

Abstract
Connective tissue cell activation is of importance during reactive conditions such as solid tumour growth, wound healing and pannus formation in rheumatoid arthritis. Here, we have compared connective tissue cells of mesenchymal origin in human tissues from these conditions and their normal counterparts using a panel of cell-type-specific markers. In particular, we investigated variations of integrin expression among connective tissue cell phenotypes. Connective tissue cell populations were defined based on their association with the microvasculature and their expression of activation markers. The phenotype of these cells varied according to the type of pathological connective tissue examined. Our morphological data from human tissues suggested that the α1β1 integrin, a collagen/laminin receptor, is involved in the differentiation of precursor cells into myofibroblasts. To mechanistically investigate this hypothesis, we employed experimental models for carcinoma growth and wound healing utilizing α1 integrin-deficient mice. The data confirmed that the α1β1 integrin is of importance not only for the differentiation of mesenchymal cells into myofibroblasts but also for the neovascularization and connective tissue organization and emphasize the importance of myofibroblasts in the pathophysiology of tissue repair, inflammation and tumour growth.
Keywords: myofibroblast, pericyte, integrin connective tissue, adenocarcinoma rheumatoid arthritis, matrigel
Introduction
Stroma formation in solid tumours, chronic inflammatory lesions and tissue repair share several features including infiltration of inflammatory cells, activation of blood vessels and angiogenesis. Of further and central pathophysiological importance, persistent activation of connective tissue cells leads to excessive extracellular matrix (ECM) deposition, dominated by collagen type I, which, in turn, leads to fibrosis and ultimately organ dysfunction [1–5]. The amount of fibrosis is not necessarily linked to the severity of inflammation, indicating mechanisms, in part, distinct from those that regulate inflammation [6]. Thus, tissue damage due to severe inflammation can, in some instances, be reversible with the reinstatement of organ architecture and function [7]. The underlying processes that result, on the one hand, in reinstatement of organ function and, on the other hand, in a chronic state resulting in fibrosis and organ dysfunction despite similar initial pathophysiology are largely unknown.
Myofibroblasts, as defined by their expression of α-smooth muscle actin (α-SMA), play a central role in the deposition and organization of ECM and thus also in the formation of fibrotic tissue [5, 8, 9]. They are related to fibroblasts and exhibit a hybrid phenotype between fibroblasts and smooth muscle cells/pericytes [5]. It has been suggested that the latter two cell types are derived from a common cell lineage [5, 10–14]. The origin of the myofibroblast is yet unclear. Resident tissue fibroblasts [5, 14], vascular cells such as smooth muscle cells and/or pericytes [10–13] and bone marrow-derived precursor cells [14] have been suggested as potential sources.
The transition to the myofibroblast phenotype in culture depends on the concerted action of cytokines such as transforming growth factor (TGF)-β and specific ECM proteins such as the fibronectin splice variant ED-A and on mechanical tension [8, 15]. Several adhesion receptors belonging to the integrin family have been implicated in the regulation of the myofibroblast phenotype in vitro. The α4β1 and α5β1 integrins promote the myofibroblast phenotype by binding to fibronectin [16]. The αvβ1 integrin binds latent TGF-β, which in concert with the αvβ6 integrin leads to TGF-β activation [17] and the development of the myofibroblast phenotype. The αvβ3 and αvβ5 integrins down-regulate α-SMA expression in myofibroblasts via binding to vitronectin [18]. In vitro studies have also suggested that the α1β1 integrin is important for the up-regulation of α-SMA in cultured fibroblasts subjected to interstitial fluid flow [19]. Thus, adhesion and the nature of that adhesion seem to be important in the orchestration of events leading to acquisition of the myofibroblast phenotype.
In the present study, tissue analysis from different human pathologies suggested that the α1 integrin subunit, which associates exclusively with the β1 integrin subunit, forming the α1β1 integrin heterodimer [20, 21], a major collagen receptor, is important for the differentiation and maintenance of the myofibroblast phenotype, as defined by the expression of α-SMA. We tested this hypothesis using a genetic approach employing mice carrying a null mutation in the gene for the integrin α1 chain [22]. Our data demonstrate a previously unrecognized importance of adhesion via the α1β1 integrin in acquisition of the myofibroblast phenotype in vivo, which, in turn, is of central importance for the neoformation of vessels and supporting connective tissue structures.
Materials and methods
Surgical specimens
Full-thickness biopsies from human tissues were taken from: (i) colorectal adenocarcinoma (CC; n= 4), (ii) adjacent normal colon (n= 2), (iii) pannus formation from synevectomies due to rheumatoid arthritis (RA; n= 3), (iv) normal synovia (n= 3), (v) 7-day-old healing cutaneous wounds (n= 3) and (vi) adjacent normal skin (n= 2). The biopsies were snap-frozen. The human ethics committee at the Uppsala Academic Hospital, Sweden, approved the present study.
Antibodies and other reagents
The following antibodies were used: monoclonal antibody (mAb) F4/80, recognizing macrophages (Serotec, Raleigh, NC, USA); polyclonal antibody (pAb) anti-NG2, recognizing pericytes and smooth muscle cells [23] (Chemicon, Temecula, CA, USA); mAb anti-perlecan, recognizing heparan sulphate proteoglycan (Chemicon); FITC-labelled mAb anti-smooth muscle α-actin (α-SMA), recognizing smooth muscle cells, myofibroblasts and pericytes [24–26] (clone 1A4; Sigma, St. Louis, MO, USA); biotinylated mAb anti-CD31 (PECAM-1), recognizing endothelial cells (Pharmingen, San Diego, CA, USA); pAb anti-platelet-derived growth factor (PDGF) β-receptor (clone 958; Santa Cruz Biotechnology, Santa Cruz, CA, USA); mAb anti-reticular fibroblast marker (RFM), recognizing fibroblasts [27] (clone ER-TR7; Cederlane laboratories, Ontario, Canada); mAb ASO2 (Thy1/CD90), recognizing fibroblasts, pericytes and smooth muscle cells [28] (Dianova, Hamburg, Germany), mAb anti-high-molecular-weight-melanoma-associated antigen (HMW-MAA), expressed on activated pericytes and smooth muscle cells [26, 29, 30] (clone 225.28; Sanbios, Uden, The Netherlands); mAb PAL-E, recognizing endothelial cells (Sanbios); mAb anti-procollagen type I C propeptide [31] (Pierce Chemical Corp., Rockford, IL, USA); mAb PDGFR-B2, recognizing activated PDGF β-receptors when used at a concentration of 1 μg/ml [32]; mAb anti-α1 integrin subunit [20] (clone TS2/7; Dr. Timothy Springer, Boston Blood Center, Boston, MA, USA); mAb anti-α2 integrin subunit (P1H5 and PIE6), mAb anti-α3 integrin subunit (P1B3) and mAb anti-α5 integrin subunit (PID6) [21, 33] (Dr. William Carter; Fred Hutchinson Cancer Research Center, Seattle, WA, USA); mAb anti-α6 integrin subunit [34] (clone GOH3; Dr. Arnoud Sonnenberg, Netherlands Cancer Institute, Amsterdam, The Netherlands) and mAb anti-α2 integrin subunit (CD49b), mAb anti-α3 integrin subunit (Cdw49c) and mAb anti-α5 integrin subunit (CDw49e) (Chemicon). The different mAbs used to detect the expression of α2β1 (PIE6, P1H5 and CD49b), α3β1 (P1B3 and Cdw49c) and α5β1 (P1D6 and CDw49e) integrins did not differ quantitatively or qualitatively (data not shown). The P1E6, P1B3 and P1D6 mAbs were used in the subsequent stainings.
The following secondary reagents were used: biotinylated (Fab)2 fragments (DAKO, Glostrup, Denmark), biotinylated IgG (Vector Labs, Burlingame, CA, USA), fluorescein- and rhodamine-conjugated IgG (Vector Labs), alexa flour 350 streptavidin- and alexa flour 350-conjugated IgG (Molecular Probes, Eugene, OR, USA) Texas Red avidin D (Vector Labs), normal serum (Sigma) and non-immune IgG (Sigma). The antibodies were diluted in PBS, pH 7.4, containing 0.1% bovine serum albumin, 150 mmol/l tranexamic acid, 20 μg/ml aprotinin (3 TIU/mg), 1.8 mmol/l ethylenediaminetetraacetic acid and 2 mmol/l iodoacetic acid. The trichrome Masson kit (Sigma) was used according to the manufacturer’s instructions.
Immunolabelling, imaging and numerical analysis
The 6-μm tissue sections were fixed in 100% acetone and subjected to immunolabelling for light and immunofluorescence microscopy, as described previously [26, 35]. From each animal and each human specimen, 10 fields of view at a magnification of ×200 were chosen randomly and digitalized. Quantification of CD31, α-SMA and RFM was performed with the NIH imaging software version 1.62 (National Institute of Health, Bethesda, MD, USA) in the animal studies. Quantification of human samples was performed with the IC 300 image-processing system from Inovision Corp. (Research Triangle Park, NC, USA), as previously described [26, 35]. Statistical analysis was performed with the Student’s t-test and considered significant at P-values below 0.01.
In vivo Matrigel assay
The α1-deficient mice [22], maintained on the 129sv/Tae background, and corresponding wild-type mice were bred at the animal facility at BMC, Uppsala, Sweden. The mice were anaesthetized by an intraperitoneal injection of 2.5% avertin (Sigma) in a volume of 200 μl saline and were then injected subcutaneously with a total of 200 μl of Matrigel™ (Costar; Fischer Scientific, Brightwaters, NY, USA), at a final concentration of 9.9 mg/ml, containing 1.4 μg/ml VEGF, 8 μg/ml FGF2 and 116 μg/ml bovine serum albumin (fatty acid-free), purchased from Sigma (St. Louis, MI, USA) and 500 nM sphingosine 1-phosphate from Biomol (Plymouth Meeting, PA, USA) [36]. The animals were killed by CO2 narcosis after 14 days. All animal experiments were approved by the Ethical Committee for Animal Experiments in Uppsala, Sweden, and Scripps Institute, San Diego, CA, USA.
Experimental tumour mouse model
CT26 colon adenocarcinoma autografts were grown from subcutaneous inoculation of 1 × 106 cells on the dorsum of syngeneic wild-type (n= 4) and α1−/− (n= 4) BALB/c mice (backcrossed to BALB/c for at least nine generations), as previously described [37].
Results
Immunofluorescence stainings, combined with computer-aided image analyses, were performed to characterize cell types with regard to the expression of particular antigens (markers) using a modification of a previously described methodology [26, 35] (Fig. 1). In a first step, immunohistochemistry was used to determine whether specific cell-type markers defined by the use of antibodies were expressed in vessels or in the interstitium (Fig. 1, step 1, and Fig. 2). In a second step, double immunofluorescence stainings (DIFS) using different combinations of antibodies directed to cell-type markers were used to determine their spatial interrelationship (Fig. 1, step 2, and Fig. 3). In a third step, the images from step 2 were analysed using computer-aided image analyses. A percentage value measuring the degree of co-localization, that is, spatial overlap between two different markers (X and Y), recognized by the respective antibodies on the same tissue section, was calculated (Fig. 1, step 3, Tables 1 and S1). A background co-localization ranging between 6% and 22%, with a mean of 19 ± 8%, was recorded for markers not expected to co-localize (Table 1) such as PAL-E (endothelial cells) and α-SMA (pericytes, smooth muscle cells and myofibroblasts). Furthermore, markers that were expected to completely co-localize such as PAL-E and PAL-E did not exceed a value of 80% due to restrictions in the technology (data not shown). These limits were in agreement with earlier studies using this technology [26, 35]. In a fourth step, data on the degree of co-localization from a number of different marker combinations, depicted as percentage values (Table 1 and Table S1), and the actual size of the individual marker populations, depicted as the number of pixels per field of vision (Table S2), was compiled and compared (Fig. 1, step 4). This procedure enabled us to determine the spatial distribution between the different markers in step 5 (Fig. 1, step 5). By including well-established cell-type-specific markers, we could correlate the spatial distribution of the markers under study in step 6 to in vivo structures/cell types of interest (Fig. 1, step 6) presented in Table 2.
Figure 1.
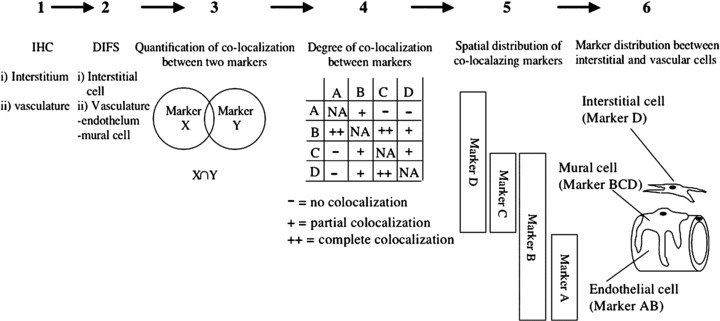
Method used to allocate expression of individual markers to different in vivo cell types. Tissues were subjected to immunohistochemical staining using different markers and analysed with light microscopy (1). Double immunofluorescent staining using combinations of markers was performed (2). Using computer-aided image analysis, co-localization between different markers was quantified (3). Co-localization between different sets of markers was analysed (4). From the analysis in steps 2 and 4, the spatial distribution and their co-localization could be determined (5). Using cell-type-specific markers, the spatial distribution of markers could be correlated to in vivo structures/cell types (6).
Figure 2.
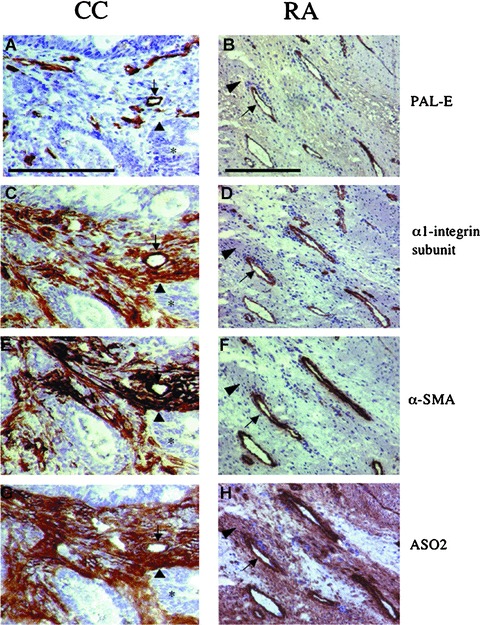
Expression of phenotypical markers in colorectal adenocarcinoma (CC) and pannus formation in rheumatoid arthritis (RA). Immunohistochemical staining was performed in 6-μm sections from CC (left column) and RA (right column) using mAbs to characterize expression profiles of phenotypical markers in relation to the vasculature for PAL-E (A, B), α1β1 (C, D), α-SMA (E, F) and ASO2 (G and H). Expression profiles were similar in microvascular structures (arrow) in both CC and RA. However, expression profiles differed in interstitial structures (arrowhead) between the two conditions. In CC, the expression profiles in interstitial structures were positive for α1β1 (C), α-SMA (E) and ASO2 (G). In contrast, the expression profiles in interstitial structures in RA were negative for α1β1 (D) and α-SMA (F), but positive for ASO2 (H). Tumour acinar structures (*). The bar represents 200 μm.
Figure 3.
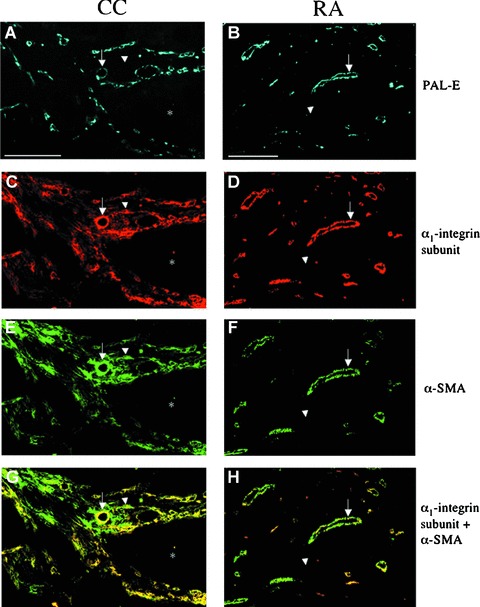
Phenotypical characterization of connective tissue cells in colorectal adenocarcinoma (CC) and rheumatoid arthritis (RA). Triple immunofluorescence staining of 6-μm sections of CC (left column) and RA (right column) was performed with mAbs to characterize the degree of co-localization as well as the spatial distribution between PAL-E (A, B), α1β1 integrin (C, D) and α-SMA (E, F). Co-localization of α1β1 integrin and α-SMA is depicted in yellow (G, H). Coexpression of α1β1 integrin and α-SMA was seen in both CC (G) and RA (H). Vascular PAL-E-positive structures (arrow) in both CC (A) and RA (B) were positive for α1β1 integrin and α-SMA (G, H). In CC, interstitial structures (arrowhead) expressed α1β1 integrin and α-SMA (G). In contrast, interstitial structures (arrowhead) in RA were negative for α1β1 integrin and α-SMA (H). Tumour acinar structures (*). The bar represents 200 μm.
Table 1.
Biopsies from tumour stroma formation in colorectal adenocarcinoma, pannus formation in rheumatoid arthritis and cutaneous healing wounds
| Percentage of smooth muscle α-actin-positive pixels that are also positive for | Colorectal adenocarcinoma | Rehumatoid arthritis | Wound healing |
|---|---|---|---|
| HMW-MAA | 28 ± 11 | 49 ± 23 | 47 ± 19 |
| PDGF β-receptor | 33 ± 14 | 47 ± 16 | 46 ± 22 |
| Integrin α1 subunit | 80 ± 11 | 72 ± 9 | 86 ± 14 |
| Integrin α5 subunit | 80 ± 11 | 72 ± 14 | 86 ± 15 |
| PAL-E | 14 ± 8 | 22 ± 11 | 20 ± 10 |
Biopsies were stained by a double immunofluorescence technique with various combinations of mAbs. Co-localization between two different markers is depicted as “percentage of pixels that are positive for both marker X and Y”. Stained sections were analysed by computer imaging processing, as detailed in Materials and methods. The recorded percentage values represent the spatial distribution of two mAb markers in relation to each other, measured in percentage of pixels that co-localize (1 pixel equals 0.9 μm × 0.9 μm) [26, 35].
Table 2.
Spatial distribution of the markers used in this study in relation to the microvasculature based on the method used in Figure 1
| Normal skin | Normal colon | Colorectal adenocarcinoma | Rheumatiod arthritis | Wound healing | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC (PAL-E) | P (α-SMA) | EC (PAL-E) | P (α-SMA) | EC (PAL-E) | P (HMW) | EC (PAL-E) | P (α-SMA) | EC (PAL-E) | P (α-SMA) | |
| α-SMA | − | NA | − | NA | − | ++ | − | NA | − | NA |
| HMW-MAA | − | − | − | − | − | NA | − | ++ | − | ++ |
| PDGF β-receptor | − | − | − | − | − | ++ | − | ++ | − | ++ |
| a1β1 integrin | ++ | ++ | ++ | + | ++ | ++ | ++ | ++ | ++ | ++ |
| a2β1 integrin | ++ | + | ++ | + | ++ | − | − | − | − | − |
| a3β1 integrin | ++ | + | ++ | + | ++ | − | ++ | − | ++ | − |
| a5β1 integrin | − | ++ | + | − | ++ | ++ | ++ | ++ | ++ | ++ |
| a6β1 integrin | ++ | − | ++ | − | ++ | − | ++ | − | ++ | − |
| Normal skin | Normal colon | Colorectal adenocarcinoma | Rheumatoid arthritis | Wound healing | |
|---|---|---|---|---|---|
| I | I | I (α-SMA) | I (HMW) | I (HMW) | |
| α-SMA | − | − | NA | − | − |
| HMW-MAA | − | − | − | NA | NA |
| PDGF α-receptor | − | − | − | ++ | ++ |
| a1β1 integrin | − | − | ++ | − | − |
| a2β1 integrin | + | + | + | − | − |
| a3β1 integrin | − | − | − | − | − |
| a5β1 integrin | − | − | ++ | ++ | ++ |
| a6β1 integrin | − | − | − | − | − |
Human tissues studied were normal skin, normal colon, tumour stroma formation in colorectal adenocarcinoma, pannus formation in rheumatoid arthritis and cutaneous healing wounds. Endothelial cells (EC) detected using PAL-E; pericytes (P) detected using α-SMA or HMW; interstitial fibroblast-like cells (I) detected using α-SMA or HMW.
Stained sections were analysed by computer imaging processing, as detailed in Materials and methods. The recorded percentage values represent the spatial distribution of two mAb markers in relation to each other, measured in percentage of pixels that co-localize (1 pixel equals 0.9 μm × 0.9 μm) [26, 35].
−, less than 30% co-localization; +, between 30% and 60% co-localization; ++, more than 60% co-localization.
Expression of β1 integrin heterodimers in colorectal adenocarcinoma, cutaneous healing wounds and pannus formation in rheumatoid arthritis
The distribution of β1 integrin-associated α-subunits expressed in reactive tissues and their normal counterparts were studied focusing on the expression patterns in the microvascular compartment, including the endothelium and pericytes, and the non-vascular interstitial compartment, including fibroblasts. The α1, α2, α3 and α5 integrin subunits associate exclusively with the β1 integrin subunit, whereas the α6 integrin subunit is capable of associating with both β1 and β4 integrin subunits [20, 21]. The results are shown in Table 2. For detection of blood vessels, the expression of PAL-E was used as an endothelial marker (Fig. 2A and B and Fig. S1A and B). To identify the non-endothelial cell type in the microvasculature, pericytes and interstitial fibroblasts, immunohistochemical staining of tissue sections using antibodies recognizing PDGF β-receptors (Fig. S1C and D), HMW-MAA (Fig. S1E and F), α-SMA (Fig. 2E and F and Fig. S1G and H) and ASO2 (Fig. 2G and H) and fluorescent multi-labelling of combinations of these markers (Fig. 3A–H and Fig. S2A–J) were employed and analysed according to the method outlined in Fig. 1.
In the tumour stroma of colorectal adenocarcinoma, endothelial cells expressed α1β1, α2β1,α3β1,α5β1 and α6 integrins (Table 2). Microvascular pericytes and interstitial fibroblasts had similar integrin expression profiles and mainly expressed α1β1 and α5β1 integrins (Table 2). The latter two cell types both expressed α-SMA but could be distinguished by their spatial relationship to the endothelium and their differential expression of HMW-MAA and PDGF β-receptors (Table 2 and Fig. S1C–F), as previously described [26]. Tumour acinar structures expressed α2β1,α3β1 and α6 integrins (data not shown).
Endothelial cells in pannus formation in rheumatoid arthritis and in tissue situated immediately adjacent to maturing granulation tissue in 7-day-old cutaneous healing wounds expressed a similar integrin subunit profile compared with colorectal adenocarcinoma with the exception of α2β1 integrin, which was not detected in the endothelium in cutaneous healing wounds or pannus formation (Table 2). Similar to what was observed in colorectal adenocarcinoma, microvascular pericytes in rheumatoid arthritis and cutaneous healing wounds expressed the α1β1 and α5β1 integrins (Table 2). In contrast to colorectal adenocarcinoma, interstitial fibroblasts, while expressing α5β1 (Fig. S2E and F), did not express the α1β1 integrin (Fig. 2C and D and Fig. 3C–H). Thus, the results suggested that the α1β1 integrin could be used as a marker to differentiate pericytes from interstitial fibroblasts in rheumatoid arthritis and cutaneous healing wounds as well as in defining interstitial fibroblast populations in the reactive conditions under study.
Phenotypical characterization of connective tissue cells in tumour stroma, pannus formation in rheumatoid arthritis and cutaneous wound healing in human beings
The possibility that the α1β1 integrin was linked to specific populations of non-endothelial connective tissue cells was investigated using the method outlined in Fig. 1. Pericytes and interstitial fibroblasts could be defined in the tumour stroma in colorectal adenocarcinoma, pannus formation in rheumatoid arthritis and cutaneous healing wounds by their spatial relationship to the microvasculature (Fig. 3A–H and Fig. S2A–J) and differential expression pattern of integrins and cell-type-specific markers (Tables 1 and 2). Microvascular pericytes were defined as cells that were juxtapositioned to the endothelium. Interstitial fibroblasts include cells that are partly dissociated from the microvascular wall or connective tissue cells situated in the interstitium. The latter cells were often accumulated in the perivascular space. However, connective tissue cells were also observed that were not overtly associated with blood vessels. Interstitial fibroblasts could be subsetted into α-SMA-positive/ASO2-negative cells (Fig. 2E and G and Fig. 3E and G), termed myofibroblasts, and α-SMA-negative/ASO2-positive cells (Fig. 2F and H and Fig. 3F and H), termed fibroblasts.
As shown in Table 2, the expression of markers in microvascular pericytes was similar in the tumour stroma, pannus formation and cutaneous healing wounds. These cells expressed the α1β1 integrin, α-SMA, PDGF β-receptors, HMW-MAA and α5β1 integrin. In addition, α2β1 integrin was detected in microvascular pericytes in cutaneous healing wounds. In the normal tissue, pericytes were negative for PDGF β-receptors and HMW-MAA, consistent with previous studies identifying these markers as activation markers [26, 30].
Connective tissue cells in the tumour stroma of colorectal adenocarcinoma not associated with vessels expressed α-SMA and integrins α1β1 and α5β1, but not the PDGF β-receptor or HMW-MAA. On the basis of their expression of α-SMA and the positioning relative to the vasculature, these cells were identified as myofibroblasts. The corresponding population of cells in rheumatoid arthritis and cutaneous healing wounds expressed α5β1 integrins, ASO2, the PDGF β-receptor and HMW-MAA. They, however, did not express α-SMA or α1β1 integrins. Because of the lack of α-SMA, these cells did not meet the criteria for myofibroblasts and were therefore identified as fibroblasts. These findings provide further support that the expression of α1β1 integrins co-distributes with that of α-SMA.
Procollagen type I C-propeptide used as a marker for collagen type I synthesis was not expressed in pericytes in any of the conditions studied. However, connective tissue cells that express procollagen type I C-propeptide were often seen to be concentrated in the surrounding perivascular space (Fig. 4A–D). In colorectal adenocarcinoma, these procollagen type I C-propeptide-expressing cells also expressed α-SMA, thus identifying them as myofibroblasts (Fig. 4C). In contrast, in rheumatoid arthritis and cutaneous healing wounds, procollagen type I C-propeptide did not co-localize with α-SMA (Fig. 4D), but was expressed in HMW-MAA- and PDGF β-receptor-positive connective tissue cells corresponding to fibroblasts (data not shown). Thus, in tumour stroma, but not in rheumatoid arthritis and cutaneous healing wounds, α-SMA expression co-distributes with collagen type I synthesis. This would suggest that the myofibroblast phenotype is not an absolute prerequisite for collagen type I neoformation in connective tissue cells.
Figure 4.
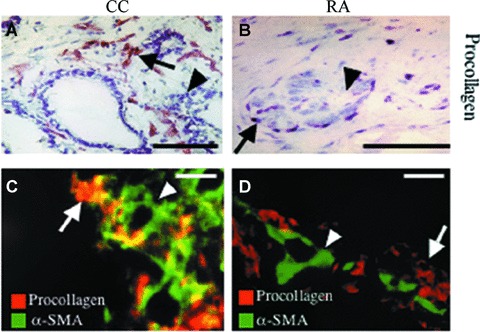
Procollagen type I expression in colorectal adenocarcinoma (CC) and pannus formation in rheumatoid arthritis (RA). Immunohistochemistry (A, B) and double immunofluorescence (C, D) staining of sections of CC (A, C) and RA (B, D) were performed with mAbs recognizing procollagen type I c-propeptide (procollagen), used as a marker for neosynthesis of collagen type I and smooth muscle α-actin (α-SMA). Co-localization is depicted in yellow. (A–D) Neosynthesis of collagen type I was not expressed in the microvasculature (arrowhead), but was largely confined to interstitial cells in the perivascular space (arrow) in both conditions. In these cellular structures, co-localization with α-SMA was high in CC (C) in contrast to RA (D), where co-localization was low. The bar represents 40 μm.
The role of α1β1 integrins in modulating connective tissue cell phenotype and function in an in vivo Matrigel plug assay
The morphological data in human tissues suggest that the α1β1 integrin co-distributes with α-SMA and raise the possibility that the α1β1 integrin is important in the acquisition of the myofibroblast phenotype. To study this correlation in detail, two experimental in vivo assays were performed with wild-type and integrin α1-deficient mice [22]. In the first set of experiments, Matrigels were inoculated subcutaneously in mice and harvested after 14 days [36].
As previously described [36], the wild-type mice displayed a substantial ingrowth of vessels into the Matrigel (Fig. 5A). Newly formed vessels were, to a large extent, surrounded by NG2- and PDGF β-receptor-expressing pericytes invested in a perlecan-positive basement membrane (data not shown and [36]). RFM-expressing fibroblasts were present in the Matrigel. These cells also expressed α-SMA. Because they were not associated with the microvasculature, they were defined as myofibroblasts (Fig. 5A and C). In the adjacent dermis, accumulations of connective tissue cells that expressed RFM and α-SMA were associated with the microvasculature, thus identifying them as pericytes. Some of these cells were also partly or completely dissociated from the vascular wall and accumulated in the perivascular interstitial space (Fig. 5E). Similar accumulations of perivascular cells were observed in the dermis adjacent to Matrigels implanted in α1-deficient animals, although to a lesser extent. These cells were positive for RFM but negative for α-SMA (Fig. 5D). The Matrigels in α1-deficient mice had a reduced density of CD31-positive structures compared with gels harvested from wild-type animals, suggesting a disturbance of angiogenesis in an α1 integrin-deficient environment (Fig. 5A and B). Furthermore, the cellular content of Matrigels inoculated in α1-deficient mice was positive for RFM but not for α-SMA (Fig. 5D). In addition, in both conditions, F4/80-expressing macrophages had infiltrated the Matrigel (data not shown).
Figure 5.
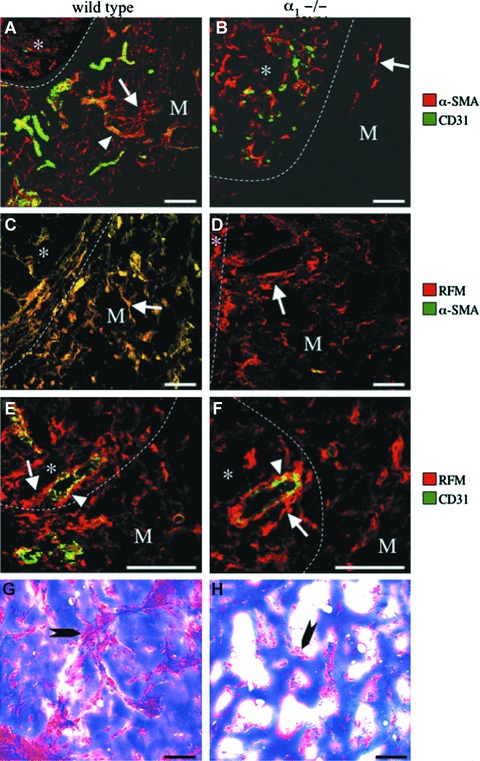
Deficient myofibroblast development and neoformation of blood vessels and supporting connective tissues in the in vivo Matrigel plug assay in α1−/− mice. Double immunofluorescence (A–F) staining using combinations of the mAbs, α-SMA, RFM and CD31, and trichrome (G–H) staining were performed on sections of Matrigel plugs together with adjacent skin that had been grown in the subcutaneous space in wild-type (WT; left column) and α1−/− (KO; right column) mice. Optical sections were obtained using confocal laser microscopy and viewed as composites (A–F). Co-localization is depicted in yellow. (A, B) Note ingrowth into the Matrigel (M) of CD31-positive microvessels (arrowhead) surrounded by α-SMA-positive myofibroblasts (arrow) in wild-type (A) but not in α1−/− mice (B). (C, D) Note ingrowth into the Matrigel (M) of myofibroblasts positive for both RFM and α-SMA in wild-type (C) mice in contrast to Matrigels in α1−/− (D) mice that contained RFM-positive fibroblasts that were negative for α-SMA (arrow). (E, F) Note RFM-expressing cells that are juxtapositioned to, partially detached (arrow) from, the CD31-positive endothelium (arrowhead) that have accumulated in the perivascular space in dermis (*) adjacent to the Matrigel (M) in wild-type (E) mice and, albeit to a lesser degree, also in α1−/− (F) mice. (G, H) Note well-developed cellular connective tissue septa (arrow) in Matrigels in wild-type (G) but not in α1−/− (H) mice, where only isolated accumulations of cells (arrow) were observed. Matrigel/dermal interface (dotted line). The bar represents 40 μm.
Quantification of blood vessel density showed a 90% reduction in the area covered by CD31-positive pixels in the Matrigels implanted in deficient animals compared with wild-type animals (Fig. 7A). In these animals, no formation of connective tissue septa in the Matrigel was observed compared with wild-type animals, where both vessels and myofibroblasts were primarily concentrated to networks of connective tissue septa extending from the normal adjacent tissue (Fig. 5G and H). However, in α1-deficient mice, the Matrigels were not acellular as both macrophages (data not shown) and RFM-expressing fibroblasts were present (Fig. 5D). A 53% reduction in area covered by RFM-positive pixels (Fig. 7A) and a 97% reduction in the area covered by α-SMA-positive pixels were observed in α1-deficient mice (Fig. 7A). Thus, RFM-expressing fibroblasts were capable of infiltrating the Matrigel, but they did not differentiate into myofibroblasts. Also, in contrast to wild-type mice, NG2-positive pericytes were absent in the gel (data not shown).
Figure 7.
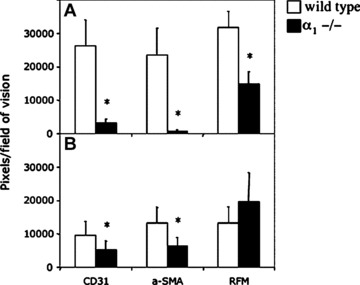
Quantification of the number of pixels positive for markers expressed in the endothelium, pericytes, fibroblasts and myofibroblasts in syngeneic colorectal adenocarcinomas (CC) grown in wild-type and α1−/− mice. Matrigels (A) or syngeneic CCs grown in the subcutaneous space (B) in wild-type (filled bars) and α1−/− (empty bars) mice were extirpated, sectioned and triple stained with regard to the markers for the endothelial cells (CD31), pericytes/smooth muscle cells and myofibroblasts (α-SMA) and pericytes and fibroblasts (RFM). Average number of positive pixels per field of view was quantified. Mean ± SD. P-value according to Student’s t-test, *P < 0.01.
The role of α1β1 integrins in modulating connective tissue cell phenotype in tumour stroma formation in experimental colorectal adenocarcinoma
In a second experimental model, syngeneic CT26 colon adenocarcinoma cells were inoculated into the subcutaneous space in wild-type and α1-deficient BALB/c mice. In the solid tumours that subsequently formed, the tumour stroma could be divided into two main compartments: a vascular compartment and a perivascular compartment. The expression of cell-type-specific markers and their spatial interrelationship were examined (Fig. 6A–J). In the tumour stroma of wild-type and α1-deficient mice, a microvascular network consisting of endothelial cells that were partially covered by pericytes (Fig. 6G and H) and enveloped in the basal lamina (data not shown) was present. In both wild-type and α1−/− mice, these microvascular pericytes expressed α-SMA (Fig. 6G and H), RFM (Fig. 6I and J) and NG2 (data not shown). Furthermore, in both conditions, the majority of cells constituting the perivascular compartment were positive for RFM (Fig. 6I and J). In contrast to α1-deficient mice, these perivascular cells also expressed α-SMA (Fig. 6I and J) in wild-type mice, thus identifying them as myofibroblasts. In α1-deficient mice, the expression of α-SMA was largely confined to the vascular space (Fig. 6J). The fine fibrillar/reticular cellular extensions creating a meshwork interconnecting the microvasculature in tumours in wild-type mice (Fig. 6G and I) were not observed in tumours in α1-deficient mice (Fig. 6H and J). Furthermore, the overall cellular density was lower in tumours in wild-type mice compared with α1-deficient mice (Fig. 6K and L).
Figure 6.
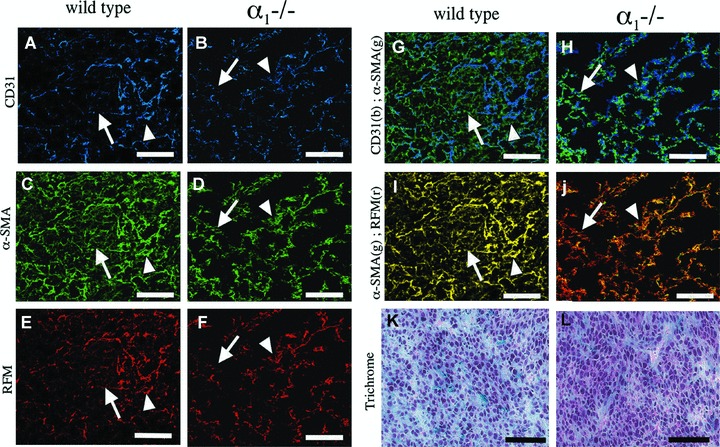
Deficient myofibroblast development in a syngeneic experimental colorectal adenocarcinoma in α1−/− mice. Triple immunofluorescence (A–J) using combinations of the mAbs, CD31 (blue (b)), α-SMA (green (g)) and RFM (red (r)), and trichrome (K–L) staining were performed on sections of biopsies from CC grown syngeneically in the subcutaneous space in wild-type (A, C, E, G, I, K) and α1−/− (B, D, F, H, J, L) mice. Optical sections obtained using confocal laser microscopy and viewed as composites (A–J). Co-localization is depicted in yellow. (G, H) Note α-SMA-positive cells associated with CD31-positive vessels (arrowhead) in tumours from both wild-type and α1−/− mice. Note α-SMA-positive interstitial myofibroblasts (arrow) in the tumour stroma in tumours from wild-type mice (G) that are not associated with vessels and the apparent lack of similar cells (arrow) in tumours from α1−/− mice (H). (I, J) Note co-localization of RFM and α-SMA in cells associated to vessels (arrowhead) in tumours from both wild-type (I) and α1−/− (J) mice. Interstitial cells (arrowhead) express both RFM and α-SMA in tumours from wild-type mice (I) while only expressing RFM in tumours from α1−/− mice (J). (K, L) Note lower cell density in tumours from wild-type (K) mice compared with α1−/− (L) mice. The bar represents 20 μm.
A quantification made for the markers above (Fig. 7B) showed a 46% reduction in the area covered by CD31-positive pixels in tumours in α1-deficient mice compared with wild-type mice. Moreover, in α1-deficient mice, a 53% reduction in the area covered by α-SMA-positive pixels compared with tumours in wild-type mice could be observed. This was not due to a decrease in fibroblasts because there was a 48% increase in the area covered by RFM-positive pixels in tumours in α1-deficient mice compared with wild-type mice.
Discussion
The results of this study demonstrate a previously unrecognized role of adhesion via the α1β1 integrin in the acquisition of the myofibroblast phenotype in vivo, which, in turn, is shown to be of central importance for the neoformation of vessels and supporting connective tissue structures. In the animal models employed, we were able to show that the α1β1 integrins plays a role in the differentiation of precursor cells into myofibroblasts and the establishment of blood vessels. Previously, in vitro experiments have shown that antibody-mediated blocking of α1β1 integrins in cultured fibroblasts reduces the expression of α-SMA when subjected to interstitial fluid flow [19]. The evidence in the present study suggests that the α1β1 integrins are also important for the development of myofibroblasts in vivo.
In the present study of human reactive tissues, interstitial fibroblasts differed in their marker expression profiles in tumour stroma compared with pannus formation and wound healing. One explanation for this might be tissue specificity or that these cells are derived from different populations of precursor cells [14]. Heterogeneity of fibrogenic cells not only between tissues but also within the same tissue has previously been shown [38–40]. For instance, in the liver, phenotypically divergent fibrogenic cells are derived from different populations of precursor cells [41, 42]. Although there are important pathophysiological similarities between stroma formation in solid tumours and wound healing, the composition of the matrix is not identical [4]. Thus, one possibility for this divergence is the differences in cytokine expression and composition of the ECM components of the microenvironment. It has been shown that spatially distinct connective tissue cell populations respond differently to different cytokines [15]. For instance, a distinct population of periportal cells in liver undergoes a PDGF-BB-dependent conversion to a pro-fibrotic phenotype distinct from myofibroblasts in septal fibrosis [43]. The current observations made in human reactive tissues suggest, and are supported by the experimental models used in this study, that interactions between the ECM and connective tissue cells modify the phenotype of the cells, and that this interaction uses the α1β1 integrin as a conduit for this purpose.
The cellular origins of fibroblasts/myofibroblasts are unclear. We have previously shown that microvascular pericytes become activated in tumour stroma, pannus in rheumatoid synovitis and healing wounds [26, 44, 45]. Upon activation, these pericytes express several markers including PDGF β-receptors, HMW-MAA and α-SMA. Thus, the activation of microvascular pericytes in these conditions are reflected in a distinct molecular marker profile. We and others have reported that pericytes, upon activation, detach from the endothelial lining and enter the perivascular space where they alter their phenotype [10, 26, 46–48]. During tumour-induced angiogenesis and cutaneous wound healing, pericytes undergo a mitotic burst that precedes cell division in the endothelium and interstitial fibroblasts, resulting in the accumulation of connective tissue cells in the perivascular space [49]. In the present study, accumulations of connective tissue cells were radially distributed and concentrated around the vasculature. Furthermore, the findings in human reactive conditions show similarities in the marker expression profile between the microvascular pericyte and the interstitial fibroblast/myofibroblast. Thus, one possibility is that fibroblasts/myofibroblasts originate from microvascular pericytes, where pericytes detach from the endothelium, proliferate and subsequently differentiate into fibroblasts/myofibroblasts. This notion is supported by previous studies in excessive dermal scarring and placenta formation [11, 13]. Furthermore, gene deletion studies in mice show that PDGF and their receptors, although important for the expansion of the pericyte population during embryogenesis, are crucial for the expansion of the fibroblast population in adult reactive tissues, suggesting a common origin of these two cell types [50, 51]. Pericytes would thereby link the processes of connective tissue formation and the microvasculature in a previously unrecognized way. However, the results of the present study do not definitively prove a pericyte origin of fibroblasts/myofibroblasts in reactive conditions and this requires further investigation.
We have previously shown that the Matrigel plugs implanted into the subcutaneous space in vivo become infiltrated by macrophages and fibroblasts that support the ingrowth of blood vessels and the neoformation of connective tissue septa [36]. In similar models, using Matrigel plugs containing VEGF-A-transfected cells and syngeneic tumour models in mice, inhibition of α1β1 and/or α2β1 integrins perturbed ingrowth of blood vessels and tumour growth [52–54]. Our present results using Matrigel plugs implanted in α1-deficient and wild-type mice show that fibroblasts are capable of populating the Matrigel but are incapable of differentiating into myofibroblasts. In concordance with the present Matrigel studies, fibroblasts are generated within tumours but are unable to undergo myofibroblast differentiation. However, in contrast to the Matrigel studies, tumour vasculature was present and was partially covered by α-SMA-expressing pericytes. The degree of reduction in vascularization in tumours grown in α1-deficient mice compared with wild-type mice is in agreement with previous studies using this model [37, 54]. The differences between the Matrigel and syngeneic colorectal adenocarcinoma models may partly be due to the more complex array of ECM and cytokines within the tumour microenvironment. Furthermore, in the tumour model, co-option and expansion of already existing vasculature may occur, limiting the need for the neoformation of vessels and surrounding stroma [55].
The differences in connective tissue cell phenotype in the Matrigels from α1-deficient mice compared with wild-type mice may be due to the differences in the inflammatory response. Monocytes/macrophages express the α1β1 integrin. Therefore one possibility is that the differences in fibroblast phenotype in the α1-deficient mice is due to the lack of inflammatory cells, which, in turn, produce cytokines that influence the fibroblast phenotype [56, 57]. In the present study using immunocompetent mice, no gross differences in macrophage infiltration/population in the Matrigel or the colorectal adenocarcinoma model were observed. This argues against the possibility that the differences in connective tissue cell phenotype was due to the lack of infiltration of α1β1 integrin-expressing inflammatory cells. The simplest explanation is that precursor cells derived from the adjacent dermis were unable to migrate/populate the Matrigel or the tumour stroma due to a lack of α1β1 integrin [41, 42]. However, in both α1-deficient and wild-type mice, connective tissue cells were able to migrate into the Matrigel and populate the non-vascular component of the tumour stroma. This would argue against the possibility that a lack of precursor cells could explain the observed differences. Taken together, our results show that, regardless of the event that triggers the transition into the myofibroblast phenotype in these conditions, it is dependent on the cellular expression of the α1β1 integrin.
Mice have been generated containing gene deletions for many of the known integrin subunits, and in some cases, double knockouts have been developed [58]. Most integrin subunits are present in only a single heterodimer, that is, the α1 integrin subunit, which only associates with the β1 subunit to form the heterodimer α1β1. A few integrins can form heterodimers with multiple partners, that is, αv, making the interpretation of gene ablation experiments difficult [59]. The lack of a phenotype may suggest overlapping functions among different proteins. It can also be due to compensatory mechanisms in response to ablation of a certain gene, that is, up-regulation of VEGF receptor 2 in β3 null mice [60, 61]. Discordance between pharmacological and genetic studies in mice adds to the complexity of interpreting the potential role of integrins in vivo[60]. The latter may be partially explained by transdominant inhibition, where occupation of one type of integrin leads to inhibition of the function of other specific integrins in the same cell [62]. Furthermore, biological processes during embryonic development differ from those in adult tissues and during pathological conditions in the adult. The expression of the α1β1 integrin is important in adult reactive tissues, but not during embryonic tissue morphogenesis. The α1-deficient mice are viable and fertile and, besides a hypocellular dermis with a low number of fibroblasts and increased collagen and collagenase synthesis, have no overt phenotype [22, 54]. Evidence supporting discrepancies in the role of the fibroblast/myofibroblast lineage in embryonic versus adult reactive tissues does exist [50, 51]. It has been shown that the α1β1 integrin plays a role in reactive tissues such as the stromal response in solid tumours and the neoformation of blood vessels with a supporting connective tissue [52–54]. One possibility is that embryonic tissue morphogenesis, compared with repair processes in the adult where damaged tissue is replaced, differs in cell–matrix interactions [7]. Furthermore, it has been shown that myofibroblasts are not a prominent feature in foetal tissue repair, a process that results in minimal scarring [7, 15]. Our results support the notion that, although the α1β1 integrin is non-essential during embryogenesis [22, 54], it is essential in modulating connective tissue cell phenotypes, and thereby plays an important role in reactive tissues in the adult.
Taken together, our results support the in vivo results in human pathologies and show that, regardless of the events that triggers the transition of mesenchymal cells into the myofibroblast phenotype, it is dependent on the cellular expression of the α1β1 integrin. Thus, modulation of the connective tissue phenotypes and their environment may shift the balance from deleterious fibrosis towards optimal tissue regeneration and repair.
Acknowledgments
We thank Dr. Timothy Springer, Dr. William Carter and Dr. Arnoud Sonnenberg for antibodies. This work was supported by grants from The Swedish Cancer Foundation, The Swedish Research Council, Children’s Cancer Foundation, Gustav V 80 Year Foundation, The Georg Wally Foundation, The Clas Groschinsky Foundation, The Swedish Society of Physicians, The UAS Cancer Foundation, Mary Åke and Hans Ländells Foundation, The Åke Wiberg Foundation, Lions Cancer Foundation, Agnes and Mac Rudbergs Foundation and Hans Jeanssons Foundation. The authors have no conflicts of interest to report.
Supporting Information
Fig. S1 Expression of phenotypical markers in colorectaladenocarcinoma (CC) and in pannus formation in rheumatoid arthritis(RA). Immunohistochemical staining was performed in sectionsfrom CC (left column) and RA (right column) using mAb?s in order tocharacterize expression profiles of phenotypical markers.Expression profiles were similar in microvascular structures(arrowhead) in both CC and RA for PAL-E (A and B),PDGF β-receptors (C and D), HMW-MAA (Eand F) and α-SMA (G and H). However,expression profiles differed in interstitial structures (arrow)between the two conditions. In CC interstitial structures werepositive for α-SMA (G), but to a lesser degree PDGFβ-receptors (C) and HMW-MAA (E). In contrast,the expression profile in interstitial structures in RA werepositive for PDGF β-receptors (D) and HMW-MAA(F), but not for α-SMA (H). Bar represents 20μm.
Fig. S2 Phenotypical characterisation of connective tissuecells in the stroma of colorectal adenocarcinoma (CC) and in pannusformation in rheumatoid arthritis (RA). Doubleimmunofluorescence-stained sections of CC (left column) and RA(right column) was performed using mAb?s in order to characterizethe degree of colocalization as well as the spatial distribution oftwo markers in relation to each other and to the microvasculature.Images were captured and analyzed using computer imaging analysis[26, 35]. Colocalization is depicted in yellow. mAbs usedrecognized PDGF β-receptors (PDGFR-β), activatedpericytes (HMW-MAA), smooth muscle α-actin (α-SMA),α1β1 integrin (α1β1) and &lapha;5β1integrin (α5β1). Expression profiles were similar inmicrovascular structures (arrowhead) in both CC and RA, and werepositive for PDGF β-receptors (A and B), HMW-MAA(C, D, G and H), α5β1 (E andF), α1β1 (G, H, I and J) andα-SMA (A?F, I and J). However, expressionprofiles differed in interstitial structures (arrow) between thetwo conditions. In CC, interstitial structures were to a lesserdegree positive for PDGF β-receptors (A) and HMW-MAA(C and G) while maintaining their expression ofα-SMA (A, C, E and I), α5β1(E) and α1β1 (G and I). In contrastthe expression profile in interstitial structures in RA were to amuch lesser degree positive for α-SMA (B, D, F andI) and α1β1 (H and J) whilemaintaining their expression of PDGF β-receptors and HMW-MAA(D and H) and α5β1 (F). Barrepresents 40 μm.
Table S1. Biopsies from tumour stroma formation in colorectal adenocarcinoma, pannus formation in rheumatoid arthritis and cutaneous healing wounds.
Biopsies were stained by a double immunofluorescence technique with various combinations of monoclonal antibodies. Co-localization between two different markers are depicted as ?percentage of pixels that are positive for both marker X and Y?. Stained sections were analysed by computer imaging processing, as detailed in Materials and methods. The recorded percentage values represent the spatial distribution of two mAb markers in relation to each other, measured in percentage of pixels that co-localize (1 pixel equals 0.9 μm× 0.9 μm) [26, 35].
Table S2. Average number of pixels per field of vision for the different markers.
Biopsies were stained by double immunofluorescence techniquewith various combinations of monoclonal antibodies. Stainedsections were analysed by computer imaging processing, as detailedin Materials and Methods. The recorded values represent the averagenumber of pixels (×103) (1 pixel = 0.9μm × 0.9 μm) present in each field of vision (300 μm× 300 μm) from stained sections of colorectaladenocarcinoma, normal colon, rheumatoid arthritis, wound healingand normal skin for the different mAb?s used in this study. Mean± S.D. (number of fields of vision).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
References
- 1.Barsky SH, Green WR, Grotendorst GR, et al. Desmoplastic breast carcinoma as a source of human myofibroblasts. Am J Pathol. 1984;115:329–33. [PMC free article] [PubMed] [Google Scholar]
- 2.Hewitt RE, Powe DG, Carter GI, et al. Desmoplasia and its relevance to colorectal tumour invasion. Int J Cancer. 1993;53:62–9. doi: 10.1002/ijc.2910530113. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 4.Dvorak HF. Tumors: wounds that do not heal: similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–5. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 5.Gabbiani F. Interpolating between cellular biophysics and computation in single neurons. Neuron. 2003;37:890–1. doi: 10.1016/s0896-6273(03)00153-3. [DOI] [PubMed] [Google Scholar]
- 6.Wynn TA. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol. 2004;4:583–94. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang GP, Lim IJ, Phan TT, et al. From scarless fetal wounds to keloids: molecular studies in wound healing. Wound Repair Regen. 2003;11:411–8. doi: 10.1046/j.1524-475x.2003.11604.x. [DOI] [PubMed] [Google Scholar]
- 8.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–37. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 9.Gabbiani G. Evolution and clinical implications of the myofibroblast concept. Cardiovasc Res. 1998;38:545–8. doi: 10.1016/s0008-6363(98)00065-0. [DOI] [PubMed] [Google Scholar]
- 10.Ronnov-Jessen L, Petersen OW, Koteliansky VE, et al. The origin of myofibroblasts in breast cancer. J Clin Invest. 1995;95:859–73. doi: 10.1172/JCI117736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sundberg C, Ivarsson M, Gerdin B, et al. Pericytes as collagen producing cells in excessive dermal scarring. Lab Invest. 1996;74:452–66. [PubMed] [Google Scholar]
- 12.Sundberg C, Kowanetz M, Brown LF, et al. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab Invest. 2002;82:387–401. doi: 10.1038/labinvest.3780433. [DOI] [PubMed] [Google Scholar]
- 13.Ivarsson M, Sundberg C, Farrokhnia N, et al. Recruitment of type I collagen producing cells from the microvasculature in vitro. Exp Cell Res. 1996;229:336–49. doi: 10.1006/excr.1996.0379. [DOI] [PubMed] [Google Scholar]
- 14.Desmouliere A, Guyot C, Gabbiani G. The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol. 2004;48:509–17. doi: 10.1387/ijdb.041802ad. [DOI] [PubMed] [Google Scholar]
- 15.Hinz B, Gabbiani G. Cell-matrix and cell-cell contacts of myofibroblasts: role in connective tissue remodeling. Thromb Haemost. 2003;90:993–1002. doi: 10.1160/TH03-05-0328. [DOI] [PubMed] [Google Scholar]
- 16.Welch MP, Odland GF, Clark RA. Temporal relationships of F-actin bundle formation, collagen and fibronectin matrix assembly, and fibronectin receptor expression to wound contraction. J Cell Biol. 1990;110:133–45. doi: 10.1083/jcb.110.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munger JS, Huang X, Kawakatsu H, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 18.Scaffidi AK, Moodley YP, Weichselbaum M, et al. Regulation of human lung fibroblast phenotype and function by vitronectin and vitronectin integrins. J Cell Sci. 2001;114:3507–16. doi: 10.1242/jcs.114.19.3507. [DOI] [PubMed] [Google Scholar]
- 19.Ng CP, Hinz B, Swartz MA. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J Cell Sci. 2005;118:4731–9. doi: 10.1242/jcs.02605. [DOI] [PubMed] [Google Scholar]
- 20.Hemler ME. VLA proteins in the integrin family: structures, functions, and their role on leukocytes. Annu Rev Immunol. 1990;8:365–400. doi: 10.1146/annurev.iy.08.040190.002053. [DOI] [PubMed] [Google Scholar]
- 21.Wayner EA, Carter WG. Identification of multiple cell adhesion receptors for collagen and fibronectin in human fibrosarcoma cells possessing unique alpha and common beta-subunits. J Cell Biol. 1987;105:1873–84. doi: 10.1083/jcb.105.4.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardner H, Kreidberg J, Koteliansky V, et al. Deletion of integrin [alpha]1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev Biol. 1996;175:301–13. doi: 10.1006/dbio.1996.0116. [DOI] [PubMed] [Google Scholar]
- 23.Levine JM, Nishiyama A. The NG2 chondroitin sulfate proteoglycan: a multifunctional proteoglycan associated with immature cells. Perspect Dev Neurobiol. 1996;3:245–59. [PubMed] [Google Scholar]
- 24.Skalli O, Ropraz P, Trzeciak A, et al. A monoclonal antibody against alpha smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol. 1986;103:2787–96. doi: 10.1083/jcb.103.6.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nehls V, Drenckhahn D. Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J Cell Biol. 1991;113:147–54. doi: 10.1083/jcb.113.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sundberg C, Ljungstrom M, Lindmark G, et al. Microvascular pericytes express platelet-derived growth factor-beta receptors in human healing wounds and colorectal adenocarcinoma. Am J Pathol. 1993;143:1377–88. [PMC free article] [PubMed] [Google Scholar]
- 27.Van Muijen GN, Ruiter DJ, Warnaar SO. Coexpression of intermediate filament polypeptides in human fetal and adult tissues. Lab Invest. 1987;57:359–69. [PubMed] [Google Scholar]
- 28.Saalbach A, Anderegg U, Bruns M, et al. Novel fibroblast-specific monoclonal antibodies: properties and specificities. J Invest Dermatol. 1996;106:1314–9. doi: 10.1111/1523-1747.ep12349035. [DOI] [PubMed] [Google Scholar]
- 29.Ziai MR, Imberti L, Nicotra MR, et al. Analysis with monoclonal antibodies of the molecular and cellular heterogeneity of human high molecular weight-melanoma associated antigen. Cancer Res. 1987;47:2474–80. [PubMed] [Google Scholar]
- 30.Schlingemann RO, Rietveld FJR, de Waal RMW, et al. Expression of the high molecular weight-melanoma associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am J Pathol. 1990;136:1393–405. [PMC free article] [PubMed] [Google Scholar]
- 31.MacDonald JA, Broekelmann TJ, Matheke ML, et al. A monoclonal antibody to the carboxyterminal domain of procollagen type I visualizes collagen-synthesizing fibroblasts: detection of an altered fibroblast phenotype in lungs of patients with pulmonary fibrosis. J Clin Invest. 1986;78:1237–44. doi: 10.1172/JCI112707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tingström A, Reuterdahl C, Lindahl P, et al. Expression of platelet-derived growth factor beta-receptors on human fibroblasts: regulation by recombinant platelet-derived growth factor-BB, IL-1 and tumor necrosis factor-alpha. J Immunol. 1992;148:546–54. [PubMed] [Google Scholar]
- 33.Wayner EA, Carter WG, Piotrowicz RS, et al. The function of multiple extracellular matrix receptors in mediating cell adhesion to extracellular matrix: preparation of monoclonal antibodies to the fibronectin receptor that specifically inhibit cell adhesion to fibronectin and react with platelet glycoprotein Ic-IIa. J Cell Biol. 1988;107:1881–91. doi: 10.1083/jcb.107.5.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sonnenberg A, Modderman PW, Hogervorst F. Laminin receptor on platelets is the integrin VLA-6. Nature. 1988;336:487–9. doi: 10.1038/336487a0. [DOI] [PubMed] [Google Scholar]
- 35.Sundberg C, Branting M, Gerdin B, et al. Tumor cell and connective tissue cell interactions in human colorectal adenocarcinoma. Transfer of platelet-derived growth factor-AB/BB to stromal cells. Am J Pathol. 1997;151:479–92. [PMC free article] [PubMed] [Google Scholar]
- 36.Goepfert C, Sundberg C, Sevigny J, et al. Disordered extracellular nucleotide-mediated cellular responses perturb angiogenesis in cd39-null mice. Circulation. 2001;104:3109–15. doi: 10.1161/hc5001.100663. [DOI] [PubMed] [Google Scholar]
- 37.Pozzi A, LeVine WF, Gardner HA. Low plasma levels of matrix metalloproteinase 9 permit increased tumor angiogenesis. Oncogene. 2002;21:272–81. doi: 10.1038/sj.onc.1205045. [DOI] [PubMed] [Google Scholar]
- 38.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–3. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 39.Sappino AP, Schurch W, Gabbiani G. Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations. Lab Invest. 1990;63:144–61. [PubMed] [Google Scholar]
- 40.Skalli O, Schurch W, Seemayer T, et al. Myofibroblasts from diverse pathologic settings are heterogeneous in their content of actin isoforms and intermediate filament proteins. Lab Invest. 1989;60:275–85. [PubMed] [Google Scholar]
- 41.Tuchweber B, Desmouliere A, Bochaton-Piallat ML, et al. Proliferation and phenotypic modulation of portal fibroblasts in the early stages of cholestatic fibrosis in the rat. Lab Invest. 1996;74:265–78. [PubMed] [Google Scholar]
- 42.Kinnman N, Francoz C, Barbu V, et al. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab Invest. 2003;83:163–73. doi: 10.1097/01.lab.0000054178.01162.e4. [DOI] [PubMed] [Google Scholar]
- 43.Shao ZM, Nguyen M, Barsky SH. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene. 2000;19:4337–45. doi: 10.1038/sj.onc.1203785. [DOI] [PubMed] [Google Scholar]
- 44.Reuterdahl C, Sundberg C, Funa K, et al. Tissue localization of beta-receptors for platelet-derived growth factor and platelet-derived growth factor B-chain during wound repair in humans. J Clin Invest. 1993;91:2065–75. doi: 10.1172/JCI116429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reuterdahl C, Tingström A, Terracio L, et al. Characterization of PDGF beta-receptor expressing cells in the vasculature of the rheumatoid synovium. Lab Invest. 1991;64:321–9. [PubMed] [Google Scholar]
- 46.Crocker DJ, Murad TM, Geer JP. The role of the pericyte in wound healing. An ultrastructural study. Exp Mol Pathol. 1970:51–65. doi: 10.1016/0014-4800(70)90084-5. [DOI] [PubMed] [Google Scholar]
- 47.Rhodin JAG, Fujita H. Capillary growth in the mesentry of normal young rats. Intravital video and electron microscope analysis. J Submicrosc Cytol Pathol. 1989;21:1–34. [PubMed] [Google Scholar]
- 48.Dore-Duffy P, Owen C, Balabanov R, et al. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res. 2000;60:55–69. doi: 10.1006/mvre.2000.2244. [DOI] [PubMed] [Google Scholar]
- 49.Ausprunk P, Folkman J. Migration and proliferation of endothelial cells in preformed and newly-formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53–65. doi: 10.1016/0026-2862(77)90141-8. [DOI] [PubMed] [Google Scholar]
- 50.Lindahl P, Betsholtz C. Not all myofibroblasts are alike: revisiting the role of PDGF-A and PDGF-B using PDGF-targeted mice. Curr Opin Nephrol Hypertens. 1998;7:21–6. doi: 10.1097/00041552-199801000-00004. [DOI] [PubMed] [Google Scholar]
- 51.Crosby JR, Tappan KA, Seifert RA, et al. Chimeric analysis reveals that fibroblasts and endothelial cells require platelet-derived growth factor receptor beta-expression for participation in reactive connective tissue formation in adults but not during development. Am J Pathol. 1999;154:1315–21. doi: 10.1016/s0002-9440(10)65384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senger DR, Claffey KP, Benes JE, et al. Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc Natl Acad Sci USA. 1997;94:13612–7. doi: 10.1073/pnas.94.25.13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Senger DR, Perruzzi CA, Streit M, et al. The alpha(1)beta(1) and alpha(2)beta(1) integrins provide critical support for vascular endothelial growth factor signaling, endothelial cell migration, and tumor angiogenesis. Am J Pathol. 2002;160:195–204. doi: 10.1016/s0002-9440(10)64363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pozzi A, Moberg PE, Miles LA, et al. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci USA. 2000;97:2202–7. doi: 10.1073/pnas.040378497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holash J, Wiegand SJ, Yancopoulos GD. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene. 1999;18:5356–62. doi: 10.1038/sj.onc.1203035. [DOI] [PubMed] [Google Scholar]
- 56.Shephard P, Martin G, Smola-Hess S, et al. Myofibroblast differentiation is induced in keratinocyte-fibroblast co-cultures and is antagonistically regulated by endogenous transforming growth factor-beta and interleukin-1. Am J Pathol. 2004;164:2055–66. doi: 10.1016/s0002-9440(10)63764-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orlidge A, D'Amore PA. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol. 1987:1455–62. doi: 10.1083/jcb.105.3.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen C, Sheppard D. Identification and molecular characterization of multiple phenotypes in integrin knockout mice. Methods Enzymol. 2007;426:291–305. doi: 10.1016/S0076-6879(07)26013-6. [DOI] [PubMed] [Google Scholar]
- 59.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 60.Hynes RO. Cell-matrix adhesion in vascular development. J Thromb Haemost. 2007;5 Suppl 1:32–40. doi: 10.1111/j.1538-7836.2007.02569.x. [DOI] [PubMed] [Google Scholar]
- 61.Reynolds AR, Reynolds LE, Nagel TE, et al. Elevated Flk1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in beta3-integrin-deficient mice. Cancer Res. 2004;64:8643–50. doi: 10.1158/0008-5472.CAN-04-2760. [DOI] [PubMed] [Google Scholar]
- 62.Diaz-Gonzalez F, Forsyth J, Steiner B, et al. Trans-dominant inhibition of integrin function. Mol Biol Cell. 1996;7:1939–51. doi: 10.1091/mbc.7.12.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item