

Abstract
SIRT1 is a member of a highly conserved gene family (sirtuins) encoding nicotinamide adenine dinucleotide (NAD)+-dependent deacetylases, originally found to deacetylate histones leading to increased DNA stability and prolonged survival in yeast and higher organisms, including mammals. SIRT1 has been found to function as a deacetylase for numerous protein targets involved in various cellular pathways, including stress responses, apoptosis and axonal degeneration. However, the role of SIRT1 in ultraviolet (UV) signalling pathways remains unknown. Using cell culture and Western blot analysis in this study we found that SIRT1 is expressed in cultured human skin keratinocytes. Both UV radiation and H2O2, two major inducers of skin cell damage, down-regulate SIRT1 in a time- and dose-dependent manner. We observed that reactive oxygen species-mediated JNK activation is involved in this SIRT1 down-regulation. SIRT1 activator, resveratrol, which has been considered as an important antioxidant, protects against UV- and H2O2-induced cell death, whereas SIRT inhibitors such as sirtinol and nicotinamide enhance cell death. Activation of SIRT1 negatively regulates UV- and H2O2-induced p53 acetylation, because nicotinamide and sirtinol as well as SIRT1 siRNA enhance UV- and H2O2-induced p53 acetylation, whereas SIRT1 activator resveratrol inhibits it. We also found that SIRT1 is involved in UV-induced AMP-activated protein kinase (AMPK) and downstream acetyl-CoA carboxylase (ACC), phosphofructose kinase-2 (PFK-2) phosphorylation. Collectively, our data provide new insights into understanding of the molecular mechanisms of UV-induced skin aging, suggesting that SIRT1 activators such as resveratrol could serve as new anti-skin aging agents.
Keywords: SIRT1, UV, p53, keratinocytes, apoptosis, skin aging
Introduction
The silent information regulator (SIR) family of genes is a highly conserved group of genes that encode nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylases, also known as class III histone deacetylases. The best characterized of these genes is Saccharomyces cerevisiae SIR2, which is involved in silencing of mating type loci, telomere maintenance, DNA damage response and cell aging [1]. SIRT1, the mammalian orthologue of SIR2, is a protein implicated in regulation of many cellular processes, including apoptosis, cellular senescence, endocrine signalling, glucose homeostasis, aging and longevity [2, 3].
Cellular targets of SIRT1 include acetylated p53 [2, 3], p300 [4], Ku70 [5], forkhead (FOXO) transcription factors [5, 6], PPARγ[7] and PPARγ coactivator-1α (PGC-1α) protein [7, 8]. Deacetylation of p53 transcription factors and FOXO represses apoptosis and increases cell survival [2, 4–6]. Deacetylation of Ku70 [9] and p300 [4] increases DNA-damage repair and promotes cell survival. Deacetylation of PPARγ and PGC-1α regulates the gluconeogenic/glycolytic pathways in the liver and fat mobilization in white adipocytes in response to fasting [7, 8]. SIRT1 remains one of the most important cell signalling components in regulation of cell death and survival.
Ultraviolet (UV) spectrum is divided into UVC (200–280 nm), UVB (280–320 nm) and UVA (320–400 nm). UVB is of environmental significance. UVB penetrates into papillary area of the dermis (∼0.2 mm) and induces DNA damage of residing dendritic cells and keratinocytes. They are perturbed both phenotypically and functionally undergoing apoptosis upon UVB radiation. A number of signals are involved in this apoptotic process, of which, reactive oxygen species (ROS) production, p53 [10], p38 [11], JNK [11, 12] are extensively studied. However, the possible involvement of SIRT1 in UV-induced skin cell damage is not fully studied.
Given that SIRT1 plays important roles in cellular apoptosis and cell survival, we undertook this study to investigate the role of SIRT1 in UV-induced cellular damage. In this study, we found for the first time that SIRT1 is functionally expressed in cultured skin keratinocytes. Both UV and H2O2, two major factors of skin cell damage, down-regulate SIRT1. ROS-mediated JNK activation is involved in UV-induced SIRT1 down-regulation. SIRT activator, resveratrol protects against UV- and H2O2-induced apoptotic cell death, whereas SIRT1 inhibitors such as sirtinol and nicotinamide as well as SIRT1 RNAi enhance apoptosis. Activation of SIRT1 deacetylates p53 after UV and H2O2 treatment, because nicotinamide and sirtinol as well as SIRT1 siRNA enhances UV- and H2O2-induced p53 acetylation. Our study provides evidence to support the notion that SIRT1 might be a novel target protecting UV/ROS-induced skin cell damage.
Materials and methods
UVB light apparatus
As previously reported [13–15], UV-irradiation apparatus used in this study consisted of four F36T12 EREVHO UV tubes. A Kodacel TA401/407 filter (International Light Inc., Newburyport, MA, USA) was mounted 4 cm in front of the tubes to remove wavelengths <290 nm. Irradiation intensity was monitored using an IL443 phototherapy radiometer and a SED240/UV/W photodetector. Before UV irradiation, cells were washed with 1 ml phosphate buffered saline (PBS) and changed to fresh 0.5 ml PBS each well. Cells were irradiated at the desired intensity without plastic dish lid. After UV irradiation, cells were returned to incubation in basal medium with treatments for various time-points prior to harvest.
Chemicals and reagents
JNK inhibitor II (SP 600125) and AMPK inhibitor (AMPKi, compound C) were from CalbioChem (San Diego, CA, USA). Bcl-XL, SIRT1 siRNA, goat anti-rabbit IgG-HRP and goat antimouse IgG-HRP antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Monoclonal mouse anti-β-actin, anti-SIRT1, sirtinol, nicotinamide, resveratrol and Hoechst33342 were obtained from Sigma (St. Louis, MO, USA). Phospho-AKT (Ser473), phospho-ERK (Tyr1068), phosphor-JNK (Thr183/185), phospho-AMPK (Thr172), p-acetyl-CoA carboxylase (ACC) (Ser 79) phospho-p38 (Thr180/Tyr182), phosphofructose kinase-2 (PFK-2) (Ser466), SAPK/JNK, p38 antibody and AKT antibody were all from Cell Signaling Technology (Bevery, MA, USA).
Cell culture
Spontaneously immortalized human keratinocytes (HaCaT cell line) were used as previously reported [15, 16]. The p53 wild-type mouse embryonic fibroblasts (MEFs) and p53 knockout MEFs were obtained from Dr. Kun-liang Guan [17]. Cells were maintained in a DMEM medium (Sigma), supplemented with a 10% foetal bovine serum (Invitrogen, Carlsbad, CA, USA), Penicillin/Streptomycin (1:100, Sigma) and 4 mM L-glutamine (Sigma), in a CO2 incubator at 37°C. For Western blot analysis, cells were reseeded in six-well plates at a density of 0.2 × 106 cells/ml with fresh complete culture medium.
Western blot analysis
As described previously [15, 16], cultured cells with or without treatments were washed with cold PBS and harvested by scraping into 150 μl of RIPA buffer with protease inhibitors. Twenty micrograms of proteins were separated by SDS-PAGE and transferred onto PVDF membrane (Millipore, Bedford, MA, USA). After blocking with 10% milk in TBS, membranes were incubated with specific antibodies in dilution buffer (2% bovine serum albumin in TBS) overnight at 4°C followed by horseradish peroxidase-conjugated anti-rabbit or antimouse IgG at appropriate dilutions and room temperature for 1 hr. Antibody binding was detected using enhanced chemiluminescence detection system from GE Biosciences (Piscataway, NJ, USA) following manufacturer’s instructions and visualized by fluorography with Hyperfilm.
Cell viability assay (MTT dye assay)
Cell viability was measured by the 3-[4,5-dimethylthylthiazol-2-yl]-2,5 diphenyltetrazolium bromide (MTT) method [15]. Briefly, cells were collected and seeded in 96-well plates at a density of 2 × 105 cells/cm2. Different seeding densities were optimized at the beginning of the experiments (data not shown). After incubation for 24 hrs, cells were exposed to fresh medium containing reagents at 37°C. After incubation for up to 24 hrs, 20 μl of MTT tetrazolium salt (Sigma) dissolved in Hank’s balanced solution at a concentration of 5 mg/ml was added to each well and incubated in CO2 incubator for 4 hrs. Finally, the medium was aspirated from each well and 150 μl of DMSO (Sigma) was added to dissolve formazan crystals and the absorbance of each well was obtained using a Dynatech MR5000 plate (Dynatech Laboratories, Alexandria, VA, USA) reader at a test wavelength of 490 nm with a reference wavelength of 630 nm.
Assessment of the percentage of apoptotic cells
To detect apoptotic cells [15], cells were stained with DNA binding dye Hoechst 33342 (Sigma). After the cells were exposed to UV and the test compounds for the allotted time periods, they were fixed with 4% formaldehyde in PBS for 10 min. at 4°C, and then washed with PBS. To stain the nuclei, cells were incubated for 20 min. with 20 μg/ml of Hoechst 33342. After washing with PBS, the cells were observed under a fluorescence microscope (Zeiss Axiophoto 2, Carl Zeiss, Thornwood, NY, USA). Cells exhibiting condensed chromatin and fragmented nuclei were scored as apoptotic cells. A minimum of 200 cells was scored from each sample.
Reactive oxygen species detection
ROS generation was detected by FACS analysis as described previously [15]. Briefly, cultured human skin keratinocytes (HaCat cells) were loaded with 1 μM of fluorescent dye dihydrorhodamine 2 hrs before UV radiation, which reacts with ROS in cells and results in a change of fluorescence. After being treated with UV and with or without inhibitors for desired time, cells were trypsinized, suspended in ice-cold PBS and fixed in 70% ethyl alcohol at −20°C. The changes in fluorescence in drug-treated cells were quantified by FACS analysis. Induction of ROS generation was expressed in arbitrary units.
SIRT1 RNA interference (RNAi) experiments
As described previously [15, 16], siRNA for SIRT1 (sc-40986) was purchased from Santa Cruz Biotechnology, HaCaT cells were cultured in complete medium that did not contain antibiotics for 4 days. Cells were seeded into a six-well plate 1 day prior to transfection and cultured to 60–70% confluence the following day. For RNAi experiments, 8 μl of Lipofectamine™ LTX together 3 μl PLUS™ Reagent (Invitrogen, Indianapolis, IN, USA) was diluted in 90 μl of DMEM for 5 min. in room temperature. Then, 12 μl SIRT1 siRNA was mixed with DMEM containing Lipofectamine together with PLUS reagent and incubated for 30 min. at room temperature for complex formation. Finally, the complex was added to the well containing 2 ml medium with the final SIRT1 siRNA concentration of 150 nM. SIRT1, protein expression was determined by Western blot after 48 hrs.
Statistical analysis
The values in the figures are expressed as the means ± standard error (S.E.). The figures in this study were representatives of more than three different experiments. Statistical analysis of the data between the control and treated groups was performed by a Student’s t-test. Values of P < 0.05 were considered as statistically significant.
Results
UV and H2O2 down-regulate SIRT1 expression in cultured skin keratinocytes
To understand the role of SIRT1 in UV-induced cell signalling processes, we first tested the expression of SIRT1 in UV- and H2O2-treated skin keratinocytes. As shown in Fig, 1A and B, UV radiation down-regulates SIRT1 in a dose-dependent manner in cultured skin keratinocytes (HaCaT cell line). SIRT1 expression begins to decrease at 10 mJ/cm2 of UV radiation with about 60–70% lost at a dose of 20 mJ/cm2 24 hrs after UV treatment. UV radiation also induces SIRT1 down-regulation in a time-dependent manner, as shown in Fig. 1C and D. SIRT1 expression begins to decrease 12 hrs after UV treatment, with about 30–40% left 24 hrs after UV radiation at the dose of 20 mJ/cm2. Furthermore, H2O2 also induces SIRT1 down-regulation in a dose (Fig. 1E and F) and a time (Fig. 1G and H) dependent manner. These results demonstrate that both UV radiation and H2O2 down-regulate SIRT1 expression, suggesting that SIRT1 down-regulation may be involved in UV- and H2O2-induced skin cell damage.
Figure 1.
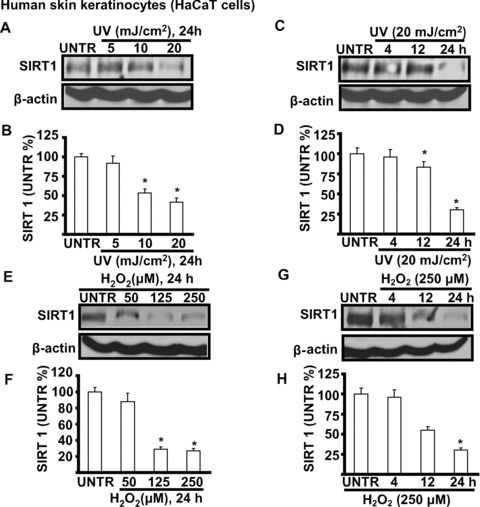
UV and H2O2 down-regulate SIRT1 expression in cultured skin keratinocytes. HaCaT cells were treated with different doses of UV (5, 10 and 20 mJ/cm2) (A and B), cells then incubated in basic medium (DMEM) for 24 hrs or treated with 20 of mJ/cm2 UV and incubated in DMEM for different time-points (4, 12 and 24 hrs) (C and D), SIRT1 and β-actin were detected by Western blot. HaCaT cells were treated with different doses of H2O2 (50, 125 and 250 μM) for 24 hrs (E and F) or treated with 250 μM of H2O2 for different time-points (4, 12 and 24 hrs), SIRT1 and β-actin were detected by Western blot (G and H). The data in figures represent mean ± S.E. of three independent experiments. The symbol ‘*’ means P < 0.05 with untreated group (lane 1).
ROS-mediated JNK activation is involved in UV- and H2O2-induced SIRT1 down-regulation
The above data showed that UV radiation and H2O2 induce SIRT1 down-regulation in cultured human skin keratinocytes, and yet cell signal transduction pathways involved in this process remain unclear. Mitogen-activated protein kinase (MAPK) and PI3K/AKT pathways are known to mediate UV-induced cellular events leading to photoaging [10, 18, 19]. To investigate whether those signalling pathways are also involved in UV-induced SIRT1 down-regulation, various pharmacological inhibitors were utilized in our experiments. Although inhibitors of p38 (SB 203580), MEK/ERK (PD 98059 and U0126) and PI3K/AKT (LY 294002 and Wortmannin) have no effects on UV- and H2O2-induced SIRT1 down-regulation (data not shown), JNK inhibitor (SP 600125, 1 μm, or JNKi) attenuates SIRT1 down-regulation (Fig. 2A–D). This result suggests that JNK activation is involved, at least in part, in UV- and H2O2-induced SIRT1 down-regulation. To further investigate the role of ROS in SIRT1 down-regulations, cells were pre-treated with antioxidant NAC (n-acetyl-l-cysteine). The results showed that NAC protects against UV- and H2O2-induced loss of SIRT1 (Fig. 2E–H). As expected, NAC pre-treatment inhibits UV-induced ROS production (Fig. 2I) and JNK activation (Fig. 2J). Collectively, our data suggest that ROS-mediated JNK activation is involved in UV- and H2O2-induced SIRT1 down-regulation.
Figure 2.
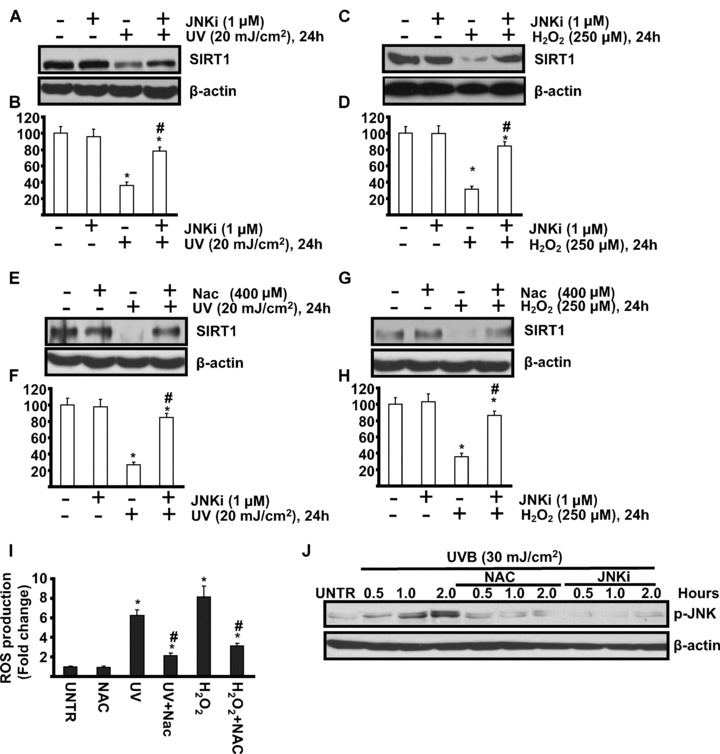
ROS-mediated JNK activation is involved in UV- and H2O2-induced SIRT1 down-regulation. HaCaT cells were pre-treated with JNK inhibitor (SP 600125, 1 μM, or JNKi) for 1 hr, followed by 20 mJ/cm2 UV radiation (A and B) or 250 μM of H2O2 (C and D) and incubated for 24 hrs, SIRT1 and β-actin expression were detected by Western blot. HaCaT cells were pre-treated with anti-oxidant NAC (n-acetyl-l-cysteine) (NAC, 400 μM) for 1 hr, followed by 20 mJ/cm2 UV radiation (E and F) or 250 μM of H2O2 (G and H) and incubated for 24 hrs, SIRT1 and β-actin expression were detected by Western blot. HaCaT cells were pre-treated with NAC for 1 hr, followed by 20 mJ/cm2 of UV radiation or 250 μM of H2O2 and incubated for 1 hr (I), ROS production was detected by FACS as described in methods. HaCaT cells were pre-treated with NAC (400 μM) or JNK inhibitor (SP 600125, 1 μM, or JNKi) for indicated time-points, p-JNK and β-actin were detected by Western blot. The data in figures represent mean ± S.E. of three independent experiments. The symbol ‘*’ means P < 0.05 with untreated group. The symbol ‘#’ means P < 0.05 with UV- or H2O2-treated group.
SIRT1 modulates UV-induced JNK activation
Because JNK MAPK, principally activated by ROS, mediates UV-induced cell death in keratinocytes, we next examined whether SIRT1 could affect UV-induced JNK phosphorylation. The results showed that treatment of keratinocytes with UV (30 mJ/cm2) leads to a rapid and time-dependent activation of MAPKs (JNK, ERK and p38). Pre-treatment with SIRT1 inhibitors such as sirtinol and nicotinamide, which alone has little effect on MAPK activation (data not shown), enhances UV-induced JNK phosphorylation (Fig. 3A), but has no effects on UV-induced AKT activation, which is known as a pro-survival signal (Fig. 3A and B). To further confirm the role of SIRT1 in UV-induced JNK activation, SIRT1 siRNA specific knockdown was used. As shown in Fig. 3C, SIRT1 siRNA knockdowns SIRT1 expression in HaCaT cells. Furthermore, SIRT1 siRNA knockdown enhances UV-induced JNK activation, whereas SIRT1 activator resveratrol inhibits it (Fig. 3D and E). These data suggest that SIRT1 inhibits UV-induced JNK activation.
Figure 3.
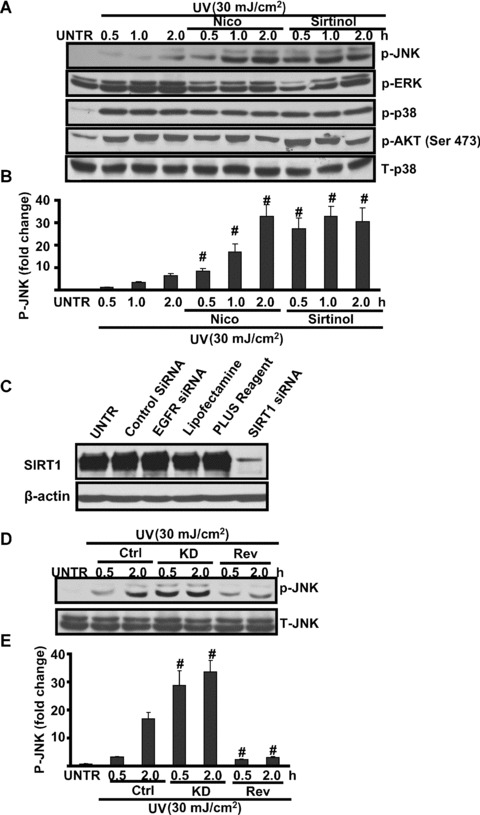
SIRT1 regulates UV-induced JNK activation. HaCaT cells were pre-treated with nicotinamide (Nico, 10 mM) or sirtinol (2 mM) for 1 hr, followed by UV radiation (25 mJ/cm2) and then incubated in DMEM for 0.5, 1.0 and 2.0 hrs, p-JNK, p-ERK, p-p38, p-AKT (473) and T-p38 were detected by Western blot (A) and JNK phosphorylation was quantified in (B). HaCaT cells with or without SIRT1 siRNA were pre-treated with resveratrol (Rev, 10 μM) for 1 hr, followed by UV radiation for indicated time-points, p-JNK and T-JNK were detected by Western blot. HaCaT cells were treated with 150 nM of SIRT1 siRNA or controls for 48 hrs, SIRT1 and β-actin were detected by Western blot (C). (D) Control or SIRT1 siRNA (150 nM) pre-treated cells were treated with UV radiation (25 mJ/cm2) for indicated time along with or without resveratrol (Rev, 10 μM, 1 hr prior to UV radiation), p-JNK and T-JNK were detected by Western blot, JNK phosphorylation was quantified in (E). The data in figures represent mean ± S.E. of three independent experiments. The symbol ‘#’ means P < 0.05 with UV-treated group.
SIRT1 negatively regulates UV- and H2O2-induced p53 acetylation
Previous studies have indicated that SIRT1 may function to promote cell survival [3, 20, 21]via direct interactions with several apoptotic proteins, including p53 [3]. We next tested the possible role SIRT1 in UV-induced p53 activation. As shown in Fig. 4A–D, UV-induced p53 acetylation is enhanced by SIRT1 inhibitor sirtinol and nicotinamide (Nico). Furthermore, pre-treatment with SIRT1 activator resveratrol (Rev) almost abolishes sirtinol or Nico’s effects. Similar results are also seen in H2O2-treated cells (Fig. 4E and F). To further confirm the involvement of SIRT1 in UV- and H2O2-induced p53 acetylation, wild-type and p53 knockout MEFs were used in our experiments. As shown in Fig. 4G and H, Nico enhances UV- and H2O2-induced p53 acetylation in wild-type but not in p53 knockout MEFs. Next we tested p53 acetylation in SIRT1 knockdown HaCaT cells. As shown in Fig. 4I, SIRT1 siRNA knockdown enhances UV- and H2O2-induced p53 acetylation in HaCaT cells. Collectively, our results suggest that SIRT1 negatively regulates p53 acetylation in UV- and H2O2-treated skin keratinocytes.
Figure 4.
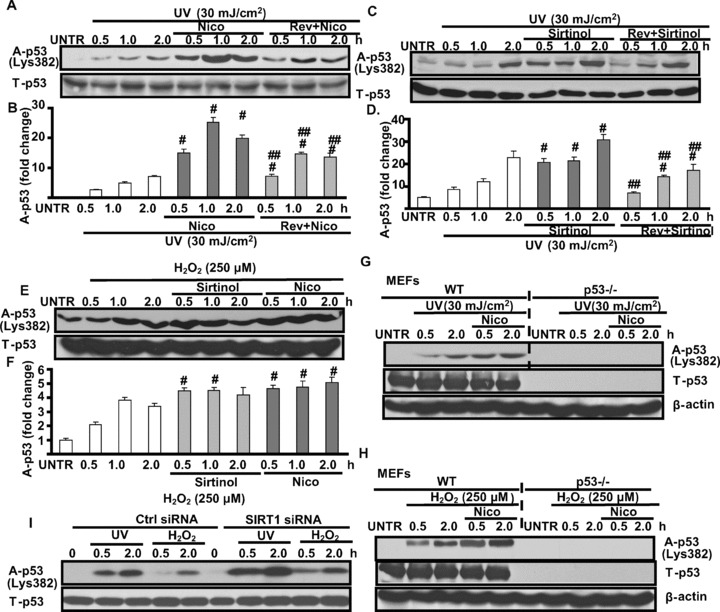
SIRT1 negatively regulates UV- and H2O2-induced p53 acetylation. HaCaT cells were pre-treated with nicotinamide (Nico, 10 mM) or resveratrol (Rev, 10 μM) plus Nico for 1 hr, followed by UV radiation (25 mJ/cm2) and incubated for 0.5, 1.0 and 2.0 hrs, acelyated p53 and T-p53 were detected by Western blot (A and B). HaCaT cells were also pre-treated with sirtinol (2 mM) or Rev plus sirtinol for 1 hr, followed by UV radiation for indicated time, acelyated p53 and T-p53 were detected by Western blot (C and D). HaCaT cells were pre-treated with Nico or sirtinol for 1 hr, followed by H2O2 (250 μM), acelyated p-53 and T-p53 were detected by Western blot (E and F). Wild-type and p53 knockout MEFs were pre-treated with Nico for 1 hr, followed by UV (G) or H2O2 (H), for indicated time-points, acetylated p53 and T-p53 were detected by Western blot. Control and SIRT1 siRNA treated HaCaT cells were treated 30 mJ/cm2 of UV radiation or 250 μM of H2O2 for indicated time-points, acetylated p53 and T-p53 were detected by Western blot (I). The data in figures represent mean ± S.E. of three independent experiments. The symbol ‘#’ means P < 0.05 with UV- or H2O2-treated group, the symbol ‘##’ means P < 0.05 with UV plus Nico or sirtinol treated group.
SIRT1 positively regulates UV-induced AMPK activation
Some of the metabolic changes caused by resveratrol, a SIRT1 activator, mimic those observed in response to AMPK activation, and our data in this study demonstrated for the first time that UV radiation induces AMPK activation in cultured skin keratinocytes (Fig. 5A and B). We next tested the possible role of SIRT1 in AMPK activation. Our results showed that SIRT1 inhibitors sirtinol and nicotinamide inhibit UV-induced AMPK (Fig. 5A and B) and downstream PFK-2 and ACC phosphorylation (Fig. 5C and D). Furthermore, SIRT1 activator resveratrol alone also induces AMPK activation, and this induction is largely impaired by SIRT1 inhibitor (nicotinamide, or Nico) or AMPK inhibitor (compound C, or AMPKi) (Fig. 5E and F). Collectively, our data suggest that SIRT1 positively regulates AMPK activation in response to UV and resveratrol. At least some of the actions of resveratrol, such as fatty acid oxidation, are mediated by AMPK activation.
Figure 5.
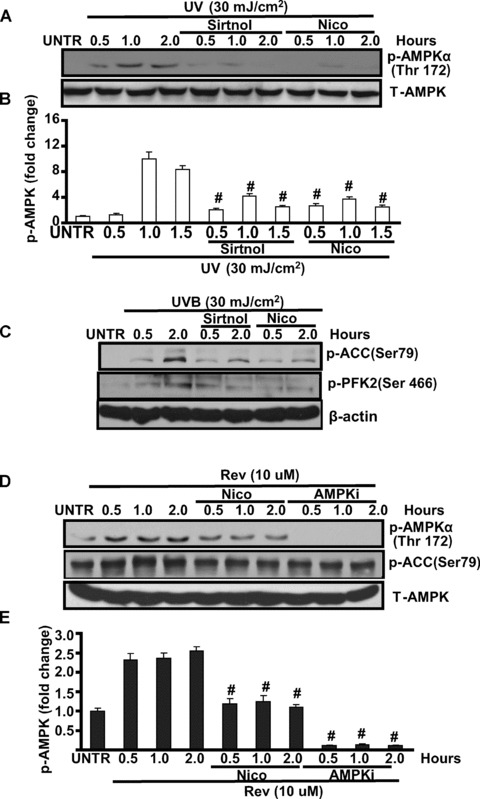
SIRT1 positively regulates AMPK activation in cultured skin keratinocytes. HaCaT cells were pre-treated with sirtinol (2 mM) or nicotinamide (Nico, 10 mM) for 1 hr, followed by 20 mJ/cm2 of UV for indicated time, p-AMPK (Thr 172) and total-AMPK activation were detected by Western blot (A and B). HaCaT cells were pre-treated with sirtinol (2 mM) or nicotinamide (Nico, 10 mM) for 1 hr, followed by 20 mJ/cm2 of UV and incubated for 0.5 and 2.0 hrs, p-ACC (Ser 79), p-PFK-2 (Ser 466) and β-actin were detected by Western blot (C), p-ACC was quantified in (D). HaCaT cells were treated with nicotinamide (Nico, 10 mM) or AMPK inhibitor Compound C (AMPKi, 10 μM) for 0.5, 2.0 hrs, followed by resveratrol (Rev, 10 μM) treatment for 0.5, 1.0 and 2.0 hrs, p-AMPK (Thr 172), p-ACC (Ser 79) and T-AMPK were detected by Western blot (E and F). The data in figures represent mean ± S.E. of three independent experiments. The symbol ‘#’ means P < 0.05 with UV- or H2O2-treated group.
SIRT1 protects against UV-radiation-induced cell death
To further test the role of SIRT1 in UV-induced cell death, we pre-treated cells with various SIRT1 inhibitors and activators and then exposed the cells with UV radiation. As shown in Fig. 6A and B, as expected, UV radiation induces HaCaT cell death in a dose-dependent manner, whereas pre-treatment with resveratrol protects against UV-induced cell death and apoptosis, nicotinamide and sirtinol enhance this process. Furthermore, nicotinamide and sirtinol pre-treatment aggravate UV-induced Bcl-xl degradation, whereas resveratrol delays the process (Fig. 6C). Next we tested the possible role of p53 in SIRT1-induced protective effects using p53 knockout cells. As demonstrated in Fig. 6D, p53 knockout MEFs are resistant to UV-induced cell death and SIRT1-induced protective effects are almost abolished in p53 knockout MEFs. To furthermore confirm these protective effects, SIRT1 siRNA was used in our experiments. As shown in Fig. 6E, SIRT1 knockdown HaCaT cells were more sensitive to UV-induced cell death. Taken all together, our data demonstrated that SIRT1 protects against UV-induced cell death, at least in part, via modulation of p53.
Figure 6.
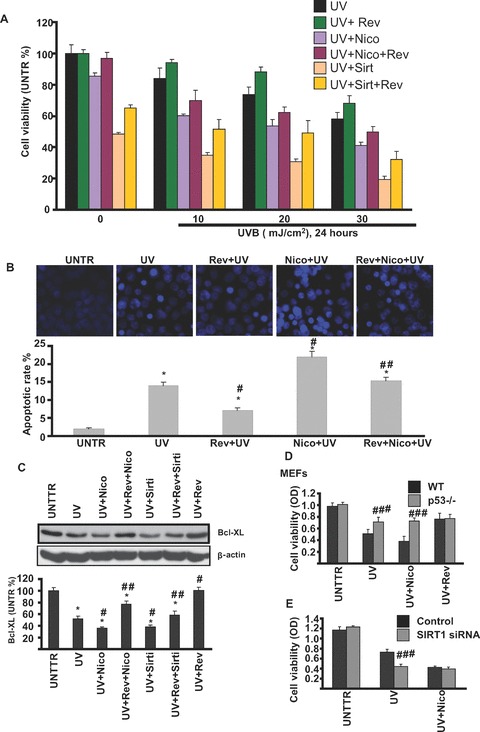
SIRT1 protects against UV-induced skin cell damage. HaCaT cells were pre-treated with resveratrol (Rev, 10 μM), sirtinol (2 mM), nicotinamide (Nico, 10 mM), Rev + Nico or Rev + sirtinol for 1 hr, followed by 10, 20 or 30 mJ/cm2 of UV radiation and incubated in DMEM for 24 hrs, cell viability was detected by MTT assay (A). HaCaT cells were pre-treated with nicotinamide, resveratrol, or Rev + Nico for 1 hr, followed by 20 mJ/cm2 of UV radiation and incubation in DMEM for 24 hrs, cell apoptosis was detected by Hoechst assay (B). HaCaT cells were treated with indicated treatments for 24 hrs, Bcl-xl and β-actin were detected by Western blot (C). Wild-type and p53 knockout MEFs were treated with UV, UV + Nico or UV + Rev and incubated in DMEM for 24 hrs, cell viability was detected by MTT assay (D). Control or SIRT1 siRNA pre-treated cells were treated with UV (20 mJ/cm2) or UV + Nico (Nico, 10 mM) for 24 hrs, cell viability were detected by MTT assay (E). The data in figures represent mean ± S.E. of three independent experiments. The symbol ‘#’ means P < 0.05 with UV- or H2O2-treated group, the symbol ‘##’ means P < 0.05 with UV plus Nico or sirtinol treated group, the symbol ‘####’ means P < 0.05 with p53 knockout or SIRT1 siRNA group.
Discussion
In response to UV radiation, p53 tumour suppressor is activated and exerts anti-proliferative effects, including growth arrest, apoptosis, and cell senescence [22]. Following DNA damage, p53 protein is protected from rapid degradation and acquires transcription-activating functions, largely as a result of post-translational modifications [23]. Activation of p53 protein as a transcription factor allows it, in turn, to up-regulate the expression of genes whose products promote cell cycle exit, such as p21WAF1 gene [24], or of genes that favour apoptosis [25]. The p53 protein is phosphorylated in response to DNA damage by ATM at residue Ser15 [26] and at residue Ser20 by Chk1/2 kinases [26].
However, recent studies suggest that Ser15 phosphorylation does not lead directly to the functional activation of p53 protein. Instead, it increases the affinity of the p300 acetylase for p53 [27]. This association leads to the acetylation of p53. Indeed, p53 is acetylated in vitro by p300 at Lys 370–373, 381, and 382 [28]. Moreover, at least two of these sites, namely residues 320 and 382, are found to be acetylated in vivo in response to DNA damage [29]. Among other factors that can affect acetylation of p53 are MDM2 protein and SIRT1, which are involved in the negative regulation of p53 [30] and are able to block acetylation of p53 protein by p300 [31].
SIRT1 is a member of a highly conserved gene family (sirtuins) encoding NAD+-dependent deacetylases. SIRT1 remains one of the most important cell signalling molecules that are associated with cell survival and longevity [2, 3]. In this study, we have evidence showing that SIRT1 plays protective role in UV-induced skin cell damage. We found that UV radiation and H2O2 induce p53 acetylation in cultured skin keratinocytes and MEFs cells (Fig. 4), SIRT1, as a deacetylase, negatively regulate UV-induced p53 acetylation (Fig. 7A), because SIRT1 inhibitors sirtinol and nicotinamide as well as SIRT1 siRNA enhance UV-induced p53 acetylation, whereas SIRT1 activator resveratrol enhances it (Fig. 4). Considering recent study showing that acetylation is indispensable for p53 activation, we suggest that SIRT1 protects from UV-induced cell death, at least in part, by negatively regulating p53 acetylation (Fig. 6D).
Figure 7.
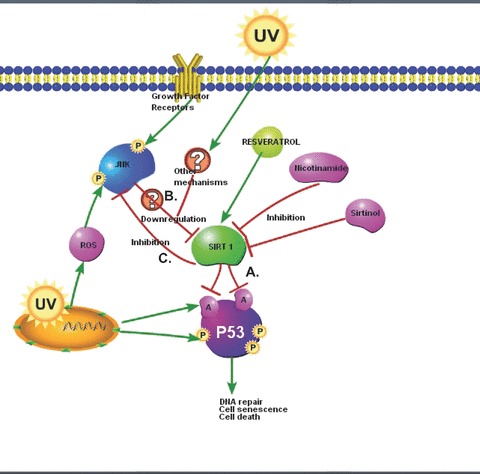
Proposed cell signalling pathways involving SIRT1 in response to UV radiation. (A) SIRT1 negatively regulates UV- and H2O2-induced p53 acetylation. (B) ROS-mediated JNK activation is involved in UV-and H2O2-induced SIRT1 down-regulation. (C) SIRT1 inhibits UV-induced JNK activation.
Previous studies have demonstrated that UV radiation induces down-regulation of a number of cellular proteins such as collagen and water channel protein aquaporin-3 or AQP-3 in both skin keratinocytes and fibroblasts [19, 32]. Interestingly, in this study, we observed that UV radiation also induces down-regulation of SIRT1 in cultured human skin keratinocytes (Fig. 1). Given the important functions of SIRT1 discussed above, one can easily envision the consequence of SIRT1 down-regulation in response to UV radiation. Systematic studies revealed that ROS-mediated JNK activation is involved in SIRT1 down-regulation in response to UV radiation. JNK inhibitor and antioxidant NAC could recover SIRT1 lost due to UV radiation (Figs. 2 and 7B). Interestingly, SIRT1 inhibits UV-induced JNK activation (Figs. 3 and 7C). Collectively, these data suggest that SIRT1 serves as a negative regulator against UV-induced JNK activation, probably by de-acetylation and inhibition of one or more of JNK upstream signals, which may serve as another mechanism to protect against UV-induced cell death. However, the detailed mechanisms through which SIRT1 negatively regulates JNK activation needs further investigation. Our data have provided more insights into understanding of the molecular mechanism through which resveratrol, the SIRT1 activator, acts as an important anti-aging agent. Our data may also help to develop better cosmetics products against UV-induced human skin photoaging and even skin cancer.
Our data also suggest that SIRT1 positively regulates AMPK activation in response to UV and resveratrol (Fig. 5). At least some of the actions of resveratrol, such as fatty acid oxidation, are mediated by AMPK activation [33]. These results are consistent with previous studies which demonstrated that resveratrol acts as an activator of AMPK in both neuron and whole brain [33], and the most recent study which indicated that AMPK activation might be involved in resveratrol’s calorie resistant effect [34]. However, the detailed mechanism through which resveratrol activates AMPK and the biological function of these effects warrant further investigation.
In summary, we found for the first time that SIRT1 is functionally expressed in cultured skin keratinocytes. Both UV and H2O2, two major factors of skin cell damage, down-regulate SIRT1 in a time and dose-dependent manner. Systematic studies revealed that ROS-mediated JNK activation is involved in UV-induced SIRT1 down-regulation (Fig. 7). SIRT activator, resveratrol which has also been considered an important antioxidant, protects against UV- and H2O2-induced apoptotic cell death, whereas SIRT1 inhibitors such as sirtinol and nicotinamide as well as SIRT1 RNAi enhance apoptosis. Our study provides evidence to support the notion that SIRT1 might be a novel target protecting UV/ROS-induced skin cell damage leading to skin photoaging and skin cancer.
Acknowledgments
This research was supported in part by a grant from NIH (P20 RR016457 from INBRE Program of the National Center for Research Resources), a grant for biomedical research from Rhode Island Foundation and a grant from Slater Center for Environmental Biotechnology.
References
- 1.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–80. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 3.Luo J, Nikolaev AY, Imai S, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–48. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 4.Bouras T, Fu M, Sauve AA, et al. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280:10264–76. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- 5.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 6.Motta MC, Divecha N, Lemieux M, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–63. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 7.Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–6. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodgers JT, Lerin C, Haas W, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 9.Jeong J, Juhn K, Lee H, et al. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med. 2007;39:8–13. doi: 10.1038/emm.2007.2. [DOI] [PubMed] [Google Scholar]
- 10.Cui R, Widlund HR, Feige E, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007;128:853–64. doi: 10.1016/j.cell.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 11.Bode AM, Dong Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci STKE. 2003;2003:RE2. doi: 10.1126/stke.2003.167.re2. [DOI] [PubMed] [Google Scholar]
- 12.Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–4. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 13.Wan YS, Wang ZQ, Shao Y, et al. Ultraviolet irradiation activates PI 3-kinase/AKT survival pathway via EGF receptors in human skin in vivo. Int J Oncol. 2001;18:461–6. doi: 10.3892/ijo.18.3.461. [DOI] [PubMed] [Google Scholar]
- 14.Fisher GJ, Kang S, Varani J, et al. Mechanisms of photoaging and chronological skin aging. Arch Dermatol. 2002;138:1462–70. doi: 10.1001/archderm.138.11.1462. [DOI] [PubMed] [Google Scholar]
- 15.Cao C, Healey S, Amaral A, et al. ATP-sensitive potassium channel: a novel target for protection against UV-induced human skin cell damage. J Cell Physiol. 2007;212:252–63. doi: 10.1002/jcp.21026. [DOI] [PubMed] [Google Scholar]
- 16.Cao C, Sun Y, Healey S, et al. EGFR-mediated expression of aquaporin-3 is involved in human skin fibroblast migration. Biochem J. 2006;400:225–34. doi: 10.1042/BJ20060816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Bi Z, Yan B, Wan Y. UVB radiation induces expression of HIF-1alpha and VEGF through the EGFR/PI3K/DEC1 pathway. Int J Mol Med. 2006;18:713–9. [PubMed] [Google Scholar]
- 18.Wan Y, Wang Z, Shao Y, et al. UV-induced expression of GADD45 is mediated by an oxidant sensitive pathway in cultured human keratinocytes and in human skin in vivo. Int J Mol Med. 2000;6:683–8. doi: 10.3892/ijmm.6.6.683. [DOI] [PubMed] [Google Scholar]
- 19.Fisher GJ, Talwar HS, Lin J, et al. Retinoic acid inhibits induction of c-Jun protein by ultraviolet radiation that occurs subsequent to activation of mitogen-activated protein kinase pathways in human skin in vivo. J Clin Invest. 1998;101:1432–40. doi: 10.1172/JCI2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 21.Cohen HY, Miller C, Bitterman KJ, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–2. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 22.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 23.Shieh SY, Ahn J, Tamai K, et al. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 24.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 25.Lin Y, Ma W, Benchimol S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet. 2000;26:122–7. doi: 10.1038/79102. [DOI] [PubMed] [Google Scholar]
- 26.Siliciano JD, Canman CE, Taya Y, et al. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–81. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lambert PF, Kashanchi F, Radonovich MF, et al. Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem. 1998;273:33048–53. doi: 10.1074/jbc.273.49.33048. [DOI] [PubMed] [Google Scholar]
- 28.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 29.Abraham J, Kelly J, Thibault P, et al. Post-translational modification of p53 protein in response to ionizing radiation analyzed by mass spectrometry. J Mol Biol. 2000;295:853–64. doi: 10.1006/jmbi.1999.3415. [DOI] [PubMed] [Google Scholar]
- 30.Oren M. Regulation of the p53 tumor suppressor protein. J Biol Chem. 1999;274:36031–4. doi: 10.1074/jbc.274.51.36031. [DOI] [PubMed] [Google Scholar]
- 31.Kobet E, Zeng X, Zhu Y, et al. MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc Natl Acad Sci USA. 2000;97:12547–52. doi: 10.1073/pnas.97.23.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao C, Wan S, Jiang Q, et al. All-trans retinoic acid attenuates ultraviolet radiation-induced down-regulation of aquaporin-3 and water permeability in human keratinocytes. J Cell Physiol. 2008;215:506–16. doi: 10.1002/jcp.21336. [DOI] [PubMed] [Google Scholar]
- 33.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci USA. 2007;104:7217–22. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–42. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]