

Abstract
Monoclonal antibodies (mAbs) and tyrosine kinase inhibitors targeting the epidermal growth factor receptor (EGFR), which is often pathogenetically overexpressed or mutated in epithelial malignancies and glioma, have been modestly successful, with some approved for human use. MAb 806 was raised against de2–7EGFR (or EGFRvIII), a constitutively active mutation expressed in gliomas, but also recognizes a subset (<10%) of wild-type (wt) EGFR when it is activated by autocrine loop, overexpression or mutation. It does not bind inactive EGFR in normal tissues like liver. Glioma xenografts expressing the de2–7EGFR treated with mAb 806 show reduced receptor autophosphorylation, increased p27KIP1 and reduced cell proliferation. Xenografts expressing the wtEGFR activated by overexpression or autocrine ligand are also inhibited by mAb 806, but the mechanism of inhibition has been difficult to elucidate, especially because mAb 806 does not prevent wtEGFR phosphorylation or downstream signalling in vitro. Thus, we examined the effects of mAb 806 on A431 xenograft angiogenesis. MAb 806 increases vascular endothelial growth factor (VEGF) and interleukin-8 production by activating NF-κB and normalizes tumour vasculature. Pharmacological inhibition of NF-κB completely abrogated mAb 806 activity, demonstrating that NF-κB activation is necessary for its anti-tumour function in xenografts. Given the increase in VEGF, we combined mAb 806 with bevacizumab in vivo, resulting in additive activity.
Keywords: EGFR, antibody therapy, angiogenesis, vascular normalization, NF-κB
Introduction
Amplification of the EGFR gene was the first reported genetic alteration described in glioma and is associated with gene rearrangements [1]. The first rearrangement to be described in detail was an extracellular domain deletion known as the de2–7 EGFR (epidermal growth factor receptor) (or EGFRvIII) [2, 3]. Numerous subsequent studies have shown this to be the most common mutation in glioma, occurring in about 50% of cases where the EGFR gene is amplified [4]. This cancer-specific EGFR mutant has a specific deletion between exons 2 and 7 of the EGFR. The truncation of exons 2–7 leads to the elimination of 267 amino acids from the extracellular domain and the insertion of a novel glycine at the fusion junction and renders the mutant EGFR unable to bind any known ligand [5, 6]. Despite this, the de2–7 EGFR is capable of a low level of constitutive signalling that is augmented by the receptor’s impaired internalization and down-regulation [7].
Monoclonal antibody (MAb) 806 is a novel EGFR antibody that was generated by immunizing with cells expressing the de2–7 EGFR, but unexpectedly, it also bound a small proportion of the wild-type (wt) EGFR in tumours and cell lines that overexpress the receptor [8]. Not surprisingly then, mAb 806 does not bind the unique junctional peptide found at the site of the de2–7 EGFR mutation; instead it binds to a short cysteine loop in the extracellular domain of the EGFR that is only transiently exposed as the wt EGFR moves from its inactive to active conformation [9]. The loop is constitutively exposed in the de2–7 EGFR, consistent with our original desire to generate a de2–7 EGFR-specific antibody. Thus, mAb 806 reactivity is restricted to cells with favourable conditions for EGFR activation, such as the presence of mutations (e.g. de2–7 EGFR), overexpression of the receptor or increased presence of EGFR ligands. In the case of EGFR overexpression, increased activation results from ligand-independent EGFR activation and from simultaneous derangements of EGFR glycosylation [10]. The conditions required for mAb 806 reactivity are common in malignant cells but rare in normal tissues, thereby allowing mAb 806 to preferentially target malignant tumours but not normal organs such as the liver. Our recent phase I clinical trial confirmed that a chimeric version of mAb 806 does not bind to normal tissue but does target a variety of cancers [11].
Some progress has been made in understanding how mAb 806 inhibits xenografts expressing the de2–7 EGFR. Treatment with mAb 806 reduces de2–7 EGFR autophosphorylation leading to induction of p27KIP1 and an inhibition of proliferation [12]. In contrast to de2–7 EGFR, mAb 806 only binds a small percentage (<10%) of the wt EGFR in tumour cells overexpressing the receptor at any given time-point; thus the bulk of EGFR not specifically interacting with mAb 806 can mask the specific effects of mAb 806 in a variety of assays. This fact, combined with mAb 806’s lack of in vitro anti-tumour activity [13], has made it difficult to examine how this antibody inhibits xenografts overexpressing the wt EGFR. One obvious difference between in vitro and in vivo models is angiogenesis. Therefore, we conducted a detailed study to analyze the effects of mAb 806 on angiogenesis using the A431 xenograft model which overexpresses wtEGFR. This model was chosen as A431 cells are considered a ‘gold standard’ for evaluation of EGFR therapeutics and are one of the few cell lines that contains an amplification of the EGFR gene [14].
Results and discussion
Treatment with mAb 806 inhibited A431 xenograft growth at day 14 after inoculation (Fig. S1), at which time tumours were collected for immunohistochemistry (Fig. S2). Two parameters were examined by immunochemistry initially: Ki67 staining, a marker of proliferation known to be reduced by mAb 806 [12] and phospho-Akt, a downstream target of EGFR not influenced by mAb 806 in A431 cells [12]. MAb 806 treatment reduced proliferation by 35% when assessed by Ki67 staining (P < 0.0001, Fig. 1a). Consistent with previous studies [12], mAb 806 did not down-regulate the level of phosho-Akt (P= 0.2, Fig.1a) in cells overexpressing the wtEGFR.
Figure 1.
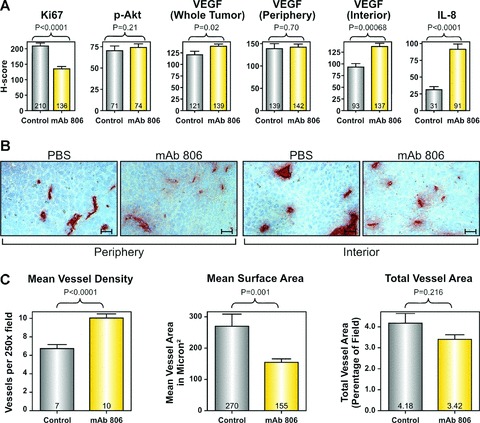
Effect of mAb 806 treatment on tumour angiogenesis (a) Bar graphs showing the mean change in H-score between mAb 806-treated and control tumours for the biological parameters shown. (b) Representative (250×) images of CD31+ blood vessels in the mAb 806-treated or control tumours, stratified by intratumoral position. scale bar: 25 μM. (c) Bar graphs showing the mean change in the MVD, MSA and total vessel area in the mAb 806-treated or control tumours.
To determine if mAb 806 might influence tumour angiogenesis, we examined the effect of mAb 806 on two representative angiogenic factors, vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8). These two factors were chosen following a qPCR screen of A431 cells designed to identify the soluble pro-angiogenic factors expressed by this cell line (Table S1). Expression of VEGF and IL-8 inA431 cells was determined by qPCR and ELISA (Fig. S3) and both were up-regulated by hypoxia (Table S1). Overall, VEGF expression was higher in the mAb 806 group than the control group (P < 0.03, Fig.1a). However, VEGF expression was also influenced by intratumoral location (periphery versus interior; anovaP < 0.002). In the control group, the interior expressed significantly less VEGF than the periphery (P < 0.01, Fig. 1a). MAb 806 treatment did not increase VEGF expression in the periphery of the tumour relative to control (P= 0.7, Fig.1a) but substantially increased VEGF expression in the interior of the tumours (P < 0.0001, Fig.1a). MAb 806 treatment resulted in a large and significant increase in IL-8 expression in all parts of the tumour (P < 0.0001, Fig.1a).
Given that mAb 806 increased the expression of two proangiogenic factors VEGF and IL-8, we analysed the effect of mAb 806 of blood vessel density and size. Mean vessel density (MVD) was significantly influenced by both mAb 806 treatment and intratumoral location (Fig. 1b and c, anova, P < 0.0001). Analysis of the entire tumour showed that control xenografts had a MVD of 6.7 vessels/field and a mean surface area (MSA) of 270 μm2 (Fig. 1c). The interior of these control tumours were significantly less vascularized than the periphery (5.3 versus 7.7 vessels/field, P < 0.01) and the vessels in the interior were also significantly larger (MSA 400 versus 185 μm2, P < 0.05). Across the whole tumour MVD was significantly higher in mAb 806 treated xenografts compared to the control xenografts (Fig. 1c, 10 versus 6.7 vessels/field, respectively, P < 0.0001). Although the MVD of the periphery of the mAb 806-treated xenografts was higher than the control group (10.2 versus 7.7 vessels/field, P < 0.0005), the increase was even more marked in the interior of mAb 806-treated tumours (9.9 versus 5.3 vessels/field in, P < 0.0001). Similar results were obtained when MVD was assessed on morphological criteria using haematoxylin and eosin stained sections (images not shown, 14.2 in mAb 806 treated xenografts versus 10.6 vessels/field in control treated xenografts, P < 0.0001).
Both treatment with mAb 806 and intratumour location affected vessel size (Fig. 1b and c, anovaP < 0.001). There was an overall decrease in MSA in the mAb 806-treated xenografts compared to the control xenografts (Fig. 1c, 155 versus 270 μm2, respectively, P < 0.001). Although vessels in the periphery of the mAb 806-treated tumours were smaller than those in the control tumours (142 versus 185 μm2, respectively, P < 0.05), the reduction in MSA was more marked in the interior of the tumour (170 versus 400 μm2, P < 0.001). The combined effect of these opposing changes in MVD and MSA results in no significant change in total vessel area (Fig. 1c, 4.18%versus 3.42%, P= 0.16). Lyve-1 staining for lymphatic vessels was very sparse, confirming that CD31+ structures were predominantly blood vessels (Fig. S4). These changes seen following treatment with mAb 806 are consistent with the model of ‘vascular normalization’ which has been described with anti-angiogenic agents including bevacizumab [15, 16] or DC101 (an antibody against VEGFR2 [17, 18]); wherein treatment causes a transient normalization of tumour vasculature, characterized by an increase in small vessels, greater coverage with pericytes, improvements in perfusion and oxygenation.
Therefore, we examined xenografts treated with mAb 806 for changes consistent with vascular normalization. Firstly, the effect of mAb 806 on pericyte coverage of tumour vasculature was determined by immunohistochemical staining for NG2 chondroitin sulphate [19]. Control A431 xenografts contained blood vessels with both complete (Fig. 2a, arrowhead) and incomplete pericyte coverage (Fig. 2a, arrow). MAb 806 significantly increased the total number of pericyte-covered vessels compared to control (Fig. 2b, 6.6 versus 4.5 vessels/field, respectively; P= 0.03) and also significantly increased the number of vessels with complete pericyte coverage compared to control (Fig. 2c, 5.2 versus 3.1 covered vessels/field, P < 0.005).
Figure 2.
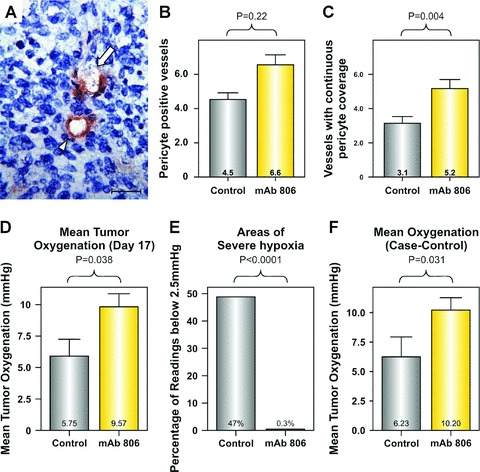
Effect of mAb 806 treatment on blood vessel function. (a) Blood vessels with continuous pericyte coverage (arrowhead) or incomplete pericyte coverage (arrow), scale bar: 20 μM. Bar graphs showing the mean change in the density of (b) vessels with any pericyte coverage and (c) vessels with complete pericyte coverage in the mAb 806-treated or control tumours. Bar graphs showing the effects of mAb 806 treatment on (d) the mean oxygenation at day 17, (e) the prevalence of severe hypoxia (less than 2.5 mmHg) and (f) the mean oxygenation in case-control analysis (i.e. matched for tumour volume).
Secondly, intratumoral oxygen was measured using the Oxford Oxylite Probe (Oxford, UK). A431 xenograft oxygenation was measured on day 17 following treatment with vehicle or mAb 806 (mean volume 1374 and 930 mm3, respectively). Mean oxygenation was significantly improved in mAb 806-treated xenografts compared to control (Fig. 2d, 5.8 ±1.3 versus 9.6 ±1.0 mmHg; P= 0.038). However, this understates the improvement in oxygenation as vehicle treated tumours had substantially more areas of severe hypoxia compared to mAb 806 treated xenografts (Fig. 2e, 47% and 0.3%, respectively, P < 0.0001). As differences in tumour volume may confound the measurement of oxygenation, we undertook a case–control analysis where tumours from the above control group were matched with tumours of equivalent volume from the mAb 806 treated group. This latter group comprised all the mice killed for oxygen measurement on day 17 as well as a cohort of mice that had were deliberately treated for an additional week for the express purposes of performing this analysis. The volumes of the matched vehicle and mAb 806 treatment groups were statistically equivalent: (1110 and 1009 mm3, respectively). This analysis showed that whilst the control group was hypoxic, the mAb 806-treated group was normoxic (Fig. 2f, 6.2 versus 10.2 mmHg, P= 0.031), demonstrating that this effect was independent of tumour volume. Taken together our data clearly show that mAb 806 mediates normalization of the tumour vasculature.
Other anti-EGFR therapeutics such as cetuximab [20–22] and gefitinib [23, 24] do not cause vascular normalization. Indeed, they mediate decreases in MVD and reduced expression of angiogenic factors such as VEGF and IL-8. To fully validate our mAb 806 analysis we also determined the effect of mAb 528, a prototypical ligand inhibitory EGFR antibody, on several of the key angiogenesic indicators described above. Treatment of A431 xenografts with mAb 528 (Fig. S5) resulted in a reduction in proliferation, a reduction in MVD, no change in MSA, a reduction in total vessel area and no change in IL-8 expression (Table 1), results which differ from those obtained following treatment with mAb 806 but consistent with the literature [25]. Thus our method of analysis provided the expected outcome for a previously described EGFR therapeutic antibody.
Table 1.
Comparison of mAb 806 and mAb 528 on proliferation and angiogenesis in A431 Xenografts
| Percentage change compared to vehicle | ||
|---|---|---|
| mAb 806 Treatment | mAb 528 Treatment | |
| Proliferation | ||
| (H-Score for Ki67) | ↓ 35% | ↓ 35% |
| Vessel density | ||
| (number per 250x field) | ↑ 49% | ↓ 36% |
| Mean vessel size | ||
| (μm2) | ↓ 35% | NS |
| Total vessel area | ||
| (Percent of 250x slide) | NS | ↓ 45% |
| IL-8 | ||
| (H-Score for IL-8) | ↑ 193% | NS |
NS = No significant difference between treatment and control groups.
The ability of mAb 806 to influence both VEGF and IL-8 expression led us to examine its effect on the NF-κB pathway, as its activation can induce both of these angiogenic factors [26]. Vehicle or mAb 806-treated A431 xenografts were harvested on day 14 (treatment schedule per Fig. S1) and NF-κB p65 DNA binding activity assayed by ELISA for NF-κB activity. There was significantly increased NF-κB p65 DNA binding activity in the nucleus of A431 xenografts treated with mAb 806 (Fig. 3a, P < 0.002). There was also significantly more p65 subunit of NF-κB in the nucleus of the mAb 806 treated group when analysed by Western blotting (Fig. 3b, 109% increase, P < 0.001). This suggests that mAb 806 activates the NF-κB pathway, thereby increasing the expression of VEGF and IL-8. The role of VEGF in angiogenesis is well established and the significance of IL-8 to this process has been recently underscored [27]. Although the changes in these two factors are almost certainly not the only ones associated with vascular normalization, they probably typify the changes in the balance of angiogenic factors related to the normalization process. Indeed, the expression of at least 10 soluble angiogenic factors by A431 cells highlights the complexity of this process (Table S1). Interestingly, vascular normalization was still seen after 14 days of treatment, suggesting that mAb 806 causes sustained normalization compared to the transient phenomenon previously reported [15–18].
Figure 3.
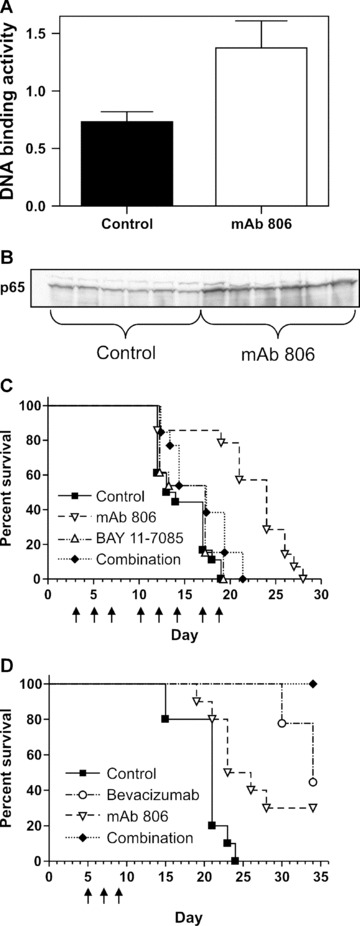
In vivo function of NF-κB and VEGF on mAb 806 anti-tumour activity. Treatment with mAb 806 significantly increases nuclear NF-κB DNA (a) as measured by ELISA or (b) Western blot of the p65 subunit. (c) Survival curves using a combined end-point of moribund condition or tumour volume reaching 1000 mm3 in mice treated with vehicle, mAb 806 (1 mg), BAY 11–7085 (100 μg) or a combination of both on the days indicated. (d) Survival curves using a combined end-point of moribund condition or tumour volume reaching 1500 mm3 in mice treated with vehicle, mAb 806 (1 mg), bevacizumab (400 μg) or a combination of both on the days indicated.
We then examined the broader role of NF-κB activation with respect to the anti-tumour effect of mAb 806 by co-administering mAb 806 with BAY 11–7085, a specific inhibitor of the NF-κB pathway that prevents the phosphorylation and degradation of IκBα[28]. Before using it in our xenograft model we demonstrated the ability of BAY 11–7085 to inhibit IL-8 expression in A431 cells (Fig. S3), confirming previous reports of this compounds capacity to inhibit IL-8 and VEGF [29, 30]. A431 xenografts were treated with mAb 806, BAY 11–7085 or combination therapy (Fig. 3c). Survival analysis showed significant differences across the four groups (Fig. 3c, P < 0.002). The median survival was 14 days (control), 17 days (BAY 11–7085), 24 days (mAb 806) and 17 days (combination). Post hoc analysis showed the mAb 806 single agent group was the only one to survive significantly longer than the control group (P < 0.0001). Hence, NF-κB blockade largely abrogates the anti-tumour effect of mAb 806 suggesting that mAb 806 activates anti-proliferative pathways down-stream of NF-κB. The modulation of the EGFR in A431 cells can cause an anti-proliferative response through induction of p21Cip1/WAF1 [31–33], the inhibitor of Cyclin Kinase 2. Importantly, this induction of p21 inA431 cells is dependent on activation NF-κB [31], firmly establishing a anti-proliferative role for NF-κB in these cells. Furthermore, a more general role for NF-κB as a tumour suppressor in some instances is now well established [34]. Interestingly, a microarray analysis of the mRNA modulated by a small molecule weight inhibitor specific to the EGFR (PD153035) showed that it reduces the invasiveness of cervical carcinoma cells and this was associated with the activation of NF-κB [35]. The in vivo consequences were not examined in this study. Finally, while seemingly unlikely, it would be interesting to determine if mAb 806 retained its vascular normalization activity in the presence of BAY11–708.
Given the increase in VEGF seen with mAb 806 therapy, we treated A431 xenografts with vehicle, mAb 806, bevacizumab (an anti-VEGF antibody) or combination therapy (Fig. 3d). Survival analysis showed a significant difference across the 4 groups (Fig. 3d, P < 0.0001 log rank). The median survivals were 21 days (control), 24 days (mAb 806), 34 days (bevacizumab) and not reached (combination). The control group did significantly worse than all other groups. The combination group did significantly better than either of the single agent’s arms (P < 0.05 for both comparisons). Hence, there is additional therapeutic benefit in combining mAb 806 with a conventional anti-angiogenic agent that potently reduces the level of VEGF and MVD [36].
MAb 806 binds a distinct epitope on the EGFR leading to distinctive outcome compared to ligand-inhibitory antibodies [37]. It inhibits tumours overexpressing wtEGFR by activating the NF-κB pathway. Consistent with NF-κB activation, mAb 806 induced expression of both VEGF and IL-8. Associated with these changes in angiogenic factors, we also observed that mAb 806 normalized tumour vasculature, profoundly reducing tumour hypoxia. This suggests the clinical development of mAb 806 could include combination with standard treatments for cancer. Pre-treatment with mAb 806 may enhance radiotherapy by reducing intratumour hypoxia or improve the penetration of chemotherapy by increasing perfusion. Although it is theoretically possible that the improved tumour vasculature and perfusion mediated by mAb 806 may increase the supply of nutrients and growth of tumours, this is negated by the direct anti-proliferative effects of mAb 806. This study also confirmed that combining of mAb 806 with bevacizumab, an approved anti-angiogenic agent, enhanced tumour inhibition.
Methods
Cell lines
A431 is a squamous carcinoma cell line from ATCC (Rockville, MD, USA) that overexpresses EGFR at levels in excess of 1 × 106 receptors per cell [38] and co-express transforming growth factor-α[38]. A431 cells have a VEGF autocrine loop and express both VEGF [20] and the VEGF receptor [39]. A431 cells were maintained in DMEM (Life Technologies, Grand Island, NY, USA) containing 10% foetal calf serum (CSL, Melbourne, Victoria, Australia), 2 mM glutamine (Sigma Chemical Co., St. Louis, MO, USA) and 2 mM penicillin/streptomycin (Life Technologies).
Antibodies
The mAb 806 (IgG2b) which recognizes a sub-set of the wt EGFR expressed on the cell surface has been described previously [8, 9, 40]. MAb 528 (IgG2a) was produced using a hybridoma obtained from ATCC (Manassas, VA, USA) [8]. Production and purification of these antibodies was performed at the Biological Production Facility (Ludwig Institute for Cancer Research, Melbourne, Australia) [41].
Xenograft models
Cells (1–3 × 106) in 100 μl of PBS were inoculated subcutaneously into both flanks of 4- to 6-week-old, female nude mice (Animal Research Centre, Perth, Australia). All studies were conducted using established tumour models as previously reported [41]. Tumours which failed to engraft properly were exclude from further analysis. Treatment commenced once tumours had reached the mean volume indicated in the appropriate figure legends. Tumour volume in mm3 was determined using the formula (length × width2)/2, where length was the longest axis and width was the perpendicular measurement. Data are expressed as mean tumour volume ± S.E. for each treatment group. These research projects were approved by the Animal Ethics Committee of the Austin Hospital.
Collection and preparation of tumour xenografts for immunohistochemistry
For fresh frozen sections, at least 5 tumour xenografts were removed from each experimental group, embedded in Tissue Tek Optimal Cutting Temperature compound (Sakura Finetek, Torrance, CA, USA), frozen in isopentone cooled in liquid nitrogen, and stored at −80°C. Sections (5 μm) were cut, fixed in ice-cold acetone for 10 min. followed by air-drying for a further 10 min. Sections were then washed in PBS and blocked in protein blocking reagent (Lipshaw Immunon, Pittsburgh, PA, USA) for 20 min. prior to incubation with the primary antibody. Antibodies used included those against Ki67 (MIB-1 clone, Dako, Glostrup, Denmark), murine CD31+ vessels (MEC 13.3, BD PharMingen, San Diego, CA, USA), human and murine VEGF (sc-507, Santa Cruz Biotechnology, Santa Cruz, CA, USA), human and murine phosphorylated (Ser473) Akt (sc-7985-R, Santa Cruz Biotechnology), human and murine NG2 Chondroitin sulphate on pericytes (AB5320, Chemicon, Victoria, Australia), Lyve-1 on murine lymph vessels (ab14917, Abcam, Cambridge, UK), human NF-κB p65 (sc-109, Santa Cruz Biotechnology) and human IL-8 (Accurate, Westbury, New York, NY, USA).
After staining with the primary antibodies described above, bound antibodies were detected with the appropriate secondary antibody. Where the primary antibody was of rat origin, slides were rinsed before application of biotinylated anti-rat secondary (ab-6851, Abcam) followed by application of Streptavidin-HRP (Dakocytomation, Carpinteria, CA, USA). Where the primary antibody was of rabbit origin, sections were rinsed before application of HRP-labelled anti-rabbit antibodies (Envision+ System, Dakocytomation). Bound antibodies were detected with AEC substrate solution (0.1 mol/l acetic acid, 0.1 mol/l sodium acetate, 0.02 mol/l AEC, and 0.03% H2O2). Slides were then counterstained in haematoxylin (BDH Laboratory, Poole, UK) and mounted. Appropriate species and isotype control staining was performed for all slides.
Analysis of vessel density and immunohistochemical staining
For each section, up to 17 images were captured by light microscopy at the 250× magnification. Images were obtained in a systematic manner to ensure homogenous and representative sampling of non-overlapping areas of viable tumour. Fifty percent of images were captured from the tumour peripheral and the rest from the interior of the tumour. Quantification of CD31+ and Lyve-1+ vessel density involved counting the number of CD31+ vessels in each (250×) image. Vessels density was also counted in haematoxylin and eosin sections. Again, up to 17 images were captured by light microscopy as described above but at 400× magnification. Rounded or tubular spaces, especially if lined by endothelial cells, were considered to be blood vessels. The cross-sectional area of CD31+ vessels (defined as all CD31+ regions and any hollow structure contained therein) was also determined by morphometric analysis using Leica QWin (North Ryde, Australia) image analysis software and expressed as a percentage of the total viable tumour area in each image [42].
For analysis of immunohistochemical staining, up to 17 images of viable tumour were captured by light microscopy at 400× magnification. For each image, an H-score for prevalence and intensity of staining was generated using Leica Qin images analysis software [43]. In brief, a composite score was obtained by multiplying each level of intensity staining (0–3) by the percentage area that it comprised (0–100%) and then adding up the resultant products. The minimum score was 0 (0 staining in 100% of the image) and the maximum scare was 300 (3+ staining in 100% of the image). Artefactual, acellular and non-viable areas were excluded from the scoring procedure.
Measurement of intratumoral oxygenation in anaesthetized mice
Intratumoral oxygen was determined in anaesthetized mice using the Oxford Oxylite Probe [44]. In brief, mice were anaesthetized then kept warm on a temperature-regulated pad. Up to three tracks were created through the equatorial plan of the tumour tissue using a 21-gauge needle. The probe was then drawn through the track(s), taking multiple oxygen measurements (in mmHg) as it traversed the track(s). The initial 5% of readings just after entry in to the tumour and the final 5% of readings just prior to exit from the tumour were discarded because of the risk of contamination by external air. Mice were killed at the end of the experiment.
Assessment of total NF-κB activity and associated p65 nuclear translocation in mAb 806 treated xenografts
Tumours were then harvested, homogenized and equal amounts of nuclear protein were subjected to Western blotting and ELISA for analysis of NF-kB p65 protein expression and NF-κB p65 DNA binding activity, respectively [45]. In brief, tissues were homogenized in 1:5 in buffer A (10 mM Hepes pH 7.8, 10 mM KCl, 2 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid [EDTA], 1 mM NaF, 1 mM Na3VO4, 10 mg/ml aprotinin, 1 mM AESBF and 5 mg/ml leupeptin) for two 20 sec. bursts. The homogenates were incubated on ice for 30 min.; Igepal was added for a final concentration of 1% (v/v), vortexed for 30 sec. and then centrifuged at 4000 ×g for 10 min. The supernatant (cytosolic fraction) was stored at −80°C. The pellet was then resuspended in buffer B (50 mM Hepes pH 7.8, 50 mM KCl, 400 mM NaCl, 20% glycerol, 0.1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 10 mg/ml aprotinin, 1 mM AESBF and 5 mg/ml leupeptin) mixed vigorously by vortexing for 15 min. and then centrifuged at 15,000 ×g for 30 min. The supernatant (nuclear fraction) was retained for Western blotting and ELISA. The protein content of tissue homogenates was determined using BCA protein assay (Pierce, Rockford, USA), using BSA as a reference standard, as previously described [45]. Nuclear NF-κB p65 DNA binding activity was measured in duplicate using a NF-κB p65 transcription factor assay kit according to manufacturer’s instructions (TransAM; Active Motif; Carlsbad, CA, USA).
Assessment of NF-κB p65 protein expression was analysed by Western blotting. Forty micrograms of tissue protein extracts were separated on a 10% polyacrylamide gel and transferred to PVDF as previously described [46]. Protein expression was identified by co-migration with a positive control and by comparison with the mobility of protein standard. Western blots were quantified using a Storm 804 Phosphoimager (Amersham Bioscience, Piscataway, NJ, USA) for analysis using the ImageQuant TL Image Analysis Software (Version 2005). Data were corrected for background, and expressed as optical density (OD/mm2).
Statistical analysis
Analyses were performed with SPSS 12.0.1 for Windows. Where required, data were tested for adherence to a normal distribution using the Shapiro–Wilk statistic and the appropriate parametric (assuming a normal distribution) or non-parametric (making no assumption regarding the distribution of the data) test was then employed. P-values of <0.05 were considered significant. All P-values were two-sided except for cases where previous experimentation or published literature had already determined a significant result for the intervention in question.
For the comparison of means, Student’s t-test or the non-parametric Mann-Whitney U-test was employed where only two groups were being considered. For comparisons between three or more group, parametric data were analysed by ANOVA and if P < 0.05, then post hoc testing was undertaken to determine which groups differed significantly. The non-parametric test employed for multiple groups was Kruskal–Wallis test and if P < 0.05, then post hoc testing was undertaken to determine which groups differed significantly. Survival analysis was analysed for significance and if the log-rank test across all groups was significantly different (P < 0.05), then post hoc testing by log-rank testing was undertaken to determine which groups differed significantly.
Acknowledgments
This work was funded in part by National Health & Medical Research Council of Australia Project grant no. 433615. We thank Steven Stacker and Marc Achen for their helpful suggestions.
Supporting Information
Fig. S1 Treatment of established A431 xenografts with mAb806. Mice (n = 4–5 mice) were treated on the daysshown (arrows) with 1 mg of mAb 806 (▿) or vehicle(▪). Mean tumor volume was 41 mm3 on day 3 whentreatment started. Data shown in all cases are mean tumor volume± S.E. The mAb 806 group was significantly smaller than thecontrol group on day 14 (mean volumes 285 mm3 and 714mm3, respectively, P ∇ 0.00012).
Fig. S2 Representative images (400x) of staining for thebiological parameters indicated in the A431 xenografts treated withmAb 806 or vehicle, scale bar: 20 μM.
Fig. S3 BAY 11-7085 inhibits IL-8 production in A431 cells in a dose-dependent manner. A431 cells were treated O/N in serum-free media with the concentration of BAY 11-7085 indicated. Next morning the Bay 11-7085 was removed and cells placed in fresh media for an additional 24 hrs, after which time the media was collected and assayed for IL-8. Data expressed as pg/ml of IL-8.
Fig. S4 A431 xenografts from the experiment described in Fig. S1 where stained with the lymph vessel marker lyve-1. Representative images are shown.
Fig. S5 Treatment of established A431 tumors with mAb528. Mice (n ∇ 5 mice) were treated on the days shown(arrows) with 1 mg of mAb 528 (▿) or vehicle control(▪). Mean tumor volume was 75 mm3 on day 4 whentreatment started. Data shown in all cases is mean tumor volume± S.E. The mAb 528 group was significantly smaller than thencontrol group on day 13 (mean volumes 241 mm3 and 709mm3, respectively, P ∇ 0.0017). Note that the tumor volumes in this experiment were equivalent to that in Fig. S1.
Table S1 Cultured cells, grown to95–100% confluency levels, were incubated underserum free conditions at 37°C for 24 hrs, either un-treated(normoxia) or within BD GasPak™ EZ Anaerobe Pouch Systembags, which produced a hypoxic environment consisting of less than1% oxygen (hypoxia). Following treatment, mRNA wereisolated from cells and cDNA prepared. The PCR reaction occurredfollowing 40 cycles consisting of the following run conditions:50°C (2 min.), 95°C (10 min.), 95°C (15 sec.) and60°C (1 min.), using the 7900HT Fast Real-Time PCR System(Applied Biosystems, Scoresby, VIC, Australia). The 18S housekeeping gene and DNAse free ddH2O were used as controlsin all cases.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
References
- 1.Libermann TA, Nusbaum HR, Razon N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–7. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 2.Sugawa N, Ekstrand AJ, James CD, et al. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA. 1990;87:8602–6. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamazaki H, Ohba Y, Tamaoki N, et al. A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Jpn J Cancer Res. 1990;81:773–9. doi: 10.1111/j.1349-7006.1990.tb02644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frederick L, Wang XY, Eley G, et al. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–7. [PubMed] [Google Scholar]
- 5.Pedersen MW, Meltorn M, Damstrup L, et al. The type III epidermal growth factor receptor mutation. Biological significance and potential target for anti-cancer therapy. Ann Oncol. 2001;12:745–60. doi: 10.1023/a:1011177318162. [DOI] [PubMed] [Google Scholar]
- 6.Wikstrand CJ, Reist CJ, Archer GE, et al. The class III variant of the epidermal growth factor receptor (EGFRvIII): characterization and utilization as an immunotherapeutic target. J Neurovirol. 1998;4:148–58. doi: 10.3109/13550289809114515. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt MH, Furnari FB, Cavenee WK, et al. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc Natl Acad Sci USA. 2003;100:6505–10. doi: 10.1073/pnas.1031790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johns TG, Stockert E, Ritter G, et al. Novel monoclonal antibody specific for the de2–7 epidermal growth factor receptor (EGFR) that also recognizes the EGFR expressed in cells containing amplification of the EGFR gene. Int J Cancer. 2002;98:398–408. doi: 10.1002/ijc.10189. [DOI] [PubMed] [Google Scholar]
- 9.Johns TG, Adams TE, Cochran JR, et al. Identification of the epitope for the epidermal growth factor receptor-specific monoclonal antibody 806 reveals that it preferentially recognizes an untethered form of the receptor. J Biol Chem. 2004;279:30375–84. doi: 10.1074/jbc.M401218200. [DOI] [PubMed] [Google Scholar]
- 10.Johns TG, Mellman I, Cartwright GA, et al. The antitumor monoclonal antibody 806 recognizes a high-mannose form of the EGF receptor that reaches the cell surface when cells over-express the receptor. FASEB J. 2005;19:780–2. doi: 10.1096/fj.04-1766fje. [DOI] [PubMed] [Google Scholar]
- 11.Scott AM, Lee F-T, Tebbutt N, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. PNAS. 2007;104:4071–6. doi: 10.1073/pnas.0611693104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perera RM, Narita Y, Furnari FB, et al. Treatment of human tumor xenografts with monoclonal antibody 806 in combination with a prototypical epidermal growth factor receptor-specific antibody generates enhanced antitumor activity. Clin Cancer Res. 2005;11:6390–9. doi: 10.1158/1078-0432.CCR-04-2653. [DOI] [PubMed] [Google Scholar]
- 13.Johns TG, Perera RM, Vernes SC, et al. The efficacy of epidermal growth factor receptor-specific antibodies against glioma xenografts is influenced by receptor levels, activation status, and heterodimerization. Clin Cancer Res. 2007;13:1911–25. doi: 10.1158/1078-0432.CCR-06-1453. [DOI] [PubMed] [Google Scholar]
- 14.Bigner SH, Humphrey PA, Wong AJ, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50:8017–22. [PubMed] [Google Scholar]
- 15.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–7. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Soffer SZ, Kim ES, et al. Vascular remodeling marks tumors that recur during chronic suppression of angiogenesis. Mol Cancer Res. 2004;2:36–42. [PubMed] [Google Scholar]
- 17.Tong RT, Boucher Y, Kozin SV, et al. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–6. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 18.Winkler F, Kozin SV, Tong RT, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–63. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Ozerdem U, Grako KA, Dahlin-Huppe K, et al. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dynam. 2001;222:218–27. doi: 10.1002/dvdy.1200. [DOI] [PubMed] [Google Scholar]
- 20.Petit AM, Rak J, Hung MC, et al. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol. 1997;151:1523–30. [PMC free article] [PubMed] [Google Scholar]
- 21.Viloria-Petit A, Crombet T, Jothy S, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090–101. [PubMed] [Google Scholar]
- 22.Bancroft CC, Chen Z, Yeh J, et al. Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF-kappaB and AP-1 activation and IL-8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int J Cancer. 2002;99:538–48. doi: 10.1002/ijc.10398. [DOI] [PubMed] [Google Scholar]
- 23.Ciardiello F, Caputo R, Bianco R, et al. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res. 2001;7:1459–65. [PubMed] [Google Scholar]
- 24.Hirata A, Ogawa S, Kometani T, et al. ZD1839 (Iressa) induces antiangiogenic effects through inhibition of epidermal growth factor receptor tyrosine kinase. Cancer Res. 2002;62:2554–60. [PubMed] [Google Scholar]
- 25.Mendelsohn J. Anti-EGF receptor monoclonal antibodies: biological studies and potential clinical applications. Trans Am Clin Climatol Assoc. 1988;100:31–8. [PMC free article] [PubMed] [Google Scholar]
- 26.Wu JL, Abe T, Inoue R, et al. IkappaBalphaM suppresses angiogenesis and tumorigenesis promoted by a constitutively active mutant EGFR in human glioma cells. Neurol Res. 2004;26:785–91. doi: 10.1179/016164104225014139. [DOI] [PubMed] [Google Scholar]
- 27.Fischbach C, Kong HJ, Hsiong SX, et al. Cancer cell angiogenic capability is regulated by 3D culture and integrin engagement. Proc Natl Acad Sci USA. 2009;106:399–404. doi: 10.1073/pnas.0808932106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O-Charoenrat P, Wongkajornsilp A, Rhys-Evans PH, et al. Signaling pathways required for matrix metalloproteinase-9 induction by betacellulin in head-and-neck squamous carcinoma cells. Int J Cancer. 2004;111:174–83. doi: 10.1002/ijc.20228. [DOI] [PubMed] [Google Scholar]
- 29.Prakash O, Swamy OR, Peng X, et al. Activation of Src kinase Lyn by the Kaposi sarcoma-associated herpesvirus K1 protein: implications for lymphomagenesis. Blood. 2005;105:3987–94. doi: 10.1182/blood-2004-07-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudack C, Steinhoff M, Mooren F, et al. PAR-2 activation regulates IL-8 and GRO-alpha synthesis by NF-kappaB, but not RANTES, IL-6, eotaxin or TARC expression in nasal epithelium. Clin Exp Allergy. 2007;37:1009–22. doi: 10.1111/j.1365-2222.2007.02686.x. [DOI] [PubMed] [Google Scholar]
- 31.Ohtsubo M, Takayanagi A, Gamou S, et al. Interruption of NFkappaB-STAT1 signaling mediates EGF-induced cell-cycle arrest. J Cell Physiol. 2000;184:131–7. doi: 10.1002/(SICI)1097-4652(200007)184:1<131::AID-JCP14>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 32.Fan Z, Lu Y, Wu X, et al. Prolonged induction of p21Cip1/WAF1/CDK2/PCNA complex by epidermal growth factor receptor activation mediates ligand-induced A431 cell growth inhibition. J Cell Biol. 1995;131:235–42. doi: 10.1083/jcb.131.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jakus J, Yeudall WA. Growth inhibitory concentrations of EGF induce p21 (WAF1/Cip1) and alter cell cycle control in squamous carcinoma cells. Oncogene. 1996;12:2369–76. [PubMed] [Google Scholar]
- 34.Das AK, Chen BP, Story MD, et al. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res. 2007;67:5267–74. doi: 10.1158/0008-5472.CAN-07-0242. [DOI] [PubMed] [Google Scholar]
- 35.Woodworth CD, Michael E, Marker D, et al. Inhibition of the epidermal growth factor receptor increases expression of genes that stimulate inflammation, apoptosis, and cell attachment. Mol Cancer Ther. 2005;4:650–8. doi: 10.1158/1535-7163.MCT-04-0238. [DOI] [PubMed] [Google Scholar]
- 36.Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–4. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 37.Li S, Schmitz KR, Jeffrey PD, et al. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–11. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 38.Laderoute KR, Murphy BJ, Short SM, et al. Enhancement of transforming growth factor-alpha synthesis in multicellular tumour spheroids of A431 squamous carcinoma cells. Br J Cancer. 1992;65:157–62. doi: 10.1038/bjc.1992.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S, Peck-Radosavljevic M, Koller E, et al. Characterization of (123)I-vascular endothelial growth factor-binding sites expressed on human tumour cells: possible implication for tumour scintigraphy. Int J Cancer. 2001;91:789–96. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1126>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 40.Luwor RB, Johns TG, Murone C, et al. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2–7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res. 2001;61:5355–61. [PubMed] [Google Scholar]
- 41.Gan HK, Walker F, Burgess AW, et al. The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor AG1478 increases the formation of inactive untethered EGFR dimers. Implications for combination therapy with monoclonal antibody 806. J Biol Chem. 2007;282:2840–50. doi: 10.1074/jbc.M605136200. [DOI] [PubMed] [Google Scholar]
- 42.Mishima K, Johns TG, Luwor RB, et al. Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res. 2001;61:5349–54. [PubMed] [Google Scholar]
- 43.Edwards J, Traynor P, Munro AF, et al. The role of HER1-HER4 and EGFRvIII in hormone-refractory prostate cancer. Clin Cancer Res. 2006;12:123–30. doi: 10.1158/1078-0432.CCR-05-1445. [DOI] [PubMed] [Google Scholar]
- 44.Zanzonico P, O’Donoghue J, Chapman JD, et al. Iodine-124-labeled iodo-azomycin-galactoside imaging of tumor hypoxia in mice with serial microPET scanning. Eur J Nucl Med Mol Imaging. 2004;31:117–28. doi: 10.1007/s00259-003-1322-y. [DOI] [PubMed] [Google Scholar]
- 45.Lappas M, Permezel M, Georgiou HM, et al. Nuclear factor kappa B regulation of proinflammatory cytokines in human gestational tissues in vitro. Biol Reproduct. 2002;67:668–73. doi: 10.1095/biolreprod67.2.668. [DOI] [PubMed] [Google Scholar]
- 46.Lappas M, Permezel M, Georgiou HM, et al. Regulation of proinflammatory cytokines in human gestational tissues by peroxisome proliferator-activated receptor-gamma: effect of 15-deoxy-Delta(12,14)-PGJ(2) and troglitazone. J Clin Endocrinol Metab. 2002;87:4667–72. doi: 10.1210/jc.2002-020613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item