

Abstract
Patients with pancreatic cancer have a poor survival rate, and new therapeutic strategies are needed. Epithelial cell adhesion molecule (EpCAM), suggested as a marker for cancer stem cells, is over-expressed on most pancreatic tumour cells but not on normal cells and may be an ideal therapeutic target. We evaluated the anti-tumour efficiency of bispecific EpCAMxCD3 antibody linking tumour cells and T lymphocytes. In NOD SCID mice, EpCAMxCD3 had a long serum half-life (t1/2∼ 7 days). EpCAMxCD3 significantly retarded growth of BxPC-3 pancreatic carcinoma xenografts. For mimicking a pancreatic cancer microenvironment in vitro, we used a three-dimensional tumour reconstruct system, in which lymphocytes were co-cultured with tumour cells and fibroblasts in a collagen matrix. In this in vivo–like system, EpCAMxCD3 potently stimulated production of the effector cytokines IFN-γ and TNF-α by extracorporally pre-activated lymphocytes. Moreover, compared with a bivalent anti-CD3 antibody, EpCAMxCD3 more efficiently activated the production of TNF-α and IFN-γ by non-stimulated peripheral blood mononuclear cells. Most excitingly, we demonstrate for the first time that EpCAMxCD3 induces prolonged contacts between lymphocytes and tumour cells, which may be the main reason for the observed anti-tumour effects. As an important prerequisite for future use in patients, EpCAMxCD3 did not alter lymphocyte migration as measured by time-lapse video microscopy. Our data may open a way to improve the immune response and treatment outcome in patients with pancreatic cancer.
Keywords: bispecific antibody, cancer stem cells, cancer immunotherapy, EpCAM, pancreatic carcinoma, lymphocytes, tumour microenvironment
Introduction
Pancreatic adenocarcinoma is an aggressive malignancy usually diagnosed at an advanced state with very limited therapeutic options. It has the worst prognosis of any major malignancy and the vast majority of patients die within the first year after diagnosis, and fewer than 1% of patients are alive after 5 years [1]. Hallmarks of pancreatic cancer are an extensive soft tissue invasion and an early systemic dissemination. Molecular mechanisms of this malignancy are poorly understood.
Epithelial cell adhesion molecule (EpCAM [CD326]) is over-expressed in the majority of epithelial tumours and derived metastases, including pancreatic adenocarcinomas [2,3]. In prostate adenocarcinoma, EpCAM over-expression occurs at an early stage of the cancer [4]. Increased EpCAM expression indicates a poor prognosis in breast and gallbladder carcinomas [5–8]. Importantly, EpCAM is expressed by tumour-initiating cells, also known as cancer stem cells, in mammary, colorectal and pancreatic carcinomas [9–11]. Thus, EpCAM targeting may evolve as an attractive anti-cancer therapeutic strategy.
Anti-EpCAM antibodies have been used in several anti-cancer studies with promising results [3, 12, 13]. The proposed mechanisms of their anti-tumour effects comprise antibody-dependent cellular cytotoxicity and the complement system activation [14]. As a radioimmunotherapeutic modality, the C215 anti-EpCAM antibody labelled with 131I delayed growth of the head and neck xenograft squamous cell carcinoma [15]. Although initial clinical trials with EpCAM-targeted immunotherapy for treatment of gastrointestinal malignancies provided controversial results [13], the recent phase I trial of adecatumumab, a fully human anti-EpCAM antibody, in patients with hormone refractory prostate cancer has proven that medication is well tolerated. Adecatumumab is currently undergoing phase II trial in prostate and breast cancer patients [16, 17].
Bispecific antibodies (bsAb), which were used in our present study, are artificial molecules with dual specificity to two different antigens. They represent a new class of immunotherapeutic agents [18, 19]. One of the concepts behind the use of bsAb in anti-cancer immunotherapy is their ability to bind to tumour-associated antigens with one arm and to specific molecules on immune cells with the other arm. This brings tumour and immune cells in close contact, triggers immune cell activation and redirects immune cells to kill tumour cells [20–23]. The most commonly used antigen for bsAb on lymphocytes is an invariant CD3 signalling complex, which induces a polyclonal T-cell activation. Several bsAb and single-chain antibody constructs against EpCAM expressed by tumour cells have been generated and tested as immunotherapeutic agents [24–29]. Quadroma-derived bsAb showed promising results in phase I/II trials [27, 30]. However, a relatively short half-life of recombinant bsAb constructs, overt production of cytokines, Fc-mediated side effects and bsAb immunogenicity limit their clinical application [19]. To overcome some of these obstacles, we developed mouse bsAb composed of mouse IgG1 and IgG2A constant heavy-chain regions, which has low affinity to human Fc receptors, to target human EpCAM on carcinoma cells and CD3 antigen on lymphocytes. This reagent, EpCAMxCD3 (HEA125xOKT3), efficiently induced tumour cell lysis in vitro, and an intraperitoneal application of bsAb reduced malignant ascites production in advanced ovarian cancer patients [31, 32].
In the present study, we investigated the anti-tumour efficiency of EpCAMxCD3 in pancreatic cancer. To begin with, we established an in vitro three-dimensional (3D) tumour reconstruct system, which closely resembles the microenvironment of pancreatic tumours. EpCAMxCD3 potently inhibited subcutaneous tumour engraftment (tumour take) and retarded growth of BxPC-3 pancreatic xenografts in NOD SCID mice. In collagen gel 3D tumour reconstructs, EpCAMxCD3 markedly increased the contact time between lymphocytes and carcinoma cells. Furthermore, EpCAMxCD3 induced production of effector cytokines TNF-α and IFN-γ more efficiently than a bivalent anti-CD3 antibody. Here, we show for the first time that increasing the contact time between CD3-positive lymphocytes and EpCAM-expressing tumour cells is a key event for EpCAMxCD3 anti-cancer efficiency.
Material and methods
Primary and established cell lines
Primary skin fibroblasts were provided by Dr. H.-J. Stark (DKFZ, Heidelberg, Germany). BxPC-3 pancreatic cancer cell line and PC-3 prostate cancer cell line were obtained from the American Type Culture Collection (Manassas, VA) and were cultured as described [33]. Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood of healthy donors by Ficoll (Inno-Train Diagnostic GmbH, Kronberg, Germany) gradient. Donor material was obtained following the University of Heidelberg Ethical Committee approval.
Antibodies
EpCAMxCD3 (HEA125xOKT3) and CD19xCD3 (HD37xOKT3) bsAb were produced by hybrid-hybridoma technique and purified by affinity chromatography over protein A-Sepharose CL-4B (Amersham Pharmacia Biotech, Freiburg, Germany) followed by HPLC purification on a Bakerbond ABx column (J.T. Baker, Phillipsburg, NJ) as described [31, 34]. Parental anti-human EpCAM hybridoma (HEA125; IgG1) was raised in our laboratory [35, 36]. Hybridoma OKT3 (IgG2A) directed against the ε-chain of the CD3 molecule was purchased from the ATCC. Both parental mAbs were purified by affinity chromatography.
In vitro collagen gel 3D tumour reconstruct system
To create 3D tumour reconstructs and to mimic tumour microenvironment, BxPC-3 cells, lymphocytes and fibroblasts were cultured in a collagen type I gel on chamber slides (Nunc, Rochester, NY). The collagen type I gel was prepared as described previously [37]. Briefly, to prepare the working collagen solution, 5.1 ml from a stock of 2.9 mg/ml of collagen type I (PureCol™, Inamed Biomaterials, Fremont, CA) were added to 798 μl of RPMI medium with 2% BSA and 330 μl of 0.34 M NaOH. BxPC-3 cells (106 cells per ml) and fibroblasts (104 cells per ml) were mixed in the collagen solution and were seeded in 8-well chamber slides in a volume of 0.5 ml. Extracorporally pre-activated lymphocytes or non-stimulated PBMCs (5 × 106 cells per gel; lymphocytes/carcinoma cells ratio 10:1) were mixed with carcinoma cells and fibroblasts in the collagen solution. This mixture formed a polymerized collagen gel after incubation at 37°C in a CO2 incubator for 1.5 hrs.
A volume of 200 μl of medium containing control parental anti-human EpCAM mAb, control irrelevant bsAb CD19xCD3 or EpCAMxCD3, respectively, at a concentration of 10 μg/ml was added on top of 3D tumour reconstructs. Gels and supernatants were harvested after 24 or 72 hrs of incubation at 37°C in a CO2 incubator. Collagen concentration and the number of carcinoma cells and fibroblasts were optimized to prevent cell-mediated collagen gel contraction naturally occurring in 3D collagen gels.
Results
EpCAMxCD3 efficiently reduces tumour engraftment and retards the growth of pancreatic carcinoma xenografts
EpCAMxCD3 has shown promising therapeutic effects in a small study with patients suffering from advanced ovarian cancer with malignant ascites formation [32], although the underlying molecular way of action remained obscure. Therefore, the present study focussed on elucidating the parameters critical for the design of an optimized therapy in patients. Firstly, we analyzed pharmacokinetic properties of EpCAMxCD3 in vivo. NOD SCID mice were injected intraperitoneally (i.p.) with EpCAMxCD3 followed by subsequent blood sampling at days 1, 2, 4, 7 and 14. The EpCAMxCD3 concentration reached its maximum at day 1 after i.p. administration. The serum half-life (t1/2) of EpCAMxCD3 was approximately 7 days (Fig. 1A).
Figure 1.

EpCAMxCD3 pharmacokinetics and in vivo xenograft BxPC-3 pancreatic tumour model. (A) NOD SCID mice (n= 5) received one i.p. injection of EpCAMxCD3 at a dose of 500 μg and peripheral blood was taken 1, 2, 4, 7, 14 days after bsAb administration. EpCAMxCD3 concentration in the serum was determined by ELISA. (B) BxPC-3 pancreatic tumour cells (5 × 106) were mixed with extracorporally pre-activated human lymphocytes (5 × 106) and s.c. engrafted to NOD SCID mice (n= 5). CD45+ cells were detected in tumour tissues by immunohistochemistry 24 days after the tumour cell/lymphocyte implantation (×400). (C) Trichrome staining of tumour tissues was used to evaluate collagen distribution (blue). Nuclei of carcinoma cells and stromal cells are coloured in red (×100). (D) Double immunofluorescence technique was used to detect EpCAM expression on tumour cells (green) and CD31+ blood vessels (red). Bar, 100 μm.
To validate in vivo effects of EpCAMxCD3 in a pancreatic xenograft tumour model, NOD SCID mice were injected subcutaneously (s.c.) either with BxPC-3 tumour cells alone or with BxPC-3 cells mixed in 1:1 ratio with extracorporally IL-2 and anti-CD3 pre-activated human peripheral blood lymphocytes. We analyzed tumour engraftment (tumour take), tumour growth kinetics and tumour morphology at day 25 after cell implantation (see Supplementary Materials and Methods). Essentially, there was no difference in tumour incidence and tumour growth kinetics between two groups (data not shown). All animals developed tumours, and tumour growth kinetics were independent of co-administration of extracorporally pre-activated human lymphocytes. CD45+ lymphocytes were detected in BxPC-3 tumours 24 days after cell implantation (Fig. 1B) suggesting that co-injected human lymphocytes survive for a long time in BxPC-3 xenografts. CD8+ and CD4+ lymphocytes are composed of the majority of immune cells in BxPC-3 tumour tissues and only single CD16+ NK cells could be detected (data not shown). Pre-activated lymphocytes did not influence BxPC-3 tumour stroma formation as seen by collagen distribution patterns (Fig. 1C), blood vessel morphology and density (Fig. 1D, data not shown). BxPC-3 tumour cells also expressed target EpCAM antigen in vivo (Fig. 1D). Therefore, this tumour model is suitable for the in vivo analysis of anti-tumour efficiency of EpCAMxCD3. These findings also present important prerequisites for a future use of ex vivo manipulated and expanded autologous lymphocytes in a bsAb therapeutic strategy in patients.
Next, we performed EpCAMxCD3 treatment experiments and injected NOD SCID mice s.c. with BxPC-3 cells mixed with extracorporally pre-activated human lymphocytes. Three days after cell transplantation, each group of animals received the first i.p. injection of EpCAMxCD3 at the doses of 0.1, 1 or 10 mg/kg, respectively. Then bsAb treatment was repeated four times with a 3-day interval. Repeated measurements of tumour growth during a time span of 24 days revealed a dose-dependent anti-tumour effect of EpCAMxCD3 (Fig. 2). Mice that received a high dose of EpCAMxCD3 (10 mg/kg) had dramatically reduced tumour take: only 50% (4 out of 8) of mice developed small tumours compared with 100% (8 out of 8) of mice in the control group. Also the control group of animals developed much larger tumours than the bsAb-treated group. The highest dose of EpCAMxCD3 retarded the BxPC-3 tumour growth compared with the animals that received control parental bivalent anti-CD3 and anti-EpCAM mAbs (Fig. 2A and B). An observed anti-tumour effect of EpCAMxCD3 at the dose of 0.1 mg/kg or 1 mg/kg was considerable compared with control Abs. Tumour take was 80% (4 out of 5 mice) in the first and second experimental groups; however, treatment efficiency in both groups was slightly less pronounced compared with the group of animals treated with the highest dose of EpCAMxCD3 (Fig. 2C and D). At day 24, the tumour weight was significantly reduced in all groups of mice treated with EpCAMxCD3 compared with the controls (Fig. 2F). Similar dose-dependent anti-tumour effects of EpCAMxCD3 were observed in an in vivo prostate carcinoma model with EpCAM-expressing PC-3 cells (Suppl. Fig. 1). In BxPC-3 tumour-bearing mice treated with 10 mg/kg of EpCAMxCD3, functionally available bsAb plasma level was 202 ± 45 ng/ml 7 days after the last injection. Body weight of tumour-bearing mice was not affected by the EpCAMxCD3 treatment (data not shown), which indicates an absence of severe toxic side effects of bsAb to normal mouse tissue. To investigate whether anti-tumour effects of EpCAMxCD3 were due to induction of apoptosis, BxPC-3 tumours were subjected to TUNEL assay and to immunohistochemistry to detect active caspase 3. The density of apoptotic cells and the percentage of active caspase 3–positive cells in BxPC-3 tumours were not different between EpCAMxCD3-treated tumours and controls (Suppl. Fig. 2). Thus, our data indicate that apoptosis induction is not the major mechanism for tumour growth retardation after EpCAMxCD3 treatment.
Figure 2.
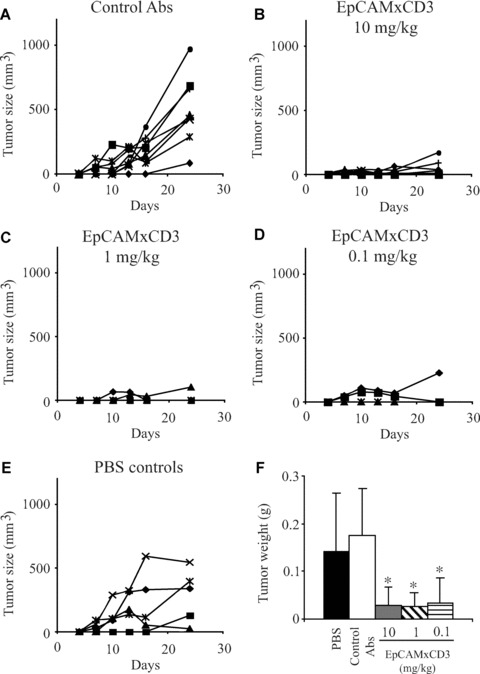
Efficiency of EpCAMxCD3 in the in vivo xenograft BxPC-3 pancreatic tumour model. NOD SCID mice were s.c. engrafted with BxPC-3 cells (5 × 106) mixed with pre-activated human lymphocytes (5 × 106) as described in Supplementary Materials and Methods. Three days later animals were randomized to five groups (five to eight animals per group) and received i.p. injections of either control parental bivalent anti-human EpCAM and anti-CD3 mAbs at a dose of 1 mg/kg (A), EpCAMxCD3 at doses 10 mg/kg (B), 1 mg/kg (C), 0.1 mg/kg (D) or PBS (E). Treatment was repeated four times with a 3-day interval. Tumour growth was measured every third day as described. Data are presented as growth kinetics of individual tumours. In (F) the weight of tumours is shown at the end-point of the bsAb treatment at day 24.
EpCAMxCD3 increases duration of the contact between lymphocytes and carcinoma cells without affecting lymphocyte migration
To investigate potential mechanisms of in vivo anti-tumour effects of EpCAMxCD3, we used a collagen gel 3D tumour reconstruct system, which closely resembled the tumour microenvironment. BxPC-3 cells and human lymphocytes were co-cultivated in the collagen matrix. Dynamic cell–cell interactions were analyzed using time-lapse video microscopy. Contact duration between lymphocytes and tumour cells was about 3 times longer in the presence of EpCAMxCD3 compared with control Abs (Fig. 3A). Pre-loading of lymphocytes with EpCAMxCD3 more efficiently increased the contact time with carcinoma cells suggesting ex vivo linking of patient-derived autologous lymphocytes to EpCAMxCD3 for a most effective anti-tumour immunotherapy. To delineate effects of EpCAMxCD3 on lymphocyte locomotion, we determined lymphocyte migration velocity and the percentage of migrating lymphocytes in 3D collagen gels. Lymphocyte locomotion was analyzed only for actively moving cells. Arrested lymphocytes following their contact with EpCAM-expressing tumour cells were excluded from an estimation of the velocity of lymphocyte migration. It appeared that EpCAMxCD3 did not influence lymphocyte velocity (Fig. 3B). Lymphocyte migration velocity in 3D gels was as high as 60 times of that of tumour cells (data not shown). The proportion of actively migrating lymphocytes was slightly decreased after exposure to EpCAMxCD3 compared with controls; however, the difference did not reach statistical significance (Fig. 3C).
Figure 3.

Effects of EpCAMxCD3 on migratory properties of lymphocytes and duration of contacts between lymphocytes and tumour cells. To evaluate the influence of EpCAMxCD3 on lymphocyte migration velocity and their interactions with tumour cells, the motility of pre-activated lymphocytes was monitored in 3D collagen gels using time-lapse video microscopy. Lymphocytes and BxPC-3 cells were mixed at a concentration of 2 × 105 cells per collagen gel. Lymphocytes were either pre-incubated with EpCAMxCD3 (10 μg/ml) or bsAb were added directly to the collagen solution prior to the 3D gel polymerization. Control irrelevant bsAb CD19xCD3 or control bivalent parental anti-human EpCAM mAbs were added to the collagen solution prior to the 3D gel polymerization. Lymphocyte migration and their interactions with carcinoma cells were monitored for a period of 1.5 hrs. (A) Pre-loading of lymphocytes with EpCAMxCD3 or an addition of EpCAMxCD3 to 3D collagen gels increased contact duration between actively migrating lymphocytes and tumour cells compared with control Abs. Contacts between a single lymphocyte and a single tumour cell were analyzed using a ‘frame to frame’ method in CapImage software as described in Materials and Methods. (B) Effects of EpCAMxCD3 on lymphocyte migration velocity and (C) the percentage of migrating lymphocytes in 3D collagen gels.
EpCAMxCD3 potently stimulates production of tumouricidal cytokines IFN-γ and TNF-α by pre-activated lymphocytes in 3D tumour reconstructs
To mimic tumour microenvironment and to elucidate the mechanisms of anti-tumour effects of EpCAMxCD3 in an easily controlled setting, we established a collagen gel 3D tumour reconstruct system. Lymphocytes were able to freely migrate in these 3D tumour reconstructs. EpCAMxCD3 was applied on top of the polymerized 3D collagen gel to allow its diffusion and to closely mimic an in vivo microenvironment (Fig. 4A). In one set of experiments, pre-activated human lymphocytes were mixed with carcinoma cells and incubated in 3D collagen gels with fibroblasts. EpCAMxCD3, control parental mAb or irrelevant bsAb were added to pre-formed 3D tumour reconstructs. EpCAMxCD3 was used at a dose of 10 μg/ml in all 3D tumour reconstruct experiments, because this concentration was found to be optimal in our previous cytotoxic T-cell assay [31]. Adding of EpCAMxCD3 to 3D tumour reconstructs with pre-activated lymphocytes significantly increased production of IFN-γ (Fig. 4B) and TNF-α (Fig. 4C) compared with control Abs. Levels of IFN-γ and TNF-α were low in control 3D tumour reconstructs without any Abs or in 3D reconstructs lacking carcinoma cells (data not shown). Also, EpCAMxCD3 did not influence secretion of cytolytic molecule granzyme B in 3D tumour reconstructs with pre-activated lymphocytes (Suppl. Fig. 3A). However, perforin levels were increased in 3D reconstructs with EpCAMxCD3 after 72 hrs of incubation compared with control Abs (Suppl. Fig. 3A). To assess apoptosis induction in 3D tumour reconstructs, a protein array system was used to detect the presence of apoptotic proteins. We could detect the presence of cleaved caspase 3, catalase, cytochrome c and HSP70 in supernatants from 3D tumour reconstructs with EpCAMxCD3 but not in control reconstructs with irrelevant bsAb CD19xCD3 after 24 hrs of incubation (Suppl. Fig. 4). To investigate whether EpCAMxCD3 induces polyclonal activation of T cells, including regulatory T cells, we analyzed secretion of inhibitory cytokines IL-10 and TGF-β1. Only trace amounts of IL-10 were detectable in 3D tumour reconstructs with pre-activated lymphocytes. The level of TGF-β1 in 3D tumour reconstructs was around 100–200 pg/ml and was not affected by EpCAMxCD3.
Figure 4.
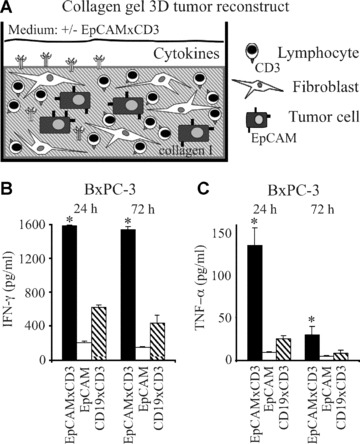
Effects of EpCAMxCD3 on the production of effector cytokines by pre-activated lymphocytes in 3D tumour reconstructs. (A) A schematic graph depicting the composition of collagen gel 3D tumour reconstructs. The production of IFN-γ (B) and TNF-α (C) in collagen gel 3D tumour reconstructs consisting of BxPC-3 carcinoma cells, pre-activated human lymphocytes and fibroblasts was analyzed by ELISA. EpCAMxCD3, control parental anti-human EpCAM mAb, or irrelevant bsAb CD19xCD3, respectively, were added to 3D tumour reconstructs (n= 4 per group) after the collagen gel polymerization. Supernatants from 3D tumour reconstructs were collected after 24 or 72 hrs of incubation of mixed cell cultures. *P < 0.05.
EpCAMxCD3 activates PBMCs in the presence of carcinoma cells
To evaluate the ability of EpCAMxCD3 to activate PBMCs in the presence of carcinoma cells, we employed 3D tumour reconstructs with non-stimulated PBMCs. Bivalent anti-CD3 mAb was used as a positive control and irrelevant bsAb CD19xCD3 or parental bivalent anti-EpCAM mAb as negative controls. Activation of PBMCs was analyzed by the production of effector cytokines IFN-γ, TNF-α and IL-2. EpCAMxCD3 induced a strong production of IFN-γ (Fig. 5A), TNF-α (Fig. 5B) and, to a lesser extent, IL-2 (Fig. 5C). EpCAMxCD3 was more efficient in stimulating TNF-α and IFN-γ production by PBMCs than a bivalent anti-CD3 mAb administered at the same dose after 24 hrs and TNF-α after 72 hrs of incubation (Fig. 5). Granzyme B level was 50% higher in 3D tumour reconstructs 24 hrs after addition of EpCAMxCD3 compared with control Abs. There was no difference in granzyme B levels between all groups after 72 hrs (Suppl. Fig. 3B). Also perforin level was increased in 3D reconstructs with EpCAMxCD3 after 24 hrs of incubation compared with irrelevant CD19xCD3 bsAb. There was no difference in perforin levels between all groups after 72 hrs (Suppl. Fig. 3B). Similarly, a loading of non-stimulated PBMCs with EpCAMxCD3 for 2 hrs prior to 3D tumour reconstruct experiments resulted in an increased production of IFN-γ and TNF-α compared with an addition of EpCAMxCD3. Thus, in 24 hrs the level of IFN-γ increased by 45% and after 72 hrs of incubation by 70%. In 24 hrs the production of TNF-α increased by 20%, compared with about 8 times increase in TNF-α production in 72 hrs following incubation (data not shown). In contrast to 3D tumour reconstructs with pre-activated lymphocytes, EpCAMxCD3 induced production of the inhibitory cytokine IL-10 in 3D tumour reconstructs with non-stimulated PBMCs (Fig. 6A). The TGF-β1 level ranged from 50 to 100 pg/ml (Fig. 6B). Because monocyte-derived IL-1β can induce the TGF-β production by fibroblasts, we analyzed its concentration in 3D tumour reconstructs. The amount of IL-1β was around 300 pg/ml in all 3D tumour reconstructs and EpCAMxCD3 did not influence IL-1β production. Concentrations of IL-10, IL-1β and TGF-β1 were not detectable in 3D tumour reconstructs of carcinoma cells co-cultured with fibroblasts without PBMCs.
Figure 5.
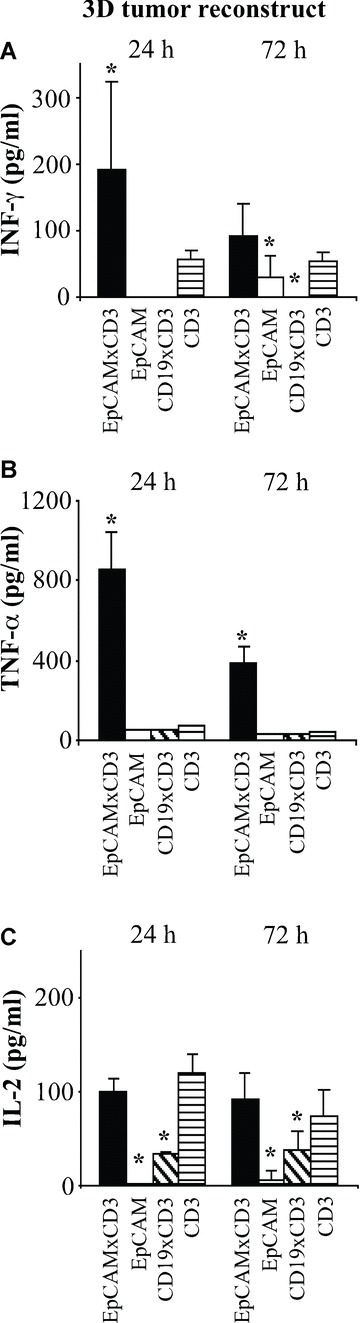
Effects of EpCAMxCD3 on the production of effector cytokines by non-stimulated PBMCs in 3D tumour reconstructs. The ability of EpCAMxCD3 to induce the production of IFN-γ (A), TNF-α (B) and IL-2 (C) by non-stimulated PBMCs was analyzed in 3D tumour reconstructs with BxPC-3 carcinoma cells. Non-stimulated PBMCs were incubated with carcinoma cells and fibroblasts in 3D collagen gels for 24 or 72 hrs (n= 4 per group). EpCAMxCD3, control parental anti-human EpCAM mAb, irrelevant bsAb CD19xCD3 or control bivalent anti-CD3 mAb, respectively, were added to 3D tumour reconstructs after the collagen gel polymerization. Cytokine levels in supernatants from 3D collagen gels were analyzed by ELISA.
Figure 6.

Effects of EpCAMxCD3 on the production of inhibitory cytokines by non-stimulated PBMCs in 3D tumour reconstructs. The ability of EpCAMxCD3 to induce the production of IL-10 (A) and TGF-β1 (B) by non-stimulated PBMCs was analyzed in 3D tumour reconstructs with BxPC-3 cells. Non-stimulated PBMCs were incubated with carcinoma cells and fibroblasts in 3D collagen gels for 24 or 72 hrs (n= 4 per group). EpCAMxCD3, control parental anti-human EpCAM mAb, irrelevant bsAb CD19xCD3 or control bivalent anti-CD3 mAb, respectively, were added to 3D tumour reconstructs after the collagen gel polymerization. Cytokine levels in supernatants from 3D collagen gels were analyzed by ELISA.
Taken together, we clearly demonstrate that EpCAMxCD3 potently stimulates pre-activated lymphocytes and non-stimulated PBMCs to secrete effector cytokines in 3D tumour reconstructs in the presence of EpCAM-expressing carcinoma cells.
Discussion
In the present work, we provide evidence that bsAb EpCAMxCD3 reduced tumour take and efficiently inhibited growth of experimental pancreatic carcinoma. EpCAMxCD3 has a long half-life in mouse serum, and EpCAMxCD3 treatment was well tolerated by tumour-bearing animals. In an in vitro collagen gel 3D tumour reconstruct system, EpCAMxCD3 induced a strong production of tumouricidal cytokines IFN-γ and TNF-α by extracorporally pre-activated human lymphocytes. It further evolved as a potent polyclonal activator of PBMCs as measured by the production of IFN-γ, TNF-α, IL-2 and IL-10. Moreover, EpCAMxCD3 substantially increased contact duration between lymphocytes and tumour cells in 3D tumour reconstructs.
In preliminary clinical studies, EpCAMxCD3 immunotherapy effectively reduced malignant ascites formation in ovarian cancer patients [27, 32, 38]. Our clinical study with EpCAMxCD3 showed that there was a dramatic increase in TNF-α concentration in ascites. However, the precise mode of EpCAMxCD3 action was not clear. To investigate anti-tumour effects of EpCAMxCD3 in more detail, we performed a set of experiments using in vitro collagen gel 3D tumour reconstructs and a BxPC-3 pancreatic carcinoma xenograft model.
For in vivo experiments, carcinoma cells were mixed with extracorporally pre-activated human lymphocytes and subsequently engrafted s.c. into NOD SCID mice. This approach was preferable to a subcutaneous engrafting of tumour cells followed by an intravenous administration of human lymphocytes because of redistribution and removal from circulation of activated lymphocytes in mouse organs. A similar in vivo tumour model was advocated by Schlereth et al.[28] and Brischwein et al.[39] studying the therapeutic effects of single-chain bsAb EpCAMxCD3 constructs MT102 and MT110 in xenograft SW480 colon tumours. Our experiments demonstrated that the administration of a mixture of pre-activated lymphocytes and tumour cells did not affect xenograft BxPC-3 tumour growth and its morphology. BxPC-3 tumours established a robust angiogenesis and a collagen containing stroma. Our findings that pre-activated lymphocytes survived in BxPC-3 tumours for more than 24 days demonstrated that our in vivo experimental model is suitable for EpCAMxCD3 anti-cancer treatment evaluation.
Both EpCAM and CD3 molecules are attractive targets for generation of bsAb and bispecific single-chain antibody constructs for anti-cancer treatment [28, 30, 39, 40]. A single-chain bsAb EpCAMxCD3, MT102, reduced growth of experimental SW480 colon tumours [28]. Because the terminal elimination half-life of MT102 is around 5.3 hrs, it had to be administered daily [28]. Importantly, we have found that our bsAb EpCAMxCD3 has a much longer half-life in mouse serum (t1/2∼ 7 days) and was detectable in tumours even 7 days after the last administration. Therefore, an increased serum half-life of EpCAMxCD3 may provide a considerable clinical advantage over single-chain bsAb constructs. In addition, attempts to generate recombinant humanized EpCAMxCD3 constructs are underway in our laboratories (Deppe et al., in preparation).
Previously, we have shown that EpCAMxCD3 brings T cells and tumour cells in a close contact redirecting T cells to kill carcinoma cells [31]. By binding to CD3 molecules on lymphocytes with one arm and to EpCAM antigen on tumour cells with the other arm, bsAb activate lymphocytes in an antigen-independent manner. This lymphocyte activation leads to the production of effector cytokines and cytolytic substances such as TNF-α, IFN-γ, granzyme B and perforin. Furthermore, Wimberger et al.[41] showed that tumour resident T cells following treatment with a single-chain bsAb EpCAMxCD3 efficiently lysed malignant cells ex vivo. Therefore, resting tumour resident lymphocytes can be potential EpCAMxCD3 targets for non-restricted stimulation of T cells. Our findings that efficient BxPC-3 tumour growth inhibition by EpCAMxCD3 was not associated with therapy-related toxic side effects in tumour-bearing mice indicate a local production of effector cytokines and an absence of TNF-α and IFN-γ in systemic circulation. BsAb EpCAMxCD3 is composed of mouse IgG1 and IgG2A constant heavy chain regions and has low affinity to human Fc receptors. This property of EpCAMxCD3 would also reduce Fc-mediated side effects. By contrast, the therapeutic efficacy of other bsAb constructs greatly relies on the Fc portion, for instance, in case of mixed rat/mouse heavy chains [21, 27].
The contact time between migrating lymphocytes and tumour cells in an in vitro 3D collagen gel system was 3 times longer in the presence of EpCAMxCD3 compared with control antibodies. The increased time of a direct contact between target and effector cells indicates that EpCAMxCD3 facilitates interactions between lymphocytes and tumour cells regardless of T-cell receptor specificity. Unfortunately, this experimental setting did not allow us to monitor the fate of tumour cells following contacts with T cells. However, it is plausible that these findings might be used in the development of approaches for site-specific delivery of anti-cancer agents.
In an in vitro 3D collagen gel system, we investigated whether binding of EpCAMxCD3 to CD3 molecules would affect migratory properties of lymphocytes. Leucocyte trafficking within 3D collagen matrices is independent of matrix remodelling by proteases [42]. Lymphocytes express collagen-binding integrins α1β1 and α2β1. Our results regarding the lymphocyte velocity in 3D collagen gels are in agreement with those of Niggemann et al.[43]. Findings that EpCAMxCD3 did not influence lymphocyte velocity on a collagen type I substrate suggest that EpCAMxCD3 binding to CD3 molecules does not block collagen receptors on lymphocytes.
EpCAMxCD3 potently stimulated IFN-γ secretion by pre-activated lymphocytes, whereas the production of TNF-α was less pronounced. In contrast, TNF-α was the predominant cytokine produced in 3D reconstructs with non-stimulated PBMCs in the presence of EpCAMxCD3. The latter findings are in agreement with the clinical data showing that TNF-α level was markedly increased in ascites from ovarian cancer patients treated with EpCAMxCD3 [32]. Noteworthy is that EpCAMxCD3 induced production of inhibitory cytokine IL-10 in reconstructs with PBMCs but not with pre-activated lymphocytes. This may indicate a non-specific activation of regulatory T cells by EpCAMxCD3. Because regulatory T cells are the major source of inhibitory cytokines in PBMCs [44, 45], we analyzed the TGF-β1 production in 3D tumour reconstructs. The TGF-β1 level was only marginally increased in 3D tumour reconstructs with EpCAMxCD3 after 72 hrs of incubation. We ruled out a possibility that monocyte-derived IL-1β could contribute to the TGF-β1 production in 3D reconstructs with non-stimulated PBMCs via stimulation of fibroblasts, because IL-1β levels were similar in all 3D reconstructs. Because the binding of bsAb containing IgG1 and IgG2A constant heavy chain regions to human monocytes is usually weak, the potential activation of T cells via binding of EpCAMxCD3 to Fc receptors on monocytes is rather unlikely in 3D tumour reconstruct experiments. Taken together, these results indicate that EpCAMxCD3 is a strong inducer of lymphocyte effector functions such as secretion of tumouricidal cytokines. The response of lymphocytes to EpCAMxCD3 is dependent on their activation state.
In conclusion, we have shown that EpCAMxCD3 possesses a potent anti-tumour activity in vivo and increases duration of the contact between migrating lymphocytes and tumour cells in an in vitro 3D tumour reconstruct system. EpCAMxCD3 stimulates secretion of effector cytokines IFN-γ and TNF-α by both pre-activated lymphocytes and non-stimulated PBMCs in the presence of EpCAM-bearing tumour cells.
Acknowledgments
We thank Dr. H.-J. Stark for providing human primary skin fibroblasts and Ms. Elvira Hallauer and Mr. Jury Gladkich for excellent technical assistance. This study was supported by grants 01GU0611 and 01GU0612 from the German Federal Ministry of Education and Research (BMBF).
Supporting Information
Fig. S1. Anti-tumor efficiency of EpCAMxCD3 in invivo xenograft PC-3 prostate tumor model. NOD SCID mice weres.c. engrafted with PC-3 prostate tumor cells (5 x106) mixed with pre-activated human lymphocytes (5 x106) as described for the BxPC-3 pancreatic tumor model.Three days later animals were randomized to 5 groups (5 animals pergroup) and received i.p. injections of either controlparental bivalent anti-human EpCAM and anti-CD3 mAbs at a dose of 1mg/kg (A), EpCAMxCD3 at doses 10 mg/kg (B), 1 mg/kg(C), 0.1 mg/kg (D), or PBS (E). Treatment wasrepeated 4 times with a three day interval. Tumor growth wasmeasured every third day as described in M&M. Data arepresented as growth kinetics of individual tumors. In (F)the weight of tumors is shown at the end-point of the bsAbtreatment.
Fig. S2. Effects of EpCAMxCD3 on apoptosis induction inxenograft BxPC-3 pancreatic tumors. To evaluate whether anti-tumoreffects of EpCAMxCD3 were due to apoptosis induction, BxPC-3 tumorswere subjected to TUNEL assay and to immunohistochemistry to detectactive caspase 3. (A) The density of apoptotic cells inBxPC-3 tumor tissues visualized by TUNEL staining (n = 4per group). The density (B) and the percentage (C) ofactive caspase 3-positive cells in BxPC-3 tumor tissues (n= 4 per group).
Fig. S3. Effects of EpCAMxCD3 on the production ofgranzyme B and perforin in 3D tumor reconstructs. The levels ofgranzyme B and perforin in collagen gel 3D tumor reconstructs bypre-activated lymphocytes (A) or by non-stimulated PBMCs(B) was analyzed by ELISA. Collagen gel 3D tumorreconstructs consisting of BxPC-3 carcinoma cells, fibroblasts andpre-activated lymphocytes or non-stimulated PBMCs were prepared asdescribed in M&M. Bispecific EpCAMxCD3 antibody, controlparental anti-human EpCAM mAb, or irrelevant bsAb CD19xCD3,respectively, were added to the 3D tumor reconstructs (n= 4 per group) after the collagen solution polymerizedto a gel. Supernatants from 3D tumor reconstructs were collectedafter 24 or 72 hours of incubation. *p < 0.05.
Fig. S4. Apoptosis detection in 3D tumor reconstructs.Supernatants of 3D tumor reconstructs were collected after 24 hoursof mixed cell cultures and apoptosis-related proteins were detectedusing a human apoptosis antibody array kit according to theinstructions from the manufacturer (R&D Systems, Abingdon, UK).(A) X-ray film images of apoptotic array membranes showpositive signals for five apoptosis-related proteins pro-caspase 3,cleaved caspase 3, catalase, cytochrome c and HSP70 in pooledsupernatants (n = 4) from 3D tumor reconstructsincubated with bsAb EpCAMxCD3 compared to pooled supernatants (n= 4) from control 3D tumor reconstructs incubated withirrelevant bsAb CD19xCD3. Three pairs of positive control spots arelocated in the corners of each array. (B) The average signalof the duplicate spots representing each apoptosis-related proteinwas quantified using ImageJ software.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
References
- 1.Neoptolemos JP, Dunn JA, Stocken DD, et al. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet. 2001;358:1576–85. doi: 10.1016/s0140-6736(01)06651-x. [DOI] [PubMed] [Google Scholar]
- 2.Went PT, Lugli A, Meier S, et al. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004;35:122–8. doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 3.Winter MJ, Nagtegaal ID, van Krieken JH, et al. The epithelial cell adhesion molecule (Ep-CAM) as a morphoregulatory molecule is a tool in surgical pathology. Am J Pathol. 2003;163:2139–48. doi: 10.1016/S0002-9440(10)63570-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poczatek RB, Myers RB, Manne U, et al. Ep-Cam levels in prostatic adenocarcinoma and prostatic intraepithelial neoplasia. J Urol. 1999;162:1462–6. [PubMed] [Google Scholar]
- 5.Gastl G, Spizzo G, Obrist P, et al. Ep-CAM overexpression in breast cancer as a predictor of survival. Lancet. 2000;356:1981–2. doi: 10.1016/S0140-6736(00)03312-2. [DOI] [PubMed] [Google Scholar]
- 6.Varga M, Obrist P, Schneeberger S, et al. Overexpression of epithelial cell adhesion molecule antigen in gallbladder carcinoma is an independent marker for poor survival. Clin Cancer Res. 2004;10:3131–6. doi: 10.1158/1078-0432.ccr-03-0528. [DOI] [PubMed] [Google Scholar]
- 7.Spizzo G, Obrist P, Ensinger C, et al. Prognostic significance of Ep-CAM and Her-2/neu overexpression in invasive breast cancer. Int J Cancer. 2002;98:883–8. doi: 10.1002/ijc.10270. [DOI] [PubMed] [Google Scholar]
- 8.Spizzo G, Went P, Dirnhofer S, et al. High Ep-CAM expression is associated with poor prognosis in node-positive breast cancer. Breast Cancer Res Treat. 2004;86:207–13. doi: 10.1023/B:BREA.0000036787.59816.01. [DOI] [PubMed] [Google Scholar]
- 9.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–63. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 12.Armstrong A, Eck SL. EpCAM: a new therapeutic target for an old cancer antigen. Cancer Biol Ther. 2003;2:320–6. doi: 10.4161/cbt.2.4.451. [DOI] [PubMed] [Google Scholar]
- 13.Chaudry MA, Sales K, Ruf P, et al. EpCAM an immunotherapeutic target for gastrointestinal malignancy: current experience and future challenges. Br J Cancer. 2007;96:1013–9. doi: 10.1038/sj.bjc.6603505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prang N, Preithner S, Brischwein K, et al. Cellular and complement-dependent cytotoxicity of Ep-CAM-specific monoclonal antibody MT201 against breast cancer cell lines. Br J Cancer. 2005;92:342–9. doi: 10.1038/sj.bjc.6602310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andratschke M, Gildehaus FJ, Johannson V, et al. Biodistribution and radioimmunotherapy of SCCHN in xenotransplantated SCID mice with a 131I-labelled anti-EpCAM monoclonal antibody. Anticancer Res. 2007;27:431–6. [PubMed] [Google Scholar]
- 16.Oberneder R, Weckermann D, Ebner B, et al. A phase I study with adecatumumab, a human antibody directed against epithelial cell adhesion molecule, in hormone refractory prostate cancer patients. Eur J Cancer. 2006;42:2530–8. doi: 10.1016/j.ejca.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 17.Kirman I, Whelan RL. Drug evaluation: adecatumumab, an engineered human anti-EpCAM antibody. Curr Opin Mol Ther. 2007;9:190–6. [PubMed] [Google Scholar]
- 18.Fischer N, Leger O. Bispecific antibodies: molecules that enable novel therapeutic strategies. Pathobiology. 2007;74:3–14. doi: 10.1159/000101046. [DOI] [PubMed] [Google Scholar]
- 19.Muller D, Kontermann RE. Recombinant bispecific antibodies for cellular cancer immunotherapy. Curr Opin Mol Ther. 2007;9:319–26. [PubMed] [Google Scholar]
- 20.Zeidler R, Reisbach G, Wollenberg B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol. 1999;163:1246–52. [PubMed] [Google Scholar]
- 21.Zeidler R, Mysliwietz J, Csanady M, et al. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer. 2000;83:261–6. doi: 10.1054/bjoc.2000.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98:2526–34. doi: 10.1182/blood.v98.8.2526. [DOI] [PubMed] [Google Scholar]
- 23.Reusch U, Sundaram M, Davol PA, et al. Anti-CD3 x anti-epidermal growth factor receptor (EGFR) bispecific antibody redirects T-cell cytolytic activity to EGFR-positive cancers in vitro and in an animal model. Clin Cancer Res. 2006;12:183–90. doi: 10.1158/1078-0432.CCR-05-1855. [DOI] [PubMed] [Google Scholar]
- 24.Ren-Heidenreich L, Davol PA, Kouttab NM, et al. Redirected T-cell cytotoxicity to epithelial cell adhesion molecule-overexpressing adenocarcinomas by a novel recombinant antibody, E3Bi, in vitro and in an animal model. Cancer. 2004;100:1095–103. doi: 10.1002/cncr.20060. [DOI] [PubMed] [Google Scholar]
- 25.Morecki S, Lindhofer H, Yacovlev E, et al. Induction of long-lasting antitumor immunity by concomitant cell therapy with allogeneic lymphocytes and trifunctional bispecific antibody. Exp Hematol. 2008;36:997–1003. doi: 10.1016/j.exphem.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Wolf E, Hofmeister R, Kufer P, et al. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. 2005;10:1237–44. doi: 10.1016/S1359-6446(05)03554-3. [DOI] [PubMed] [Google Scholar]
- 27.Burges A, Wimberger P, Kumper C, et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: a phase I/II study. Clin Cancer Res. 2007;13:3899–905. doi: 10.1158/1078-0432.CCR-06-2769. [DOI] [PubMed] [Google Scholar]
- 28.Schlereth B, Fichtner I, Lorenczewski G, et al. Eradication of tumors from a human colon cancer cell line and from ovarian cancer metastases in immunodeficient mice by a single-chain Ep-CAM-/CD3-bispecific antibody construct. Cancer Res. 2005;65:2882–9. doi: 10.1158/0008-5472.CAN-04-2637. [DOI] [PubMed] [Google Scholar]
- 29.Amann M, Brischwein K, Lutterbuese P, et al. Therapeutic window of MuS110, a single-chain antibody construct bispecific for murine EpCAM and murine CD3. Cancer Res. 2008;68:143–51. doi: 10.1158/0008-5472.CAN-07-2182. [DOI] [PubMed] [Google Scholar]
- 30.Sebastian M, Passlick B, Friccius-Quecke H, et al. Treatment of non-small cell lung cancer patients with the trifunctional monoclonal antibody catumaxomab (anti-EpCAM x anti-CD3): a phase I study. Cancer Immunol Immunother. 2007;56:1637–44. doi: 10.1007/s00262-007-0310-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strauss G, Guckel B, Wallwiener D, et al. Without prior stimulation, tumor-associated lymphocytes from malignant effusions lyse autologous tumor cells in the presence of bispecific antibody HEA125xOKT3. Clin Cancer Res. 1999;5:171–80. [PubMed] [Google Scholar]
- 32.Marme A, Strauss G, Bastert G, et al. Intraperitoneal bispecific antibody (HEA125xOKT3) therapy inhibits malignant ascites production in advanced ovarian carcinoma. Int J Cancer. 2002;101:183–9. doi: 10.1002/ijc.10562. [DOI] [PubMed] [Google Scholar]
- 33.Zhang C, Kolb A, Buchler P, et al. Corticosteroid co-treatment induces resistance to chemotherapy in surgical resections, xenografts and established cell lines of pancreatic cancer. BMC Cancer. 2006;6:61. doi: 10.1186/1471-2407-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Csoka M, Strauss G, Debatin KM, et al. Activation of T cell cytotoxicity against autologous common acute lymphoblastic leukemia (cALL) blasts by CD3xCD19 bispecific antibody. Leukemia. 1996;10:1765–72. [PubMed] [Google Scholar]
- 35.Moldenhauer G, Momburg F, Moller P, et al. Epithelium-specific surface glycoprotein of Mr 34,000 is a widely distributed human carcinoma marker. Br J Cancer. 1987;56:714–21. doi: 10.1038/bjc.1987.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Momburg F, Moldenhauer G, Hammerling GJ, et al. Immunohistochemical study of the expression of a Mr 34,000 human epithelium-specific surface glycoprotein in normal and malignant tissues. Cancer Res. 1987;47:2883–91. [PubMed] [Google Scholar]
- 37.Ryschich E, Kerkadze V, Lizdenis P, et al. Active leukocyte crawling in microvessels assessed by digital time-lapse intravital microscopy. J Surg Res. 2006;135:291–6. doi: 10.1016/j.jss.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 38.Heiss MM, Strohlein MA, Jager M, et al. Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer. 2005;117:435–43. doi: 10.1002/ijc.21165. [DOI] [PubMed] [Google Scholar]
- 39.Brischwein K, Schlereth B, Guller B, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43:1129–43. doi: 10.1016/j.molimm.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 40.Brischwein K, Parr L, Pflanz S, et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J Immunother. 2007;30:798–807. doi: 10.1097/CJI.0b013e318156750c. [DOI] [PubMed] [Google Scholar]
- 41.Wimberger P, Xiang W, Mayr D, et al. Efficient tumor cell lysis by autologous, tumor-resident T lymphocytes in primary ovarian cancer samples by an EP-CAM-/CD3-bispecific antibody. Int J Cancer. 2003;105:241–8. doi: 10.1002/ijc.11056. [DOI] [PubMed] [Google Scholar]
- 42.Wolf K, Muller R, Borgmann S, et al. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood. 2003;102:3262–9. doi: 10.1182/blood-2002-12-3791. [DOI] [PubMed] [Google Scholar]
- 43.Niggemann B, Maaser K, Lu H, et al. Locomotory phenotypes of human tumor cell lines and T lymphocytes in a three-dimensional collagen lattice. Cancer Lett. 1997;118:173–80. doi: 10.1016/s0304-3835(97)00328-5. [DOI] [PubMed] [Google Scholar]
- 44.Roncarolo MG, Bacchetta R, Bordignon C, et al. Type 1 T regulatory cells. Immunol Rev. 2001;182:68–79. doi: 10.1034/j.1600-065x.2001.1820105.x. [DOI] [PubMed] [Google Scholar]
- 45.Roncarolo MG, Gregori S, Battaglia M, et al. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item