

Abstract
Evidence shows that women have lower tumour necrosis factor-α (TNF-α) levels and lower incidences of heart dysfunction and sepsis-related morbidity and mortality. To identify the cardioprotective effects and precise cellular/molecular mechanisms behind estrogen and estrogen receptors (ERs), we investigated the effects of 17β-estradiol (E2) and estrogen receptor α (ERα) on LPS-induced apoptosis by analyzing the activation of survival and death signalling pathways in doxycycline (Dox)-inducible Tet-On/ERα H9c2 myocardial cells and ERα-transfected primary cardiomyocytes overexpressing ERα. We found that LPS challenge activated JNK1/2, and then induced IκB degradation, NFκB activation, TNF-α up-regulation and subsequent myocardial apoptotic responses. In addition, treatments involving E2, membrane-impermeable BSA-E2 and/or Dox, which induces ERα overexpression, significantly inhibited LPS-induced apoptosis by suppressing LPS-up-regulated JNK1/2 activity, IκB degradation, NFκB activation and pro-apoptotic proteins (e.g. TNF-α, active caspases-8, t-Bid, Bax, released cytochrome c, active caspase-9, active caspase-3) in myocardial cells. However, the cardioprotective properties of E2, BSA-E2 and ERα overexpression to inhibit LPS-induced apoptosis and promote cell survival were attenuated by applying LY294002 (PI3K inhibitor) and PI3K siRNA. These findings suggest that E2, BSA-E2 and ERα expression exert their cardioprotective effects by inhibiting JNK1/2-mediated LPS-induced TNF-α expression and cardiomyocyte apoptosis through activation of Akt.
Keywords: myocardial cell apoptosis, 17β-estradiol, estrogen receptor-α, PI3K/Akt, NFκB, IκB, JNK, lipopolysacchride
Introduction
Heart failure is a worldwide health problem. Heart diseases, such as pathologic hypertrophy, dilated cardiomyopathy and coronary artery disease, eventually lead to heart failure. Accumulating evidence suggests that apoptosis in cardiac myocytes contributes to the progression of heart failure [1]. Cardiomyocytes undergoing apoptosis have been found in a variety of pathological conditions, such as dilated cardiomyopathy, ischaemia/reperfusion injury, myocardial infarction and end-stage heart failure [2].
Studies have showed that death-receptor–induced apoptotic responses are involved in the pathogenesis of cardiac diseases [3]. The death-receptor–mediated apoptotic pathway is triggered by the binding of death-receptor ligands, such as tumour necrosis factor-α (TNF-α), to membrane-bound death receptors, such as TNF-α receptor 1 (TNF-R1). The interaction subsequently leads to activation of caspase-8. Activated caspase-8 then cleaves Bid into t-Bid, which promotes instability of the outer mitochondrial membrane, resulting in the release of cytochrome c into the cytosol. Once in the cytosol, cytochrome c activates caspase-9, which then further cleaves and activates caspase-3, causing the induction of apoptosis [4].
Lipopolisaccharide (LPS) from gram-negative bacteria stimulates an inflammatory response by inducing the up-regulation and release of cytokines [5]. Many studies have suggested that LPS contributes to cardiovascular collapse. Administration of LPS has been demonstrated to mediate vascular inflammation in atherosclerosis [6]. Furthermore, studies have demonstrated that LPS directly decreases contractility [7] and dramatically induces TNF-α expression [8] in cardiomyocytes through binding to its specific toll-like receptor-4 (TLR-4). Clinical studies have shown that severe and potentially fatal hypotension and cardiac contractile dysfunction are common symptoms in patients with sepsis [9].
It is well established today that the prevalence of cardiovascular diseases is lower in pre-menopausal women than in age-matched men [9] and that estrogen-replacement therapy contributes to a low incidence of heart disease after menopause [10]. Clinical studies of patients with sepsis have revealed that the mortality rate and the levels of TNF-α are lower in women than in men [11]. In addition, it has been shown that estrogen replacement reduces both myocardial infarct size and ventricular arrhythmias induced by ischaemia/reperfusion in both female and male rabbits [12]. Furthermore, 17β-estradiol (E2) has been shown to reduce pathological cardiac hypertrophy and heart failure by up-regulating the PI3K–Akt signalling pathway and by suppressing the calcineurin-NFAT3 signalling pathway in female rats [13]. In the present study, we investigated whether E2 and/or ERα inhibits LPS-induced apoptosis in Dox-inducible Tet-On H9c2 myocardial cells and primary cardiomyocytes that overexpress ERα. We then identified the precise molecular and cellular mechanisms behind the cardioprotective effects of E2 and ERα. Our findings suggest that E2 and ERα can significantly protect myocardial H9c2 cells and cardiomyocytes from LPS-induced apoptosis. Moreover, we found that the PI3K–Akt pathway is required for E2 and ERα to block LPS-up-regulated JNK1/2 activation, IκB degradation, NFκB activation as well as to block LPS-induced increases in pro-apoptotic proteins, thereby inhibiting LPS-induced myocardial cell damage.
Materials and methods
Cells, antibodies, reagents and enzymes
Heart-derived H9c2 myocardial cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 100 μg/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, 1 mM HEPS buffer and 10% Clontech fetal bovine serum specially prepared for the Tet-On system. 17β-estradiol (E2), β-estradiol-17-hemisuccinate:BSA (BSA-E2), doxycycline (Dox), melatonin (Mel), lipopolysacchride (LPS) and TNF-α were purchased from Sigma (Sigma Chemical Co., St. Louis, MO). The PI3-K inhibitor LY294002, MEK1/2 inhibitor U0126, p38 MAPK inhibitor SB203680, JNK inhibitor SP600125 and ER antagonist ICI 182,780 (ICI) were purchased from TOCRIS (Ellisville, MO). 6-Amino-4-(4-phenoxyphenylethylamino) quinazoline (QNZ), NFκB activation inhibitor, and N-(R)-[2-(hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-L-naphthylalanyl-L-alanine amide (TAPI-0), and TNF-α converting enzyme (TACE) inhibitor were purchased from Peptides International, Louisville, KY. We used the following antibodies against phosphorylated Akt (p-Akt) and phosphorylated JNK1/2 (p-JNK1/2) (BioSource International, Inc., Camarillo, CA); Akt and NFκBp65 (BD Biosciences, San Jose, CA); ERα, TNF-α, IκBα, IκBβ, c-jun, JNK1/2, phosphorylated p38 (p-p38), p38, active caspase-3, active caspase-8, active caspase-9, β-TrCP/HOS (E3 ligase), tBid and Bax (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); phosphated IkBa (Ser32), phosphated NFkBp65 (Ser536), phosphated c-jun, TLR4 and cytochrome c (Cell Signaling Technology, Inc., Beverly, MA). α-tubulin (Lab Vision Corporation, Fremont, CA) was used as the loading control. Goat anti-mouse IgG antibody conjugated to horseradish peroxidase and goat anti-rabbit IgG horseradish peroxidase conjugate were purchased from Santa Cruz Biotechnology.
Establishment of the double-stable Tet-On/ERα H9c2 cell line (supplementary data 1)
The double-stable Tet-On/ERα H9c2 cell line, which grows well in the presence of both G418 and hygromycin, was established by plasmid transfection using the LipofectAMINE method. Briefly, the primary Tet-On H9c2 cell line was generated by transfecting H9c2 cells with 10 μg pTet-On (CLONTECH Laboratories, Inc., Worcester, MA), a regulator plasmid encoding the G418-resistant gene. The primary Tet-On H9c2 cells were then transfected with 10 μg of pTRE2/ERα plasmid encoding the hygromycin-resistant gene. Double-stable cells were selected with 700 μg/ml G418 and 100 μg/ml hygromycin, and further screened for ERα mRNA levels and protein expression by RT-PCR and Western blot using anti-ERα antibody.
Cardiomyocyte culture
Neonatal cardiomyocytes were isolated and cultured using the commercial Neonatal Cardiomyocyte Isolation System Kit according to manufacture’s directions (Cellutron Life Technology, Highland Park, NJ). Ventricular cardiomyocytes were isolated and cultured in DMEM containing 10% fetal bovine serum, 100 μg/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamine. After 3–4 days, cells were incubated in serum-free essential medium overnight before treatment with indicated agents.
Cotransfection of SiRNA duplexes and plasmids
Primary ventricular cardiomyocytes were seeded at a density of 2 to 3 × 105 cells in 60-mm culture dishes. Cotransfection of siRNAs and plasmids was carried out with DharmaFECT Duo transfection reagent (Dharmacon, Inc., Lafayette, CO) according to manufacture’s directions. Specific silencing was confirmed by at least three independent immunoblotting with cellular extracts 72 hrs after transfection.
Total RNA extraction and reverse transcription and PCR amplification
Total RNA was extracted using the Ultraspec RNA isolation System (Biotecx Laboratories, Houston, TX) according to directions supplied by the manufacturer. The RNA was quantified and checked for purity and condition by spectrophotometry at a wavelength of 260 nm. The extract integrity was assessed by 1.5% agarose gel electrophoresis and RNA was visualized by ethidium bromide staining.
An aliquot of total RNA (0.5 μg) was reverse transcribed using 0.5 μM oligo d(T) primers in a reaction solution (50 μl) containing 75 mM KCl, 50 mM Tris–HCl (pH 8.3), 3 mM MgCl2, 10 mM DTT, 10 U RNase inhibitor (Promega, Madison, WI), 0.8 mM total dNTPs and 200 U of Moloney murine leukemia virus reverse transcriptase (Promega). The RT product (2 μl) was diluted with the PCR buffer (50 mM KCl,10 mM Tris–HCl and 2 mM MgCl2) to a final volume of 50 μl, containing 0.5 μM dNTPs (final concentration, 0.8 mM) and 0.5 U of Taq DNA polymerase. PCR was initiated with a hot start (5 min. at 95°C); the samples were then subjected to 32 cycles at 95°C for 1 min., 58°C for 1 min. and 72°C for 2 min. The annealing temperature for the TLR4, CD14, IκBα and NFκBp65 primers was 58°C; the annealing temperature for the TNF-α and GAPDH primers was 54°C. This was followed by a final extension step at 72°C for 20 min. Primers were as follows: rat TLR4 forward primer CATGGCATTGTTCCTTTCCT, reverse primer CATGGAGCCTAATTCCCTGA; rat CD14 forward primer GTGGACACGGAAGC AAATCT, reverse primer CGAGATCAGTCCTTTCTCGC; rat IκBα forward primer AACCTGCAGCAGACTCCACT, reverse primer CTCCTCATCCTCACTCTCGG; rat NFκBp65 forward primer AAGATCAATGGCTACACGGG, reverse primer ATCTTGAGCTCGGCAGTGTT; rat TNF-α forward primer CCTCTTCTCATTCCT GCTCG, reverse primer GGTATGAAATGGCAAATCGG; rat GAPDH forward primer GGGTGTGAACCACGAGAAAT, reverse primer CCACAGTCTTCTGAGT GGCA (MDBio, Taipei, Taiwan).
Immunoblotting and immunoprecipitation
Cells were re-suspended in lysis buffer (50 mM Tris, pH 7.5; 0.5 M NaCl; 1.0 mM EDTA, pH 7.5; 10% glycerol; 1 mM BME; 1% IGEPAL-630 and a proteinase inhibitor cocktail [Roche Molecular Biochemicals, Mannheim, Germany]). Samples containing equal amounts of total protein (60 μg) were separated by 12% SDS-PAGE and transferred onto PVDF membranes (Millipore, Belford, MA). The membranes were then incubated with primary antibodies in the blocking buffer in an orbit shaker at 4°C overnight. Following incubation with the primary antibodies, membranes were incubated with horseradish peroxidase–linked secondary antibodies. For cytosolic cytochrome c separation, cells were suspended in a buffer (50 mM Tris, pH 7.5; 0.5 M NaCl; 1.0 mM EDTA, pH 8.0; 10% glycerol and proteinase inhibitor cocktail tablet (Roche) for 3 min. on ice, homogenized with 40 strokes in a Dounce homogenizer, and centrifuged at 12,000g for 15 min. The supernatant was the cytosol fraction, and the pellet was resolved in lysis buffer as the mitochondrial fraction.
Cells were re-suspended in RIPA buffer (50 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1% IGEPAL-630; 0.5% sodium deoxycholate; 0.1% SDS and proteinase inhibitor cocktail [Roche]). Samples containing equal amounts of protein (100 μg) were incubated overnight with 20 μl of protein G beads and 2 μg of primary antibodies at 4°C on a rotating wheel. Samples were centrifuged at 12,000g for 5 min. at 4°C. The precipitated protein G beads were washed five times with RIPA buffer and analyzed using immunoblotting.
Nuclear extraction
Cells were re-suspended with 200 μl ice-cold BUFFER-I (10 mM Hepes, pH 8.0; 1.5 mM MgCl2; 10 mM KCl; 1 mM dithiothreitol and proteinase inhibitor cocktail [Roche]) and incubated for 15 min. on ice to allow cells to swell, followed by adding 20 μl IGEPAL-CA630. The supernatant (cytoplasmic fraction) was carefully aspirated and the pellet was re-suspended with ice-cold BUFFER-II (20 mM Hepes, pH 8.0; 1.5 mM MgCl2; 25% glycerol; 420 mM NaCl; 0.2 mM EDTA; 1 mM dithiothreitol and proteinase inhibitor cocktail [Roche]) and vigorously vortexed. The supernatants (nuclear extracts) were stored at −80°C. The protein concentration was determined by the colorimetric assay (Bio-Rad, Richmond, CA, USA).
Electrophoretic mobility shift assay (EMSA)
EMSA was carried out with a commercially available LightShift Chemiluminescent EMSA kit (Pierce, Rockford, IL) according to the manufacturer’s protocol. The binding reactions were performed in a solution containing 15 μg nuclear extract protein, 10× binding buffer (100 mM Tris, 500 mM KCl, 10 mM dithiothreitol; pH 7.5), 1 μg poly(dI-dC) and 2 μM of biotin-labelled DNA. Biotin end-labelled DNA duplex of sequence 5′-AGTTGAGGGGACTTTCC CAGGC-3′ and unlabelled oligonucleotide sequences 3′-TCAACTCCCCTGAAA GGGTCCG-5′ containing a putative binding site for NFκB were used for EMSA. DNA–protein (NFκB) complexes were subjected to 6% native polyacrylamide gel in a 0.5× Tris borate–EDTA buffer at 100 V for 3 hrs and then transferred onto a positively charged nylon membrane (Hybond-N+) in 0.5× Tris borate–EDTA buffer at 100 V for 1 hr. Competition EMSA was also performed. The nuclear extracts obtained from the myocardial cells were incubated with a biotin-labelled NFκB probe, a 2× unlabelled NFκB probe, a 10× unlabelled NFκB probe or 2 μg of anti-NFκBp65 antibody raised against DNA-binding motif (Santa Cruz Biotechnology).
Immunofluorescence
Cells subjected to various treatments were subsequently fixed with 4% paraformaldehyde at room temperature for 30 min. Cells were permeabilized with 0.5% Triton X-100 for 10 min. at 4°C. Non-specific binding of the fixed cells was blocked with PBS containing 2% bovine serum albumin at 37°C for 30 min., followed by incubation with primary NFκBp65 antibody overnight at 4°C. After washing, cells were incubated with anti-rabbit FITC-conjugated antibody at 37°C for 1 hr. Cells were stained with FITC-conjugated antibody alone as a negative control. The fluorescence was visualized using a fluorescence microscope coupled with an image analysis system.
TUNEL assay
Detection of apoptosis using the TUNEL (TdT-mediated digoxigenin–dUTP nick-end labelling) method was carried out with a commercially available in situ apoptosis detection kit (Roche). TUNEL-positive cells were identified with a fluorescence microscope using an excitation wavelength in the range of 450–500 nm and a detection wavelength in the range of 515–565 nm (green). Three independent experiments were then averaged and statistically analyzed.
Results
LPS induces Tet-On/ERα H9c2 myocardial cell apoptosis
To identify the cytotoxic effect induced by LPS in myocardial cells, the inducible Tet-On/ERα H9c2 myocardial cells established in this study were incubated with LPS, a specific TLR4 agonist, and subjected to TUNEL analysis. After incubation with various concentrations of LPS (0–1000 ng/ml) for 24 hrs, we observed a significant dose-dependent reduction in cell viability. Few positively stained cells were noted in control cultures; however, in cultures treated with LPS (50–1000 ng/ml) for 24 hrs, there was a dose-dependent increase in the number of cells undergoing apoptosis (Fig. 1A). Immunoblotting assay was used to determine the expression levels of pro-apoptotic proteins, including TNF-α, active caspase-8, truncated Bid (tBid), released cytochrome c, active caspase-9 and active caspase-3 in Tet-On/ERα H9c2 myocardial cells after LPS (0–5 μg/ml) treatment. The results demonstrated that LPS dramatically induced the expression and activation of these pro-apoptotic proteins (Fig. 1B), suggesting that LPS indeed executed the apoptotic effects on myocardial cells by binding to TLR4.
Figure 1.
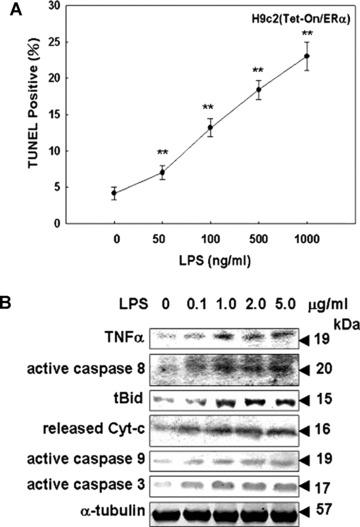
The apoptotic effects of LPS on Tet-On/ERα H9c2 cells. (A) Tet-On/ERα H9c2 cells were incubated for 24 hrs with the indicated concentrations of LPS (0–1000 ng/ml). LPS-induced apoptosis of Tet-On/ERαH9c2 cells were determined by TUNEL assay. (B) Tet-On/ERα H9c2 cells were treated with vehicle or LPS (0–5 μg/ml) for 24 hrs. Cell extracts were analyzed by immunoblotting with antibodies against proteins as indicated. *P < 0.05 versus control: **P < 0.01 versus control (mean ± S.E., n= 3).
E2, BSA-E2 and ERα overexpression inhibit LPS-induced nuclear localization of NFκB
We further tried to identify the association between IκB and NFκB following administrations of E2, BSA-E2 and/or Dox, which induces ERα overexpression, in the presence of LPS in myocardial cells. The immunoprecipitation assay showed that administrations of E2 (10−8 M), Dox (1 μg/ml), E2 (10−8 M) plus Dox (1 μg/ml) or BSA-E2 (E2= 10 ng/ml) enhanced the interaction between IκB and NFκB proteins in the presence of LPS (1 μg/ml), relative to the LPS-treated group (lane 2) (Fig. 2A). The amounts of NFκB in the nuclear and cytosolic fractions were further determined by immunoblotting analysis. Increased levels of NFκBp65 were observed in the nuclear fractions after LPS (1 μg/ml) treatment in myocardial cells (Fig. 2B and C). Nuclear translocation of NFκBp65 by LPS was further confirmed by immunofluorescence in myocardial cells (Fig. 2D). In contrast, treatments involving E2 (10−8 M), Dox (1 μg/ml), E2 (10−8 M) plus Dox (1 μg/ml) or BSA-E2 (E2= 10 ng/ml) significantly inhibited LPS-induced nuclear translocation of NFκBp65. However, the effect of E2, BSA-E2 and ERα overexpression on reducing the level of nuclear translocation of NFκB subunit p65 was blocked when cells were exposed to ICI 182 780 (0.5 μM) and melatonin (1 μM) (Fig. 2C and D). At the same time, DNA-binding activity of NFκB transcription factor was detected by the electrophoretic mobility shift assay (EMSA). Two apparent forms of NFκB were detected with EMSA analysis following LPS exposure for 24 hrs (lanes 2 and 3). However, LPS (1 μg/ml)-induced NFκB–DNA binding was significantly inhibited by administrations of E2 (10−8 M), Dox (1 μg/ml), E2 (10−8 M) plus Dox (1 μg/ml) and BSA-E2 (E2= 10 ng/ml) in myocardial cells. Competition EMSA assay for the formation of NFκB–DNA complexes was performed using 2× cold probes, 10× cold probes and 2 μg of anti-p65 antibody raised against p65 DNA-binding motif. Application of cold probes and p65 antibody significantly suppressed binding interaction between NFκB and biotin-labelled DNA (Fig. 2E).
Figure 2.
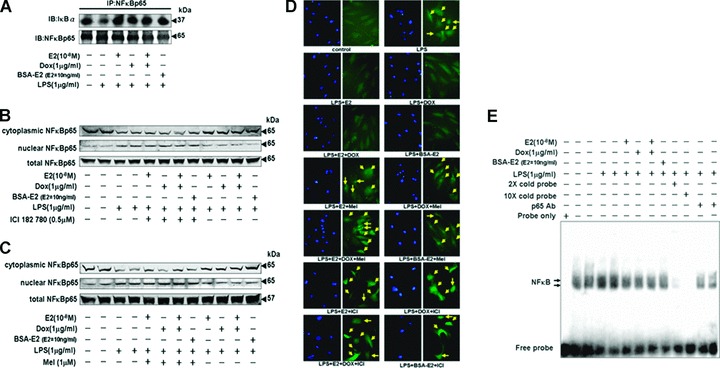
E2, BSA-E2 and ERα overexpression inhibited the LPS-induced nuclear localization of NFkB p65 through ERα. (A) Tet-On/ERα H9c2 cells were incubated with E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS (1 μg/ml) for 24 hrs. NFκBp65 was immunoprecipitated with anti-NFκBp65 antibody, subjected to immunoblotting assay with anti-IκBα and NFκBp65 antibodies. (B and C) Tet-On/ERα H9c2 cells were pre-incubated with ICI 182,780 (0.5 μM) or melatonin (1 μM) for 1 hr, followed by incubation with E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS (1 μg/ml) for 24 hrs. Cell lysates and nuclear extracts were prepared, and the levels of cytoplasmic p65, nuclear p65 and total p65 were determined by immunoblotting analysis. (D) Cells pre-incubated with ICI 182,780 (0.5 μM) or melatonin (1 μM) were treated with E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS (1 μg/ml) for 24 hrs. Cells were then fixed, and the immunofluorescence staining with antibody against p65 was performed and visualized with a fluorescence microscope coupled with an image analysis system. (E) The specific DNA-binding activity for nuclear NFκB was analyzed by EMSA using a probe corresponding to the NFκB-binding site. The arrows indicate the NFκB-DNA complexes. Competition EMSA was also performed using 2× cold probe, 10× cold probe or 2 μg of anti-p65 antibody raised against DNA-binding motif.
E2, BSA-E2 and ERα overexpression inhibit LPS-induced TNF-α expression and TNF-α-downstream proapoptotic protein activation
Tet-On/ERα H9c2 myocardial cells were administrated various concentrations of TNF-α (0, 1, 2 and 5 ng/ml) for 24 hrs and subjected to immunoblotting and TUNEL assays. The immunoblotting assay showed that TNF-α significantly activated caspase-3 in a dose-dependent manner. TNF-α–induced myocardial cell apoptosis was confirmed by the TUNEL assay (Fig. 3A). NFκB activation inhibitor (QNZ) and TNF-α–converting enzyme (TACE) inhibitor (TAPI-0) suppressed the LPS-triggered signalling pathway, and completely inhibited LPS-activated caspase-3 in myocardial cells (Fig. 3B). We further examined whether E2 and ERα overexpression suppress LPS-induced TNF-α expression and TNF-α–induced expression of pro-apoptotic proteins. The results from RT-PCR and immunoblotting assays revealed that administrations of E2 (10−8 M), Dox (1 μg/ml), E2 (10−8 M) plus Dox (1 μg/ml) or BSA-E2 (E2= 10 ng/ml) significantly inhibited LPS-up-regulated TNF-α expression (Fig. 3C), and hence inhibited TNF-α–induced expression of pro-apoptotic proteins such as active caspase-8, tBid, Bax, released cytochrome c, active caspase-9 and active caspase-3 (Fig. 3D).
Figure 3.
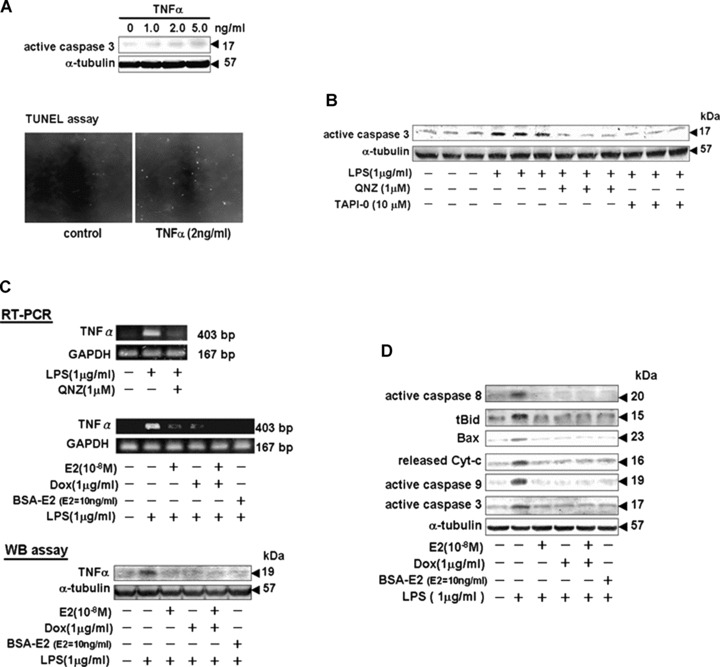
E2, BSA-E2 and ERα overexpression inhibited LPS-induced TNF-α expression and TNF-α-downstream pro-apoptotic protein activation. (A) Tet-On/ERα H9c2 cells were cultured with TNF-α (0–5 ng/ml) for 24 hrs. Cells were harvested and detected by immunoblotting using a specific antibody against active caspase 3. Percentage of myocardial cell apoptosis by TNF-α (2 ng/ml) treatment was determined by the TUNEL assay. (B) Myocardial cells pre-incubated with 6-Amino-4-(4-phenoxyphenylethylamino) quinazoline (QNZ) and N-(R)-[2-(Hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-L-naphthylalanyl- L-alanine Amide (TAPI-0) were treated with LPS (1 μg/ml) for 24 hrs and harvested for immunoblotting analysis with a specific anti-active caspase-3 antibody. (C) After indicated treatments, cells were harvested and lysed for RT-PCR and immunoblotting analysis. RT-PCR was performed with the indicated primers of TNF-α and GAPDH. Immunoblotting was performed using a TNF-α antibody. Equal loading was assessed with an anti-α-tubulin antibody. (D) Cells were incubated with or without E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS (1 μg/ml) for 24 hrs. Pro-apoptotic proteins including tBid, Bad, released cytochrome c, active caspase-8, -3 and -9 were detected by immunoblotting using specific antibody.
Selective JNK1/2 inhibitor SP600125 inhibits LPS-induced IκBα degradation and TNF-α expression in myocardial cells
We assessed the suppressive effects of inhibitors of p38 MAPK (SB203580), JNK1/2 (SP600125) and ERK1/2 (U0126) on LPS-induced phosphorylation and degradation of IκBα in myocardial cells. Tet-On/ERα H9c2 cells were pre-treated with inhibitors SB203580, SP600125 or U0126 for 1 hr prior to the administration of LPS for 1 hr or 24 hrs, and subsequently subjected to immunoblotting assay. The findings revealed that pre-treatment of the JNK1/2 inhibitor SP600125 significantly inhibited LPS-induced increases in phosphorylation and degradation of IκBα. SP600125 also specifically inhibited the expression of phosphorylated c-jun, the downstream target of LPS-activated JNK1/2 in myocardial cells (Fig. 4A and B). Furthermore, we observed that JNK1/2 mediated the expression of TNF-α in myocardial cells that had been pre-treated with SP600125 for 1 hr, followed by incubation with LPS for 3 or 15 hrs (Fig. 4C). The result suggests that JNK1/2 may mediate LPS-induced phosphorylation and degradation of IκBα, and the subsequent activation of NFκB to modulate TNF-α expression in myocardial cells.
Figure 4.
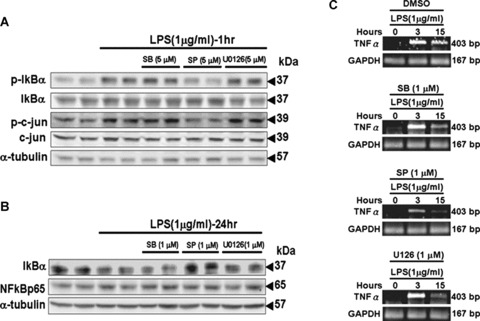
Selective JNK inhibitor SP600125 blocked LPS-induced IκBα degradation and TNF-α expression in myocardial cells. (A and B) Tet-On/ERα H9c2 cells were pre-incubated with the selective p38 MAPK inhibitor SB203680, the selective JNK inhibitor SP600125 and the MEK1/2 inhibitor U0126 for 30 min., followed by LPS (1 μg/ml) treatment for 1 hr and 24 hrs. Cells were harvested, and immunoblotting analysis was performed with antibodies against proteins as indicated. (C) Cells pre-incubated with SB203680 (1 μM), SP600125 (1 μM) or U0126 (1 μM) were treated with LPS (1 μg/ml) for 0–15 hrs. Cells were harvested and lysed at the indicated time points for RT-PCR analysis to detect TNF-α expression.
Activation of the PI3K–Akt pathway suppresses JNK1/2 activity, allowing for E2, BSA-E2 and ERα to inhibit LPS-induced IκBα degradation and nuclear localization of NFκB
To examine whether E2 and/or ERα inhibit LPS-induced IκB degradation and nuclear localization of NFκB by suppressing JNK1/2 activity, we detected the level of activation/phosphorylation of JNK1/2 in Tet-On/ERα H9c2 myocardial cells that had been administrated E2 (10−8 M), BSA-E2 (E2= 10 ng/ml) or Dox (1 μ/ml) in the presence of LPS (1 μg/ml) for 30 min. We observed that JNK1/2 activity was significantly inhibited by E2, BSA-E2 and ERα. However, the reduction in JNK1/2 activity was reversed by LY294002 (PI3K inhibitor) treatment (Fig. 5A–C). Several studies have shown that β-tranducin repeats containing protein (β-TrCP) mediates ubiquitination/proteasome-dependent degradation of IκB [14]; thus, we further examined the association between IκB and β-TrCP in myocardial cells (Fig. 5D). The results of the immunoprecipitation assay showed that administrations of E2 (10−8 M), Dox (1 μg/ml), E2 (10−8 M) plus Dox (1 μg/ml) or BSA-E2 (E2= 10 ng/ml) inhibited the interaction between IκB and β-TrCP proteins, compared with the LPS-treated group (lanes 3 and 4).
Figure 5.
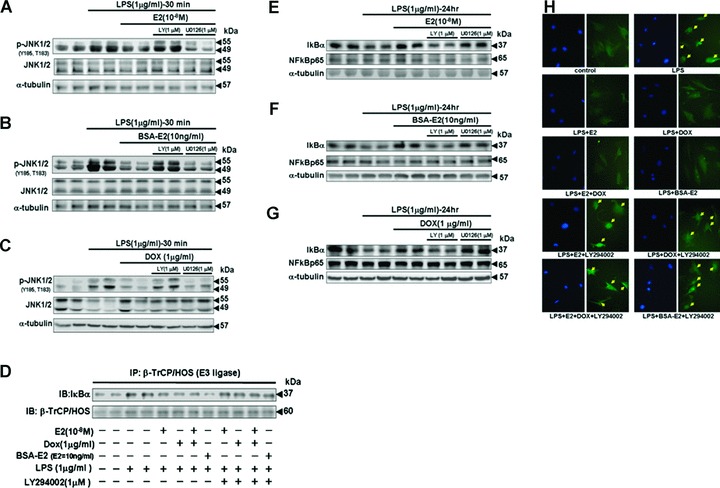
The activity of the PI3K–Akt pathway was required for E2, BSA-E2 and ERα overexpression to inhibit LPS-induced JNK activity and subsequent IκBα degradation and nuclear localization of NFκB p65. (A, B and C) Tet-On/ERα H9c2 cells were pre-incubated with LY294002 (1 μM), PI3-K inhibitor or U0126 (1 μM), MEK1/2 inhibitor for 30 min., followed by treatment with E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS (1 μg/ml) for 30 min. Immunoblotting analysis was performed with antibodies against phosphorylated JNK1/2 (Y185, T183) and JNK1/2. (D) Cells were pre-incubated with LY294002 (1 μM) for 30 min., followed by incubation with E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS for 24 hrs. β-TrCP/HOS was immunoprecipitated with anti-β-TrCP/HOS antibody, subjected to immunoblotting analysis with anti-IκBα and anti-β-TrCP/HOS antibodies. (E, F and G) Cells were pre-incubated with LY294002 (1 μM) or U0126 (1 μM) for 30 min., followed by treatment with E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS (1 μg/ml) for 24 hrs. Cell protein extract was detected by immunoblotting using antibodies against IκBα and NFκB p65 antibodies. (H) Cells were pre-incubated with LY294002 (1 μM) 30 min. prior to the treatments involving E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2), followed by LPS (1 μg/ml) for 24 hrs. Cells were then fixed. Immunofluorescence staining with antibody against p65 was performed and visualized with a fluorescence microscope coupled with an image-analysis system.
However, LY294002 treatment suppressed the protective effects of E2 and ERα, resulting in formation of the IκB-β-TrCP complex (Fig. 5D), degradation of IκBα (Fig. 5E–G) and nuclear localization of NFκB (Fig. 5H). These findings suggested that E2 and ERα inhibit LPS-induced IκBα degradation and nuclear localization of NFκB by suppressing JNK1/2 activity in the PI3K–Akt pathway.
PI3K SiRNA inhibits the cardioprotective effects of E2, ERα and BSA-E2 in LPS-treated Tet-On/ERα H9c2 myocardial cells
To confirm whether Akt is involved in E2- and ERα-mediated cardioprotective mechanisms, we applied small interfering RNA (siRNA)-mediated specific knockdown of PI3-kinase proteins in order to inhibit the activation of downstream Akt. SiGLO siRNA, a non-functional and non-targeting siRNA, was used as the control siRNA. Tet-On/ERα H9c2 myocardial cells were pre-transfected with PI3K siRNA or siGLO siRNA for 36 hrs, followed by treatment with E2 (10−8 M), Dox (1 μg/ml), E2 (10−8 M) plus Dox (1 μg/ml) or BSA-E2 (E2= 10 ng/ml) in the presence of LPS (1 μg/ml) for 24 hrs. The results of our immunoblotting assay showed that PI3K siRNA blocked the cardioprotective properties of E2, ERα overexpression and BSA-E2 by promoting the expression of TNF-α and the activation of caspase-3 in Tet-On/ERα H9c2 myocardial cells (Fig. 6A). The results of the TUNEL assay further confirmed that E2, ERα overexpression or BSA-E2 could inhibit LPS-induced apoptosis in myocardial cells, and that this inhibition could be reversed by PI3K siRNA (Fig. 6B). These findings demonstrate that E2, ERα overexpression and membrane-impermeable BSA-E2 inhibited LPS-induced cell apoptosis and promoted cell survival through the PI3K–Akt signalling pathway.
Figure 6.
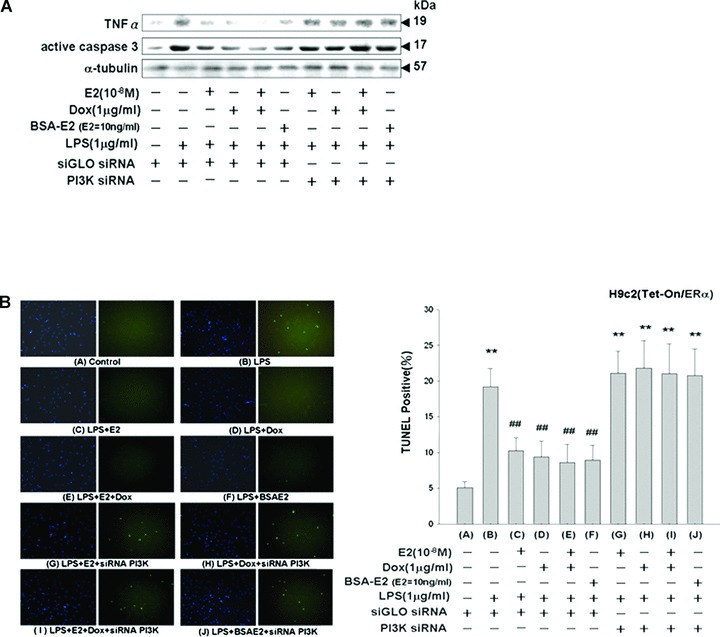
Inhibition of the cardioprotective effects of E2, ERα and BSA-E2 on LPS-treated Tet-On/ERα H9c2 myocardial cells by PI3K siRNA treatment. (A) Thirty-six hours after being transfected with siRNA (100 nM siGLO siRNA or PI3K siRNA), Tet-On/ERα H9c2 cells were incubated with vehicle, E2 (10−8 M), Dox (1 μg/ml) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS for 24 hrs. Cells were harvested and analyzed by immunoblotting using antibodies against TNF-α and active caspase 3. (B) TUNEL staining was performed according to the manufacturer’s protocol PI3K siRNA abolished the reduction of apoptosis induced by E2, ERα and BSA-E2 in LPS-treated cells. **P < 0.01 versus control (lane 1); ##P < 0.01 versus LPS treatment only (lane 2) (mean ± S.E., n= 3).
PI3K SiRNA inhibits the cardioprotective effects of E2, ERα and BSA-E2 in LPS-treated primary cardiomyocytes
We further confirm the functional role of Akt in mediating the protective effects of E2 and ERα on primary ventricle cardiomyocytes. Seventy-two hours after siRNA transfection, ventricle cardiomyocytes were harvested and levels of PI3K were assessed using immunoblotting analysis with PI3K-specific antibody. Treatment with siRNAs significantly suppressed PI3K expression and blocked downstream activation of Akt in the presence of E2 (10−8 M) (supplementary data 4). Thirty-six hours after cotransfection of ERα-encoding plasmid and PI3K siRNA, we treated the transfected cardiomyocytes with E2 or BSA-E2 in the presence of LPS (1 μg/ml) for 24 hrs. Loss of Akt activation by PI3K siRNA significantly suppressed the inhibitory effects of E2, ERα and BSA-E2 on LPS-induced TNF-α expression, caspase-3 activation and ventricle cardiomyocyte apoptosis (Fig. 7A–C).
Figure 7.
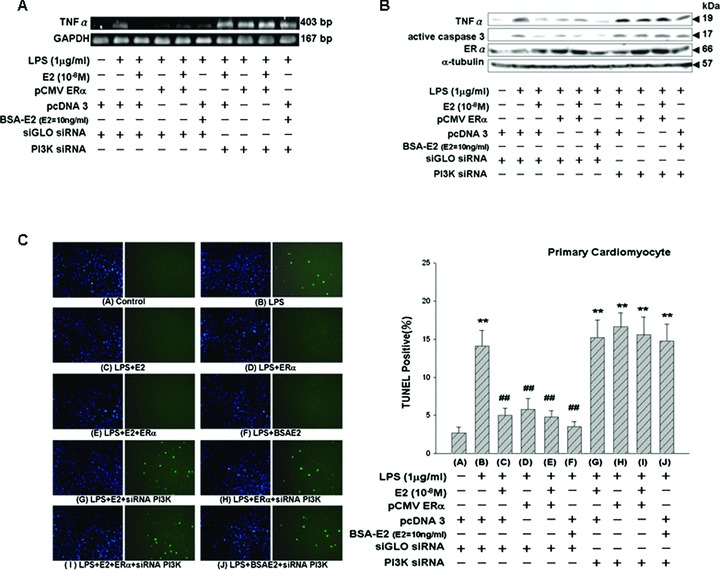
Inhibition of the cardioprotective effects of E2, ERα and BSA-E2 on LPS-treated ventricular cardiomyocytes by PI3K siRNA treatment. (A and B) Thirty-six hours after being cotransfected with plasmids (3.5 μg ERα or pcDNA3) and siRNA (100 nM siGLO siRNA or PI3K siRNA), ventricular cardiomyocytes were incubated with vehicle, E2 (10−8 M) or BSA-E2 (equivalent to 10 ng/ml free E2) in the presence of LPS for 24 hrs. Cells were harvested and analyzed by RT-PCR to detect TNF-α expression and by immunoblotting using antibodies against TNF-α, ERα and active caspase 3. (C) TUNEL staining of the ventricular cardiomyocytes was performed according to the manufacturer’s protocol PI3K siRNA abolished the reduction of apoptosis induced by E2, ERα and BSA-E2 in LPS-treated ventricular cardiomyocytes. **P < 0.01 versus control (lane 1); ##P < 0.01 versus LPS treatment only (lane 2) (mean ± S.E., n= 3).
Discussion
In this study, we found that LPS directly induces myocardial cell apoptosis via the TNF-α death-receptor–dependent (TNF-α, active caspases-8 and -3) pathway and by activating t-Bid to further induce mitochondria-dependent (Bax, released cytochrome c and active caspases-9 and -3) apoptotic pathways. Administrations of E2, BSA-E2 and/or Dox, which induces ERα overexpression, significantly inhibited LPS-induced phosphorylation of IκB, degradation of IκB, phosphorylation/activation of NFκB, nuclear translocation of NFκB and DNA-binding activity of NFκB. We also found that E2, BSA-E2 and ERα overexpression exerted cardioprotective properties by dramatically suppressing LPS-up-regulated TNF-α, active caspases-8, t-Bid, Bax, released cytochrome c, active caspase-9, active caspase-3 and apoptosis of myocardial cells. We also found that incubation of LPS-treated myocardial cells with the JNK1/2 inhibitor SP600125 resulted in marked inhibition of LPS-induced phosphorylation of IκB, degradation of IκB and up-regulation of TNF-α. This suggests that JNK1/2 may be the key mediator of the downstream signal when LPS binds to TLR-4, thereby modulating LPS-induced degradation of IκB, up-regulation of TNF-α and subsequent myocardial apoptotic responses. These findings suggest a novel role for JNK1/2 in LPS-induced apoptotic responses of myocardial cells, particularly in patients with sepsis. Finally, our results show that E2, BSA-E2 and ERα inhibit LPS-induced IκB degradation and nuclear translocation of NFκB by suppressing JNK1/2 activity in the PI3K–Akt pathway. Activated Akt further contributes to the cardioprotective effects of E2, BSA-E2 and ERα by inhibiting LPS-induced TNF-α and active caspase-3 expression as well as apoptosis in myocardial cells (Fig. 8).
Figure 8.
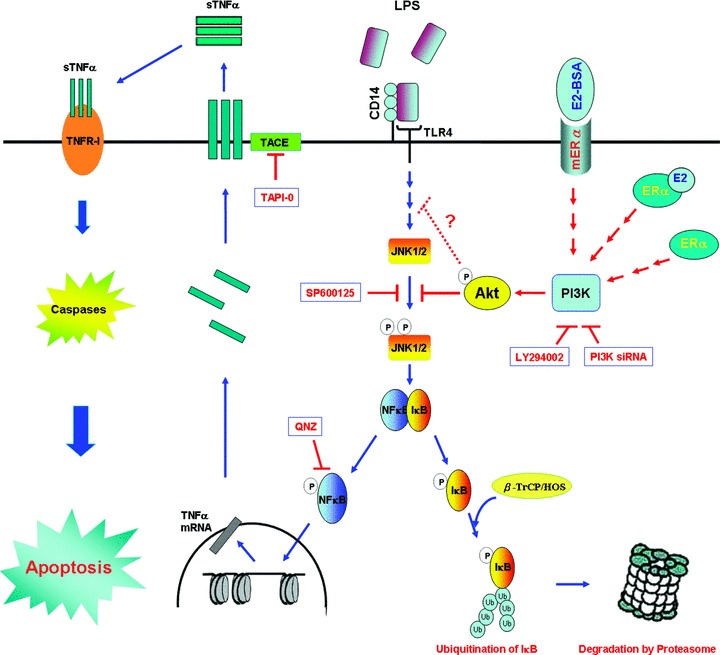
A schematic representation showing how E2, BSA-E2 and ERα expression inhibit JNK1/2-mediated LPS-induced TNF-α expression and cardiomyocyte apoptosis through activation of Akt. Activation of toll-like receptor 4 (TLR4) by specific LPS binding induces JNK1/2 activation, which in turn leads to the activation of transcription factor NFκB. Therefore, phosphated IκB marked with a polyubiquitin chain by β-TrCP/HOS (E3, an ubiquitin ligase) is targeted for proteasomal degradation. After transcription and translation, trimeric TNF-α is inserted into the cell membrane. TNF-α converting enzyme (TACE) cleaves the pro-form of TNF-α off the cell membrane, which yields a soluble form of TNF-α (sTNF-α) that then binds to TNF receptor-I (TNFR-I) through the autocrine loop to activate downstream caspases, thereby executing cardiomyocyte apoptosis. E2, acting through cytosolic ERα or membrane ERα, and ERα overexpression alone inside the cells block LPS-induced JNK1/2 activation by activating the PI3K–Akt pathway. This blockade helps to maintain the association between IκB and NFκB and to inhibit nuclear localization of NFκB, thereby inhibiting cardiomyocyte apoptosis.
Evidence suggests that plasma concentrations of LPS are raised in patients with chronic heart failure (CHF) and an activated immune system [15]. LPS is a potent stimulator of proinflammatory cytokine (PIC) production in cardiomyocytes [16]. The elevation of circulating PICs, such as IL-1 [17] and IL-6 [18], is associated with worsening of symptoms and even mortality [19] in patients with CHF. PICs have been shown to cause cardiac dysfunction by suppressing myocardial contractility [20]. In cardiomyocytes, TLR-4 specifically recognizes LPS [21]. Studies have suggested that TLR-4–mediated cytokine expression plays a role in immune-mediated progression of atherosclerosis [22], in heart dysfunction during bacterial sepsis [23] and in myocardial ischaemia/reperfusion (I/R) damage [24]. Our findings suggest that LPS-induced IκB degradation and NFκB activation in myocardial cells contributes to cardiac dysfunction and/or poor prognosis in patients with sepsis by inducing apoptotic responses via the TNF-α death-receptor–dependent (TNF-α, active caspases-8 and -3) and mitochondria-dependent (tBid, Bax, released cytochrome c and active caspases-9 and -3) apoptotic pathways.
An accumulating body of evidence shows that stimuli of cardiomyocyte apoptosis, such as hypoxia, oxidative stress and ischaemia/reperfusion damage result in the activation of protein kinase cascades, which in turn activate MAPKs such as p38 MAPK, JNK1/2 and ERK1/2 [25]. These serine-threonine kinases have been showed to phosphorylate important downstream mediators that participate in cardiac pathologies, such as heart failure, hypertensive cardiac hypertrophy and cardiomyopathies [25]. Therefore, in the present study we used SB203580 (p38 MAPK inhibitor), SP600125 (JNK1/2 inhibitor) and U0126 (ERK1/2 inhibitor) in order to further identify the roles that p38 MAPK, JNK1/2 and ERK1/2 play in LPS-induced apoptosis in myocardial cells. We observed that SP600125 significantly inhibited LPS-induced phosphorylation of IκB, degradation of IκB and up-regulation of TNF-α. However, pre-treatment with SB203580 or U0126 showed no suppressive effects on the above modulations. These findings suggest that JNK1/2 modulates LPS-induced apoptotic responses in myocardial cells by mediating the downstream signal when LPS binds TLR-4.
Studies have shown that the incidence of sepsis or multi-organ dysfunction after trauma and burns is lower among women than among men [26, 27]. In a clinical study involving patients with sepsis, Schroder et al. reported that the mortality rate and the levels of TNF-α were lower in women than in men [11]. After burn trauma injury, women tend to have lower levels of cytokine production, lower myocardial inflammatory responses and better myocardial function [28]. Deshpande et al. showed that estradiol inhibited LPS-induced NFκB-binding activity and TNF-α expression in macrophages in an animal model [29]. In addition, the cardiovascular protective effects of estrogen administration in vivo have been shown in a left anterior descending (LAD) coronary artery ischaemia/reperfusion model [30], in a coronary artery occlusion model [12] and in an abdominal aorta coarctation model [13]. In the present study, we observed that administration of E2, BSA-E2 and/or Dox, which induces ERα overexpression, significantly provided cardioprotective effects by suppressing LPS-induced IκB degradation, NFκB phosphorylation/activation, nuclear translocation of NFκB, DNA-binding activity of NFκB and subsequent apoptotic responses, including increases in pro-apoptotic proteins (e.g. TNF-α, active caspase-8, t-Bid, Bax, released cytochrome c, active caspase-9 and active caspase-3) and TUNEL-positive cells in myocardial cells.
Studies have indicated that the PI3K–Akt pathway is involved in the anti-apoptotic effects of certain stimuli and that it plays a central role in cellular survival in many different cell types [31]. Activation of Akt by growth factor is known to mediate the anti-apoptotic effects and to contribute to the cardioprotection and cell survival [32]. Camper-Kirby et al. found that females have higher nuclear localization of phosphor-Akt and higher Akt activity in myocardium [33]. Akt activation by 17β-estradiol has been shown by Wu et al. to reduce pathological cardiac hypertrophy and heart failure in an abdominal aorta coarctation model [13]. In the results of this study, we observed that Akt deactivation by LY294002 (PI3K inhibitor) and PI3K siRNA blocked E2-, BSA-E2- and/or ERα-mediated cardioprotective properties during LPS stimulation. Akt activation was required for E2, BSA-E2 and/or ERα to significantly inhibit LPS-induced JNK1/2 phosphorylation/activation, which then suppresses LPS-increased the association of IκB with β-TrCP proteins, IκB degradation, and nuclear translocation of NFκB in myocardial cells. In the detection of myocardial apoptosis, loss of Akt activation by PI3K siRNA significantly reversed E2-, BSA-E2- and ERα-mediated cardioprotective effects by increasing TNF-α expression, caspase-3 activation and TUNEL-positive myocardial cells during LPS treatment. Furthermore, we found that ERα overexpression alone significantly contributed to the cardioprotective effects against LPS challenge via PI3K–Akt pathway. This finding is similar to that in Fowler’s work in which ERα overexpression alone promoted cellular responses in MCF-7 cells, such as increasing transcription activity, cell proliferation and cell growth in the absence of E2[34]. We speculate that ERα overexpression may stabilize receptors in an active conformation, efficiently increasing the association with activators, thereby hindering repressors from blocking ERα activation by mass action effects in myocardial cells.
The incidence and mortality of cardiovascular disease are very low in pre-menopausal women but significantly higher in menopausal women, suggesting estrogen may act as a cardiovascular system protector. However, results from the Women’s Health Initiative suggest that estrogen therapy used in post-menopausal women has no beneficial impact against the development of cardiomyopathy such as arteriosclerotic coronary artery disease and myocardial infarction [35]. The controversy may be explained by the findings that estrogen prevents the development of early atherosclerotic lesions by reducing lipid deposits when hormone therapy is initiated in the perimenopausal period or before the development of atherosclerosis; however, estrogen can promote MMP expression, inflammatory activation and plaque instability once atherosclerosis has been established [36]. However, these previous studies mainly focussed on vasculature and differ from the processes being explored in our study. In the present study, we investigated the cardioprotective effects and mechanisms provided by E2 and ERα in myocardial cells exposed to LPS. The present study clearly shows that the cardioprotective effects and mechanisms of E2 and ERα involve the inhibition of LPS-induced TNF-α death-receptor–dependent and mitochondria-dependent apoptotic responses in myocardial cells. These findings may explain why women have lower rates of mortality and heart dysfunction and less acute inflammatory responses than men, particularly in those with sepsis.
In conclusion, we found that LPS up-regulates the expression of pro-apoptotic proteins such as TNF-α, active caspases-8, t-Bid, Bax, released cytochrome c, active caspase-9 and active caspase-3 through the TLR-4–JNK1/2–NFκB pathway, leading to large-scale apoptosis in myocardial cells. In addition, using Dox-induced Tet-On H9c2 myocardial cells and ERα-transfected cardiomyocytes to overexpress ERα, we observed that activation of the PI3K–Akt signalling pathway is required for E2, BSA-E2 and/or ERα to significantly abolish the LPS-induced JNK1/2 activation, IκB degradation, NFκB activation and subsequent increases in pro-apoptotic proteins and apoptosis in myocardial cells.
Acknowledgments
We are grateful to Dr. Benita S. Katzenellenbogen in University of Illinois at Urbana, USA, for providing the pCMV-ERs.
References
- 1.Mani K, Kitsis RN. Myocyte apoptosis: programming ventricular remodeling. J Am Coll Cardiol. 2003;41:761–4. doi: 10.1016/s0735-1097(02)02958-3. [DOI] [PubMed] [Google Scholar]
- 2.Narula J, Kolodgie FD, Virmani R. Apoptosis and cardiomyopathy. Curr Opin Cardiol. 2000;15:183–8. doi: 10.1097/00001573-200005000-00011. [DOI] [PubMed] [Google Scholar]
- 3.van Empel VP, Bertrand AT, Hofstra L, et al. Myocyte apoptosis in heart failure. Cardiovasc Res. 2005;67:21–9. doi: 10.1016/j.cardiores.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–77. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 5.Yang RB, Mark MR, Gray A, et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature. 1998;395:284–8. doi: 10.1038/26239. [DOI] [PubMed] [Google Scholar]
- 6.Stoll LL, Denning GM, Weintraub NL. Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:2227–36. doi: 10.1161/01.ATV.0000147534.69062.dc. [DOI] [PubMed] [Google Scholar]
- 7.Boyd JH, Mathur S, Wang Y, et al. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res. 2006;72:384–93. doi: 10.1016/j.cardiores.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998;274:R577–95. doi: 10.1152/ajpregu.1998.274.3.R577. [DOI] [PubMed] [Google Scholar]
- 9.Baker L, Meldrum KK, Wang M, et al. The role of estrogen in cardiovascular disease. J Surg Res. 2003;115:325–44. doi: 10.1016/s0022-4804(03)00215-4. [DOI] [PubMed] [Google Scholar]
- 10.Ylikorkala O. HRT as secondary prevention of cardiovascular disease. Maturitas. 2004;47:315–8. doi: 10.1016/j.maturitas.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 11.Schroder J, Kahlke V, Staubach KH, et al. Gender differences in human sepsis. Arch Surg. 1998;133:1200–5. doi: 10.1001/archsurg.133.11.1200. [DOI] [PubMed] [Google Scholar]
- 12.Hale SL, Birnbaum Y, Kloner RA. Estradiol, administered acutely, protects ischemic myocardium in both female and male rabbits. J Cardiovasc Pharmacol Ther. 1997;2:47–52. doi: 10.1177/107424849700200106. [DOI] [PubMed] [Google Scholar]
- 13.Wu CH, Liu JY, Wu JP, et al. 17beta-estradiol reduces cardiac hypertrophy mediated through the up-regulation of PI3K/Akt and the suppression of calcineurin/NF-AT3 signaling pathways in rats. Life Sci. 2005;78:347–56. doi: 10.1016/j.lfs.2005.04.077. [DOI] [PubMed] [Google Scholar]
- 14.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 15.Niebauer J, Volk HD, Kemp M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353:1838–42. doi: 10.1016/S0140-6736(98)09286-1. [DOI] [PubMed] [Google Scholar]
- 16.Wagner DR, Combes A, McTiernan C, et al. Adenosine inhibits lipopolysaccharide-induced cardiac expression of tumor necrosis factor-alpha. Circ Res. 1998;82:47–56. doi: 10.1161/01.res.82.1.47. [DOI] [PubMed] [Google Scholar]
- 17.Blum A, Sclarovsky S, Rehavia E, et al. Levels of T-lymphocyte subpopulations, interleukin-1 beta, and soluble interleukin-2 receptor in acute myocardial infarction. Am Heart J. 1994;127:1226–30. doi: 10.1016/0002-8703(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 18.Anker SD, von Haehling S. Inflammatory mediators in chronic heart failure: an overview. Heart. 2004;90:464–70. doi: 10.1136/hrt.2002.007005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rauchhaus M, Doehner W, Francis DP, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102:3060–7. doi: 10.1161/01.cir.102.25.3060. [DOI] [PubMed] [Google Scholar]
- 20.Kumar A, Haery C, Parrillo JE. Myocardial dysfunction in septic shock. Crit Care Clin. 2000;16:251–87. doi: 10.1016/s0749-0704(05)70110-x. [DOI] [PubMed] [Google Scholar]
- 21.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 22.Methe H, Kim JO, Kofler S, et al. Expansion of circulating toll-like receptor 4-positive monocytes in patients with acute coronary syndrome. Circulation. 2005;111:2654–61. doi: 10.1161/CIRCULATIONAHA.104.498865. [DOI] [PubMed] [Google Scholar]
- 23.Baumgarten G, Knuefermann P, Nozaki N, et al. In vivo expression of proinflammatory mediators in the adult heart after endotoxin administration: the role of toll-like receptor-4. J Infect Dis. 2001;183:1617–24. doi: 10.1086/320712. [DOI] [PubMed] [Google Scholar]
- 24.Li C, Ha T, Kelley J, et al. Modulating toll-like receptor mediated signaling by (1–>3)-beta-D-glucan rapidly induces cardioprotection. Cardiovasc Res. 2004;61:538–47. doi: 10.1016/j.cardiores.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Baines CP, Molkentin JD. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38:47–62. doi: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Oberholzer A, Keel M, Zellweger R, et al. Incidence of septic complications and multiple organ failure in severely injured patients is sex specific. J Trauma. 2000;48:932–7. doi: 10.1097/00005373-200005000-00019. [DOI] [PubMed] [Google Scholar]
- 27.Cumming J, Purdue GF, Hunt JL, et al. Objective estimates of the incidence and consequences of multiple organ dysfunction and sepsis after burn trauma. J Trauma. 2001;50:510–5. doi: 10.1097/00005373-200103000-00016. [DOI] [PubMed] [Google Scholar]
- 28.Horton JW, White DJ, Maass DL. Gender-related differences in myocardial inflammatory and contractile responses to major burn trauma. Am J Physiol Heart Circ Physiol. 2004;286:H202–13. doi: 10.1152/ajpheart.00706.2003. [DOI] [PubMed] [Google Scholar]
- 29.Deshpande R, Khalili H, Pergolizzi RG, et al. Estradiol down-regulates LPS-induced cytokine production and NFkB activation in murine macrophages. Am J Reprod Immunol. 1997;38:46–54. doi: 10.1111/j.1600-0897.1997.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 30.Delyani JA, Murohara T, Nossuli TO, et al. Protection from myocardial reperfusion injury by acute administration of 17 beta-estradiol. J Mol Cell Cardiol. 1996;28:1001–8. doi: 10.1006/jmcc.1996.0093. [DOI] [PubMed] [Google Scholar]
- 31.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 32.Chao W, Matsui T, Novikov MS, et al. Strategic advantages of insulin-like growth factor-I expression for cardioprotection. J Gene Med. 2003;5:277–86. doi: 10.1002/jgm.347. [DOI] [PubMed] [Google Scholar]
- 33.Camper-Kirby D, Welch S, Walker A, et al. Myocardial Akt activation and gender: increased nuclear activity in females versus males. Circ Res. 2001;88:1020–7. doi: 10.1161/hh1001.090858. [DOI] [PubMed] [Google Scholar]
- 34.Fowler AM, Solodin N, Preisler-Mashek MT, et al. Increases in estrogen receptor-alpha concentration in breast cancer cells promote serine 118/104/106-independent AF-1 transactivation and growth in the absence of estrogen. FASEB J. 2004;18:81–93. doi: 10.1096/fj.03-0038com. [DOI] [PubMed] [Google Scholar]
- 35.Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women’s health initiative randomized controlled trial. JAMA. 2002;288:321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 36.Ouyang P, Michos ED, Karas RH. Hormone replacement therapy and the cardiovascular system lessons learned and unanswered questions. J Am Coll Cardiol. 2006;47:1741–53. doi: 10.1016/j.jacc.2005.10.076. [DOI] [PubMed] [Google Scholar]