

Abstract
In neurons alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are heteromeric cation channels composed of different sub-units, including GluA1-GluA4. When expressed without GluA2, AMPA receptors function as Ca2+-permeable cation channels. In erythrocytes, activation of Ca2+-permeable cation channels triggers suicidal erythrocyte death or eryptosis, which is characterized by cell shrinkage and cell membrane scrambling with subsequent exposure of phosphatidylserine at the cell surface. Activators of the channels and thus eryptosis include removal of extracellular Cl− (replaced by gluconate) and energy depletion (removal of glucose). The present study explored whether GluA1 is expressed in human erythrocytes and whether pharmacological AMPA receptor inhibition modifies Ca2+ entry and suicidal death of human erythrocytes. GluA1 protein abundance was determined by confocal microscopy, phosphatidylserine exposure was estimated from annexin V binding, cell volume from forward scatter in FACS analysis, cytosolic Ca2+ concentration from Fluo3 fluorescence and channel activity by whole-cell patch-clamp recordings. As a result, GluA1 is indeed expressed in the erythrocyte cell membrane. The AMPA receptor antagonist NBQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide) inhibited the cation channels following Cl− removal and the eryptosis following Cl− removal or energy depletion. The present study reveals a novel action of AMPA receptor antagonists and raises the possibility that GluA1 or a pharmacologically related protein participates in the regulation of Ca2+ entry into and suicidal death of human erythrocytes.
Keywords: cell volume, annexin, eryptosis, calcium, phosphatidylserine
Introduction
Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are tetramers composed of four sub-units GluA1–GluA4 [1, 2]. They are responsible for generating excitatory synaptic responses [1]. If expressed without GluA2, GluA1 may form Ca2+-permeable cation channels [3]. Excessive activation of AMPA receptors may lead to neuronal apoptosis [3–11] and may suppress glial cell proliferation [12, 13]. Glutamate receptors are expressed in non-neuronal cells [14, 15], in which they similarly may influence apoptosis [14].
Erythrocytes express Ca2+-permeable cation channels [16–21]. Excessive activation of those channels triggers suicidal erythrocyte death or eryptosis [22], which is characterized by exposure of phosphatidylserine (PS) at the erythrocyte surface [23–26]. PS exposure results from phospholipid scrambling of the cell membrane [27, 28]. The Ca2+-permeable cation channels are activated by osmotic shock, oxidative stress and energy depletion [17–19, 29]. Besides triggering cell membrane scrambling, Ca2+ activates Ca2+-sensitive K+ channels [30, 31], leading to exit of KCl with osmotically obliged water and thus to cell shrinkage [32]. The shrinkage fosters cell membrane scrambling [33]. Effects of cytosolic Ca2+ on phospholipid scrambling are potentiated by ceramide [34].
Experiments aiming at the molecular identification of the Ca2+ entry mechanism have shown the contribution of TRPC6 [35]. However, albeit blunted, Ca2+ entry into erythrocytes is still observed in TRPC6 knockout mice [35]. Thus, additional channel proteins must contribute to the observed Ca2+ entry. The present study explored whether GluA1 is expressed in human erythrocytes and whether AMPA receptor blockage influences Ca2+ entry into and suicidal death of erythrocytes.
Materials and methods
Volunteers
Four to six different leukocyte-depleted erythrocyte concentrates provided by the blood bank of the University of Tübingen were studied. For retrieval of the concentrates, citrate was used as anticoagulant. Prior to our experiments, the concentrates were stored at 4°C in the commonly used SAG mannitol solution (0.41–0.26 ml/ml concentrate) with CPD stabilizer solution (0.015–0.007 ml/ml concentrate). Hundred milliltres of SAG mannitol solution contained 0.877 g NaCl, 0.9 g glucose, 0.0169 g adenosine and 0.525 g mannitol. Hundred milliltres of CPD stabilizer solution contained 0.327 g citric acid monohydrate, 2.63 g sodium citrate, 2.55 g glucose monohydrate and 0.251 g sodium dihydrogenphosphate. The erythrocyte concentrates were 7–20 days old when starting the experiment. Furthermore, erythrocytes from EDTA blood freshly retrieved from volunteers were investigated. The study is approved by the Ethical Commission of the University of Tübingen.
Solutions
The experiments were performed at 37°C in Ringer solution containing (in mM) 125 NaCl, 5 KCl, 1 MgSO4, 32 N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES)/NaOH (pH 7.4), 5 glucose, 1 CaCl2. Where indicated, Cl− was substituted for gluconate or glucose was removed. The AMPA receptor inhibitor NBQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide) and CNQX (6-cyano-7-nitroquinoxaline-2,3-dione) were obtained from Sigma (Schnelldorf, Germany).
Phosphatidylserine exposure and forward scatter
After incubation, erythrocytes were washed once in Ringer solution containing 5 mM CaCl2[36]. The cells were then stained with Annexin V-Fluos (Roche, Mannheim, Germany) at a 1:500 dilution [37]. After 15 min., samples were measured by flow cytometric analysis (FACS-Calibur from Becton Dickinson, Heidelberg, Germany). Cells were analyzed by forward scatter, and annexin V fluorescence intensity was measured in fluorescence channel FL-1 with an excitation wavelength of 488 nm and an emission wavelength of 530 nm.
Measurement of intracellular Ca2+
After incubation, erythrocytes were washed in Ringer solution and then loaded with Fluo-3/AM (Calbiochem, Bad Soden, Germany) in Ringer solution containing 5 mM CaCl2 and 2 μM Fluo-3/AM [38]. The cells were incubated at 37°C for 20 min. and washed twice in Ringer solution containing 5 mM CaCl2. The Fluo-3/AM-loaded erythrocytes were re-suspended in 200 μl Ringer. Then, Ca2+-dependent fluorescence intensity was measured in fluorescence channel FL-1 in FACS analysis [39].
Patch-clamp recordings
Patch pipettes made of borosilicate glass (150 TF-10, Clark Medical Instruments, Pangbourne, UK) were pulled using a horizontal DMZ puller (Zeitz, Martinsried, Germany). Pipettes with high resistance from 8 to 12 M were connected via a Ag–AgCl wire to the headstage of an EPC 9 patch-clamp amplifier (HEKA, Lambrecht/Pfalz, Germany). Data acquisition and data analysis were controlled by a computer equipped with an ITC 16 interface (Instrutech, Lambrecht/Pfalz, Germany) and by using Pulse software (HEKA) as already described [17]. For current measurements (room temperature), erythrocytes were held at a holding potential (Vh) of –30 mV, and 200 msec. pulses from –100 to +100 mV were applied in increments of 20 mV. The original whole-cell current traces are depicted without filtering (acquisition frequency of 5 kHz). The currents were analyzed by averaging the current values measured between 90 and 190 msec. of each square pulse (I–V relationship). The applied voltages refer to the cytoplasmic face of the membrane with respect to the extracellular space. The offset potentials between both electrodes were zeroed before sealing. The liquid junction potentials between bath and pipette solution and between the bath solutions and the salt bridge (filled with NaCl bath solution) were calculated according to Barry and Lynch [40]. Data were corrected for liquid junction potentials. The pipette solution contained (in mM) 125 Na-gluconate, 10 NaCl, 1 MgATP, 1 EGTA and 10 HEPES/NaOH (pH 7.4). The recorded cells were superfused with standard NaCl bath solution containing (in mM) 145 NaCl, 5 KCl, 10 HEPES/NaOH, 5 d-glucose, 2 MgCl2 and 1 CaCl2 (pH 7.4). In Na-gluconate bath solution, Cl− was substituted for gluconate. NMDG-gluconate bath solution contained (in mM) 180 N-methyl-d-glucamine (NMDG)-gluconate, 10 HEPES/NMDG, 1 MgCl2 and 1 CaCl2 (pH 7.4).
Preparation of erythrocyte ghosts
Ten microliters of banked erythrocytes were lysed at 4°C in 2 ml hypotonic buffer containing 10 mM HEPES/NaOH (pH 7.4) and a protease inhibitor cocktail tablet (Roche). Ghost membranes were pelleted at 17,000g for 15 min. at 4°C and re-suspended in 10 μl Ringer solution. The ghost membranes were then subjected to confocal microscopy.
Confocal microscopy
Fresh EDTA whole blood or erythrocyte ghosts prepared from banked erythrocyte concentrates were taken and suspended in PBS (EDTA blood) or Ringer (ghosts) at a cell density of 5 × 107 cells/ml. Ten to 20 μl of the suspension were smeared onto a glass slide that was air dried for 30 min. and then fixed with methanol for 2 min. After four washing steps with PBS for 10 min., the specimen was blocked by incubation with 10% goat serum. Following three washing steps with PBS for 5 min., the specimen was incubated with rabbit GluR1 antibody (1:200; Millipore, Billerica, MA) at 4°C overnight. The slide was washed again three times for 5 min. and then incubated with Cy3-conjugated Affinipure goat anti-rabbit antibody (Jackson Immuno Research, Hamburg, Germany) at room temperature for 1.5 hrs. Then, the specimen was mounted using Prolong® Gold antifade reagent (Invitrogen, Karlsruhe, Germany). Images were taken on a Zeiss LSM 5 EXCITER confocal laser scanning microscope (Carl Zeiss MicroImaging GmbH, Göttingen, Germany) with a water immersion Plan-Neofluar 40 or 63/1.3 NA DIC. As a control for the specificity of the primary antibody, erythrocytes from mice lacking GluA1 (GluA1−/–) and from their wild-type littermates (GluA1+/+) were retrieved (EDTA blood) and similarly analyzed in confocal microscopy.
Statistics
Data are expressed as arithmetic means ± S.E.M., and statistical analysis was made by ANOVA using Tukey’s test as post hoc test or by two-tailed t-test, as appropriate. P < 0.05 was considered as statistically significant.
Results
Confocal microcopy was used to explore whether GluA1 is expressed in human erythrocytes. As illustrated in Fig. 1, upper panels, a preparation of human whole blood indeed revealed the expression of GluA1 in the cell membrane of human erythrocytes. Similarly, GluA1 could be detected in the membranes of erythrocyte ghosts (Fig. 1, middle panel). To check for the specificity of the antibody against GluA1, the antibody was probed against erythrocytes from GluA1-deficient mice (gluA1–/–) and from their wild-type littermates (gluA1+/+). As shown in Fig. 1, lower left panel, GluA1 could be readily detected in gluA1+/+ erythrocytes, whereas no signal was observed in gluA1–/– erythrocytes (Fig. 1, lower right panel).
Figure 1.
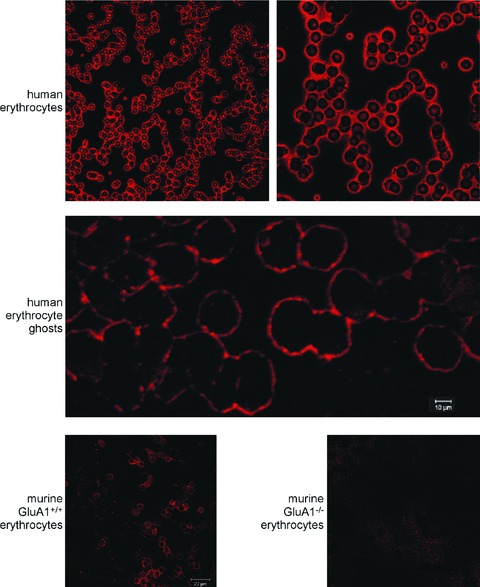
Expression of GluA1 in erythrocytes. (A) Examination of GluA1 expression in different erythrocyte preparations. The two upper panels show GluA1-dependent fluorescence in human erythrocytes. The middle panel depicts GluA1-dependent fluorescence in human erythrocyte ghosts. The lower panels illustrate GluA1-dependent fluorescence in murine gluA1+/+ (left panel) and gluA1–/– (right panel) erythrocytes.
Whole-cell patch-clamp recordings were performed to elucidate the sensitivity of the Ca2+-permeable cation channels of human erythrocytes for the AMPA receptor blocker NBQX. In confirmation of earlier observations, a cation channel could be observed in the absence of Cl− (Fig. 2). NBQX added to the bath solution at a concentration of 10 μM significantly decreased the cation current (Fig. 2). However, a residual cation conductance was observed even in the presence of NBQX. Consequently, substitution of Na+ by impermeable NMDG+ in the bath solution led to a prominent decrease of the rest inward current and to the shift of the reversal potential (Fig. 2). The patch-clamp experiments thus reveal that NBQX-sensitive cation channels contribute to the cation conductance of human erythrocytes.
Figure 2.
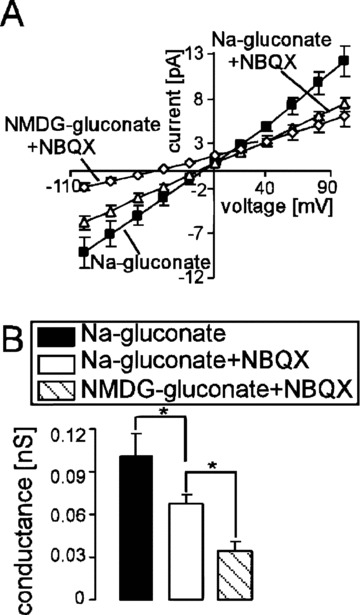
Inhibition of the non-selective cation channels by NBQX in human erythrocytes. (A) Mean current voltage (I–V) relationships (± S.E.M., n= 6) of human erythrocytes recorded in Na+-gluconate (closed squares), then in Na-gluconate + 10 μM NBQX (open triangles) and then in NMDG-gluconate + 10 μM NBQX (open diamonds) bath solutions. (B) Mean conductance of the inward currents (± S.E.M., n= 6) recorded as in (A) calculated by linear regression between –100 mV and –40 mV in Na+-gluconate (closed bar), Na-gluconate + 10 μM NBQX (open bar) and NMDG-gluconate + 10 μM NBQX (striped bar). * (P < 0.05) indicates significant difference (one-way anova).
Further experiments were performed to elucidate whether the channel modifies the intracellular Ca2+ concentration. According to Fluo3 fluorescence, Cl− deficiency (replacement of Cl− by gluconate) markedly increased the cytosolic Ca2+ activity in human erythrocytes (Fig. 3). Similarly, energy depletion (incubation of erythrocytes in glucose-free solution) was followed by a significant increase in the intracellular Ca2+ concentration (Fig. 3). Exposure to NBQX (10 or 50 μM) significantly blunted the increase in the intracellular Ca2+ concentration in erythrocytes following Cl− removal and glucose depletion. The effect of 50 μM NBQX was more pronounced than the effect of 10 μM NBQX (Fig. 3).
Figure 3.
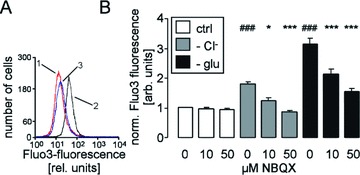
Cytosolic Ca2+ concentration in human erythrocytes. (A) Histogram of Fluo3 fluorescence in a representative experiment of human erythrocytes exposed for 48 hrs to plain Ringer (1, red line) or to Cl−-depleted Ringer without (2, black line) or with AMPA receptor blocker NBQX (50 μM, 3, blue line). (B) Arithmetic means ± S.E.M. (n= 16) of the normalized Fluo3 fluorescence in human erythrocytes exposed for 48 hrs to plain Ringer (white bars), to Cl−-depleted Ringer (grey bars) or to glucose-free Ringer (black bars) in the presence of 0–50 μM NBQX. ### indicates significant difference from plain Ringer (anova, P < 0.001). *, *** indicate significant difference from the absence of NBQX (anova, P < 0.05, P < 0.001).
Stimulation of Ca2+ entry and the resulting increase in the cytosolic Ca2+ activity are expected to activate Ca2+-sensitive K+ channels with following cell shrinkage [32]. The forward scatter of human erythrocytes as a measure of cell volume was indeed decreased by both Cl− removal and glucose depletion (Fig. 4). Treatment with the AMPA receptor blocker NBQX (50 μM) significantly blunted the decrease of forward scatter following Cl− removal (Fig. 4). Lower concentrations of NBQX (10 μM) tended to increase the forward scatter in the absence of Cl− or glucose, an effect, however, not reaching statistical significance.
Figure 4.
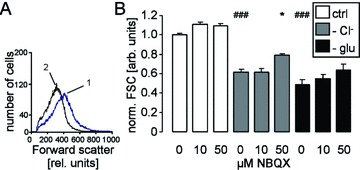
Forward scatter in human erythrocytes. (A) Histogram of forward scatter in a representative experiment of human erythrocytes exposed for 48 hrs to Cl−-depleted Ringer without (1, blue line) or with AMPA receptor blocker NBQX (2, black line). (B) Arithmetic means ± S.E.M. (n= 12–16) of the normalized forward scatter of human erythrocytes exposed for 48 hrs to plain Ringer (white bars), to Cl−-depleted Ringer (grey bars) or to glucose-free Ringer (black bars) in the presence of 0–50 μM NBQX. ### indicates significant difference from plain Ringer (anova, P < 0.001). * indicates significant difference from the absence of NBQX (anova, P < 0.05).
Stimulation of Ca2+ entry further triggers cell membrane scrambling with subsequent PS exposure [23–26, 41]. As illustrated in Fig. 5, annexin V binding of human erythrocytes was indeed enhanced by both Cl− removal and glucose depletion. The presence of NBQX (10 or 50 μM) significantly blunted the increase in annexin V binding following Cl− removal and glucose depletion (Fig. 5). In a further series of experiments, we compared the inhibitory potency of NBQX in freshly drawn blood with that in banked erythrocytes. As a result, following Cl− removal for 48 hrs, the percentage of annexin V–binding banked erythrocytes approached 39.0 ± 5.9% in the absence and 16.9 ± 0.8% in the presence of 50 μM NBQX (n= 8). Exposure of freshly drawn erythrocytes to Cl−-free Ringer resulted in 36.9 ± 4.6% annexin V–binding cells in the absence and in 11.1 ± 2.2% annexin V–binding cells in the presence of 50 μM NBQX (n= 4–6). Thus, NBQX was similarly effective in freshly drawn blood and banked erythrocytes.
Figure 5.
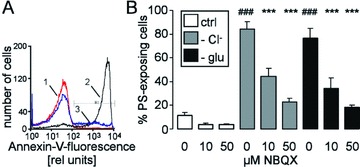
Annexin V binding of human erythrocytes (A) Histogram of annexin V binding in a representative experiment of human erythrocytes exposed for 48 hrs to plain Ringer (1, red line) or to Cl−-depleted Ringer without (2, black line) or with AMPA receptor blocker NBQX (50 μM, 3, blue line). (B) Arithmetic means ± S.E.M. (n= 16) of the percentage of annexin V–binding human erythrocytes exposed for 48 hrs to plain Ringer (white bars), to Cl−-depleted Ringer (grey bars) or to glucose-free Ringer (black bars) in the presence of 0–50 μM NBQX. ### indicates significant difference from plain Ringer (anova, P < 0.001), *** indicates significant difference from the absence of NBQX (anova, P < 0.001).
Additional experiments explored whether the anti-eryptotic efficacy of NBQX is shared by the other AMPA receptor blocker CNQX. As a result, in the presence of 50 μM CNQX, the percentage of annexin V binding following Cl− removal for 48 hrs was significantly (P < 0.05) decreased from 31.9 ± 5.2% (n= 16) to 14.0 ± 2.8% (n= 16).
Discussion
The present study reveals a novel element in the regulation of erythrocyte survival. Both genetic and pharmacological evidence suggest that GluA1 contributes to the Ca2+-permeable cation channels, Ca2+ entry, cell shrinkage and phosphatidylserine (PS) exposure of mature erythrocytes. Accordingly, GluA1 or a pharmacologically similar channel presumably participates in the orchestration leading to suicidal death of erythrocytes.
PS-exposing erythrocytes are bound to PS receptors on macrophages [42], which engulf and degrade PS-exposing cells [43]. Accordingly, PS-exposing erythrocytes are rapidly cleared from circulating blood [44]. Therefore, enhanced eryptosis has been observed in a variety of clinical conditions associated with anaemia [45]. Moreover, eryptosis may affect the microcirculation, as PS-exposing erythrocytes may bind to the vascular wall and participate in blood clotting [46–52]. As a matter of fact, suicidal erythrocytes have been proposed to participate in vascular injury of metabolic syndrome [53]. Finally, oxidative stress may limit the life span of stored erythrocytes [54].
According to our observations, AMPA receptor triggering does not only affect the survival of neurons [3–5, 7–11] and glial cells [12, 13] but may similarly affect erythrocyte survival. It should be kept in mind, though, that NBQX and CNQX could exert effects other than blocking AMPA receptors.
Parallel death of erythrocytes and neurons has led to the term neuroacanthocytosis, which is characterized by nervous system abnormalities in association with acanthocytosis in the patients’ blood. The disorder may be caused by a defect of the cytoskeletal protein 4.1 [55]. The protein regulates the surface expression and activity of GluA1 [56–58].
Neuronal and erythrocyte survival are further affected in parallel by a mutation within GLUT-1 turning the glucose carrier into a cation channel [59]. Affected individuals of the family suffered from exertion-induced dyskinesia, epilepsy, mild developmental delay, reduced CSF glucose levels and haemolytic anaemia with echinocytosis [59].
AMPA receptors are further regulated by phospholipase A2 [60], which similarly participates in the regulation of erythrocyte cation channels and erythrocyte survival [61].
Most recently, expression of AMPA receptor sub-units have been discovered in platelets and shown to participate in blood coagulation [15]. Accordingly, in mice lacking GluA1 the time required to complete thrombosis is prolonged [15]. It should be kept in mind that PS-exposing erythrocytes adhere to the vascular wall [46, 48, 49, 53] and thus may contribute to haemostasis [46].
In conclusion, Ca2+ entry into erythrocytes is blunted by an AMPA receptor antagonist. The present observations disclose a completely novel effect of AMPA-modulating drugs and unravel a novel parallelism between erythrocyte and neuronal survival.
Acknowledgments
The authors acknowledge the technical assistance of V. Schnorr and the meticulous preparation of the manuscript by Tanja Loch. This study was supported by the Deutsche Forschungsgemeinschaft, Nr. La 315/4–3 and La 315/6–1 and the Carl-Zeiss-Stiftung.
References
- 1.Biou V, Bhattacharyya S, Malenka RC. Endocytosis and recycling of AMPA receptors lacking GluR2/3. Proc Natl Acad Sci USA. 2008;105:1038–43. doi: 10.1073/pnas.0711412105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panicker S, Brown K, Nicoll RA. Synaptic AMPA receptor subunit trafficking is independent of the C terminus in the GluR2-lacking mouse. Proc Natl Acad Sci USA. 2008;105:1032–7. doi: 10.1073/pnas.0711313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizuno T, Zhang G, Takeuchi H, et al. Interferon-{gamma} directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-{gamma} receptor and AMPA GluR1 receptor. FASEB J. 2008;22:1797–806. doi: 10.1096/fj.07-099499. [DOI] [PubMed] [Google Scholar]
- 4.Beart PM, Lim ML, Chen B, et al. Hierarchical recruitment by AMPA but not staurosporine of pro-apoptotic mitochondrial signaling in cultured cortical neurons: evidence for caspase-dependent/independent cross-talk. J Neurochem. 2007;103:2408–27. doi: 10.1111/j.1471-4159.2007.04937.x. [DOI] [PubMed] [Google Scholar]
- 5.Das A, Sribnick EA, Wingrave JM, et al. Calpain activation in apoptosis of ventral spinal cord 4.1 (VSC4.1) motoneurons exposed to glutamate: calpain inhibition provides functional neuroprotection. J Neurosci Res. 2005;81:551–62. doi: 10.1002/jnr.20581. [DOI] [PubMed] [Google Scholar]
- 6.Denes L, Szilagyi G, Gal A, et al. Talampanel a non-competitive AMPA-antagonist attenuates caspase-3 dependent apoptosis in mouse brain after transient focal cerebral ischemia. Brain Res Bull. 2006;70:260–2. doi: 10.1016/j.brainresbull.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 7.Kim SJ, Han Y. Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase B (Akt) J Neural Transm. 2005;112:179–91. doi: 10.1007/s00702-004-0163-6. [DOI] [PubMed] [Google Scholar]
- 8.Klimaviciusa L, Safiulina D, Kaasik A, et al. The effects of glutamate receptor antagonists on cerebellar granule cell survival and development. Neurotoxicology. 2008;29:101–8. doi: 10.1016/j.neuro.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Molz S, Decker H, Dal Cim T, et al. Glutamate-induced toxicity in hippocampal slices involves apoptotic features and p38 MAPK signaling. Neurochem Res. 2008;33:27–36. doi: 10.1007/s11064-007-9402-1. [DOI] [PubMed] [Google Scholar]
- 10.Segura Torres JE, Chaparro-Huerta V, Rivera Cervantres MC, et al. Neuronal cell death due to glutamate excitotocity is mediated by p38 activation in the rat cerebral cortex. Neurosci Lett. 2006;403:233–8. doi: 10.1016/j.neulet.2006.04.063. [DOI] [PubMed] [Google Scholar]
- 11.Zou S, Li L, Pei L, et al. Protein-protein coupling/uncoupling enables dopamine D2 receptor regulation of AMPA receptor-mediated excitotoxicity. J Neurosci. 2005;25:4385–95. doi: 10.1523/JNEUROSCI.5099-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Groot JF, Piao Y, Lu L, et al. Knockdown of GluR1 expression by RNA interference inhibits glioma proliferation. J Neurooncol. 2008;88:121–33. doi: 10.1007/s11060-008-9552-2. [DOI] [PubMed] [Google Scholar]
- 13.Ishiuchi S, Yoshida Y, Sugawara K, et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci. 2007;27:7987–8001. doi: 10.1523/JNEUROSCI.2180-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoogduijn MJ, Hitchcock IS, Smit NP, et al. Glutamate receptors on human melanocytes regulate the expression of MiTF. Pigment Cell Res. 2006;19:58–67. doi: 10.1111/j.1600-0749.2005.00284.x. [DOI] [PubMed] [Google Scholar]
- 15.Morrell CN, Sun H, Ikeda M, et al. Glutamate mediates platelet activation through the AMPA receptor. J Exp Med. 2008;205:575–84. doi: 10.1084/jem.20071474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernhardt I, Weiss E, Robinson HC, et al. Differential effect of HOE642 on two separate monovalent cation transporters in the human red cell membrane. Cell Physiol Biochem. 2007;20:601–6. doi: 10.1159/000107543. [DOI] [PubMed] [Google Scholar]
- 17.Duranton C, Huber SM, Lang F. Oxidation induces a Cl(−)-dependent cation conductance in human red blood cells. J Physiol. 2002;539:847–55. doi: 10.1113/jphysiol.2001.013040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duranton C, Huber S, Tanneur V, et al. Electrophysiological properties of the Plasmodium falciparum-induced cation conductance of human erythrocytes. Cell Physiol Biochem. 2003;13:189–98. doi: 10.1159/000072421. [DOI] [PubMed] [Google Scholar]
- 19.Huber SM, Gamper N, Lang F. Chloride conductance and volume-regulatory nonselective cation conductance in human red blood cell ghosts. Pflugers Arch. 2001;441:551–8. doi: 10.1007/s004240000456. [DOI] [PubMed] [Google Scholar]
- 20.Kaestner L, Christophersen P, Bernhardt I, et al. The non-selective voltage-activated cation channel in the human red blood cell membrane: reconciliation between two conflicting reports and further characterisation. Bioelectrochemistry. 2000;52:117–25. doi: 10.1016/s0302-4598(00)00110-0. [DOI] [PubMed] [Google Scholar]
- 21.Kaestner L, Bernhardt I. Ion channels in the human red blood cell membrane: their further investigation and physiological relevance. Bioelectrochemistry. 2002;55:71–4. doi: 10.1016/s1567-5394(01)00164-5. [DOI] [PubMed] [Google Scholar]
- 22.Lang KS, Lang PA, Bauer C, et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem. 2005;15:195–202. doi: 10.1159/000086406. [DOI] [PubMed] [Google Scholar]
- 23.Berg CP, Engels IH, Rothbart A, et al. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 2001;8:1197–206. doi: 10.1038/sj.cdd.4400905. [DOI] [PubMed] [Google Scholar]
- 24.Bratosin D, Estaquier J, Petit F, et al. Programmed cell death in mature erythrocytes: a model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ. 2001;8:1143–56. doi: 10.1038/sj.cdd.4400946. [DOI] [PubMed] [Google Scholar]
- 25.Daugas E, Cande C, Kroemer G. Erythrocytes: death of a mummy. Cell Death Differ. 2001;8:1131–3. doi: 10.1038/sj.cdd.4400953. [DOI] [PubMed] [Google Scholar]
- 26.Lang PA, Warskulat U, Heller-Stilb B, et al. Blunted apoptosis of erythrocytes from taurine transporter deficient mice. Cell Physiol Biochem. 2003;13:337–46. doi: 10.1159/000075121. [DOI] [PubMed] [Google Scholar]
- 27.Dekkers DW, Comfurius P, Bevers EM, et al. Comparison between Ca2+-induced scrambling of various fluorescently labelled lipid analogues in red blood cells. Biochem J. 2002;362:741–7. doi: 10.1042/0264-6021:3620741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woon LA, Holland JW, Kable EP, et al. Ca2+ sensitivity of phospholipid scrambling in human red cell ghosts. Cell Calcium. 1999;25:313–20. doi: 10.1054/ceca.1999.0029. [DOI] [PubMed] [Google Scholar]
- 29.Lang KS, Duranton C, Poehlmann H, et al. Cation channels trigger apoptotic death of erythrocytes. Cell Death Differ. 2003;10:249–56. doi: 10.1038/sj.cdd.4401144. [DOI] [PubMed] [Google Scholar]
- 30.Bookchin RM, Ortiz OE, Lew VL. Activation of calcium-dependent potassium channels in deoxygenated sickled red cells. Prog Clin Biol Res. 1987;240:193–200. [PubMed] [Google Scholar]
- 31.Brugnara C, de Franceschi L, Alper SL. Inhibition of Ca(2+)-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J Clin Invest. 1993;92:520–6. doi: 10.1172/JCI116597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lang PA, Kaiser S, Myssina S, et al. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am J Physiol Cell Physiol. 2003;285:C1553–60. doi: 10.1152/ajpcell.00186.2003. [DOI] [PubMed] [Google Scholar]
- 33.Schneider J, Nicolay JP, Foller M, et al. Suicidal erythrocyte death following cellular K+ loss. Cell Physiol Biochem. 2007;20:35–44. doi: 10.1159/000104151. [DOI] [PubMed] [Google Scholar]
- 34.Lang KS, Myssina S, Brand V, et al. Involvement of ceramide in hyperosmotic shock-induced death of erythrocytes. Cell Death Differ. 2004;11:231–43. doi: 10.1038/sj.cdd.4401311. [DOI] [PubMed] [Google Scholar]
- 35.Foller M, Kasinathan RS, Koka S, et al. TRPC6 contributes to the Ca(2+) leak of human erythrocytes. Cell Physiol Biochem. 2008;21:183–92. doi: 10.1159/000113760. [DOI] [PubMed] [Google Scholar]
- 36.Bentzen PJ, Lang E, Lang F. Curcumin induced suicidal erythrocyte death. Cell Physiol Biochem. 2007;19:153–64. doi: 10.1159/000099203. [DOI] [PubMed] [Google Scholar]
- 37.Nicolay JP, Gatz S, Liebig G, et al. Amyloid induced suicidal erythrocyte death. Cell Physiol Biochem. 2007;19:175–84. doi: 10.1159/000099205. [DOI] [PubMed] [Google Scholar]
- 38.Foller M, Shumilina E, Lam R, et al. Induction of suicidal erythrocyte death by listeriolysin from Listeria monocytogenes. Cell Physiol Biochem. 2007;20:1051–60. doi: 10.1159/000110715. [DOI] [PubMed] [Google Scholar]
- 39.Attanasio P, Shumilina E, Hermle T, et al. Stimulation of eryptosis by anti-A IgG antibodies. Cell Physiol Biochem. 2007;20:591–600. doi: 10.1159/000107542. [DOI] [PubMed] [Google Scholar]
- 40.Barry PH, Lynch JW. Liquid junction potentials and small cell effects in patch-clamp analysis. J Membr Biol. 1991;121:101–17. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- 41.Brand VB, Sandu CD, Duranton C, et al. Dependence of Plasmodium falciparum in vitro growth on the cation permeability of the human host erythrocyte. Cell Physiol Biochem. 2003;13:347–56. doi: 10.1159/000075122. [DOI] [PubMed] [Google Scholar]
- 42.Fadok VA, Bratton DL, Rose DM, et al. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- 43.Boas FE, Forman L, Beutler E. Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc Natl Acad Sci USA. 1998;95:3077–81. doi: 10.1073/pnas.95.6.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kempe DS, Lang PA, Duranton C, et al. Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J. 2006;20:368–70. doi: 10.1096/fj.05-4872fje. [DOI] [PubMed] [Google Scholar]
- 45.Lang F, Gulbins E, Lerche H, et al. Eryptosis, a window to systemic disease. Cell Physiol Biochem. 2008:373–80. doi: 10.1159/000185448. [DOI] [PubMed] [Google Scholar]
- 46.Andrews DA, Low PS. Role of red blood cells in thrombosis. Curr Opin Hematol. 1999;6:76–82. doi: 10.1097/00062752-199903000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Chung SM, Bae ON, Lim KM, et al. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler Thromb Vasc Biol. 2007;27:414–21. doi: 10.1161/01.ATV.0000252898.48084.6a. [DOI] [PubMed] [Google Scholar]
- 48.Closse C, Dachary-Prigent J, Boisseau MR. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br J Haematol. 1999;107:300–2. doi: 10.1046/j.1365-2141.1999.01718.x. [DOI] [PubMed] [Google Scholar]
- 49.Gallagher PG, Chang SH, Rettig MP, et al. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood. 2003;101:4625–7. doi: 10.1182/blood-2001-12-0329. [DOI] [PubMed] [Google Scholar]
- 50.Pandolfi PP, Di Pietro N, Sirolli V, et al. Mechanism of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J Cell Physiol. 2007;213:699–709. doi: 10.1002/jcp.21138. [DOI] [PubMed] [Google Scholar]
- 51.Wood BL, Gibson DF, Tait JF. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: flow-cytometric measurement and clinical associations. Blood. 1996;88:1873–80. [PubMed] [Google Scholar]
- 52.Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–88. doi: 10.1007/s00018-005-4527-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zappulla D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: adaptations to CO2 increases. J Cardiometab Syndr. 2008;3:30–4. doi: 10.1111/j.1559-4572.2008.07263.x. [DOI] [PubMed] [Google Scholar]
- 54.Kriebardis AG, Antonelou MH, Stamoulis KE, et al. Progressive oxidation of cytoskeletal proteins and accumulation of denatured hemoglobin in stored red cells. J Cell Mol Med. 2007;11:148–55. doi: 10.1111/j.1582-4934.2007.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orlacchio A, Calabresi P, Rum A, et al. Neuroacanthocytosis associated with a defect of the 4.1R membrane protein. BMC Neurol. 2007;7:4. doi: 10.1186/1471-2377-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baines AJ, Keating L, Phillips GW, et al. The postsynaptic spectrin/4.1 membrane protein “accumulation machine”. Cell Mol Biol Lett. 2001;6:691–702. [PubMed] [Google Scholar]
- 57.Coleman SK, Cai C, Mottershead DG, et al. Surface expression of GluR-D AMPA receptor is dependent on an interaction between its C-terminal domain and a 4.1 protein. J Neurosci. 2003;23:798–806. doi: 10.1523/JNEUROSCI.23-03-00798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen L, Liang F, Walensky LD, et al. Regulation of AMPA receptor GluR1 subunit surface expression by a 4. 1N-linked actin cytoskeletal association. J Neurosci. 2000;20:7932–40. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weber YG, Storch A, Wuttke TV, et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest. 2008;118:2157–68. doi: 10.1172/JCI34438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Menard C, Chartier E, Patenaude C, et al. Calcium-independent phospholipase A(2) influences AMPA-mediated toxicity of hippocampal slices by regulating the GluR1 subunit in synaptic membranes. Hippocampus. 2007;17:1109–20. doi: 10.1002/hipo.20343. [DOI] [PubMed] [Google Scholar]
- 61.Lang PA, Kempe DS, Myssina S, et al. PGE(2) in the regulation of programmed erythrocyte death. Cell Death Differ. 2005;12:415–28. doi: 10.1038/sj.cdd.4401561. [DOI] [PubMed] [Google Scholar]