

Abstract
Endostatin is a well-characterized endogenous inhibitor of angiogenesis that affects cell proliferation and migration by inhibiting integrin and Wnt-mediated signalling pathways. Here, we show that endothelial cells treated with native and P125A-endostatin activate autophagy. Because autophagy can either be protective or induce programmed cell death, experiments were carried out to understand the signalling pathways leading to autophagy in endothelial cells. P125A-endostatin treatment increased the levels of Beclin 1, a crucial molecule in vesicle nucleation and autophagy. The treatment also reduced the levels of Bcl-2, Bcl-xL and β-catenin; however, progressively increasing amounts of Bcl-2 and Bcl-xL were found to be complexed with Beclin 1. Increased β-catenin and Wnt-mediated signalling reduced Beclin 1 levels and rescued endothelial cells from endostatin-induced autophagy. Finally, knocking down Beclin 1 levels by RNA interference decreased autophagy and accelerated caspase activation in endostatin-treated cells. These studies suggest that endothelial cells may initiate autophagy as a survival response to limit the effects of angiogenesis inhibitors. Thus, interfering with autophagy can potentiate the effects of endostatin by promoting a switch to apoptosis.
Keywords: endostatin, autophagy, apoptosis, angiogenesis, Beclin 1, Bcl-2, Bcl-xL, Beta-catenin, Wnt-pathway
Introduction
Both physiological and pathological angiogenesis are regulated by a delicate balance between angiogenic growth factors and endogenous inhibitors [1]. Tumour growth and metastasis are promoted when this balance is shifted towards angiogenesis. Angiogenesis inhibitors can be used to reverse this trend [2]. Endostatin, a 20-kD fragment of the non-collagenous 1 domain of collagen XVIII-α1, is a well-characterized endogenous angiogenesis inhibitor [1, 3, 4] and is known to inhibit endothelial cell proliferation, and cell migration [5], and to induce apoptosis [6]. Recently, a mutant form of endostatin with a proline to alanine substitution at position 125 was identified and characterized [7]. P125A-endostatin showed higher binding to endothelial cell surface and exhibited stronger inhibitory effects on both cell proliferation and cell migration [7, 8]. Integrin α5β1 and glypican-1 are the major targets of endostatin [9]. Endostatin binding is known to induce dynamic changes in integrins and associated stress fibres that ultimately lead to inhibition of cell migration [10]. In addition, endostatin has also been shown to antagonize the Wnt-signalling pathway [11, 12] and target β-catenin for degradation. Concomitantly, endostatin-treated cells also show increased dephosphorylation of the anti-apoptotic protein Bcl-2, affecting its intracellular levels [6]. Bcl-2 is known to be associated with human vascular endothelial growth factor (VEGF)-mediated angiogenesis and tumour growth [13, 14]. Bcl-2 and another family member, Bcl-xL, have also been found to regulate autophagy [15, 16], a self-consumption process involving bulk degradation of long-lived proteins and defective or superfluous organelles, which plays a major role in cell survival. Bcl-2 and Bcl-xL regulate autophagy via Beclin 1, a pivotal initiator of autophagy. The identification of Beclin 1 as a binding partner of Bcl-2 and Bcl-xL not only suggests a communication channel between apoptosis and autophagy [15] but also establishes the connection between autophagy and oncogenesis [17].
The autophagic response in endothelial cells treated with angiogenesis inhibitors has hitherto been largely undefined. In an earlier study, endostatin was found to induce autophagy in Eahy926 human endothelial cell line, which is derived from the fusion between HUVEC and an epithelial lung cancer cell line [18]. In the present study, we have investigated the possible mechanism by which primary cultures of endothelial cells initiate autophagic survival responses during endostatin treatment. Our studies suggest that Beclin 1 levels and autophagic vesicle formation are regulated by Bcl-2, Bcl-xL and the Wnt–β-catenin signalling pathway.
Materials and methods
Materials
MitoTracker Deep red 633 and Alexa Fluor 488 anti-rabbit IgG antibody were from Molecular Probes (Eugene, OR). Rapamycin, 3-methyladenine (3-MA) and β-actin antibody were obtained from Sigma (St. Louis, MO). Oligofectamine 2000 was from Invitrogen (San Diego, CA). Protein A/G Plus-Agarose, Bcl-2 polyclonal antibody, Bcl-xL polyclonal, β-catenin polyclonal antibody, MAP LC3 antibody, integrin α5 siRNA and integrin β1 siRNA were from Santa Cruz (Santa Cruz, CA). Silencer® pre-designed siRNA to β-catenin and control was from Ambion (Austin, TX). Vinculin antibody was from Abcam (Cambridge, MA). Human integrin α5β1 mAb was obtained from Chemicon (Temecula, CA). Beclin 1 mAb was from BD Transduction Laboratories (Lexington, KY). Recombinant hVEGF (VEGF165) was from R&D Systems (Minneapolis, MN). Ad-CMV–β-catenin and Ad-CMV–GFP were acquired from Vector Biolabs (Philadelphia, PA). Ad-Wnt and pcDNA dominant negative β-catenin were generated in the laboratory of Dr. Randall Moon, University of Washington. Pichia pastoris native endostatin was from Calbiochem (San Diego, CA). P. pastoris–derived P125A-endostatin was purified as previously described [19]. LAMP1-GFP (lysosomal associated membrane protein 1) construct was a generous gift from Dr. Dell’Angelica, University of California, Los Angeles. LC3-GFP construct was kindly provided by T. Yoshimori (National Institute of Genetics, Shizuoka-ken, Japan) and N. Mizushima (The Tokyo Metropolitan Institute of Medical Science, Japan).
Cell culture
MA148 ovarian cancer cells, human foreskin fibroblasts and human umbilical vein cells (HUVECs) were cultured as previously described [7, 20]. Endothelial cells were cultured in chamber slides and stimulated with VEGF (20 ng/ml) prepared in 5% FBS containing M199 medium.
Confocal and immunofluorescence microscopy
HUVECs, MA148 or human foreskin fibroblasts were transfected with either LAMP1-GFP or LC3-GFP and cultured as previously described [20]. Transfected cells were treated with different concentrations (2.5–20 μg/ml) of endostatin for 24 hrs or as otherwise stated. Mitotracker Red or DAPI (Vector Laboratories, Burlingame, CA) was used to visualize mitochondria and the nucleus, respectively. Confocal images were recorded at 600× magnification (PlanApo N 60×/1.24 oil, ∞/0.17/FN26.5) using a Fluoview 1000 System (Olympus, Irving, TX) via a FV1000 software Ver.01.06. Fields were chosen randomly from various sections to ensure objectivity of sampling. Digital images were processed to determine the number of autophagic vesicles per cell [20]. β-Catenin distribution in cells treated with endostatin was monitored by staining the cells with mouse anti-human β-catenin antibody linked to phycoerythrin. Cells were counterstained with DAPI and observed using a Fluoview 1000, Olympus, inverted microscope.
Western blotting
HUVECs were treated with either P125A-endostatin (20 μg/ml) or rapamycin (100 ng/ml) with or without E64d (10 μg/ml), a protease inhibitor and pepstatin A (10 μg/ml) for 24 hrs in complete medium supplemented with 20 ng/ml of recombinant VEGF-A (R&D Systems). Control and treated cells were then lysed and about 10 μg of lysate proteins were used for Western blotting as previously described [20].
Flow cytometry
Endothelial cells were co-transfected with either scrambled or shRNA specific for Beclin 1 and a DsRed expression construct. Subsequently, cells were treated with P125A-endostatin (20 μg/ml). Caspase activation in transfected cells treated with endostatin was assessed by flow cytometry using carboxyfluorescein FLICA apoptosis detection kit (Immunochemistry Technologies, LLC, Bloomington, MN, USA) as previously described [20]. Briefly, treated cells were labelled with green fluorescent-labelled inhibitor of caspases (FLICA) and analyzed by flow cytometer (BD Biosciences, Rockville, MD) according to the manufacturer’s protocol. Transfected HUVECs were gated for DsRed+ cell populations and scored for FAM-VAD-FMK+ cells FAM-VAD-FMK, a carboxyfluorescein (FAM) derivative of benzyloxycarbonyl-valine-alanine-aspartic acid–fluoromethyl ketone (zVAD-FMK), irreversibly binds to activated caspases. Caspase activation in apoptotic cells can then be determined by the amount of cellular FAM-VAD-FMK retention.
Statistical analysis
The results are given as the mean standard error. Statistical analysis was performed by using Student’s t-test. Differences with P values <0.05 were considered significant.
Results
Both native endostatin and P125A-endostatin induce autophagy in endothelial cells
At the outset, we compared the effects of native and P125A-endostatin on endothelial cell autophagy. HUVECs were transfected with either LC3-GFP, a biomarker for autophagy [21, 22], or LAMP1-GFP, a marker for lysosomes, autolysosomes, late endosomes and multi-vesicular bodies [23–25], and exposed to both native endostatin and P125A-endostatin in the presence of VEGF (20 ng/ml). LC3, microtubule-associated protein–light chain 3 (MAP-LC3), typically exhibits diffuse cytosolic distribution. Representative confocal images, shown in the lower panel (Fig. 1A), confirm that the treatment of endostatin led to the redistribution of modified LC3 to punctate structures and increased number of LC3-GFP-positive vesicles in the endostatin-treated cells. Although both native endostatin and P125A-endostatin induced autophagy in endothelial cells (Fig. 1A and Supplementary S1A), the number of autophagic vesicles per cell was higher in P125A-endostatin–treated cells. The mean number of LC3-GFP–positive vesicles in the control, untreated endothelial cells was 24 ± 5. In the presence of native endostatin, the LC3-GFP–positive vesicles increased to 42 ± 15, which is statistically significant (P < 0.05). P125A-endostatin treatment under similar conditions further increased the number of autophagic vesicle number to 68 ± 7 per cell (P < 0.001). P125A-endostatin has been previously shown to have better anti-angiogenic activity than the native endostatin [7, 8]. Subsequent studies were therefore carried out with P125A-endostatin. A concentration-dependent increase in LC3-GFP–positive vesicles was observed in P125A-treated HUVECs (Fig. 1B). Similarly, the number of LAMP1-GFP–positive vesicles were also increased in treated HUVECs plateaued at 10 μg/ml of P125A-endostatin (Supplementary S1B). The treatment of human microvascular endothelial cells with P125A-endostatin also resulted in a >2.5-fold increase in autophagic vesicles compared with control (data not shown). P125A-endostatin, however, failed to induce autophagic vesicles in human foreskin fibroblast (primary culture). In contrast, as a positive control, etoposide induced an increase in the number of autophagic vesicles in the latter (Fig. 1C and Supplementary S1C). P125A-endostatin also did not induce any autophagic response in MA148 cells, an ovarian cancer cell line (data not shown).
Figure 1.

Endostatin induces autophagy in endothelial cells. (A) Representative confocal images of LC3-GFP–transfected HUVECs treated with P125A-endostatin (20 μg/ml). Native endostatin (20 μg/ml) was used under similar conditions. (B) LC3-GFP–transfected HUVECs were treated with different concentrations of P125A-endostatin (□)(n= 3,**P < 0.001) or recombinant, native endostatin (20 μg/ml) (█), P < 0.05. Rapamycin (▴) was used as a positive control. Cells were treated for 24 hrs. Results are shown as mean ± S.E. (C) LC3-GFP–transfected fibroblasts were treated with P125A-endostatin (20 μg/ml). Etoposide (10 μg/ml) was used as a positive control. (D) Transmission electron microscope image of HUVECs treated with P125A-endostatin were collected at two different time-points (12 and 24 hrs). Images were recorded using a JEOL 1200 EX transmission electron microscope (Tokyo, Japan) as previously described [43]. Double-membrane autophagosomes and single-membrane autolysosomes that contained disintegrated materials clustering at the perinuclear sites are shown (open arrow or filled arrows, respectively). (E) Whole-cell lysates of HUVECs treated with rapamycin (100 ng/ml), P125A-endostatin (20 μg/ml) in the presence and absence of E64d (10 μg/ml) and pepstatin A (10 μg/ml) for 24 hrs, in medium supplemented with 20 ng/ml of recombinant VEGF-A, were evaluated for LC3-I and LC3-II levels by Western blotting. β-Actin was used to normalize the values of LC3-II.
Transmission electron microscopy was also used to confirm the nature of P125A-endostatin–induced vesicles (Fig. 1D). An increased accumulation of large vesicles within the cytoplasm of treated cells was observed after a 24-hr treatment compared with that at the earlier time-points (0 and 12 hrs). Ultrastructural images showed both double- and single-membrane containing vesicles enclosing intact and disintegrating mitochondria. A few single-membrane vesicles were also observed at the edge of control cells. The presence of these vesicles in the control-treatment can be attributed to serum restriction.
Autophagosome formation is known to correlate with the conversion of LC3-I to LC3-II that involves proteolytic cleavage and lipidation [26]. As further confirmation of autophagy induced by endostatin, cell lysates were analyzed for the conversion of LC3-I to LC3-II. In these experiments E64d, a membrane-permeable inhibitor of cathepsins B, H and L, and pepstatin A, an inhibitor of cathepsins D and E, in combination with treatment groups [27] were also used in order to determine the contribution of lysosomal degradation to LC3-II levels in endothelial cells. Data in Fig. 1E show an increase in LC3-II upon endostatin treatment compared with control cells. As expected, rapamycin-treated cell lysates also showed an increase in the level of LC3-II. Treatment of endostatin or rapamycin in combination with the inhibitors further enhanced the levels of LC3-II, suggesting an active lysosomal turnover induced by endostatin. Finally, because trichloroacetic acid (TCA) precipitates proteins but not free amino acids and peptides, an increase in the ratio of TCA-soluble/TCA-insoluble radioactivity was used to determine whether endostatin treatment affected the intracellular long-lived protein degradation and turnover, one of the hallmarks of autophagy [28]. Endothelial cells treated with endostatin in the presence or absence of 3-MA (an autophagy inhibitor) showed a 30% increase in TCA-soluble radioactivity (data not shown). Collectively, these studies demonstrate that P125A-endostatin selectively induces autophagy in endothelial cells.
Effect of integrin knockdown on autophagic response
Because endostatin binds to the integrins α5β1[9, 10, 29] with high affinity, we next determined whether autophagy induced by endostatin is dependent on integrin binding. Figure 2A shows that autophagic response was partially inhibited by function-blocking antibody against integrin α5β1 in endothelial cells. In the next series of experiments, siRNA specific to α5 and β1 were used to reduce the levels of integrin sub-units (Supplementary S2C). P125A-endostatin treatment dramatically increased the number of autophagic vesicles in scrambled siRNA–transfected cells (Fig. 2B). In contrast, knockdown of α5 and β1 integrin with specific siRNA reduced by up to 45% the number of autophagic vesicles induced by P125A-endostatin. In a parallel experiment using LAMP1-GFP–transfected HUVECs, similar changes were observed (Supplementary S2A and S2B). These studies suggest that P125A-endostatin binding to α5β1 integrin is important for the endothelial autophagic response.
Figure 2.
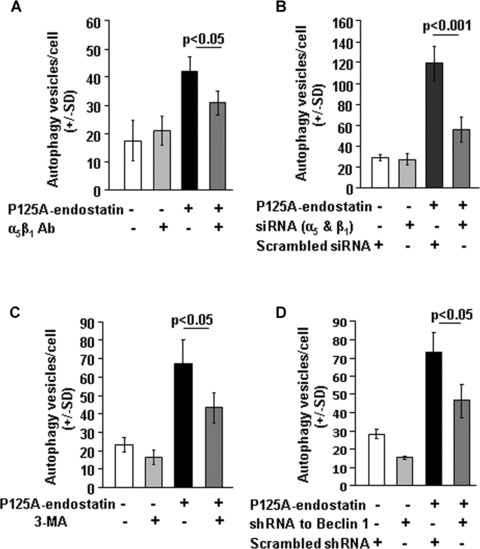
Effect of integrin antibodies and down-regulation of integrin sub-units on endostatin-induced autophagy. (A) LC3-GFP–transfected HUVECs were treated with P125A-endostatin (20 μg/ml) for 24 hrs in the presence of monoclonal antibody against integrin α5β1 (15 μg/ml)(n= 3). LC3-GFP–transfected HUVECs treated with isotype matched, normal IgG was used as a control for the study. Notice that autophagy induction was partially inhibited by integrin α5β1 antibody. (B) HUVECs were transfected with siRNA specific to integrin α5 and integrin β1 at a concentration of 40 nM. After 36 hrs of transfection, HUVECs were further transfected with LC3-GFP. Cells were subsequently treated with P125A-endostatin for 24 hrs and assessed for autophagic vesicles per cell (n= 3). (C) Effect of 3-MA on P125A-endostatin induced autophagy in HUVECs. LC3-GFP–transfected HUVECs were treated with P125A-endostatin (20 μg/ml) for 24 hrs. 3-MA (5 mM) was added to LC3-GFP–transfected HUVEC culture 12 hrs into the experiment (n= 3). (D) LC3-GFP was co-transfected with shRNA specific to Beclin 1 or control scrambled shRNA to HUVECs [20, 43]. Results are shown as mean ± S.E. Statistical significance was determined using Student’s t-test.
P125A-endostatin–induced autophagy is Beclin 1–PI-3 kinase dependent
Class III PI-3 kinase–p150–Beclin 1 complex is essential for the vesicle nucleation step in autophagy [30]. To evaluate whether autophagic induction by P125A-endostatin can be inhibited by interfering with the activity of PI-3kinase, 3-MA, a widely used PI-3kinase inhibitor [31], was added to LC3-GFP–transfected HUVEC cultures at a non-toxic concentration. Figure 2C shows that 3-MA treatment reduced the P125A-endostatin–induced autophagy by about 30% compared with control. Similarly, reducing Beclin 1 using shRNA [32] (Supplementary S2F) also resulted in reduction in autophagic response. P125A-endostatin treatment induced up to 2.4-fold increase in the number of autophagic vesicles when compared with untreated control endothelial cells, and knocking down Beclin 1 led to a 36% reduction in the number of vesicles per cell under similar conditions (Fig. 2D). Endothelial cells transfected with LAMP1–GFP showed similar results (Supplementary S2D and S2E), suggesting that Beclin 1 and PI-3 kinase complex play important roles in endostatin-induced autophagy of endothelial cells.
P125A-endostatin up-regulates Beclin 1 levels
The interaction of Beclin 1 and Bcl-2 in the Beclin 1–Bcl-2 complex has been implicated as a rheostat that regulates the homeostasis of autophagic capacity [15, 32]. In addition, Beclin 1 was also found to interact with a conserved hydrophobic groove in Bcl-xL, a close counterpart of Bcl-2, through a BH3-like domain [16]. Western blot analysis showed gradual increase in Beclin 1 levels that reached a plateau after 16 hrs of P125A-endostatin treatment. In contrast, a reduction in total levels of Bcl-2 was observed over the course of 24-hr P125A-endostatin treatment (Fig. 3A). A similar effect was observed when HUVECs were treated with native endostatin and rapamycin (data not shown). To determine whether endostatin-induced autophagy in endothelial cells involved interactions between Beclin 1 and Bcl-2 family members, endogenous Beclin 1–Bcl-2 or Beclin 1–Bcl-xL complexes were immunoprecipitated with antibodies against Beclin 1 (Fig. 3B). A steady increase in the levels of Bcl-2 and Bcl-xL co-immunoprecipitating with Beclin 1 was observed over 24 hrs. Levels of Beclin 1 complexed with Bcl-2 and Bcl-xL reached a steady state after 16 hrs of treatment (Fig. 3B). Endostatin treatment reduced the levels of Bcl-2 as has been previously shown [6] (Fig. 3C). Changes in Bcl-2 levels concomitantly reduced the amount of Beclin 1 that was complexed with Bcl-2 (Fig. 3C). Similar observation was found in immunoprecipitation studies with Bcl-xL. Substantial reduction in Bcl-xL levels was accompanied by distinct dissociation of Bcl-xL from the Beclin 1–Bcl-xL complex after 16 hrs of treatment with P125A-endostatin (Fig. 3D). Together, these data suggest that P125A-endostatin induces autophagy in endothelial cells by up-regulating Beclin 1 expression. The up-regulation disrupts the balance between Beclin 1 and Bcl-2 or Bcl-xL interaction.
Figure 3.
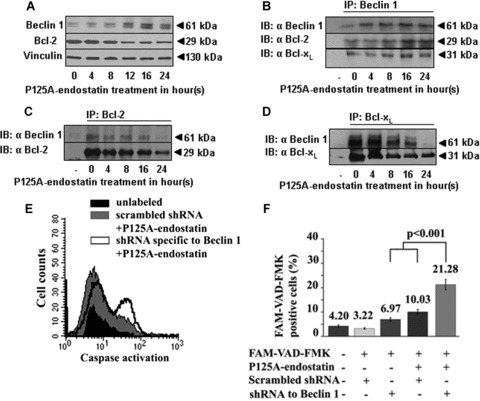
P125A-endostatin induced changes in Beclin 1-Bcl-2/Bcl-xL association. HUVECs were treated with P125A-endostatin (20 μg/ml). (A) Western blot analysis of Beclin 1, Bcl-2 and vinculin expression in HUVEC. Total cell lysates were collected at different time-points of P125A-endostatin treatment (n= 3). (B) Representative of co-immunoprecipitation blot of Beclin 1, Bcl-2 and Bcl-xL in HUVEC. (C) Representative of co-immunoprecipitation blot of Bcl-2 and Beclin 1 from whole-cell lysates of P125A-endostatin–treated HUVECs. (D) Representative of co-immunoprecipitation blot of Beclin 1 and Bcl-xL in treated HUVECs. Immunoprecipitation using IgG was used as a control for all studies. (E) Flow cytometric analysis showing Beclin 1 down-regulation in HUVECs potentiates P125A-endostatin–induced apoptosis. HUVECs were co-transfected with a transposon-expressing DsRed 2 plasmid and shRNA specific to Beclin 1 or a scrambled shRNA using Lipofectamine 2000. After 36 hrs, transfected HUVECs were treated with P125A-endostatin (20 μg/ml). Treated cells were labelled with green fluorescent-labelled inhibitor of caspases (FLICA) and analyzed by FACS. Transfected HUVECs were gated for DsRed-positive cell population and scored for FAM-VAD-FMK–positive cells. Histogram shows FAM-VAD-FMK–positive cells in the control, scrambled shRNA–transfected and shRNA specific to Beclin 1–transfected HUVECs. (F) Summary of the data from the flow cytometric analyses is shown as a percentage (n= 3, mean ±S.E.) of transfected HUVECs positive for the activation of caspase (FAM-VAD-FMK–positive cells).
Down-regulation of Beclin 1 in endothelial cells potentiates the apoptotic effect of P125A-endostatin
P125A-endostatin treatment induces the up-regulation of Beclin 1 in endothelial cells and simultaneously impinges on the availability of anti-apoptotic proteins Bcl-2 and Bcl-xL to bind the protein. This prompted us to investigate whether the pro-apoptotic effect of endostatin could be unmasked by knocking down Beclin 1. HUVECs, transiently co-transfected with shRNA specific to Beclin 1 and DsRed2 marker plasmid, were treated with P125A-endostatin (Fig. 3E and supplementary S3). Figure 3F summarizes the flow cytometric analysis of caspase activation in HUVECs. P125A-endostatin treatment of scrambled shRNA–transfected cells showed a marginal increase in caspase-positive cells, whereas down-regulation of Beclin 1 increased active caspase-positive cells to 21%. Together, these data suggest that inhibition of endostatin-induced autophagy increases apoptosis in endothelial cells.
β-Catenin modulates P125A-endostatin–induced autophagy in HUVECs
Studies described above suggest that endostatin induces autophagy by modulating Beclin 1, Bcl-2 and Bcl-xL levels and the Beclin 1–Bcl-2–Bcl-xL complex in treated endothelial cells. Endostatin treatment is also known to perturb the Wnt–β-catenin pathway. Specifically, native endostatin has been shown to reduce the endogenous levels of β-catenin [11]. To further explore the possible cross-talk between Wnt–β-catenin pathway and autophagy in endostatin-treated endothelial cells, we first confirmed that the levels of β-catenin were greatly reduced by both P125A-endostatin (Fig. 4A-confocal image panels and Supplementary S4-A) and native endostatin (data not shown). Modulation of β-catenin by siRNA treatment show reduced endogenous β-catenin levels that were further lowered upon P125A-endostatin treatment (Fig. 4A). Concomitant increase in Beclin 1 levels was observed. β-Catenin knockdown led to a small increase in autophagic vesicle formation that was enhanced by P125A-endostatin exposure (Fig. 4B). Similar observations were found in LAMP1-GFP–transfected HUVECs following a 24-hr P125A-endostatin treatment (Supplementary S4A). These results suggest that the Wnt–β-catenin pathway regulates the induction of autophagy in endothelial cells. To confirm this observation, dominant negative β-catenin was expressed in HUVEC. Treatment with endostatin did not affect the turnover rate of the dominant negative β-catenin. After 24-hr endostatin treatment, increased expression of dominant negative β-catenin was accompanied by increased levels of Beclin 1 and autophagic vesicles (Supplementary S4B and S4C). Collectively, these data demonstrate the involvement of the Wnt–β-catenin signalling pathway in the induction of autophagy by P125A-endostatin in endothelial cells.
Figure 4.

Effects of modulation of β-catenin levels on P125A-endostatin-induced autophagy. (A) Upper panel: Representative confocal images of HUVECs that were cultured in complete medium, 10% FBS supplemented with VEGF-A (20 ng/ml) or complete medium containing P125A-endostatin (20 μg/ml) are shown. β-Catenin localization on the cell membrane and in the cytoplasm is indicated by yellow and green arrows, respectively. Notice the reduction in the cytoplasmic β-catenin levels in P125A-endostatin-treated cells. Lower panel: HUVECs were transfected with either siRNA specific to β-catenin or scrambled control siRNA (20 pmols per reaction). Whole-cell lysates from different treatments were evaluated for β-catenin, Beclin 1 and β-actin levels. (B) HUVECs were co-transfected with LC3-GFP and siRNA specific to β-catenin or scrambled control siRNA. The number of autophagic vesicles per siRNA or scrambled siRNA–treated cell was quantified as described previously. Scrambled siRNA–transfected cells were used as a control for the study (n= 3). (C) Expression of β-catenin. HUVECs were infected with 500 MOI of either Ad-CMV-β-catenin (solid dark line) or Ad-CMV-GFP (solid grey line). Infected cells were treated with 20 μg/ml of P125A-endostatin (abbr. E) and assessed by FACS analysis for β-catenin levels. (D) Expression levels of Beclin 1 in the corresponding treatments were measured by flow cytometry. Fold changes in Beclin 1 were calculated by comparing with Beclin 1 levels in Ad-CMV-β-catenin–transduced HUVECs treated with P125A-endostatin and that of Ad-CMV-GFP–infected control cells (n= 3).
To confirm that β-catenin expression modulates Beclin 1 levels in HUVECs, cells were infected with a replication-deficient adenovirus that over-expressed β-catenin (Ad-CMV-β-catenin). Ad-CMV-β-catenin virus infection decreased the levels of Beclin 1 in HUVECs in a concentration-dependent manner compared with the control Ad-CMV-GFP construct (Supplementary S5B). Endostatin treatment significantly reduced the levels of β-catenin in the Ad-CMV-GFP–infected cells but did not alter the levels in Ad-CMV-β-catenin–infected cells (Fig. 4C). Ad-CMV-β-catenin–infected cells, however, showed reduced levels of Beclin 1 that remained low even in the presence of P125A-endostatin (Fig. 4D) as compared with controls that showed an increase. These results indicate a negative correlation between β-catenin and Beclin 1 levels and raise the possibility that β-catenin may be one of the regulators of autophagy.
The Wnt–β-catenin canonical pathway modulates the induction of autophagy in endothelial cells
To determine whether P125A-endostatin–induced changes in β-catenin plays an essential role in the induction of autophagy, we attempted to over-ride the effect of endostatin by exogenous activation of the Wnt pathway. As shown in Fig. 5A and B, HUVECs either transfected with pcDNA-expressing Wnt or infected with a replication-deficient adenovirus-expressing Wnt (Ad-Wnt) led to marked increase in β-catenin levels when compared with that of the control pcDNA vector–transfected cells or adenovirus-expressing β-galactosidase (Ad-β-Gal)-infected cells, respectively. The treatment of P125A-endostatin significantly reduced the β-catenin levels in the control, empty vector–transfected cells or Ad-β-Gal–infected cells, while progressively increasing the levels of Beclin 1 over a period of 24 hrs. In contrast, in Wnt over-expressing HUVECs, endostatin treatment did not alter either β-catenin or Beclin 1 levels after 24 hrs of P125A-endostatin treatment. These results further confirm a negative correlation between β-catenin and Beclin 1 levels during P125A-endostatin treatment. Likewise, the autocrine stimulation of Wnt did not greatly affect the basal level of autophagy vesicles in control group (Fig. 5E and F). Addition of P125A-endostatin to control cells induced >2-fold increase in autophagic vesicles. These results suggest that Wnt–β-catenin pathway is important for endostatin-mediated autophagy of endothelial cells.
Figure 5.

Effects of modulating canonical Wnt–β-catenin pathway on P125A-endostatin–induced autophagy. (A) HUVECs were co-transfected with pcDNA-expressing Wnt or pcDNA control (500 ng). Cells were treated with 20 μg/ml P125A-endostatin and analyzed for the levels of β-catenin, Beclin 1 or β-actin at the indicated time by Western blot. (B) HUVECs were infected with the Ad-Wnt at 1000 MOI. Ad-β-Gal was used as a negative control. P125A-endostatin–treated and control cells were lysed at different time-points and analyzed for β-catenin, Beclin 1 and β-actin levels by Western blot. (C) Densitometric analysis of Beclin 1 and β-catenin levels in control, pcDNA-expressing Wnt and pcDNA control–transfected cells following P125A-endostatin treatment. (D) Densitometric analysis of Beclin 1 and β-catenin levels in control, Ad-β-Gal and Ad-Wnt–infected cells following P125A-endostatin treatment. (E) The number of autophagic vesicles in HUVECs transfected with pcDNA-expressing Wnt and pcDNA control. Vesicles were quantified after 24 hrs of P125A-endostatin treatment (n= 3). (F) The number of autophagic vesicles in HUVECs infected with Ad-Wnt and Ad-β-Gal. Vesicles were quantified after 24 hrs of P125A-endostatin treatment (n= 3).
Figure 6.

Schematic illustration of the pathway for autophagy induction by endostatin in endothelial cells. Endostatin treatment reduces Bcl-2, BclxL and β-catenin levels while increasing the levels of Beclin 1. These events disrupt the physiological Bcl-2 (or Bcl-xL)/Beclin 1 ratio resulting in the induction of autophagy. In addition, β-catenin levels negatively correlate with Beclin 1 levels, and it is speculated that β-catenin regulates Beclin 1 either by altering its stability or by transcriptional repression.
Discussion
Autophagy has been characterized as a survival response as well as a pathway culminating in cell death. This pathway is normally induced under conditions of stress, such as nutritional or growth factor deprivation [30]. Our studies show that native and P125A-endostatin selectively induced autophagic responses in endothelial cells independent of nutritional stress. Normal fibroblast and even ovarian cancer cells, which are known to bind endostatin [7, 38], failed to elicit an autophagic response upon endostatin treatment. Therefore, binding of endostatin to integrin alone is not sufficient to induce autophagy. Downstream signalling pathways following integrin ligation may differ between the endothelial and tumour cells, which may account for the differences seen in the autophagic response to endostatin.
Endostatin is known to affect two cellular signalling pathways. High-affinity binding to the integrin α5β1 has been shown to induce clustering of endostatin–integrin complex in lipid rafts leading to Src kinase–dependent activation of p190RhoGAP. Subsequent inhibition of RhoA activity disrupts focal adhesion and ultimately affects endothelial cell migration [33]. The second pathway affected by endostatin involves low-affinity binding to glypican and interruption of Wnt-mediated signalling, resulting in the degradation of β-catenin independent of glycogen synthase kinase-3β (GSK-3β) [11, 34].
Because sustained autophagy can lead to cell death, the pathway is also referred to as programmed cell-death type II (PCD II) [30]. Alternatively, inhibiting apoptosis (PCD I) can also activate autophagy and vice versa [35]. Fewer than 10% of endothelial cells undergo apoptosis in response to endostatin (data not shown) suggesting that the majority of the endothelial cells resist death by activating a survival pathway. Endothelial cells possess many redundant survival pathways. For example, endothelial cells treated with chemotherapeutic agents such as carboplatin induce expression of VEGF, a survival growth factor for endothelial cells [36]. Recent studies using conditional knock-out of VEGF in endothelial cells have elucidated the importance of stress-induced autocrine stimulation of endothelial cells by VEGF [37]. Our studies provide evidence for another survival mechanism, autophagy, that is initiated in angiogenesis inhibitor–treated endothelial cells. In this respect, the effect of endostatin parallels the previous studies with kringle-5 (K5) of human plasminogen [20].
Although K5 and endostatin bind to distinct sets of target molecules and affect different signalling systems, both of them increased Beclin 1 levels in endothelial cells. Beclin 1 is involved in vesicle nucleation along with PI-3 kinase and is believed to be an early initiator of autophagy [23]. How Beclin 1 levels are modulated by endostatin treatment is not clear. Real-time PCR studies showed that the transcript levels change after endostatin treatment (data not shown). Although transcriptional activation can increase Beclin 1 levels, it does not rule out alternate mechanisms, such as increased protein stability. Because Beclin 1 is known to interact with Bcl-2 and Bcl-xL, we determined its association with these two anti-apoptotic proteins by immunoprecipitation. Bcl-2, Bcl-xL and Mcl-1 inhibit caspase activation and subsequent downstream apoptotic events by preventing the release of cytochrome c from the mitochondrial intermembrane space [38–40]. Although our data show that Beclin 1 sequestered Bcl-2 and Bcl-xL in the early stages of endostatin treatment, a distinct dissociation of Bcl-2 and Bcl-xL from the Beclin 1 complex was observed after 16 hrs, suggesting that endothelial cells may switch from a protective autophagic response to programmed cell death upon sustained exposure. This hypothesis is further supported by the increased caspase activation upon Beclin 1 knockdown using shRNA. The mediator of this conversion, however, remains unknown. Recent studies suggest that the balance between proapoptotic BH3-only proteins and anti-apoptotic Bcl-2 proteins determines the final outcome of programmed cell death. For example, a BH3-mimetic, ABT737, not only inhibited the interaction between Beclin 1 and Bcl-2–Bcl-xL but also induced apoptosis when the intracellular level of Beclin 1 was reduced [16]. It is tempting to speculate that angiogenesis inhibitors impinge upon signalling pathways that regulate the interaction of Beclin 1 with Bcl-2–Bcl-xL. This regulation may further control the switch between autophagy and apoptosis in endothelial cells.
Endostatin is known to affect the Wnt-mediated signalling pathway by down-regulating β-catenin levels via an unidentified GSK-3β–independent mechanism [11]. GSK-3β plays a critical role in cell proliferation, differentiation and apoptosis. In addition, GSK-3β also regulates the degradation of gene expression regulators, particularly, β-catenin, one of the main effectors of the Wnt signalling pathway. The binding of Wnt to Frizzled activates Dishevelled (Dsh), often antagonizes GSK-3β activities, and leads to the stabilization of β-catenin [41]. Although endostatin directly inhibited the Wnt–β-catenin signalling pathway, the effect was found to be independent from GSK-3β regulations [11]. Therefore, our studies focussed on the correlation between the expression levels of β-catenin and autophagy in endostatin-treated cells. Consistent with previous studies, we found that the treatment of endothelial cells with endostatin (P125A-endostatin and native endostatin) reduced β-catenin levels in endothelial cells. Furthermore, we showed that β-catenin levels negatively correlated with Beclin 1 levels and autophagy. It is not clear whether β-catenin directly binds Beclin 1 and modulates its levels in the cytoplasm. This is currently under investigation. Exogenous stimulation of Wnt pathway, however, increased β-catenin levels and prevented endostatin-induced increase in Beclin 1. As a consequence, the activation of Wnt pathway significantly inhibited autophagy. Furthermore, siRNA to β-catenin was found to potentiate the effects of endostatin by increasing Beclin 1 levels and autophagy. Elucidating the mechanisms behind this inverse relationship between β-catenin and Beclin 1 could reveal novel targets that can be modulated to improve the effects of angiogenesis inhibitors. In conclusion, our studies suggest that endothelial cells mount an autophagic survival response to endostatin, and inhibition of autophagy is required to channel them towards apoptotic cell death. Thus, concomitant inhibition of autophagy may be a useful strategy to increase the potency and efficacy of endostatin treatment.
Acknowledgments
We thank Dr. Dell’Angelica for the LAMP1-GFP plasmid. We also thank John Oja, Jerry Sedgewick, Julia Nguyen, Dr. Sabita Roy, Dr. Robert Hafner, Dr. Yumi Yokoyama, Dr. Michael Olin and Dr. Paul Marker for technical help and discussion. This work was supported in part by a grant from the NIH CA114340, NIH DA11806, Academic Health Center of the University of Minnesota and Sparboe Endowment for Women’s Cancer Research (SR). A.K. was supported by a grant from the University of Minnesota Cancer Center and the Regis Foundation.
Supporting Information
Fig. S1 Endostatin induces autophagy in endothelialcells. (A) Representative confocal images ofLAMP1-GFP–transfected HUVECs (upper panel) andLC3-GFP–transfected HUVECs (lower panel) treated withP125A-endostatin (20 μg/ml). Native endostatin (20 μg/ml) wasused under similar conditions. (B)LAMP1-GFP–transfected HUVECs were treated with differentconcentrations of P125A-endostatin (▴) (n= 6,*P <0.05,**P < 0.001) or recombinant, nativeendostatin (20 μg/ml) (□). Rapamycin (█) wasused as a positive control. Results are shown as mean ± S.E.(C) LAMP1-GFP–transfected fibroblasts were treatedwith P125A-endostatin (20 μg/ml). Etoposide (10 μg/ml) wasused as a positive control.
Fig. S2 Effect of integrin antibodies and down-regulationof integrin sub-units on endostatin-induced autophagy. (A)The number of LAMP-1-positive vesicles per cell treated withcontrol IgG was used as the basal level to calculate fold change inthe antibody-treated groups. Autophagy induction was partiallyinhibited by monoclonal antibody against integrinα5β1 (15 μg/ml)(n= 3,*P < 0.05). (B) HUVECs weretransfected with LAMP1-GFP and siRNA specific to integrinα5 and integrin β1. Transfectedcells were treated with P125A-endostatin for 24 hrs and assessedfor autophagic vesicles per cell (n = 3, P< 0.001). (C) Effect of integrin antibodies and integrindown-regulation on endostatin-induced autophagy. FACS analysis dataof α5β1 levels in HUVECstransfected with siRNA α5 and β1.Cells were transfected and analyzed as previously described[33]. Mouse IgG1 was used as a control.(D) Effect of 3-MA on P125A-endostatin–inducedautophagy in HUVECs. 3-MA (5 mM) was added toLAMP1-GFP–transfected HUVECs culture 12 hrs into theexperiment (n = 3, *P < 0.05,**P < 0.001). (E) LAMP1-GFP wasco-transfected with shRNA specific to Beclin 1 or control scrambledshRNA to HUVECs [20, 43]. Results are shown asmean ± S.E. Statistical significance was determined usingStudent’s t-test (*P < 0.05,**P < 0.001). (F)P125A-endostatin–induced autophagy in HUVECs is Beclin 1/PI-3kinase dependent. Knocking down Beclin 1 expression in HUVECs isshown. HUVECs were transfected with shRNA specific to Beclin 1 orcontrol scrambled shRNA as previously described [20,32]. Total cell lysates were evaluated for Beclin 1expression.
Fig. S3 Beclin 1 down-regulation in HUVECs potentiatesP125-endostatin–induced apoptosis. Transfected HUVECs weregated for DsRed-positive cell populations and scored forFAM-VAD-FMK–positive cells. (A) Histogram showsFAM-VAD-FMK–positive cells in the control, scrambledshRNA–transfected and shRNA specific to Beclin1–transfected HUVECs. (B) Histogram ofP125A-endostatin–treated HUVECs that were transfected witheither shRNA specific to Beclin 1 or scrambled shRNA.
Fig. S4 (A) The number of LAMP1-GFP vesicles persiRNA or scrambled siRNA–treated cell was quantified asdescribed previously. Data are shown as fold changes in the numberof autophagic vesicles in treated cells compared with scrambledsiRNA–transfected cells (n = 3). The effectof a dominant negative β-catenin construct onP125A-endostatin–induced autophagy is shown. (B)HUVECs were co-transfected with pcDNA control (□) orpcDNA-expressing dominant negative β-catenin (█).Cells were treated with 20 μg/ml P125A-endostatin for 24 hrs andanalyzed for β-catenin, Beclin 1 or β-actin levels byWestern blot. (C) The number of autolysosomes in treatedcells was quantified as described previously. Data were presentedas fold increase in the number of vesicles per cell in controltransfectant or dominant negative construct of β-catenintransfectant as compared with control cells. Data represent mean± S.E. (*P = 0.033).
Fig. S5 Effect of modulation of β-catenin levels onP125A-endostatin–induced autophagy. (A) The effect ofP125A-endostatin on β-catenin levels in HUVECs. Lysates fromtreated HUVECs were collected at different time-points and analyzedfor β-catenin levels by Western blot. (B) HUVECs wereinfected with 50 or 500 multiplicity of infection (MOI) ofAd-CMV-β-catenin and subsequently treated withP125A-endostatin. Ad-CMV-GFP was used as a negative control.Expression levels of Beclin 1 were measured by flow cytometry aftera 24-hr treatment. Fold increase in β-catenin levels wascalculated by comparing β-catenin levels ofAd-CMV-β-catenin–transduced HUVECs againstAd-CMV-GFP–transduced cells.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
References
- 1.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–86. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 3.O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 4.Abdollahi A, Hahnfeldt P, Maercker C, et al. Endostatin’s antiangiogenic signaling network. Mol Cell. 2004;13:649–63. doi: 10.1016/s1097-2765(04)00102-9. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi N, Anand-Apte B, Lee M, et al. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J. 1999;18:4414–23. doi: 10.1093/emboj/18.16.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dhanabal M, Ramchandran R, Waterman MJ, et al. Endostatin induces endothelial cell apoptosis. J Biol Chem. 1999;274:11721–6. doi: 10.1074/jbc.274.17.11721. [DOI] [PubMed] [Google Scholar]
- 7.Yokoyama Y, Ramakrishnan S. Improved biological activity of a mutant endostatin containing a single amino-acid substitution. Br J Cancer. 2004;90:1627–35. doi: 10.1038/sj.bjc.6601745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokoyama Y, Ramakrishnan S. Binding of endostatin to human ovarian cancer cells inhibits cell attachment. Int J Cancer. 2007;121:2402–9. doi: 10.1002/ijc.22935. [DOI] [PubMed] [Google Scholar]
- 9.Sudhakar A, Sugimoto H, Yang C, et al. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci USA. 2003;100:4766–71. doi: 10.1073/pnas.0730882100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 10.Wickstrom SA, Alitalo K, Keski-Oja J. Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res. 2002;62:5580–9. [PubMed] [Google Scholar]
- 11.Hanai J, Gloy J, Karumanchi SA, et al. Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol. 2002;158:529–39. doi: 10.1083/jcb.200203064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoppler S, Kavanagh CL. Wnt signalling: variety at the core. J Cell Sci. 2007;120:385–93. doi: 10.1242/jcs.03363. [DOI] [PubMed] [Google Scholar]
- 13.Nor JE, Christensen J, Mooney DJ, et al. Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol. 1999;154:375–84. doi: 10.1016/S0002-9440(10)65284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nor JE, Christensen J, Liu J, et al. Up-Regulation of Bcl-2 in microvascular endothelial cells enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res. 2001;61:2183–8. [PubMed] [Google Scholar]
- 15.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pattingre S, Levine B. Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res. 2006;66:2885–8. doi: 10.1158/0008-5472.CAN-05-4412. [DOI] [PubMed] [Google Scholar]
- 18.Chau YP, Lin SY, Chen JH, et al. Endostatin induces autophagic cell death in EAhy926 human endothelial cells. Histol Histopathol. 2003;18:715–26. doi: 10.14670/HH-18.715. [DOI] [PubMed] [Google Scholar]
- 19.Yokoyama Y, Dhanabal M, Griffioen AW, et al. Synergy between angiostatin and endostatin: inhibition of ovarian cancer growth. Cancer Res. 2000;60:2190–6. [PubMed] [Google Scholar]
- 20.Bui Nguyen TM, Subramanian IV, Kelekar A, et al. Kringle 5 of human plasminogen, an angiogenesis inhibitor, induces both autophagy and apoptotic death in endothelial cells. Blood. 2007;109:4793–802. doi: 10.1182/blood-2006-11-059352. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–9. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004;313:453–8. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 23.Kelekar A. Autophagy. Ann N Y Acad Sci. 2006;1066:259–71. doi: 10.1196/annals.1363.015. [DOI] [PubMed] [Google Scholar]
- 24.Eskelinen EL, Tanaka Y, Saftig P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13:137–45. doi: 10.1016/s0962-8924(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 25.Kirkegaard K, Taylor MP, Jackswon WT. Cellular autography: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–14. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanida I, Minematsu-Ikeguchi N, Ueno T, et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 28.Brown CR, McCann JA, Hung GG, et al. Vid22p, a novel plasma membrane protein, is required for the fructose-1,6-bisphosphatase degradation pathway. J Cell Sci. 2002;115:655–66. doi: 10.1242/jcs.115.3.655. [DOI] [PubMed] [Google Scholar]
- 29.Rehn M, Veikkola T, Kukk-Valdre E, et al. Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci USA. 2001;98:1024–9. doi: 10.1073/pnas.031564998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 31.Hendil KB, Lauridsen AM, Seglen PO. Both endocytic and endogenous protein degradation in fibroblasts is stimulated by serum/amino acid deprivation and inhibited by 3-methyladenine. Biochem J. 1990;272:577–81. doi: 10.1042/bj2720577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abedin MJ, Wang D, McDonnell MA, et al. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–10. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- 33.Wickstrom SA, Alitalo K, Keski-Oja J. Endostatin associates with lipid rafts and induces reorganization of the actin cytoskeleton via down-regulation of RhoA activity. J Biol Chem. 2003;278:37895–901. doi: 10.1074/jbc.M303569200. [DOI] [PubMed] [Google Scholar]
- 34.Karumanchi SA, Jha V, Ramchandran R, et al. Cell surface glypicans are low-affinity endostatin receptors. Mol Cell. 2001;7:811–22. doi: 10.1016/s1097-2765(01)00225-8. [DOI] [PubMed] [Google Scholar]
- 35.Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 36.Wild R, Dings RP, Subramanian I, et al. Carboplatin selectively induces the VEGF stress response in endothelial cells: potentiation of antitumor activity by combination treatment with antibody to VEGF. Int J Cancer. 2004;110:343–51. doi: 10.1002/ijc.20100. [DOI] [PubMed] [Google Scholar]
- 37.Lee S, Chen TT, Barber CL, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–32. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 39.Kluck RM, Bossy-Wetzel E, Green DR, et al. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–6. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 40.Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 41.Taketo MM. Shutting down Wnt signal-activated cancer. Nat Genet. 2004;36:320–2. doi: 10.1038/ng0404-320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item