti-phospho AKT (clone J1–223.371, threonine 308, BD Biosciences Pharmingen) antibody were added. For additional details concerning flow cytometry analysis see online Supporting Information.
Western blot analysis
BM-MNCs from normoglycaemic and DM mice were separated by Ficoll gradient and incubated with SDF-1 (100 ng/ml) at 37°C for 1, 5 and 10 min. in serum-free RPMI. Western blotting was performed following standard procedures using 1: 1000 dilution of a primary anti-phospho-AKT antibody (cod. 9271S, serine 473, Cell Signalling Technology, Danvers, MA, USA) for 2 hrs at room temperature or overnight at 4°C followed by secondary antibody incubation and ECL, followed by autoradiography.
Statistical analysis
Statistical analysis was performed on at least three independent observations in each experimental group and the results were analysed either by Student’s t-test, 1-way or 2-way anova according to the experimental design. If the overall anova P-value was significant, pairwise comparisons were performed by Student–Newman–Keuls (NK) or Bonferroni post hoc tests. The GraphPad Prism software (version 5.00 for Windows, GraphPad Software, San Diego, CA, USA, http://www.graphpad.com) was used for computer analysis. The results are expressed as mean ± S.E.M. The threshold for statistical significance was set as P-value less than 0.05.
Results
Effect of DM on recovery of perfusion, circulating c-kit+ cell number and SDF-1 levels in hindlimb ischemia
Initial experiments were aimed at establishing whether there was a difference in blood flow recovery following acute hindlimb ischemia in normoglycaemic versus DM mice. In agreement with prior studies [19, 28–30], it was found that recovery of perfusion index in the ischemic limb was delayed in DM. The rescue of hindlimb perfusion in normoglycaemic animals started at day 14 after surgery and became significantly higher than in DM mice at later time-points, i.e. 21 and 28 days after femoral artery dissection (Fig. 1A and B, Fig. S1A). To unravel whether BM stem cell mobilization differed between DM and normoglycaemic mice, c-kit+ cells in the systemic circulation and in the BM were quantified by flow cytometry analysis. Prior to ischemia, c-kit+ cells in the systemic circulation represented 0.55 ± 0.09% of total PB-MNCs in control (n= 12) and 0.20 ± 0.07% in DM (n= 11) mice (P < 0.05). Moreover, after surgery the transient increase in PB-c-kit+ cells was more sustained in normoglycaemic than in DM mice (Fig. 1C). In contrast, c-kit+ cell number in the BM was similar in control and DM mice both under baseline conditions and at different times after acute ischemia (not shown). Neither normoglycaemic nor DM sham-operated animals showed evidence of c-kit+ cell mobilization (Fig. S2). Interestingly, we found no difference between control and DM mice in SDF-1 plasma and skeletal muscle protein levels although, as shown in Fig. S3, they were both elevated after induction of ischemia. Thus, DM mice had fewer circulating c-kit+ cells under baseline conditions and exhibited an increase of these cells in the bloodstream following the induction of acute and severe hindlimb ischemia. In addition, the return of c-kit+ cells to basal levels was faster in DM than in control animals. These differences could not be attributed to different SDF-1 levels either in the muscle or in the systemic circulation between normoglycaemic and DM mice. It has been suggested that both CD34 and Flk-1/KDR (VEGFR2) antigens, as well as c-kit, may characterize EPC populations [31]. We therefore determined the expression of these markers in BM-derived c-kit+ cells from normal mice and found c-kit+ cell subfractions coexpressing either CD34 or KDR (Fig. 2A, Fig. S1B). We evaluated DM effect on BM and PB CD34+, Flk-1+/KDR+(VEGFR2+) and CD34+/KDR+ mononuclear cell number; both in the BM and PB of DM mice there was a trend towards fewer CD34+, KDR+ and CD34+/KDR+ cells than in control animals; however, a statistical difference was found only in the case of PB KDR+ cells (Fig. 2B and C). Previous studies have described two EPCs types, namely CFU-ECs (early EPCs) and ECFCs (late EPCs). CFU-EC and ECFCs express stem cell markers such as CD34 and (in human beings) CD133. However, CFU-ECs can be distinguished from ECFCs on the basis of haematopoietic lineage markers expression such as CD45 [32, 33]. Therefore, to discriminate between CFU-EC and ECFC phenotypes, the expression of haematopoietic lineage markers (CD3ε, CD11b, CD45R/B220, Ly76 and Gr-1 markers) in circulating KDR+ and c-kit+ cells was investigated by flow cytometry. The results showed that, in normal mice, the percentage of c-kit+/lin− and c-kit+/lin+ cells was, respectively, 0.035 ± 0.034 and 0.87 ± 0.45 and that the percentage of KDR+/lin− and KDR+/Lin+ cells was, respectively 0.037 ± 0.031 and 0.71 ± 0.21 (mean ± S.E., n= 4; Fig. S4). In DM mice, it was not possible to determine the number of KDR+/lin−, KDR+/lin+, c-kit+/lin− and c-kit+/lin− cells as this value was below detection limits, at least under our experimental conditions (not shown). We conclude that circulating KDR+ and c-kit+ cells have a phenotype resembling CFU-EC EPC type, and that DM reduces the number of these circulating progenitors.
Figure 1.
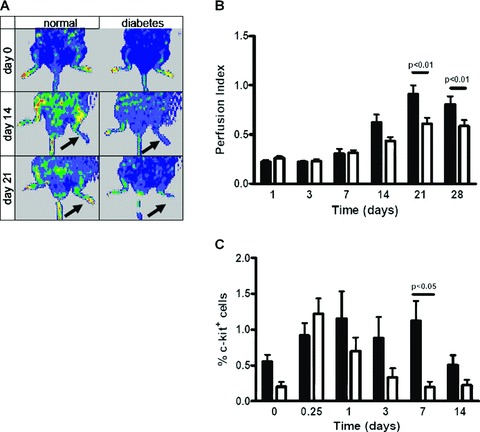
Diabetes impairs blood flow recovery in response to hindlimb ischemia and c-kit+ cell number in the systemic circulation. (A) Laser Doppler perfusion imaging of normoglycaemic and DM mice before and at 14 and 21 days following acute hindlimb ischemia. Perfusion index was calculated by normalizing the colour intensity of the ischemic versus non ischemic limb in the same animal. Arrows indicate the ischemic limb. Note the impaired perfusion recovery in DM. (B) Average perfusion index evaluated by laser Doppler perfusion imaging and expressed as the ratio between ischemic and non-ischemic paw in normoglycaemic (black bars; n= 3–7) and DM (open bars; n= 3–12) mice. The response profile was significantly different between normal and DM mice (anovaP < 0.01). Post hoc analysis for pairwise comparisons between DM and control animals demonstrated significant differences in perfusion index at day 21 (P < 0.05) and day 28 (P < 0.05). Student’s t-test showed significant differences at 14, 21 and 28 days after ischemia (P < 0.05) (C) C-kit+ cells in the systemic circulation expressed as% of PB-MNCs. Prior to femoral artery dissection there were more c-kit+ cells in normoglycaemic (black bars; n= 12) than in DM (open bars; n= 11) mice (Student’s t-test; P < 0.05). Moreover, after acute ischemia the transient increase in c-kit+ cells was more pronounced and sustained in normoglycaemic (n= 8–14 at each time-point) than in DM (n= 8–14 at each time-point) mice (anovaP < 0.01); post hoc analysis for pairwise comparisons demonstrated a significant difference at day 7 (P < 0.05). PB c-kit+ cells were expressed as percentage of 5 × 104 PB-MNCs as evaluated by flow cytometry. Prior to femoral artery dissection total PB-MNCs number was 2.85 × 106± 1.27 × 106 in normoglycaemic (n= 4) and 2.81 × 106± 0.59 × 106 in DM (n= 4) mice (P= n.s.); at different time-points after ischemia there continued to be no difference in PB-MNCs number between normoglycaemic and DM mice.
Figure 2.
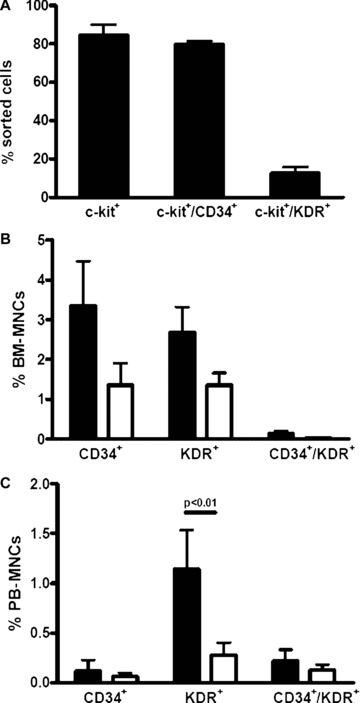
CD34+ and KDR+ cells in the bone marrow and peripheral blood of control and diabetic mice. (A) Percentages of BM-derived c-kit+, c-kit+/CD34+ and c-kit+/KDR+ cells purified for c-kit antigen by MACS (n= 3). (B) Histogram showing percentage of CD34+, KDR+ and CD34+/KDR+ in the BM of control (black bars; n= 9) and DM (open bars; n= 11) mice. (C) Histogram showing percentage of CD34+, KDR+ and CD34+/KDR+ in the PB of control (black bars; n= 10) and DM (open bars; n= 9) mice. Result of statistical analysis by anova and post hoc test is indicated by P-value above bars.
Effects of DM on c-kit+ cell differentiation
Diabetes has been reported to inhibit human EPCs differentiation [12, 17] and defects in CXCR4 signalling are known to jeopardize EPCs’ angiogenic properties [34, 35]. Therefore, we examined the effect of diabetes on SDF-1-directed EPC differentiation into endothelial cells, and tested whether culture in hyperglycaemia mimics DM effects on c-kit+ cell differentiation. We have previously described that SDF-1 enhances mouse BM c-kit+ cells endothelial differentiation through increased stem cell adhesion to FN and collagen I [11]. Under our experimental conditions the majority (>95%) of adherent cells differentiated and expressed factor VIII (vWF), KDR, CD31 and were also positive for acetylated LDL-DiI uptake [11] (Fig. S5). Therefore, an increase in differentiation was indicated by a higher number of cells adherent to the FN-coated glass chamber slide and not by an increase in the number of cells positive for endothelial cell markers. It was then quantitatively examined the adherence/differentiation of c-kit+ cells into endothelial cells as determined by acetylated LDL-DiI uptake [36]. In the absence of exogenous SDF-1, the basal level of endothelial adhesion/differentiation was similar in c-kit+ cells isolated from both experimental groups. In contrast, upon exposure to SDF-1 adhesion/differentiation was significantly higher in c-kit+ cells from normal than from DM mice. Moreover, stimulation by inactivated SDF-1 (SDF-1 B) failed to induce adhesion/differentiation of cells isolated both from normoglycaemic and DM mice (Fig. 3A).
Figure 3.
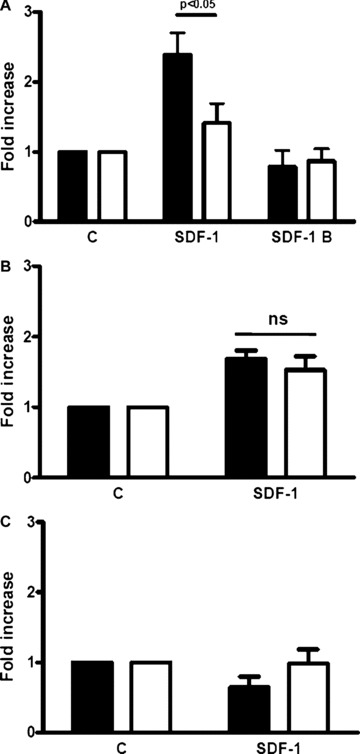
DM reduces BM-derived c-kit+ cell adhesion/differentiation into endothelial cell. BM-derived c-kit+ cell adhesion/differentiation into endothelial lineage was determined by Ac-LDL-DiI uptake. (A) SDF-1 enhanced c-kit+ cells adhesion/differentiation into endothelium when cells were obtained from normoglycaemic mice (black bars; n= 7, Student’s t-test P < 0.05) but had no effect when cells were obtained from DM mice (open bars; n= 7, Student’s t-test, P= n.s.). Inactive SDF-1 (SDF-1 B) failed to induce cell adhesion/differentiation in both experimental groups. The response to SDF-1 differed between normoglycaemic- and DM-derived cells (P-value for anova and post hoc test is shown in the figure above the bar graph). (B) C-kit+ cells from DM mice (open bars), expanded for 1 week in stem span medium supplemented with IL-3, IL-6, Flt3-L and SCF, and subsequently kept in differentiation medium for another week, augmented their adhesion/differentiation into endothelial cells in the presence of SDF-1 (n= 8, Student’s t-test P < 0.05). A similar response to SDF-1 was observed in c-kit+ cells from normoglycaemic mice (black bars) (n= 8, Student’s t-test P < 0.05). Interestingly, under these experimental conditions, the response to SDF-1 was similar between the two experimental groups (P-value [n.s.] for anova and post hoc test is shown). (C) C-kit+ cells from control (black bars; n= 4) and DM mice (open bars; n= 4) were expanded for 1 week in hyperglycaemic medium and then shifted to hyperglycaemic differentiation medium for another week (see ‘Materials and methods’). C-kit+ cells from normoglycaemic mice exposed to hyperglycaemia failed to enhance adhesion/differentiation towards the endothelial lineage in response to 100 ng/ml SDF-1 and this effect was comparable to that of cells from DM mice.
A modified differentiation potential of c-kit+ cells may reflect a modulation of their haematopoietic stem cell properties due to DM. However, flow cytometry analysis showed that CXCR4, Sca-1, VLA-4 and CD34 marker expression was similar in sorted c-kit+ cells from normoglycaemic and DM mice (Fig. S6A–C). Furthermore, there were no differences in haematopoietic clonogenicity (Fig. S7A) of c-kit+ cells from control and DM mice. In agreement with prior studies SDF-1 markedly enhanced c-kit+ cell migration in a modified Boyden chamber assay; however, there were no differences in the migratory response of c-kit+ cells from normoglycaemic and DM mice (Fig. S7B). Finally, culturing c-kit+ cells for 1 week in high glucose did not modify the percentage of c-kit+, c-kit+/CD34+ and c-kit+/KDR+ cells compared to normal culture or culture in the presence of the iso-osmotic control (Fig. S7C).
In additional experiments, we examined whether the impairment in SDF-1-induced c-kit+ cell differentiation into endothelial cells was reversible upon cell culture in normal glucose. BM-derived c-kit+ cells were obtained from control and DM mice and expanded in normoglycaemic liquid culture for 1 week. Thereafter, differentiation assays were performed and we found no difference between normal and DM mice in c-kit+ cells ability to differentiate in response to SDF-1 (Fig. 3B). This result indicates that DM ability to impair SDF-1-induced c-kit+ cell differentiation into endothelial cells is reversible. It is noteworthy that c-kit+ cells from both control and DM mice cultured for 1 week in high glucose exhibited impaired SDF-1-induced c-kit+ cell differentiation into endothelial cells (Fig. 3C).
PI3K/AKT pathway is involved in human EPCs differentiation [5] and represents an intracellular signalling cascade activated by SDF-1 in haematopoietic progenitors [37, 38]. Thus, we tested whether, under our experimental conditions, PI3K/AKT pathway activity was linked to SDF-1-induced endothelial differentiation. BM-derived c-kit+ cells from normoglycaemic mice were cultured for 1 week either in the presence of SDF-1, the selective PI3K inhibitor LY294002 (LY) or both SDF-1 and LY. Interestingly, LY abolished SDF-1-mediated c-kit+ cell adhesion/differentiation into Ac-LDL-DiI+ endothelial cells (Fig. 4A). In additional experiments, we examined SDF-1 ability to induce PI3K/AKT phosphorylation in BM-derived mononuclear cells. In cells obtained from normoglycaemic mice AKT phosphorylation increased as early as 1 min. upon SDF-1 treatment and remained elevated up to 10 min. thereafter (Fig. 4B, left panel and Fig. 4C). In contrast, SDF-1 failed to induce AKT phosphorylation in BM-derived mononuclear cells from DM mice (Fig. 4B, right panel and Fig. 4C). It is noteworthy that these experiments could not be performed on c-kit+ cells alone and the whole mononuclear cell fraction was used in order to have enough material for Western blot analysis. In order to clearly establish whether AKT phosphorylation was modulated in c-kit+ cells, BM-derived mononuclear cells were obtained from normal and DM mice and flow cytometry analysis was performed by double staining cells for c-kit and pAKT. The number of c-kit+/pAKT+ cells was evaluated before and after 10 min. exposure to SDF-1. It was found that SDF-1-induced AKT phosphorylation of c-kit+ cells was significantly impaired in DM compared to normal mice (Fig. 4D and E). Altogether, the results of these experiments show that SDF-1-mediated c-kit+ cells differentiation into endothelial cells involves the PI3K/AKT pathway and that DM strongly reduces SDF-1-induced AKT activation as well as differentiation towards the endothelial phenotype.
Figure 4.
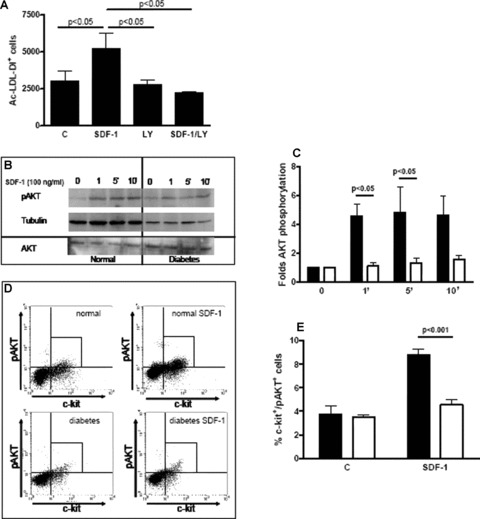
Diabetes impairs SDF-1-induced c-kit+ cell adhesion/differentiation into endothelium and AKT phosphorylation. (A) BM-derived c-kit+ cells were obtained from normoglycaemic mice and cultured in RPMI containing 5 mM glucose for 7 days. SDF-1 enhanced Ac-LDL-DiI+ cell number and this effect was abolished by the PI3K/AKT inhibitor LY294002 (n= 3 for each group). This effect was comparable to that of DM shown in Fig. 3A. Statistical significance was evaluated by anova and post hoc analysis; P-values are reported above the bar graph. (B, C) Western blot analysis shows that SDF-1 enhanced AKT phosphorylation in BM-derived mononuclear cells obtained from normoglycaemic mice (black bars in C); this effect was already evident at 1 min. and remained elevated up to 10 min. after the exposure to the chemokine (Student’s t-test P < 0.05). In contrast, SDF-1 had no effect on AKT phosphorylation in BM-derived mononuclear cells obtained from DM mice (open bars in C). Significance was evaluated also by anova and post hoc analysis; n= 4 for each group, P-values are reported above the bar graph. (D, E) SDF-1 effect on AKT phosphorylation in c-kit+ cells from normal and DM mice. (D) shows representative flow cytometry plots of pAKT levels in c-kit+ cells. SDF-1 enhanced c-kit+/pAKT+ cell fraction in the control population (normal); in contrast it had no effect on cells obtained from DM mice (diabetes). Average results are shown in (E). SDF-1 increased c-kit+/pAKT+ cell percentage in total BM cells from normal mice after 10 min. of exposure to the chemokine (n= 3, Student’s t-test P < 0.05). In contrast, AKT phosphorylation was impaired in c-kit+ cells from DM mice (n= 3, Student’s t-test, P= n.s.). The response to SDF-1 differed between the two experimental groups (ANOVA, P < 0.001; post hoc P-value is indicated above the bar graph).
Discussion
Ischemia causes transient mobilization of BM-derived EPCs into the systemic circulation and homing in the ischemic tissue where these cells play a role in angiogenesis both by differentiating into endothelial cells[3] and by producing angiogenic cytokines that stimulate pre-existing endothelial cells to proliferate and differentiate [4, 39]. Both SDF-1 and VEGF have been involved in BM-derived EPC mobilization, homing in the ischemic tissue and differentiation into endothelial cells. In diabetic patients, as well as in animal models of DM, the angiogenic response to ischemia is inhibited; further, in human beings it has been shown that DM lowers the number [12] of circulating EPCs and their ability to form endothelial colonies in vitro[17, 40]. This phenomenon, at least in part, may be due to enhancement of oxidative stress related to hyperglycaemia [41]. In the present work, we used a mouse model of hindlimb ischemia to examine whether DM impairs SDF-1 effects on EPCs.
Initially, it was confirmed that DM inhibits blood flow recovery in the ischemic limb following femoral artery dissection and we found that c-kit+ cell number in the systemic circulation was lower in DM than in normoglycaemic mice both under control conditions and at different time-points following acute hindlimb ischemia. Interestingly, c-kit+ cell number in the BM was similar in control and DM mice. In order to establish whether in DM mice SDF-1/CXCR4 axis was impaired several end-points were evaluated: (i) CXCR4 expression in BM-derived c-kit+ cells, (ii) SDF-1 plasma levels, (iii) SDF-1 protein levels in the adductor skeletal muscle and (iv) c-kit+ cell response to SDF-1. DM had no effect on BM-derived c-kit+ cell expression of the SDF-1 receptor CXCR4. Further, SDF-1 levels in the plasma and in the adductor skeletal muscle of the ischemic limb were similar between control and DM mice prior to and at different times after femoral artery dissection. These results suggest that under our experimental conditions the lower c-kit+ cell number in the systemic circulation as well as the inhibited recovery of blood flow in the ischemic limb of DM mice could not be attributed to lower SDF-1 systemic or tissue levels and/or to lower CXCR4 expression on c-kit+ cells. It is noteworthy that a prior work in a rat model of hindlimb ischemia/reperfusion showed that DM inhibits SDF-1 transient increase in plasma SDF-1 [22]. This discrepancy with the present study may be due to the difference in species and ischemic injury, i.e. permanent ischemia versus ischemia/reperfusion.
In additional experiments, it was evaluated the effect of DM on SDF-1-induced c-kit+ cells clonogenic ability on methylcellulose, migration and differentiation into endothelial cells. DM had no effect on CFUs’ number both in the absence and presence of SDF-1, nor did it modulated SDF-1-directed c-kit+ cell migration. The latter result is in contradiction with findings reporting that hyperglycaemia impairs human EPCs migration [42]; experimental conditions, species and cell type differences may account for the discrepancy. In contrast with these negative results, it was found that DM inhibited SDF-1 ability to induce c-kit+ cell adhesion/differentiation into endothelium.
Recent studies have shown that PI3K/AKT plays a key role in EPC response to ischemia [6] and that DM impairs some EPCs functions [22, 43]. Similar results have been obtained with EPCs obtained from non-diabetic patients cultured in high glucose conditions [42, 44]. Since SDF-1 binds its receptor CXCR4 and via this mechanism activates PI3K-dependent signalling [37, 45, 46] leading to AKT phosphorylation [47, 48] it was examined whether SDF-1 ability to induce c-kit+ cell differentiation towards the endothelial lineage was related to AKT and, eventually, whether SDF-1-induced AKT phosphorylation was inhibited in DM. It was found that LY294022, a selective PI3K inhibitor, abolished SDF-1 ability to induce c-kit+ cell adhesion/differentiation into endothelial cell and that DM abrogated SDF-1 induced AKT-phosphorylation in this cell type. Therefore, inhibition of SDF-1 signalling appears to be a key mechanism for the impairment of SDF-1-induced c-kit+ cell differentiation into endothelial cells, their mobilization into the systemic circulation found in DM and, analogous to pancreatic β-Cell [49], EPC survival.
Glucotoxicity was likely the cause of this defect, as cells from DM mice expanded under normoglycaemic conditions recovered the ability to respond to SDF-1 and their response was similar to that of cells from control mice. Furthermore, c-kit+ cells from control mice kept in hyperglycaemia failed to respond to SDF-1 and their behaviour was similar to that of cells from DM mice. Our results are in line with reports by other groups that identified the PI3K-AKT axis as one of the most affected intracellular pathways in DM [50]. Possible mechanisms underlying the observed reduction of SDF-1-elicited AKT phosphorylation include the hyperglycaemia-associated up-regulation of PTEN phosphatase in c-kit+ cells as a consequence of intracellular reactive nitrogen species accumulation [51], and possible changes in c-kit+ cell stem differentiation caused by enhanced oxidative stress [52] and/or enhanced activity of FOXO transcription factors, as a result of diminished PI3K/AKT activity in DM [53, 54].
In summary, the present study did not identify differences between DM and control mice in SDF-1 plasma and skeletal muscle levels, neither in normoperfused nor in ischemic mice. Further, DM had no effect on CXCR4, Sca-1, VLA-4 and CD34 expression on c-kit+ cells, on CFUs number both in the absence and presence of SDF-1, nor it modulated SDF-1-directed c-kit+ cell migration. In contrast, DM inhibited SDF-1-induced c-kit+ cell differentiation into endothelial cell as well as AKT phosphorylation. The role of BM-derived c-kit+ cells in revascularization after hindlimb ischemia has been previously established [6, 11, 22] and EPCs appear to contribute significantly to the angiogenic response to ischemia [1]; however, EPC function is inhibited in DM [17, 44]. The findings of the present study identify a mechanism for EPC functional impairment in DM and suggest that decreased SDF-1-induced c-kit+ cell differentiation into endothelial cells and AKT phosphorylation may play a role in the inhibition of the angiogenic response to ischemia in DM.
Acknowledgments
This work has been supported by the Italian Ministry of Health, Program Grants: RFS contract no. 186/2000 issued to M.C.C. and M.P.); contract no. 164/2003 issued to M.P. and EU funded Project ‘Ulcer Therapy’ contract no: LSHB-CT-2005–512102 issued to M.C.C. and M.P.
Supporting Information
Fig. S1 Scatter plots of physical properties and gating of PB cells from control and DM mice assessed by flow cytometry analysis.
Fig. S2 Surgical manipulation does not inducec-kit+ cell mobilization.
Fig. S3 Plasma and skeletal muscle SDF-1 levels.
Fig. S4 Flow cytometry determination of lineage markers expression into PB from non-diabetic animals.
Fig. S5 Effect of SDF-1 on immunophenotypicalcharacterization and Ac-LDL-DiI uptake of culturedc-kit+ cells.
Fig. S6 DM does not alter stem cell markers expression inBMderived c-kit+ cells.
Fig. S7 DM does not modulate BM-derivedc-kit+ cells clonogenicity and migration in response to SDF-1.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
References
- 1.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi T, Kalka C, Masuda H, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–8. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 3.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–8. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 4.Tateishi-Yuyama E, Matsubara H, Murohara T, et al. Therapeutic angiogenesis for patients with limb ischaemia by autologous transplantation of bone-marrow cells: a pilot study and a randomised controlled trial. Lancet. 2002;360:427. doi: 10.1016/S0140-6736(02)09670-8. [DOI] [PubMed] [Google Scholar]
- 5.Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–7. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madeddu P, Kraenkel N, Barcelos LS, et al. Phosphoinositide 3-kinase gamma gene knockout impairs postischemic neovascularization and endothelial progenitor cell functions. Arterioscler Thromb Vasc Biol. 2008;28:68–76. doi: 10.1161/ATVBAHA.107.145573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright DE, Bowman EP, Wagers AJ, et al. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195:1145–54. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hidalgo A, Sanz-Rodriguez F, Rodriguez-Fernandez JL, et al. Chemokine stromal cell-derived factor-1alpha modulates VLA-4 integrin-dependent adhesion to fibronectin and VCAM-1 on bone marrow hematopoietic progenitor cells. Exp Hematol. 2001;29:345–55. doi: 10.1016/s0301-472x(00)00668-8. [DOI] [PubMed] [Google Scholar]
- 9.Peled A, Kollet O, Ponomaryov T, et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95:3289–96. [PubMed] [Google Scholar]
- 10.Sanz-Rodriguez F, Hidalgo A, Teixido J. Chemokine stromal cell-derived factor-1alpha modulates VLA-4 integrin-mediated multiple myeloma cell adhesion to CS-1/fibronectin and VCAM-1. Blood. 2001;97:346–51. doi: 10.1182/blood.v97.2.346. [DOI] [PubMed] [Google Scholar]
- 11.De Falco E, Porcelli D, Torella AR, et al. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood. 2004;104:3472–82. doi: 10.1182/blood-2003-12-4423. Epub. [DOI] [PubMed] [Google Scholar]
- 12.Loomans CJ, de Koning EJ, Staal FJ, et al. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–9. doi: 10.2337/diabetes.53.1.195. [DOI] [PubMed] [Google Scholar]
- 13.Waltenberger J. Impaired collateral vessel development in diabetes: potential cellular mechanisms and therapeutic implications. Cardiovasc Res. 2001;49:554–60. doi: 10.1016/s0008-6363(00)00228-5. [DOI] [PubMed] [Google Scholar]
- 14.Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest. 2007;117:1219–22. doi: 10.1172/JCI32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hazarika S, Dokun AO, Li Y, et al. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ Res. 2007;101:948–56. doi: 10.1161/CIRCRESAHA.107.160630. [DOI] [PubMed] [Google Scholar]
- 16.Tepper OM, Galiano RD, Capla JM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–6. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 17.Hill JM, Zalos G, Halcox JP, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 18.Fadini GP, Miorin M, Facco M, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. Journal of the American College of Cardiology. 2005;45:1449–57. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 19.Tamarat R, Silvestre JS, Le Ricousse-Roussanne S, et al. Impairment in ischemia-induced neovascularization in diabetes: bone marrow mononuclear cell dysfunction and therapeutic potential of placenta growth factor treatment. Am J Pathol. 2004;164:457–66. doi: 10.1016/S0002-9440(10)63136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Awad O, Jiao C, Ma N, et al. Obese diabetic mouse environment differentially affects primitive and monocytic endothelial cell progenitors. Stem Cells. 2005;23:575–83. doi: 10.1634/stemcells.2004-0185. [DOI] [PubMed] [Google Scholar]
- 21.Segal MS, Shah R, Afzal A, et al. Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes. 2006;55:102–9. [PubMed] [Google Scholar]
- 22.Fadini GP, Sartore S, Schiavon M, et al. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia. 2006;49:3075–84. doi: 10.1007/s00125-006-0401-6. [DOI] [PubMed] [Google Scholar]
- 23.Gallagher KA, Liu ZJ, Xiao M, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117:1249–59. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly DJ, Zhang Y, Gow RM, et al. Cells expressing the stem cell factor receptor, c-kit, contribute to neoangiogenesis in diabetes. Diab Vasc Dis Res. 2005;2:76–80. doi: 10.3132/dvdr.2005.013. [DOI] [PubMed] [Google Scholar]
- 25.Kunjathoor VV, Wilson DL, LeBoeuf RC. Increased atherosclerosis in streptozotocin-induced diabetic mice. J Clin Invest. 1996;97:1767–73. doi: 10.1172/JCI118604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Couffinhal T, Silver M, Zheng LP, et al. Mouse model of angiogenesis. Am J Pathol. 1998;152:1667–79. [PMC free article] [PubMed] [Google Scholar]
- 27.Pesce M, Orlandi A, Iachininoto MG, et al. Myoendothelial differentiation of human umbilical cord blood-derived stem cells in ischemic limb tissues. Circ Res. 2003;93:e51–62. doi: 10.1161/01.RES.0000090624.04507.45. [DOI] [PubMed] [Google Scholar]
- 28.Rivard A, Silver M, Chen D, et al. Rescue of diabetes-related impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. Am J Pathol. 1999;154:355–63. doi: 10.1016/S0002-9440(10)65282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebrahimian TG, Tamarat R, Clergue M, et al. Dual effect of angiotensin-converting enzyme inhibition on angiogenesis in type 1 diabetic mice. Arterioscler Thromb Vasc Biol. 2005;25:65–70. doi: 10.1161/01.ATV.0000149377.90852.d8. [DOI] [PubMed] [Google Scholar]
- 30.Schiekofer S, Galasso G, Sato K, et al. Impaired revascularization in a mouse model of type 2 diabetes is associated with dysregulation of a complex angiogenic-regulatory network. Arterioscler Thromb Vasc Biol. 2005;25:1603–9. doi: 10.1161/01.ATV.0000171994.89106.ca. [DOI] [PubMed] [Google Scholar]
- 31.Peichev M, Naiyer AJ, Pereira D, et al. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–8. [PubMed] [Google Scholar]
- 32.Prater DN, Case J, Ingram DA, et al. Working hypothesis to redefine endothelial progenitor cells. Leukemia. 2007;21:1141–9. doi: 10.1038/sj.leu.2404676. [DOI] [PubMed] [Google Scholar]
- 33.Timmermans F, Van Hauwermeiren F, De Smedt M, et al. Endothelial outgrowth cells are not derived from CD133+ cells or CD45+ hematopoietic precursors. Arterioscler Thromb Vasc Biol. 2007;27:1572–9. doi: 10.1161/ATVBAHA.107.144972. [DOI] [PubMed] [Google Scholar]
- 34.Walter DH, Haendeler J, Reinhold J, et al. Impaired CXCR4 signaling contributes to the reduced neovascularization capacity of endothelial progenitor cells from patients with coronary artery disease. Circ Res. 2005;97:1142–51. doi: 10.1161/01.RES.0000193596.94936.2c. [DOI] [PubMed] [Google Scholar]
- 35.Seeger FH, Haendeler J, Walter DH, et al. p38 mitogen-activated protein kinase downregulates endothelial progenitor cells. Circulation. 2005;111:1184–91. doi: 10.1161/01.CIR.0000157156.85397.A1. [DOI] [PubMed] [Google Scholar]
- 36.Gill M, Dias S, Hattori K, et al. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167–74. doi: 10.1161/01.res.88.2.167. [DOI] [PubMed] [Google Scholar]
- 37.Wang JF, Park IW, Groopman JE. Stromal cell-derived factor-1alpha stimulates tyrosine phosphorylation of multiple focal adhesion proteins and induces migration of hematopoietic progenitor cells: roles of phosphoinositide-3 kinase and protein kinase C. Blood. 2000;95:2505–13. [PubMed] [Google Scholar]
- 38.Wysoczynski M, Reca R, Ratajczak J, et al. Incorporation of CXCR4 into membrane lipid rafts primes homing-related responses of hematopoietic stem/progenitor cells to an SDF-1 gradient. Blood. 2005;105:40–8. doi: 10.1182/blood-2004-04-1430. [DOI] [PubMed] [Google Scholar]
- 39.Kamihata H, Matsubara H, Nishiue T, et al. Implantation of bone marrow mononuclear cells into ischemic myocardium enhances collateral perfusion and regional function via side supply of angioblasts, angiogenic ligands, and cytokines. Circulation. 2001;104:1046–52. doi: 10.1161/hc3501.093817. [DOI] [PubMed] [Google Scholar]
- 40.Krankel N, Adams V, Linke A, et al. Hyperglycemia reduces survival and impairs function of circulating blood-derived progenitor cells. Arterioscler Thromb Vasc Biol. 2005;25:698–703. doi: 10.1161/01.ATV.0000156401.04325.8f. [DOI] [PubMed] [Google Scholar]
- 41.Thum T, Fraccarollo D, Schultheiss M, et al. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes. 2007;56:666–74. doi: 10.2337/db06-0699. [DOI] [PubMed] [Google Scholar]
- 42.Chen YH, Lin SJ, Lin FY, et al. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes. 2007;56:1559–68. doi: 10.2337/db06-1103. [DOI] [PubMed] [Google Scholar]
- 43.Capla JM, Grogan RH, Callaghan MJ, et al. Diabetes impairs endothelial progenitor cell-mediated blood vessel formation in response to hypoxia. Plast Reconstr Surg. 2007;119:59–70. doi: 10.1097/01.prs.0000244830.16906.3f. [DOI] [PubMed] [Google Scholar]
- 44.Marchetti V, Menghini R, Rizza S, et al. Benfotiamine Counteracts Glucose Toxicity Effects on Endothelial Progenitor Cell Differentiation via Akt/FoxO Signaling. Diabetes. 2006;55:2231–7. doi: 10.2337/db06-0369. [DOI] [PubMed] [Google Scholar]
- 45.Hiasa K, Ishibashi M, Ohtani K, et al. Gene transfer of stromal cell-derived factor-1alpha enhances ischemic vasculogenesis and angiogenesis via vascular endothelial growth factor/endothelial nitric oxide synthase-related pathway: next-generation chemokine therapy for therapeutic neovascularization. Circulation. 2004;109:2454–61. doi: 10.1161/01.CIR.0000128213.96779.61. [DOI] [PubMed] [Google Scholar]
- 46.Petit I, Goichberg P, Spiegel A, et al. Atypical PKC-zeta regulates SDF-1-mediated migration and development of human CD34+ progenitor cells. J Clin Invest. 2005;115:168–76. doi: 10.1172/JCI21773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang Z, Brooks J, Willard M, et al. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem Biophys Res Commun. 2007;359:716–22. doi: 10.1016/j.bbrc.2007.05.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng H, Fu G, Dai T, et al. Migration of endothelial progenitor cells mediated by stromal cell-derived factor-1alpha/CXCR4 via PI3K/Akt/eNOS signal transduction pathway. J Cardiovasc Pharmacol. 2007;50:274–80. doi: 10.1097/FJC.0b013e318093ec8f. [DOI] [PubMed] [Google Scholar]
- 49.Yano T, Liu Z, Donovan J, et al. Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta-cell survival by activation of the prosurvival kinase Akt. Diabetes. 2007;56:2946–57. doi: 10.2337/db07-0291. [DOI] [PubMed] [Google Scholar]
- 50.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 51.Song P, Wu Y, Xu J, et al. Reactive nitrogen species induced by hyperglycemia suppresses Akt signaling and triggers apoptosis by upregulating phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) in an LKB1-dependent manner. Circulation. 2007;116:1585–95. doi: 10.1161/CIRCULATIONAHA.107.716498. [DOI] [PubMed] [Google Scholar]
- 52.Ingram DA, Krier TR, Mead LE, et al. Clonogenic endothelial progenitor cells are sensitive to oxidative stress. Stem Cells. 2007;25:297–304. doi: 10.1634/stemcells.2006-0340. [DOI] [PubMed] [Google Scholar]
- 53.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–50. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 54.Coffer PJ, Burgering BM. Stressed marrow: FoxOs stem tumour growth. Nat Cell Biol. 2007;9:251–3. doi: 10.1038/ncb0307-251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item