

Abstract
Hyaluronan (HA) is a key molecule of the extracellular matrix that is thought to be critically involved in both atherosclerosis and restenosis. Recently, it has been demonstrated that the cyclooxygenase (COX) products, prostacyclin and prostaglandin E2, induce HA synthesis in vitro by transcriptional up-regulation of HA-synthase 2 (HAS2) and HAS1. The relative roles in atherosclerotic and restenotic artery disease of tissue specifically expressed COX-1 and COX-2 are still under debate. Thus, the present study aimed to investigate the effect of COX isoform inhibition on HA-accumulation and regulation of HAS isoform expression in two models of pathologic artery remodelling in vivo. Firstly, ApoE-deficient mice were treated with a prototypic isoform non-selective inhibitor, indomethacin or with a prototypic COX-2 selective inhibitor, rofecoxib, for 8 weeks. Aortic HAS mRNA expression and HA-accumulation in atherosclerotic aortic root lesions were analyzed. Secondly, neointimal hyperplasia was induced by carotid artery ligation in ApoE-deficient mice on a high fat diet and the effects of the COX inhibitors were determined after 4 weeks of treatment. Intimal HA-accumulation was markedly reduced in both models by indomethacin and rofecoxib. This coincided with a strong inhibition of HAS1 mRNA expression in both models and with decreased HAS2 mRNA in the aorta of ApoE-deficient mice. HAS3 was not affected. The repression of HA-accumulation by both COX-2 selective and non-selective COX inhibition implicates COX-2 in the regulation of HA synthesis via stimulation of HAS1 and HAS2 expression in vivo. Modulation of vascular HA-accumulation might play a role in chronic effects of COX inhibitors on the progression of atherosclerosis.
Keywords: hyaluronan, extracellular matrix, vascular remodelling, cyclooxygenase inhibitors, atherosclerosis, neointimal hyperplasia
Introduction
Hyaluronic acid (HA) is an unbranched, unsulfated carbohydrate component of the extracellular matrix that is synthesized at the plasma membrane by three HA-synthase isoforms (HAS1,-2,-3). HA mediates cellular signalling via HA-receptors such as RHAMM, CD44, LYVE-1 and toll-like receptors [1]. During the last decade, strong evidence evolved that HA is a key regulator of tumour growth and metastasis [2]. In arterial blood vessels, the expression of HA is mainly confined to the adventitia and the endothelial glycocalyx. During atherosclerosis, atherothrombosis and restenosis, HA is strongly induced and associates with proliferation of vascular smooth muscle cells (VSMC), neointimal expansion and possibly inflammation [3, 4]. HA is therefore thought to promote atherogenesis and neointimal hyperplasia [5]. Although many factors have been shown to stimulate HA synthesis in vitro[6, 7], little is known about the key factors regulating HA-accumulation in vivo and effects of drugs on cardiovascular HA-accumulation have not been studied yet. With respect to the specific functions of HAS-isoforms, it is known from HAS2-deficient mice that HAS2-mediated HA synthesis is critical for heart development and that deletion of HAS2 causes embryonic lethality [8]. In contrast, HAS1- and HAS3-deficient mice are viable. In adults, it is not known yet whether the three HAS-isoforms serve specific functions in the cardiovascular system and/or the pathophysiology of cardiovascular disease. We have recently observed that prostacyclin (PGI2) and prostaglandin E2 (PGE2) markedly induce HAS2 and HAS1 expression in cultured human VSMC [9, 10]. Cyclooxygenase 1 (COX-1) and COX-2 are constitutively expressed in endothelial cells, whereas COX-2 is strongly induced in VSMC by many of the major pro-atherogenic mediators such as PDGF-BB, cytokines, thrombin and oxidized LDL [11]. Therefore, we hypothesize that prostaglandins could indeed be key regulators of sustained neointimal HA-synthesis.
Because non-steroidal anti-inflammatory drugs (NSAID) that inhibit COX-dependent prostaglandin synthesis are widely used, this regulatory pathway might be of clinical relevance. Furthermore, in the light of the ongoing discussion about adverse cardiovascular effects of COX-2 inhibition, it will be important to consider also chronic effects on plaque remodelling [12]. Therefore, the role of COX products specifically in vascular HA synthesis was assessed in murine models of accelerated atherosclerosis and neointimal hyperplasia using the two prototypic non-isoform selective and COX-2-selective inhibitors, indomethacin and rofecoxib.
Materials and methods
Animals and experimental design
Male ApoE–/– mice were obtained from Taconic M&B (Denmark) and kept on normal chow diet with or without 3 mg indomethacin or 50 mg rofecoxib per kg and day. Indomethacin from Sigma (Deisenhofen, Germany) and rofecoxib (Vioxx® tablets) were pelleted into the chow. ApoE-deficient mice were used in two disease models. First, HA-synthesis in atherosclerosic lesions was analyzed in ApoE-deficient mice receiving indomethacin or rofecoxib from 15 weeks to 23 weeks of age on normal chow (Fig. 1A). Second, ApoE-deficent mice underwent ligation of the left common carotid artery [13] to induce neointimal hyperplasia. Following carotid artery ligation, these mice were fed a Western diet (21% butter fat and 0.15% cholesterol) with or without the COX inhibitors for 4 weeks (Fig. 1B). All experiments were performed according to the guidelines for the use of experimental animals as given by the Deutsches Tierschutzgesetz and the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health.
Figure 1.
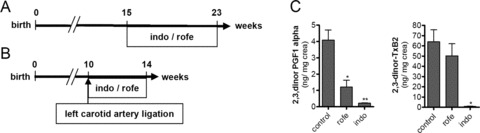
Experimental design. (A) ApoE-deficient mice were treated with indomethacin (3 mg/kg/day) or rofecoxib (50 mg/kg/day) for 8 weeks beginning at 15 weeks of age on normal chow. (B) Neointimal hyperplasia in the left carotid artery was induced by permanent ligation at the age of 10 weeks in ApoE-deficient mice. Starting with the ligation animals were fed Western diet and treated with indomethacin or rofecoxib as described in (A). (C) Urinary excretion of the prostacyclin (PGI2) metabolite (2,3-dinor-6-keto PGF1α) and the thromboxane (Tx) A2 metabolite (2,3-dinor TxB2) was measured by tandem mass spectroscopy to monitor COX-2 and platelet COX-1 activity, respectively.
Urinary prostaglandin metabolites
Circulating PGI2 is rapidly converted into 2,3-dinor-6-keto PGF1α and TxA2 is rapidly hydrolyzed to form TxB2. The predominant urinary metabolite in mice is the β oxidation product 2,3-dinor TxB2. TxB2 and 2,3-dinor-6-keto PGF1α were measured by isotope dilution liquid chromatography tandem mass spectrometry (LC-MS/MS).
Urine of the mice was collected over a period of 48 hrs. A 200 μl sample of urine was spiked with 4 ng of d3–2,3-dinor-6-keto PGF1α and 2/ng of 18O-2,3-dinor TXB2 and the methoximine derivative was formed by addition of 125 mg methoxamine hydrochloride in water (30 min., RT). The sample was purified by solid phase extraction (SPE) cartridge (StrataX 8B-S100-TAK, Phenomenex, Torrance, CA). The chromatography was performed on a Hypersil 3 μm C18 (150 × 2.0 mm) analytical column (Phenomenex) using water and acetonitrile/methanol (95:5) as the mobile phase. A gradient of 10–38% mobile phase B was run over 30 min. at a flow rate of 0.200 ml/min. Tandem mass spectrometry (Quantum Ultra, ThermoFinnigan, San Jose, CA) was performed using negative atmospheric pressure ionization and multiple reaction monitoring of the ion transitions m/z 370→155 (2,3-dinor TxB2), m/z 374→155 (18O2–2,3-dinor TxB2), m/z 370→232 (2,3-dinor-6-keto PGF1α) and m/z 373→235 (d3–2,3-dinor-6-keto PGF1α). Quantification of the endogenous metabolites used the ratio of the peak areas of the analyte and internal standard. Data were corrected for urinary creatinine (Oxford Biomedical Research, Oxford, MI).
RNA isolation
The total thoracic and abdominal aorta was separated from adventitia and frozen in liquid nitrogen. The whole common carotid artery was dissected between the aortic arch and the suture 4 weeks after ligation and also separated from adhering tissue. Total RNA was prepared using TRI-Reagent (Sigma). The RNA was quantified by spectroscopic analysis at 260 nm.
Characterization of mRNA expression
The expression levels were analyzed by quantitative real-time RT-PCR. Aliquots of total RNA (1000 ng) were applied for cDNA-synthesis using Superscript-III First-Strand synthesis system for RT-PCR (Invitrogen, Karlsruhe, Germany). Real-time RT-PCR analysis of mouse HAS1–3 and GAPDH was performed as described previously [14].
Immunohistochemical analysis
Whole hearts were fixed in Lillie’s neutral buffered 4% formaline solution and 3 μm paraffin sections of the aortic root were prepared for immunohistochemical stainings. Sections were stained for HA with biotinylated HA binding protein (2 ng/μl; Seikagaku, Tokyo, Japan) and HRP-conjugated streptavidin (1:200, Sigma) or FITC-conjugated streptavidin (1:200, Dako, Hamburg, Germany). SMC were detected with a monoclonal mouse anti-α-SM-actin clone 1A4 (1:1000, Sigma) and macrophages by an antibody against mac2 (1:400, Cedarlane, Burlington, Canada) and a rhodamine red™-X-conjugated goat anti-rat IgG (preabsorbed to rodent, 1:400, Jackson ImmunoResearch, Suffolk, UK) used as a secondary antibody. Non-fluorescent detection was performed using diaminobenzidine (Zytomed, Berlin, Germany) as a chromogen.
Histochemical stainings
Three-micrometre paraffin sections of the aortic root were stained for collagen accumulation by picrosirius red. Qualitative analysis of collagen deposition was performed using polarized light microscopy.
Image analysis
Bright-field images were taken using a ColorViewII and AnalySIS 3.2 software (Soft Imaging System, Münster, Germany) and analyzed by ImageJ 1.37v software (NIH) using the colour deconvolution technique and threshold values as described earlier [14].
Statistical analysis
Data are presented as the mean ± S.E.M. Statistical analysis was performed by unpaired t-test or one-way anova followed by comparison of selected pairs (Bonferroni). A value of P <0.05 was considered significant.
Resuπlts
HA-accumulation in atherosclerotic plaques
Mass spectrometric quantitation of urinary thromboxane A2 (TxA2) metabolite (2,3-dinor-TxB2), an index of platelet COX-1 activity, revealed complete depression by indomethacin (P < 0.05) and no effect of rofecoxib (Fig. 1C). PGI2 biosynthesis as assessed by quantitation of its urinary metabolite, 2,3-dinor-6-keto-PGF1α, was depressed by >90% by indomethacin (P < 0.05) and by 75% by rofecoxib (P < 0.05). Roughly, 70% of PGI2 formation is COX-2 dependent in mice [15]. Thus, rofecoxib acted, indeed, as selective inhibitor of COX-2 at our dosing regimen, whereas indomethacin inhibited – as expected – both isoenzymes (Fig. 1C). The overall condition of mice, including body weight and plasma levels of IL6, MCP1 and hsCRP, was not affected by indomethacin or rofecoxib (data not shown).
Plaque size at the aortic root was not changed by treatment with rofecoxib and indomethacin (not shown). However, treatment with both rofecoxib and with indomethacin resulted in decreased levels of HA in atherosclerotic plaques as determined by affinity histochemistry (Fig. 2A–D). Quantitative real-time RT-PCR (qRT-PCR) revealed significant inhibition of HAS1 mRNA and HAS2 mRNA expression in the thoracic aorta by indomethacin and rofecoxib (Fig. 2E). Furthermore, although HAS3 expression is not responsive to prostaglandins, a trend towards reduced mRNA expression was observed in response to both COX inhibitors. The reduction of HA-accumulation, HAS1 and HAS2 expression was paralleled by changes in the cellular composition of plaques. Alpha-SM-positive SMC in the caps of aortic root lesions were increased and mac2-positive macrophages were significantly decreased after indomethacin but not rofecoxib treatment (Fig. 3), as would be expected with COX-1 inhibition [16].
Figure 2.
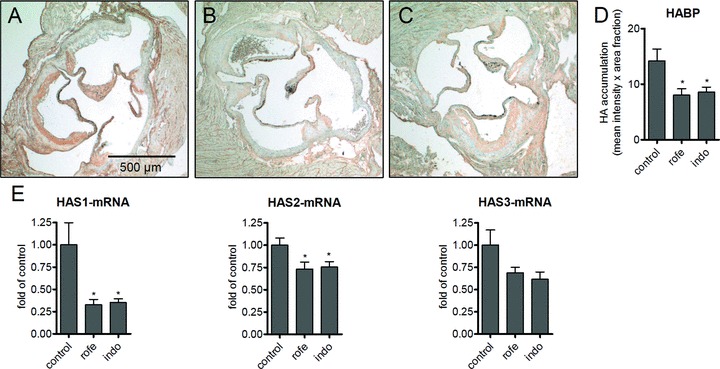
Aortic root lesions of ApoE-deficient mice stained for HA (A–D). (A) Control; (B) indomethacin (8 weeks, 3 mg/kg/day); (C) rofecoxib (8 weeks, 50 mg/kg/day); (D) densitometric quantitation of HA affinity histochemistry; (E) aortic mRNA expression of HAS isoforms; n= 15–12, mean ± S.E.M., *P < 0.05.
Figure 3.
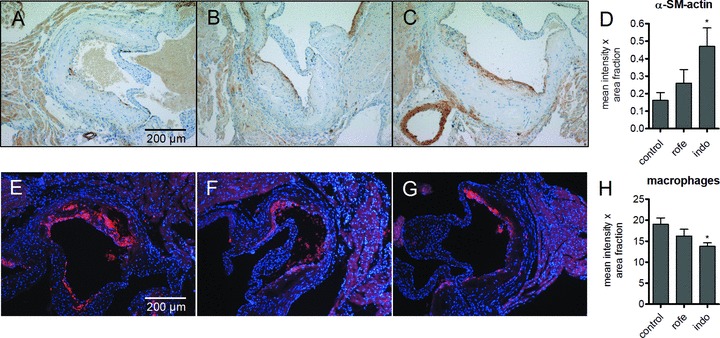
VSMC (α-SM actin, A–D) and macrophage (mac2, E–H) content of aortic root lesions of ApoE-deficient mice after treatment with COX inhibitors. A, E, controls; B, F indomethacin (8 weeks, 3 mg/kg/day); C, G, rofecoxib (8 weeks, 50 mg/kg/day); densitometric quantitation of D, α-SM actin and H, mac2; n= 12, mean ± S.E.M., *P < 0.05.
To investigate whether other matrix molecules were regulated as well by inhibition of COX-1 and COX-2 collagen accumulation was analyzed by picrosirius red staining. In aortic root plaques, collagen abundance was not affected by the treatment. As an indication for the arrangement and packing of collagen fibrils the picrosirius red staining was also analyzed and quantified by birefringence analysis under polarized light (Fig. 4). However, arrangement of collagen was also not changed in response to COX inhibitors, suggesting that the observed changes in HA-accumulation and HAS-isoform expression are indeed specific responses and not a general effect on matrix remodelling.
Figure 4.
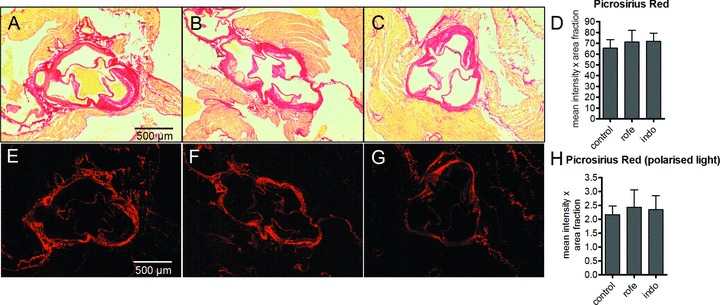
Collagen content of aortic root lesions of ApoE-deficient mice indicated by picrosirius red staining after treatment with COX inhibitors. Total collagen was evaluated by light microscopy (A–D) and collagen arrangement was analyzed by birefringence analysis using polarized light (E–H). A, E, controls; B, F indomethacin (8 weeks, 3 mg/kg/day); C, G, rofecoxib (8 weeks, 50 mg/kg/day); densitometric quantitation of D, picrosirius red staining, light microscopy and H, picrosirius red staining under polarized light to visualize densely packed collagen (bright red); n= 12, mean ± S.E.M., *P < 0.05.
HA-accumulation after ligation injury
Rapid induction of neointimal hyperplasia occurs in ApoE-deficient mice after ligation of the carotid artery [13]. In this model, neointimal hyperplasia is driven mainly by SMC proliferation and migration and was used in comparison with the chronic model of atherogenesis as detailed earlier. Neointimal hyperplasia was significantly reduced by indomethacin that corresponded to decreased frequency of thrombotic occlusion of ligated carotid arteries (not shown). Arteries with thrombus formation were excluded from further analysis. In contrast, rofecoxib had no effect on neointima formation (not shown).
HA-accumulation was detected in the luminal part of the neointima and found to be strongly reduced by treatment with rofecoxib and indomethacin (Fig. 5A–D). Furthermore, only HAS1 mRNA expression was dramatically reduced by both COX inhibitors (Fig. 5E), whereas HAS2 was not changed. Moreover, indomethacin resulted in increased staining of alpha SM-actin in neointimal hyperplasia (Fig. 5F–I) similar to the effects observed in atherosclerotic lesions (compare to Fig. 3). Macrophage accumulation as evidenced by immunohistochemistry of mac2 was not responsive to COX inhibition in the carotid injury model (Fig. 6). Similar to the results in atherosclerotic plaques, the amount collagen deposited in the neointima was not affected (not shown).
Figure 5.
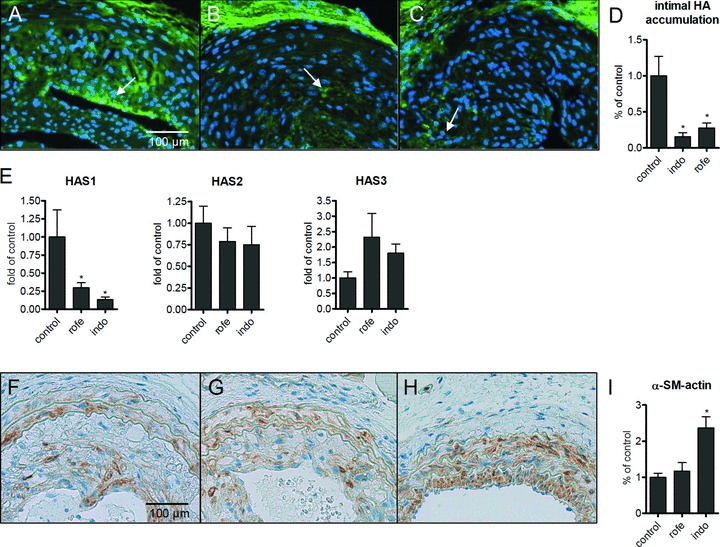
HA-accumulation in neointimal hyperplasia 4 weeks after ligation of the left carotid artery of ApoE-deficient mice on a Western diet. (A) Control; (B) indomethacin (4 weeks, 3 mg/kg/day); (C) rofecoxib (4 weeks, 50 mg/kg/day); (D) densitometric quantitation of HA-affinity histochemistry in the neointima; (E) mRNA expression of HAS isoforms in ligated carotid arteries; (F–H) α-SM-actin staining; densitometric quantification of α-SM-actin; n= 4–7; mean ± S.E.M., *P < 0.05.
Figure 6.
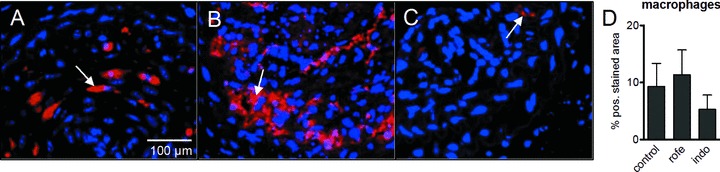
Macrophage staining (mac2) in neointimal tissue 4 weeks after ligation of the left internal carotid artery in ApoE-deficient mice on Western diet. (A) Control; (B) indomethacin (4 weeks, 3 mg/kg/day); (C) rofecoxib (4 weeks, 50 mg/kg/day); (D) densitometric quantitation of mac2 within the neointima; n= 4–7; mean ± S.E.M., *P < 0.05.
Discussion
The biology of the COXs in vascular lesions is not well understood. Multiple cell types such as macrophages, VSMC and endothelial cells expressing COX-1, COX-2 or both interact during the development of atherosclerosis. These cell types are thought to have distinct prostaglandin biosynthesis profiles depending on the presence of, for example, prostaglandin synthase, thromboxane A2 synthase and prostacyclin synthase. Furthermore, the response to these prostanoids is dependent on the prostanoid receptor sub-type expression in the respective cell types. Although COX-1–derived TxA2 accelerates atherogenesis in mice [17], COX-2 deletion or inhibition may accelerate [18], retard [19] or leave lesion progression unaltered [20] in mice. These conflicting observations may perhaps be explained by the contrasting cardiovascular effects of COX-2 products derived from the distinct cellular sources as detailed earlier and their relative importance in distinct phases of the disease. The specific aim of the present study was to evaluate whether COX inhibition affects HA synthesis and HA accumulation in vascular lesions. HA is widely induced during the development of atherosclerosis and also after vascular injury in association with proliferating VSMC [3, 21, 22]. Therefore, both atherosclerotic lesions and experimentally induced neointimal hyperplasia were analyzed in ApoE–/– mice. Vascular HAS1 expression and HA-accumulation were markedly inhibited by treatment with both COX inhibitors at biochemically defined doses after both chronic treatment in ApoE–/– mice and after ligation injury to the carotid artery. Aortic HAS2 mRNA levels were significantly reduced only in ApoE–/– mice after 8 weeks of rofecoxib and indomethacin treatment, however, to a lesser extent compared with HAS1. Furthermore, HAS2 was not significantly reduced in the ligation injury model, despite the marked induction of HAS2 by PGs in VSMC in vitro[9, 10]. The present data therefore suggest that HAS2 expression is less dependent on COX function in vivo compared with HAS1. HAS2 is known to be strongly up-regulated by a variety factors present during atherogenesis and arterial injury such as PDGF-BB, thrombin, TGFβ1 and EGF [7, 10, 23, 24]. Therefore, the smaller effect of COX inhibition on HAS2 mRNA levels might reflect the relative importance of the other factors sustaining HAS2 expression during atherogenesis. Thus, the combined evidence supports that prostaglandins are indeed regulators of HA synthesis and particularly important for HAS1 expression during lesion formation. The repression of HA synthesis by both non-selective (indomethacin) and selective inhibition of COX-2 (rofecoxib) to almost the same extent suggests that COX-2–mediated prostaglandin synthesis is critical. Because the blood pressure of mice on a C57Bl6 background is not responsive to COX-2 deletion [25], an increase in blood pressure is unlikely as an additional factor involved in the decrease of HA-accumulation after pharmacologic COX inhibition.
HA-accumulation and HA synthesis have been associated with cellular functions and plaque morphology that favoured a pro-atherosclerotic role of HA during atherosclerosis. This is supported by the finding that over-expression of HAS2 accelerates atherosclerosis in ApoE-deficient mice [26]. Therefore, inhibition of HAS1 and HAS2 expression might cause a change in HA content that might be anti-atherogenic in the long term. Of note, in both models used in this study increased αSM-actin staining suggested increased numbers of VSMC and/or more differentiated VSMC within the lesions after treatment with indomethacin. Furthermore, macrophages were reduced in ApoE–/–-mice by indomethacin possibly reflecting an anti-inflammatory effect that was paralleled by a trend towards reduced HAS3 mRNA expression observed in response to both COX inhibitors. Because HAS3 is responsive to macrophage-derived cytokines such as interleukin-1β[10], it might be of interest to investigate the role of macrophages in HAS3-mediated HA-synthesis during atherosclerosis in future studies.
The present findings support strongly the conclusion that chronic administration of COX inhibitors interfere with ECM remodelling and specifically with HA matrix synthesis and accumulation in atherosclerotic lesions. These effects on HA might affect progression and stability of atherosclerotic lesions and should be considered as a potential additional facet of chronic COX inhibitor treatment. Clearly, further studies are needed to address the roles of HAS1/-2 and the COXs during atherosclerosis in detail. This seems particularly important given that a large-scale trial to address potential cardioprotective effects of a COX-2 inhibitor is ongoing – the Prospective Randomized Evaluation of Celecoxib Integrated Safety vs. Ibuprofen or Naproxen (PRECISION) (ClinicalTrials.gov Identifier: NCT00346216).
Acknowledgments
Funded by Deutsche Forschungsgemeinschaft, SFB 612, TPB9.
References
- 1.Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435–61. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 2.Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–39. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 3.Riessen R, Wight TN, Pastore C, et al. Distribution of hyaluronan during extracellular matrix remodeling in human restenotic arteries and balloon-injured rat carotid arteries. Circulation. 1996;93:1141–7. doi: 10.1161/01.cir.93.6.1141. [DOI] [PubMed] [Google Scholar]
- 4.Cuff CA, Kothapalli D, Azonobi I, et al. The adhesion receptor CD44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. J Clin Invest. 2001;108:1031–40. doi: 10.1172/JCI12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toole BP, Wight TN, Tammi MI. Hyaluronan-cell interactions in cancer and vascular disease. J Biol Chem. 2002;277:4593–6. doi: 10.1074/jbc.R100039200. [DOI] [PubMed] [Google Scholar]
- 6.Fischer JW, Schrör K. Regulation of hyaluronan synthesis by vasodilatory prostaglandins. Implications for atherosclerosis. Thromb Haemost. 2007;98:287–95. [PubMed] [Google Scholar]
- 7.Evanko SP, Johnson PY, Braun KR, et al. Platelet-derived growth factor stimulates the formation of versican-hyaluronan aggregates and pericellular matrix expansion in arterial smooth muscle cells. Arch Biochem Biophys. 2001;394:29–38. doi: 10.1006/abbi.2001.2507. [DOI] [PubMed] [Google Scholar]
- 8.Camenisch TD, Spicer AP, Brehm-Gibson T, et al. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106:349–60. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sussmann M, Sarbia M, Meyer-Kirchrath J, et al. Induction of hyaluronic acid synthase 2 (HAS2) in human vascular smooth muscle cells by vasodilatory prostaglandins. Circ Res. 2004;94:592–600. doi: 10.1161/01.RES.0000119169.87429.A0. [DOI] [PubMed] [Google Scholar]
- 10.van den Boom M, Sarbia M, von Wnuck Lipinski K, et al. Differential regulation of hyaluronic acid synthase isoforms in human saphenous vein smooth muscle cells: possible implications for vein graft stenosis. Circ Res. 2006;98:36–44. doi: 10.1161/01.RES.0000199263.67107.c0. [DOI] [PubMed] [Google Scholar]
- 11.Pritchard KA, Jr, O’Banion MK, Miano JM, et al. Induction of cyclooxygenase-2 in rat vascular smooth muscle cells in vitro and in vivo. J Biol Chem. 1994;269:8504–9. [PubMed] [Google Scholar]
- 12.Funk CD, FitzGerald GA. COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol. 2007;50:470–9. doi: 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- 13.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–44. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 14.Dai G, Freudenberger T, Zipper P, et al. Chronic ultraviolet B irradiation causes loss of hyaluronic acid from mouse dermis because of down-regulation of hyaluronic acid synthases. Am J Pathol. 2007;171:1451–61. doi: 10.2353/ajpath.2007.070136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudic RD, Brinster D, Cheng Y, et al. COX-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005;96:1240–7. doi: 10.1161/01.RES.0000170888.11669.28. [DOI] [PubMed] [Google Scholar]
- 16.Cyrus T, Yao Y, Ding T, et al. Thromboxane receptor blockade improves the antiatherogenic effect of thromboxane A2 suppression in LDLR KO mice. Blood. 2007;109:3291–6. doi: 10.1182/blood-2006-08-044990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi T, Tahara Y, Matsumoto M, et al. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114:784–94. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rott D, Zhu J, Burnett MS, et al. Effects of MF-tricyclic, a selective cyclooxygenase-2 inhibitor, on atherosclerosis progression and susceptibility to cytomegalovirus replication in apolipoprotein-E knockout mice. J Am Coll Cardiol. 2003;41:1812–9. doi: 10.1016/s0735-1097(03)00304-8. [DOI] [PubMed] [Google Scholar]
- 19.Burleigh ME, Babaev VR, Oates JA, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002;105:1816–23. doi: 10.1161/01.cir.0000014927.74465.7f. [DOI] [PubMed] [Google Scholar]
- 20.Pratico D, Tillmann C, Zhang ZB, et al. Acceleration of atherogenesis by COX-1-dependent prostanoid formation in low density lipoprotein receptor knockout mice. Proc Natl Acad Sci USA. 2001;98:3358–63. doi: 10.1073/pnas.061607398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 22.Libby P, Schwartz D, Brogi E, et al. A cascade model for restenosis. A special case of atherosclerosis progression. Circulation. 1992;86:III47–52. [PubMed] [Google Scholar]
- 23.Saavalainen K, Pasonen-Seppanen S, Dunlop TW, et al. The human hyaluronan synthase 2 gene is a primary retinoic acid and epidermal growth factor responding gene. J Biol Chem. 2005;280:14636–44. doi: 10.1074/jbc.M500206200. [DOI] [PubMed] [Google Scholar]
- 24.Samuel SK, Hurta RA, Spearman MA, et al. TGF-beta 1 stimulation of cell locomotion utilizes the hyaluronan receptor RHAMM and hyaluronan. J Cell Biol. 1993;123:749–58. doi: 10.1083/jcb.123.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang T, Huang YG, Ye W, et al. Influence of genetic background and gender on hypertension and renal failure in COX-2-deficient mice. Am J Physiol Renal Physiol. 2005;288:F1125–32. doi: 10.1152/ajprenal.00219.2004. [DOI] [PubMed] [Google Scholar]
- 26.Chai S, Chai Q, Danielsen CC, et al. Overexpression of hyaluronan in the tunica media promotes the development of atherosclerosis. Circ Res. 2005;96:583–91. doi: 10.1161/01.RES.0000158963.37132.8b. [DOI] [PubMed] [Google Scholar]