

Abstract
Phenoxodiol (PXD) is a synthetic analogue of the plant isoflavone genistein with improved anticancer efficacy. Various properties and mechanisms of action have been attributed to the drug, the most important being its ability to sensitize resistant tumour cells to chemotherapy, which led to its fast track FDA approval for phase II/III clinical trials. In this study, we examined the effects of PXD on human peripheral blood mononuclear cells (PBMC) and its potential role in regulating immune responses. We show that PXD, at concentrations ≥1 μg/ml (4 μM), inhibited proliferation and reduced the viability of healthy donor-derived PBMC. In contrast, lower PXD concentrations (0.05–0.5 μg/ml) augmented, upon 3-day incubation, PBMC cytotoxicity. Experiments with purified CD56+ lymphocytes revealed that PXD enhanced the lytic function of natural killer (NK) cells by directly stimulating this lymphocytic subpopulation. Furthermore, in an in vivo colon cancer model, Balb/C mice administered low-dose PXD, exhibited significantly reduced tumour growth rates and prolonged survival (in 40% of the animals). Ex vivo results showed that PXD stimulated both NK and tumour-specific cell lytic activity. We conclude that PXD, when administered at low concentrations, can act as an immunomodulator, enhancing impaired immune responses, often seen in cancer-bearing individuals.
Keywords: phenoxodiol, isoflavones, anticancer drug, immune responses, NK cytotoxicity, in vivo model
Introduction
The soybean isoflavones, genistein (GEN) and daidzein (DZ), attracted scientific interest in the mid 1980s due to epidemiological studies showing lower incidence of sex-related tumours in Asian than in Western populations [1]. However, second generation Asians living in the United States showed probability of cancer development similar to US citizens. This observation led to the hypothesis that nutritional factors, related to dietary soy products, had some beneficial anticancer effect and extended research towards the exploitation of the pharmaceutical potential of soy compounds [2].
Phenoxodiol (2H-1-benzopyran-7–0, 3-(hydroxylphenyl); PXD) is a synthetic analogue of the plant isoflavone GEN, being 5–20 times more effective in inhibiting cancer cell growth than the latter. Existing literature attributes several roles to PXD, related to significant anticancer activities. PXD was initially shown to inhibit DNA topoisomerase II [3] and reduce mammary carcinogenesis in vivo[4]. These intriguing data were followed by a series of studies aiming to investigate the relevance of PXD to chemotherapy. In two different reports, PXD was shown to restore sensitivity of resistant epithelial ovarian cancer cells to chemotherapy [5, 6], suggesting that this molecule acts as a chemosensitizer. Further investigations on its mechanism of action showed that PXD can inhibit cancer cell proliferation by inducing G1 cell cycle arrest [7] and apoptosis in cancer cells, both in a caspase-dependent and -independent manner [8]. Three studies attributed the antitumour activity of PXD to degradation of the caspase inhibitor XIAP and disruption of the expression of the caspase antagonist FLIP on cancer cells [9–11]. Recently, a molecular target protein of PXD was identified, the tumour-specific surface molecule tNOX, inhibition of which, following PXD ligation [12], compromises plasma membrane electron transport (PMET) and drives actively proliferating cells to apoptosis [13]. Moreover, PXD possesses antiangiogenic properties [14] and protects cells from cisplatin-induced neurite toxicity in vitro[15]. In vivo animal tumour models have demonstrated the ability of PXD to act either as a chemopreventive agent in DMBA-induced mammary tumour-bearing rats [4] or as a chemosensitizer along with various chemotherapeutic agents (e.g. carboplatin, gemcitabine, topotecan) against xenografted epithelial ovarian tumours [5, 16].
Based on the aforementioned in vitro and in vivo reports, the drug was granted a fast track approval by FDA in 2004 and entered human clinical trials. Results from two different phase I clinical studies in 2006, where PXD was administered intravenously in late stage solid cancer patients, showed that the drug was well tolerated at doses up to 30 mg/kg with minor side effects, such as fatigue, emesis, rash [17] or grade III lymphocytopenia, the latter not showing any correlation with increasing dose levels of PXD [18]. In both studies, no objective responses were recorded; however, some patients experienced stabilization of their disease up to 6 months after treatment. PXD has now entered phase II/III clinical trials, administered in principle in patients with hormone related cancers, such as ovarian, prostate and cervical.
Although PXD in principle is considered to preferentially target cancer cells, a most elegant recent report by Herst et al.[13], provided a possible explanation of the development of lymphocytopenia in PXD-treated patients; the drug through PMET and sphingosine kinase inhibition, induces apoptosis of rapidly growing cells, such as cancer cells and activated T cells.
In order to directly assess the effect of PXD monotherapy on immune cells, we used in vitro and in vivo assays initially aiming to precisely determine the dose toxicity of PXD to lymphocytes. We report that PXD at low concentrations (0.05–0.5 μg/ml), enhances in vitro peripheral blood mononuclear cell (PBMC) cytotoxic responses both of normal donors and cancer patients. This effect can be most likely attributed to the selective activation of the cytotoxic natural killer (NK) cell subpopulation. Compared with its parental molecules, GEN and DZ, PXD enhanced the killing capacity of NK cells several fold. Most importantly, low concentrations of PXD reduced the tumour burden and prolonged survival in a mouse colon cancer model. We therefore conclude that although high concentrations of PXD arrest cancer development, low concentrations can enhance the ability of certain subsets of the immune system to eliminate cancer cells in the body.
Materials and methods
Cells and reagents
The cell lines K562 (human, leukaemic) CT-26.WT (murine, colon carcinoma) and YAC-1 (murine, leukaemic) were propagated in RPMI-1640 (Gibco-BRL, Grand Island, NY, USA) containing 10% heat-inactivated foetal calf serum (Gibco-BRL), 2 mM L-glutamine (Sigma Chemical Co., St. Louis, MO, USA), 10 mM HEPES (Gibco-BRL) and 1% penicillin-streptomycin (Gibco-BRL) (referred to hereafter as complete medium) at 37°C in a 5% CO2 incubator.
PXD, GEN and DZ (provided by NOVOGEN, North Ryde, NSW, Australia) were suspended in DMSO at 10 mg/ml (stock solution) and stored at –80°C. Serial dilutions of the stock solution were freshly prepared in complete medium and were kept for less than 4 weeks at –20°C. In all cultures, the final concentration of DMSO in the medium was ≤0.1%.
Cell isolation and separation
Peripheral blood was collected from 25 healthy donors (11 male and 14 female; age 21–65 years; median 48.5 years) and 16 cancer patients (7 male and 9 female; age 22–86 years; median 55.8 years) with various solid tumours: lung (n= 5), ovarian (n= 4), breast (n= 3) and prostate cancer (n= 4). Cancer patients were free from any anticancer therapy for at least 2 months prior to blood collection. All patients gave their written informed consent, which was approved by the ‘Alexandra’ Hospital Institutional Review Board.
Blood (10–20 ml) was collected in heparinized tubes. PBMC were isolated by centrifugation over Ficoll-Histopaque (Sigma) density gradients, suspended in complete medium and adjusted to 1 × 106 cells/ml or as indicated. Highly purified (>96%) CD56+ cells were obtained using an immunomagnetic isolation procedure. In brief, PBMC (5–7 × 106 cells/ml) were incubated for 1 hr at 4°C with 0.5 μg of anti-CD56 monoclonal antibody (mAb) conjugated to magnetic beads (Miltenyi Biotec, Auburn, CA, USA). Magnetic cell separation was performed by positive selection using an MS column (MACS, Miltenyi Biotec), according to the manufacturers’ instructions. Purity of the isolated cell populations was tested by flow cytometry (FACSCalibur, Becton Dickinson, Mountain View, CA, USA) using anti-CD3 and anti-CD56 mAbs conjugated with FITC and PE, respectively (BD Pharmingen, San Diego, CA, USA). PBMC and NK cell viability was determined by Trypan blue exclusion test and was always over 95%.
3H-thymidine incorporation assay
PBMC were seeded in 96-well U-bottom plates (Costar, Corning Inc., NY, USA) and incubated with different concentrations of each compound for 90 hrs. Cells incubated in parallel with complete medium (non-exposed culture) or in the presence of interleukin-2 (IL-2; 500 IU/ml) were used as controls. Eighteen hours before culture termination 1 μCi 3H-thymidine (Amersham, Biosciences Ltd, Buckinghamshire, UK) was added per well and cells were harvested in a semi-automatic cell harvester (Skatron Inc., Tranby, Norway). The amount of incorporated radioactivity, proportional to DNA synthesis, was measured in a liquid scintillation counter (Wallac, Turku, Finland) and expressed as counts per minute (cpm). The resulting cpm were then transformed in percentage of isotope incorporation using the formula: (average cpm of sample / average cpm of non-exposed culture) × 100. Similarly treated PBMC were also used to determine cell viability by Trypan blue exclusion. After 90 hrs, cells were counted in a Neubauer haemocytometer and percentage of viable cells was determined as: (mean viable cells of sample / mean viable cells of non-exposed culture) × 100.
NK cell cytotoxicity
PBMC, magnetically isolated CD56+ and CD56− cells were activated in 24-well flat bottom plates (Costar) in the presence of flavonoids. Following 3-day incubation at 37°C, 5% CO2, cells were harvested and tested for their cytotoxicity against the NK-sensitive tumour target cells K562 (106 cells) pre-radiolabelled with 51Cr (Amersham) as described [19]. After 18 hrs co-incubation at 37°C, 5% CO2, at effector to target (E:T) ratios ranging from 5:1 to 40:1, 100 μl of supernatant was removed from each well and isotope was counted in a γ-counter (1275 Mini-gamma LKB Wallac, Turku, Finland). To determine maximal and spontaneous isotope release, target cells were incubated with 3 N HCl and in complete medium alone, respectively. Spontaneous isotope release did not exceed 12% of maximal release in any experiment. All cultures were set in triplicate. Percentage of specific cytotoxicity was calculated according to the formula: ([cpm experimental – cpm spontaneous]/[cpm maximal – cpm spontaneous]) × 100.
Analysis for perforin content
PBMC were activated as for the cytotoxicity assay, harvested and stained for perforin, CD3 and CD56 expression. Perforin was detected intracellularly according to Skopeliti et al.[19]. In brief, prior to labelling, to enhance intracellular fluorescence, protein secretion was inhibited by the addition of 10 μl (50 μg/ml) Brefeldin A (Sigma, B7651). After 5 hrs incubation, PBMC were washed, resuspended in 500 μl permeabilizing solution (Becton Dickinson) and incubated for 12 min. at 25°C. Cells were washed with PBS containing 0.5% bovine serum albumin and 0.1% NaN3, and incubated with PE-conjugated anti-perforin mAb (1 μg/ml; BD Pharmingen) for 30 min. in the dark at 25°C. Staining for surface markers was subsequently performed using FITC-labelled anti-CD3 or anti-CD56 at saturating concentrations for 30 min. on ice. The same cells were stained with unrelated FITC- and PE-conjugated anti-mouse IgG1 mAb (BD Pharmingen; isotype controls). A total of 1-2 × 104 cells were acquired and flow cytometric data were analysed using CellQuest (Becton Dickinson) software. The percentage of cell subsets (CD3+ or CD56+ lymphocytes) expressing perforin on gated lymphocytes was determined.
Experimental animal tumour model
The murine colon carcinoma cell line CT-26.WT (ATCC CRL-2638) was grown in complete medium and was subcutaneously (sc) inoculated in 20 g, 7–8-week-old female Balb/C mice (purchased from the Hellenic Pasteur Institute Animal Facility and maintained in accordance with Law 2015/27.2.1992, Presidential Decree 160/3.5.1991 and the Directive 86/609/EEC/24.11.1986 of the Council of Europe on Animal Welfare), following the protocol: on day 0 all mice received 0.5 × 106 CT-26 and were randomly assigned to five groups. Each group consisted of 12 mice; 10 to monitor tumour growth and survival and 2 to sacrifice for ex vivo estimation of splenocyte cytotoxicity. On days 6 (day of palpable tumour formation), 8, 10, 12 and 14 after cancer cell inoculation, mice were injected intraperitoneally (i.p.) with PXD, GEN, DZ (all diluted in PBS) or plain PBS. GEN and DZ were administered at 20 mg/kg (0.8 mg/ml), while the effect of PXD was assessed at doses 20 mg/kg (0.8 mg/ml; PXD20) and 10 mg/kg (0.4 mg/ml; PXD10). All injections were performed at a total volume of 0.5 ml. Tumour growth was monitored every other day, using a digital vernier to measure the two axes of the ellipse-like tumour formed. The measurements were transformed in actual tumour volume using the formula: tumour volume (cm3) = major axis × minor axis2× 0.5. For each group, tumour growth rate and overall murine survival were recorded. For ethical reasons mice were euthanased when their tumour volume exceeded 6 cm3.
On day 30, two mice from each group were sacrificed, spleens were aseptically removed, homogenized and murine splenocytes were isolated following red blood cell lysis with ammonium chloride/Tris-HCl buffer pH 7.2 for 3 min. at 37°C. Cells were washed twice and resuspended in complete medium. To determine the splenocytes’ cytotoxic anticancer activity upon therapeutic administration of the flavonoids, spleen cells were used as effectors in cytotoxicity assays against CT-26 and YAC-1 (murine leukaemia; NK sensitive) tumour cells at an E:T ratio of 50:1.
Statistical analysis
The data were analysed by the Student’s t-test and statistical significance was presumed at significance level of 5% (P < 0.05).
Results
PXD inhibits PBMC proliferation and reduces PBMC viability
Previous reports [5, 11, 13] demonstrated that PXD was cytotoxic to tumour cells. Herein, we assessed the effect of PXD and parental isoflavones GEN and DZ on human lymphocytes. Healthy donor-derived PBMC (n= 15) were incubated with the flavonoids for 90 hrs at concentrations ranging from 0.05 to 10 μg/ml. Ten μg/ml of all compounds significantly reduced PBMC proliferation, whereas this effect was less pronounced at lower concentrations (Fig. 1A). Specifically, 10 μg/ml of PXD inhibited (by 89%) PBMC proliferation. However, at concentrations 0.05–0.5 μg/ml, PXD weakly affected PBMC proliferation, indicating that its effect on lymphocyte proliferation is dose dependent. GEN and DZ showed somewhat reduced inhibition of PBMC proliferation at concentrations corresponding to those of PXD. Assessment of PBMC viability by Trypan blue exclusion at 90 hrs, revealed that high concentrations of PXD resulted in significant loss of viable cells (>60%) and suggest that PXD doses >1 μg/ml are toxic to PBMC (Fig. 1B). Similar results were obtained when the flavonoids were titrated against cancer patient-derived PBMC (data not shown).
Figure 1.
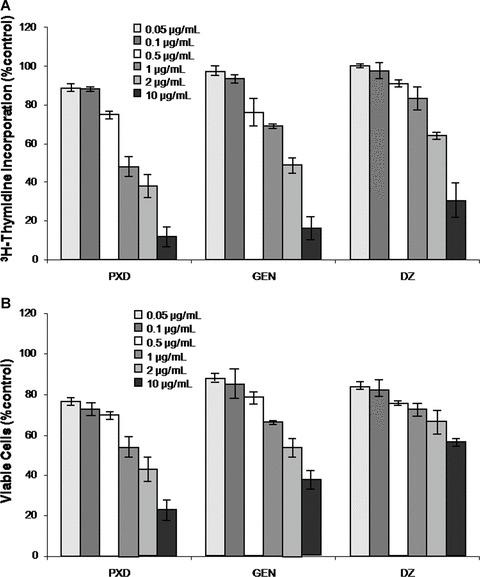
3H-Thymidine incorporation (A) and cell viability (B) induced by phenoxodiol (PXD), genistein (GEN) and daidzein (DZ). PBMC from healthy donors were incubated with various concentrations of flavonoids for 90 hrs. Pooled data from n= 10 (A) and n= 5 (B) individuals are presented as percentage of control cultures ± S.D.
PXD enhances PBMC and NK (CD56+) cell cytotoxicity
We next assessed the ability of PXD, in comparison to GEN and DZ, to induce cytolytic PBMC responses in vitro. Normal donor-derived mononuclear cells (n= 20) as well as cancer patient-derived PBMC (n= 14) were stimulated with 0.025–1 μg/ml of flavonoids for 3 days and tested for their cytotoxic activity against the NK-sensitive K562 cancer cells. To eliminate individual donor responsiveness, percentage of specific NK cell lysis was normalized according to each donor’s basal cytotoxicity and the median fold increase was calculated (Fig. 2). Median NK cytotoxicity of unstimulated (control) healthy donor- and cancer patient-derived PBMC was low (net values of specific lysis were 30% and 10%, respectively), whereas that of IL-2 (500 IU/ml)-stimulated cells was up to two- and three-fold higher, respectively (Fig. 2, grey bars). These results are in agreement with previous reports showing that high IL-2 concentration can augment cancer patient deficient cytotoxic responses in vitro[19]. PXD significantly enhanced normal donor PBMC cytotoxicity for K562 cells at 0.1–0.2 μg/ml (Fig. 2A) and that of cancer patients at 0.05–0.5 μg/ml (Fig. 2B). In both cases, maximal effect (1.5- and 1.8-fold increase, for healthy donors and cancer patients, respectively) was recorded at PXD concentrations between 0.1 and 0.2 μg/ml. GEN and DZ did not show any immunoenhancing potential on PBMC cytotoxicity when tested at the same concentrations as PXD, exhibiting fold-increase cytotoxicity values similar to those of the respective controls (maximal fold-increase ≤1.1 in all cases).
Figure 2.
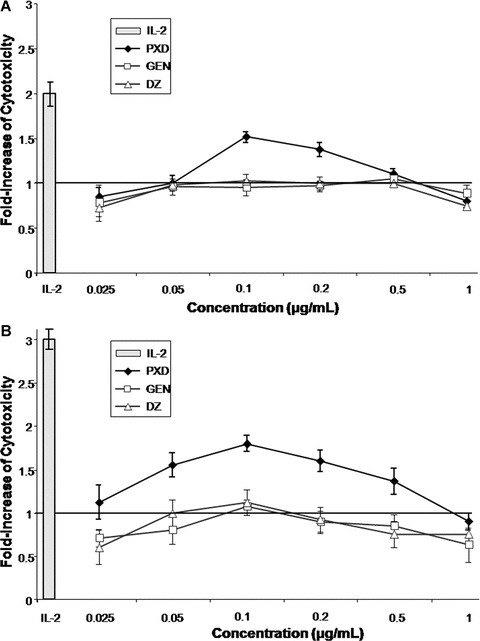
Immunoenhancing effect of isoflavones on PBMC cytotoxicity. Healthy donor- (n= 20; (A)) and cancer patient- (n= 14; (B)) derived PBMC were incubated for 3 days with phenoxodiol (PXD), genistein (GEN) and daidzein (DZ) at the indicated concentrations and were tested as effectors against K562 targets. In all experiments, the effector:target ratio was 40:1. Data are presented as mean fold increase of cytotoxicity compared to the control for each donor ± S.D. Grey bars represent the mean fold increase of the positive control (PBMC incubated with 500 IU/ml interleukin-2) ± S.D. The horizontal line is the level of non-stimulated cultures.
In order to study whether PXD exerts its effect on a specific PBMC cytotoxic subpopulation, purified CD56+ cells from five healthy donors were incubated with optimal PXD cytoenhancing concentrations (0.1 and 0.2 μg/ml), as determined in the previous experiment. Two simultaneous sets of cytotoxicity assays were performed using as effectors: (i) CD56+ and CD56-depleted (CD56−) cells isolated from PBMC prior to incubation with PXD (Fig. 3A) and (ii) CD56+ and CD56− cells isolated after PBMC incubation with PXD (Fig. 3B). Similarly isolated cells incubated with complete medium or with 500 IU/ml IL-2 were used as controls. As shown in Fig. 3, PXD enhanced basal mean percentage cytotoxicity of PBMC (28.6%) by ca. two-fold (63.5% and 54.4%, for 0.1 and 0.2 μg/ml PXD, respectively). Moreover, K562 target lysis by the CD56+ subpopulation resembled that of total PBMC. This pattern was similar for CD56+ cells isolated prior to or after incubation with PXD, suggesting that CD56+ NK cells are the principle cytotoxic subpopulation activated by PXD. On the contrary, the CD56− PBMC fraction showed significantly reduced cytotoxicity levels compared to those of CD56+ and of total PBMC. In all groups assayed, maximal cytotoxicity enhancement was demonstrated upon incubation with the lower PXD concentration (0.1 μg/ml), although percentage values did not significantly differ from those incubated with 0.2 μg/ml PXD. The enhanced PXD-induced NK cell cytotoxicity was associated with significant up-regulation of the intracellular perforin content in the CD56+ subset (Table 1). In contrast, the NK cell perforin content of cells treated with GEN was much lower compared to PXD and no significant changes were observed with DZ (in all cases ≤2.1%). Finally, the percentage of CD3+perforin+ cells was not increased at any PXD, GEN or DZ concentration tested and remained at the levels of non-stimulated control T cells (≤7%; data not shown).
Figure 3.
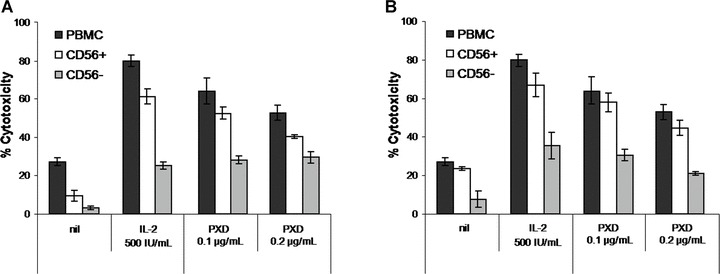
Effect of phenoxodiol (PXD) on NK cell cytotoxicity. CD56+ and CD56− cells were isolated from healthy donor-derived PBMC prior (A) or after (B) 3-day incubation with PXD. Cytotoxicity was assessed against K562 targets at an effector:target ratio of 40:1 for PBMC, 5:1 for CD56+ and 20:1 for CD56− cells. PXD was tested at the optimal cytoenhancing concentrations (0.1 and 0.2 μg/ml). Data are presented as mean percentage cytotoxicity ± S.D. from five healthy individuals. nil: PBMC incubated in plain medium; IL-2: interleukin-2.
Table 1.
Intracellular perforin expression in healthy donor-derived CD56+ cells stimulated with phenoxodiol (PXD), genistein (GEN) and daidzein (DZ) for 3 days
| Concentration of compound (μg/ml) | PXD | GEN | DZ |
|---|---|---|---|
| - | 1.1 ± 0.3* | 1.1 ± 0.3 | 1.1 ± 0.3 |
| 0.025 | 1.2 ± 0.4 | 1.15 ± 0.6 | 0.7 ± 0.2 |
| 0.05 | 2.6 ± 0.6 | 1.3 ± 0.7 | 0.5 ± 0.2 |
| 0.1 | 5.4 ± 1.5 | 1.85 ± 0.4 | 1.0 ± 0.4 |
| 0.2 | 4.3 ± 1.1 | 1.95 ± 0.5 | 1.1 ± 0.5 |
| 0.5 | 2.7 ± 1.3 | 2.1 ± 0.4 | 1.55 ± 0.8 |
| 1 | 2.6 ± 1.2 | 2.05 ± 0.6 | 1.4 ± 0.8 |
Mean percentage of CD56+perforin+ cells on gated lymphocytes ± S.D. from pooled data (n= 5).
PXD reduces tumour mass and prolongs survival of cancer-bearing mice in vivo
To further extend our in vitro studies, we investigated the effect of flavonoids in an in vivo murine cancer model. PXD, GEN and DZ were administrated therapeutically in Balb/C mice, bearing syngeneic colon carcinomas. In detail, on day 0 mice received CT-26 colon cancer cells sc and by day 6 they all developed palpable tumours. On that day (day 6) and every other day for 10 consecutive days, mice were administered i.p. PBS (control) or flavonoids at predetermined concentrations (total of five doses per animal) and were followed until day 38 for tumour growth and overall survival. As shown in Fig. 4A, the control group tumours rapidly progressed after day 6 and all mice were euthanased by day 31 (Fig. 4B). The four experimental groups exhibited different kinetics compared to the control, showing, in general, reduced tumour growth rates and prolonged survival. Specifically, mice receiving GEN (20 mg/kg) showed relatively slower tumour progression and reached, by day 33, a tumour volume of 2.8 cm3; similar growth rates were recorded for mice receiving DZ (20 mg/kg) and PXD (20 mg/kg; PXD20), all of which developed tumours of ca. 3 cm3 by day 33. Interestingly, the lower PXD dose (10 mg/kg) was the most effective (Fig. 4A, PXD10), as it restricted tumour size to 1.6 cm3 by day 33. The mean tumour volume measured with these animals did not increase by the end of the monitoring period (day 38), suggesting the in vivo activation of antitumour mechanisms capable of efficiently eliminating progressive cancer cell growth. Tumour growth results paralleled overall survival (Fig. 4B); 30% of mice (i.e. 3 of 10 animals) administered GEN or PXD20 survived until day 38, whereas 40% of mice receiving 10 mg/kg PXD (PXD10) were still alive at the end of the 38-day monitoring period.
Figure 4.
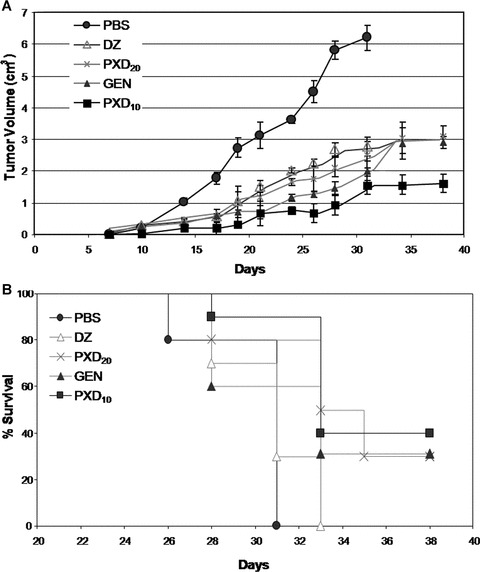
Tumour volume (A) and overall survival (B) of CT-26 tumour-bearing mice treated with flavonoids. Mice received five i.p. doses of the compounds genistein (GEN), daidzein (DZ) or phenoxodiol (PXD20) at 20 mg/kg or PXD at 10 mg/kg (PXD10). Control mice received PBS. Tumour growth and survival were monitored every 2–3 days over a total of 38 days after cancer cell inoculation and tumour volume was calculated as described in ‘Materials and methods’. Pooled data from 10 Balb/C mice per group are shown.
PXD enhances in vivo antitumour responses
To evaluate the direct in vivo effect of flavonoids in inducing tumour-specific responses, two mice from each group were sacrificed 30 days after cancer cell inoculation and spleen cells were isolated. Immediately (i.e. without ex vivo sensitization) splenocytes were used as effectors against the murine NK-sensitive YAC-1 cells, as well as the inoculated syngeneic CT-26 tumour cells. As shown in Fig. 5, splenocytes from control mice killed 7% and 12% of YAC-1 or CT-26 cells, respectively. Spleen cells from the GEN and DZ groups also showed reduced lytic ability against YAC-1 targets (2% and 2.4%, respectively), indicating that neither compound can augment NK cell responses in vivo, and this is in agreement with our in vitro data in humans (Fig. 2). However, a slight but not statistically significant increase in CT-26 cell killing was induced by GEN (14.5% compared to 12% of the control). PXD administered at 20 mg/kg significantly improved in vivo spleen cell cytotoxicity, reaching 17% and 20% specific lysis for YAC-1 and CT-26 targets, respectively. Finally, splenocytes from mice administered 10 mg/kg PXD showed the most potent cytotoxic ability, as they exhibited statistically significantly higher specific lysis of YAC-1 and the syngeneic CT-26 cells (22% and 24%, respectively) compared to the respective control values (P < 0.05).
Figure 5.
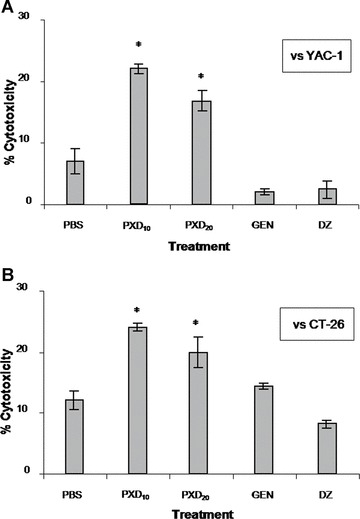
Ex vivo cytotoxic responses of isoflavone-treated murine spleen cells. Splenocytes were isolated on day 30 and used as effectors versus YAC-1 (NK sensitive) and CT-26 cells at an effector:target ratio of 50:1. Data represent mean percentage specific cytotoxicity ± S.D. from spleen cells of two mice from the groups described in the legend of Fig. 4. *: P < 0.05 compared to PBS.
Discussion
Immune suppression associated with cancer may augment tumour growth and represents an important target for anticancer therapy. In this report, we investigated the effect of the chemotherapeutic molecule PXD on specific immune responses. Using PXD concentrations lower (<1 μg/ml) than those used to date [5–11, 16] and shown not to inhibit PBMC proliferation or induce cell death, we show that in addition to its antitumour effect, PXD increases NK cell cytotoxicity in vitro and in vivo. Our data suggest that PXD, when given at appropriate dosages, can act as an immunomodulator in addition to its anticancer effects.
To our knowledge, this is the first study investigating the potential immunological role of PXD on human lymphocytes, in comparison to its ‘parental’ molecules GEN and DZ. Contradictory data on the effect of PXD on non-malignant/normal cells have been published. Being 20 times more potent than GEN [4, 11, 12], PXD was shown to selectively target cancer cells [3, 10–12, 20]. However, high PXD concentrations stimulated apoptosis in normal trophoblasts [9], exerted antiproliferative effects on normal endothelial cells [14] and induced toxicity in neurites [15]. Moreover, activated murine T cells exposed to 10 μM PXD died within 48 hrs [13]. The latter data are in accordance with our results, where PXD at concentrations >1 μg/ml greatly reduced both PBMC proliferation and cell viability (Fig. 1). Interestingly, GEN and DZ, at concentrations equal to those of PXD showed less, but still significant antiproliferative effects and viable cell loss.
Previous reports on GEN and DZ have shown that both compounds, at nutritionally relevant concentrations 10-fold lower than those used to inhibit tumour cell growth, enhanced human NK cell cytotoxicity in vitro[21]. In addition, GEN fed by oral gavage increased interferon-γ production, CTL activation and NK cytotoxicity in mice [22, 23]. These results prompted us to investigate whether PXD could affect specific lymphocyte functions. Indeed, PXD significantly enhanced (by 1.5-fold) healthy donor-derived PBMC cytotoxicity against NK-sensitive targets (K562) and this effect was more pronounced with cancer patient-derived PBMC (Fig. 2). In contrast, the same concentrations of GEN or DZ only marginally increased PBMC cytotoxicity. In our experiments, all flavonoids were tested in the absence of IL-2. GEN and DZ have been reported to act synergistically with IL-2 in augmenting NK activity [21], implying a correlation between IL-2-induced prostaglandin down-regulation and GEN or DZ effect. To investigate whether PXD directly regulated the lytic programme of NK cells, we assayed its activity on highly purified CD56+ NK cells. Our results suggest that PXD selectively stimulates NK cells (Fig. 3A) and up-regulates their perforin content (Table 1), independent of the concomitant presence of other PBMC subpopulations, such as T lymphocytes, monocytes and macrophages. The mechanism of action of PXD on NK cells is unknown. It would be of interest to investigate if PXD triggers NK cell-specific activation receptors, such as those of the NCRs or NKG2D families [24], or TLR [25], or, like glucans, mediates NK cell killing via CR3 [26]. It would also help unify the two modes of action of PXD, as a chemosensitizer (via tNOX) [12, 13] and as a modulator of NK cell activity.
We further extended our results in an in vivo therapeutic animal tumour model. Although PXD was used for treating drug-resistant ovarian cancer [5, 11, 16], we inoculated immune competent Balb/C mice with syngeneic CT-26 colon cancer cells, as they form accurately measurable sc tumours and can be used as targets for ex vivo evaluation of cytotoxicity [27]. We administered PXD at the dose of 20 mg/kg per mouse based on the protocol of Kamsteeg et al.[11], but we treated mice with five doses of the isoflavene every other day to prolong the duration of therapy (9 days) and eliminate possible toxic side-effects. In contrast to most in vivo studies where animals received PXD per os [4, 5, 16], we injected the compound i.p. to precisely determine the dose administered. Finally, based on our in vitro observations, we treated a group of animals with the half dose of PXD (10 mg/kg). Our results show that all flavonoids arrested tumour growth, whereas PXD at 10 mg/kg further reduced tumour volume (by 50% compared to GEN, DZ and PXD20). Although not directly comparable to our in vivo model, the efficacy of PXD to inhibit tumour growth in animals has been demonstrated previously. DMBA-treated rats fed with PXD had a lower incidence of mammary tumour development [4] and PXD combined with chemotherapeutics arrested chemoresistant ovarian cancer expansion [5, 16]. However, and in agreement with our results, lower PXD doses given as monotherapy were also effective [5].
Ex vivo cytotoxic responses correlated with the observed inhibition of tumour growth. Spleen cells from mice treated with PXD efficiently lysed NK-sensitive YAC-1 and CT-26 targets, indicating that PXD stimulated both NK and tumour-specific T cells in vivo. GEN followed another spleen cell-lytic pattern, stimulating CT-26-specific cells but not NK cells; however, this CT-26 cell killing was not statistically significant compared to the control. In support of this, Guo et al.[22] showed that basal NK activity was not affected by GEN, whereas cytotoxic T cells were greatly activated. Finally, DZ had no effect on either targets and this low cytotoxic response, although it does not fully explain, might have contributed to the early death (by day 33) of all mice belonging to this group.
In 2004, PXD gained fast track approval from the FDA and has already been tested in phase II/III trials in humans [28] with various types of primary and metastatic cancers. Lymphocytopenia has been associated with PXD use, and Herst et al.[13] showed evidence of the possible toxicity of PXD on murine lymphocytes. Herein, we directly proved that PXD at concentrations higher than 1 μg/ml, (4 μM), can severely damage peripheral blood lymphocyte responses in vitro. Most importantly, although most studies attribute to PXD a chemosensitizing anticancer role [28 and references therein], we add to this information that PXD, if given at sub-therapeutic doses, can also act as an immunoenhancer, improving specific immune cell functions. This finding further supports the use of this agent in future therapeutic interventions aiming to activate tumour-reactive lymphocytes.
Acknowledgments
This study was supported by the General Secretariat for Research and Technology, Hellenic Ministry of Development, Bilateral Grant 2408/2004 (to O.E.T) and Cyprus Research Promotion Foundation, KY-EL0603/24 (to A.I.C.).
References
- 1.Lee MM, Gomez SL, Chang JS, et al. Soy and isoflavone consumption in relation to prostate cancer risk in China. Cancer Epidemiol Biomarkers Prev. 2003;12:665–8. [PubMed] [Google Scholar]
- 2.Usui T. Pharmaceutical prospects of phytoestrogens. Endocr J. 2006;53:7–20. doi: 10.1507/endocrj.53.7. [DOI] [PubMed] [Google Scholar]
- 3.Constantinou AI, Husband A. Phenoxodiol (2H-1-benzopyran-7–0,1,3-(4-hydroxyphenyl)), a novel isoflavone derivative, inhibits DNA topoisomerase II by stabilizing the cleavable complex. Anticancer Res. 2002;22:2581–5. [PubMed] [Google Scholar]
- 4.Constantinou AI, Mehta R, Husband A. Phenoxodiol, a novel isoflavone derivative, inhibits dimethylbenz[a]anthracene (DMBA)- induced mammary carcinogenesis in female Sprague–Dawley rats. Eur J Cancer. 2003;39:1012–8. doi: 10.1016/s0959-8049(03)00124-2. [DOI] [PubMed] [Google Scholar]
- 5.Alvero AB, O’Malley D, Brown D, et al. Molecular mechanism of phenoxodiol-induced apoptosis in ovarian carcinoma cells. Cancer. 2006;106:599–608. doi: 10.1002/cncr.21633. [DOI] [PubMed] [Google Scholar]
- 6.Sapi E, Alvero AB, Chen W, et al. Resistance of ovarian carcinoma cells to docetaxel is XIAP dependent and reversible by phenoxodiol. Oncol Res. 2004;14:567–78. doi: 10.3727/0965040042707943. [DOI] [PubMed] [Google Scholar]
- 7.Aguero MF, Facchinetti MM, Sheleg Z, et al. Phenoxodiol, a novel isoflavone, induces G1 arrest by specific loss in cyclin-dependent kinase 2 activity by p53-independent induction of p21WAF/IP1. Cancer Res. 2005;65:3364–73. doi: 10.1158/0008-5472.CAN-04-2429. [DOI] [PubMed] [Google Scholar]
- 8.Yu F, Watts RN, Zhang XD, et al. Involvement of BH3-only proapoptotic proteins in mitochondrial-dependent phenoxodiol-induced apoptosis of human melanoma cells. Anticancer Drugs. 2006;17:1151–61. doi: 10.1097/01.cad.0000231484.17063.9a. [DOI] [PubMed] [Google Scholar]
- 9.Straszewski-Chavez SL, Abrahams VM, Funai EF, et al. X-linked inhibitor of apoptosis (XIAP) confers human trophoblast cell resistance to Fas-mediated apoptosis. Mol Hum Reprod. 2004;10:33–41. doi: 10.1093/molehr/gah001. [DOI] [PubMed] [Google Scholar]
- 10.Kluger HM, McCarthy MM, Alvero AB, et al. The X-linked inhibitor of apoptosis protein (XIAP) is up-regulated in metastatic melanoma, and XIAP cleavage by phenoxodiol is associated with carboplatin sensitization. J Transl Med. 2007;5:6–20. doi: 10.1186/1479-5876-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamsteeg M, Rutherford T, Sapi E, et al. Phenoxodiol – an isoflavone analog – induces apoptosis in chemoresistant ovarian cancer cells. Oncogene. 2003;22:2611–20. doi: 10.1038/sj.onc.1206422. [DOI] [PubMed] [Google Scholar]
- 12.Morré DJ, Chueh PJ, Yagiz K, et al. ECTO-NOX target for the anticancer isoflavene phenoxodiol. Oncol Res. 2007;16:299–312. doi: 10.3727/000000006783980973. [DOI] [PubMed] [Google Scholar]
- 13.Herst PM, Petersen T, Jerram P, et al. The antiproliferative effects of phenoxodiol are associated with inhibition of plasma membrane electron transport in tumour cell lines and primary immune cells. Biochem Pharmacol. 2007;74:1587–95. doi: 10.1016/j.bcp.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 14.Gamble JR, Xia P, Hahn CN, et al. Phenoxodiol, an experimental anticancer drug, shows potent antiangiogenic properties in addition to its antitumour effects. Int J Cancer. 2006;118:2412–20. doi: 10.1002/ijc.21682. [DOI] [PubMed] [Google Scholar]
- 15.Klein R, Brown D, Turnley AM. Phenoxodiol protects against cisplatin induced neurite toxicity in a PC-12 cell model. BMC Neurosci. 2007;8:61–7. doi: 10.1186/1471-2202-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvero AB, Brown D, Montagna M, et al. Phenoxodiol-topotecan co-administration exhibit significant anti-tumor activity without major adverse side effects. Cancer Biol Ther. 2007;6:612–7. doi: 10.4161/cbt.6.4.3891. [DOI] [PubMed] [Google Scholar]
- 17.Choueiri TK, Mekhail T, Hutson TE, et al. Phase I trial of phenoxodiol delivered by continuous intravenous infusion in patients with solid cancer. Ann Oncol. 2006;17:860–5. doi: 10.1093/annonc/mdl010. [DOI] [PubMed] [Google Scholar]
- 18.De Souza PL, Liauw W, Links M, et al. Phase I and pharmacokinetic study of weekly NV06 (Phenoxodiol™), a novel isoflav-3-ene, in patients with advanced cancer. Cancer Chemother Pharmacol. 2006;58:427–33. doi: 10.1007/s00280-006-0189-6. [DOI] [PubMed] [Google Scholar]
- 19.Skopeliti M, Voutsas IF, Klimentzou P, et al. The immunologically active site of prothymosin α is located at the carboxy-terminus of the polypeptide. Evaluation of its in vitro effects in cancer patients. Cancer Immunol Immunother. 2006;55:1247–57. doi: 10.1007/s00262-005-0108-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yagiz K, Wu LY, Kuntz CP, et al. Mouse embryonic fibroblast cells from transgenic mice overexpressing tNOX exhibit an altered growth and drug response phenotype. J Cell Biochem. 2007;101:295–306. doi: 10.1002/jcb.21184. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Song TT, Cunnick JE, et al. Daidzein and genistein glucuronides in vitro are weakly estrogenic and activate human natural killer cells at nutritionally relevant concentrations. J Nutr. 1999;129:399–405. doi: 10.1093/jn/129.2.399. [DOI] [PubMed] [Google Scholar]
- 22.Guo TL, McCay JA, Zhang LX, et al. Genistein modulates immune responses and increases host resistance to B16F10 tumor in adult female B6C3F1 mice. J Nutr. 2001;131:3251–8. doi: 10.1093/jn/131.12.3251. [DOI] [PubMed] [Google Scholar]
- 23.Guo TL, Chi RP, Hernandez DM, et al. Decreased 7,12-dimethylbenz[a]anthracene-induced carcinogenesis coincides with the induction of antitumor immunities in adult female B6C3F1 mice pretreated with genistein. Carcinogenesis. 2007;28:2560–6. doi: 10.1093/carcin/bgm223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Connor GM, Guinan KJ, Cunningham RT, et al. Functional polymorphism of the KIR3DL/1 receptor on human NK cells. J Immunol. 2007;178:235–41. doi: 10.4049/jimmunol.178.1.235. [DOI] [PubMed] [Google Scholar]
- 25.Schröder M, Bowie AG. TLR3 in antiviral immunity: key player or bystander. Trends Immunol. 2005;26:462–8. doi: 10.1016/j.it.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Xia Y, Ross GD. Generation of recombinant fragments of CD11b expressing the functional beta-glucan-binding lectin site of CR3 (CD11b/CD18) J Immunol. 1999;162:7285–93. [PubMed] [Google Scholar]
- 27.Jiang HR, Gilham DE, Mulryan K, et al. Combination of vaccination and chimeric receptor expressing T cells provides improved active therapy of tumors. J Immunol. 2006;177:4288–98. doi: 10.4049/jimmunol.177.7.4288. [DOI] [PubMed] [Google Scholar]
- 28.Mor G, Fu HH, Alvero AB. Phenoxodiol, a novel approach for the treatment of ovarian cancer. Curr Opin Investig Drugs. 2006;7:542–8. [PubMed] [Google Scholar]