

Abstract
Most poxvirus proteins are either highly conserved and essential for basic steps in replication or less conserved and involved in host interactions. Homologs of the CPXV219 protein, encoded by cowpox virus, are present in nearly all chordopoxvirus genera and some species have multiple copies. The CPXV219 homologs have estimated masses of greater than 200 kDa, making them the largest known poxvirus proteins. We showed that CPXV219 was expressed early in infection and cleaved into N- and C-terminal fragments that remained associated. The protein has a signal peptide and transited the secretory pathway where extensive glycosylation and proteolytic cleavage occurred. CPXV219 was located by immunofluorescence microscopy in association with the endoplasmic reticulum, Golgi apparatus and plasma membrane. In non-permeabilized cells, CPXV219 was accessible to external antibody and biotinylation. Mutants that did not express CPXV219 replicated normally in cell culture and retained virulence in a mouse respiratory infection model.
Graphical Abstract
Introduction
Poxviruses are large DNA viruses that reproduce in the cytoplasm of infected cells and have been widely studied because of their impact on human health, zoonotic spread, historic role as the first live virus vaccine, modern development as recombinant vaccine vectors, and use as model systems to investigate virus replication and host interactions (Damon, 2013; Moss, 2013). Of the 200 or more proteins encoded by poxviruses, nearly 100 are conserved in all members of the chordopoxvirus subfamily and about half of those are also conserved in entomopoxviruses (Upton et al., 2003). The majority of the highly conserved proteins have essential roles in virus replication, whereas most of the less well-conserved proteins of chordopoxviruses are concerned with modulating host interactions (Haller et al., 2014).
The goal of the present study was to characterize an unusual protein that is widely conserved among the chordopoxvirus genera except for members of the parapoxvirus genus, suggesting an evolutionarily conserved but not strictly essential function. Curiously, vaccinia virus is the sole member of the orthopoxvirus genus that lacks an open reading frame (ORF) encoding this protein. With predicted molecular weights of more than 200 kDa, these conserved proteins are larger than any other known poxvirus proteins. Here we characterize the cowpox virus (CPXV) homolog encoded by open reading frame CPXV219 (UniProt Q8QMM9). CPXV is of particular interest because it may have been the original smallpox vaccine, is the cause of an increasing number of zoonoses, contains the largest and most complete genome and has the broadest host range of all known orthopoxviruses (Dabrowski et al., 2013; Gubser et al., 2004). CPXV219 was expressed early during infection, trafficked through the secretory pathway, was N-glycosylated and cleaved into two major fragments and exposed on the plasma membrane. Additional studies indicated that the CPXV219 gene was dispensable for growth in tissue culture and unnecessary for virulence in mice.
Results
CPXV219 is conserved in most chordopoxviruses
The CPXV219 ORF of the Brighton red strain of CPXV is located near the right end of the genome and is predicted to encode a 1,919 amino acid protein with a N-terminal signal peptide (SP), a near C-terminal transmembrane (TM) domain, two additional hydrophobic domains, and sites of N-linked glycosylation (Fig. 1A–C). With the exception of VACV, all sequenced orthopoxviruses have similar length orthologs. In VACV strain WR, the absence of DNA adjacent to the expected site of the CPXV219 ortholog suggests the occurrence of a large deletion. The amino acid sequence identities of CPXV219 homologs of the orthopoxviruses ectromelia virus, variola virus, monkeypox virus and horsepox virus compared to CPXV are 93%, 93%, 85% and 86%. Except for parapoxviruses, at least one strain of all other chordopoxvirus genera encodes a CPXV219 ortholog with sequence identity of 30 to 45% relative to CPXV219. The highest sequence conservation of CPXV219 orthologs is in the C-terminal region and some sheeppox strains have a N-terminal deletion. Avipoxvirus strains contain 5 to 6 duplicated full-length orthologs in the center of the genome and crocodilepox virus has 3 copies near the left end of the genome. No homologs have been recognized in entomopoxviruses, non-poxvirus species or prokaryotic or eukaryotic organisms.
Fig. 1.

Predicted features of CPXV219. (A) Diagram of CPXV219 ORF. Top shows the position of CPXV219 (blue) flanked by ORFs of unknown function (pink) near the right end of the CPXV genome. CPXV Serp123 corresponds to VACV C12L. Within the gene is a 20 amino acid N-terminal signal peptide (SP) predicted by the SignalP program (http://www.cbs.dtu.dk/services/SignalP/), a strongly predicted C-terminal transmembrane (TM) domain and hydrophobic domains (HD) that were more weakly predicted TMs by the DAS TM prediction server (http://www.sbc.su.se/~miklos/DAS/). (B) Hydrophobicity plot of CPXV219 predicted by the Protscale program using a window of 9 (http://web.expasy.org/cgi-bin/protscale/protscale.pl Kyle & Doolittle). (C) N-glycosylation sites predicted by the NetNGlyc program (http://www.cbs.dtu.dk/services/NetNGlyc/).
CPXV219 is non-essential for replication
The absence of a CPXV219 homolog in VACV suggested that the protein is not essential for replication and a recent study determined that CPXV219 could be deleted from CPXV, though the growth properties of the mutant were not described (Xu et al., 2014). To more fully analyze the requirement of the 219 protein for CPXV replication, homologous recombination was used to make a deletion mutant (CPXV 219delGFP) by replacing the entire approximately 6,000 bp ORF in CPXV with one encoding the green fluorescent protein regulated by a VACV promoter in the same orientation as CPXV219. Recombinant virus plaques were identified by fluorescence microscopy and clonally purified by repeated plaque isolation. In addition, the GFP of CPXV 219delGFP was replaced with the original CPXV219 gene in order to make the control virus CPXV 219Rev and with one containing stop codons near the N-terminus to make the mutant CPXV 219Stop. PCR and DNA sequencing confirmed the genomic modifications (data not shown). In addition, we confirmed that the stop codons arrested translation by constructing and testing a recombinant CPXV that had the same stop codons in the CPXV219 gene with a C-terminal epitope tag (shown below). The CPXV 219delGFP and CPXV 219Stop viruses replicated under one step growth conditions and formed plaques as well as the parent and CPXV 219Rev control viruses in BS-C-1 cells (Fig. 2A, B) or HeLa, RK13, BHK21 and MRC5 cell lines (data not shown), demonstrating that the 219 protein was not required for CPXV replication in cell culture.
Fig. 2.

CPXV219 is not essential for virus replication. (A) One step growth curve. HeLa cells were infected with 3 PFU/cell of the parent CPXV strain Brighton (CPX-BR) and CPXV 219delGFP, 219Rev and 219Stop and the cells plus media were collected at the indicated times. The cells were lysed by repeated freeze-thawing and the virus titers were determined by plaque assay on BS-C-1 cells. (B) Plaque formation. BS-C-1 cells were infected with the indicted viruses, overlaid with semi-solid medium and stained with crystal violet after 72 h.
CPXV219 is expressed early in infection as full-length and N- and C-terminal fragments
Initial Western blotting experiments employing a recombinant CPXV with a FLAG tag at the C-terminus of CPXV219 revealed a minor band with an apparent mass greater than the 260 kDa marker, a major band of about 160 kDa and additional more rapidly migrating products (data not shown). To further investigate synthesis and apparent processing of CPXV219, we constructed a recombinant CPXV containing specific epitope tags at both the N- and C-termini. Because of the large size of the ORF, a stepwise green-to-white and white-to-green fluorescence screen was used to first replace the C-terminus with GFP and then replace the GFP with the C-terminus of CPXV219 containing an HA tag just before the termination codon to form CPXV 219-HA. A similar two-step procedure was used to replace the N-terminus of 219-HA with GFP and then with the N-terminus of CPXV219 containing a V5 tag downstream of the predicted SP to form CPXV V5-219-HA. In this manner, we obtained a recombinant virus with a specific tag at each end of CPXV219 (Fig. 3A). The addition of the epitope tags did not affect plaque formation or growth kinetics (not shown).
Fig. 3.
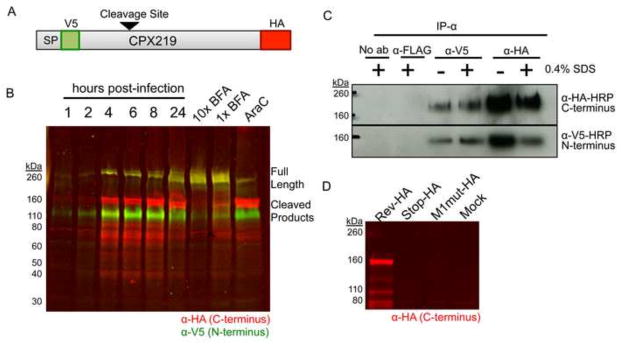
Expression of CPXV219. (A) Diagram of the CPXV219 protein containing a V5 epitope tag immediately after the predicted SP and an HA epitope tag just before the termination codon expressed by CPXV V5-219-HA. The arrow indicates the predicted cleavage site. (B) Western blots. HeLa cells were infected with 3 PFU per cell and whole cell lysates were collected at the indicated hours. In parallel, cells were pretreated and infected with 5 μg/ml (1X) or 50 μg/ml (10X) of BFA or 40 μg/ml AraC for 24 h. The infected cells were lysed with β-mercaptoethanol and SDS and the proteins were resolved by SDS-PAGE. The proteins were transferred to a membrane, probed with antibodies to the V5 (green) and HA (red) epitopes and visualized by infrared fluorescent imaging. Yellow indicates coincidence of V5 and HA antibodies. (C) Association of N- and C-terminal fragments. HeLa cells were infected with 5 PFU per cell of CPXV V5-219-HA for 24 h, harvested in non-denaturing lysis buffer, and incubated with anti-V5 or anti-HA MAb in the presence or absence of 0.4% SDS. Antibody complexes were bound to magnetic protein G Dynabeads and then analyzed by SDS-PAGE and Western blotting. (D) Absence of internal initiation. BS-C-1 cells were infected with 5 PFU per cell of CPXV 219RevHA, CPXV 219Stop-HA and CPXV M1mut-HA. After 24 h, whole cell lysates were analyzed by SDS-PAGE and Western blotting using antibody to the HA epitope tag, and visualized by infrared fluorescence. The positions of marker proteins are shown on the left of each panel.
To analyze protein synthesis, HeLa cells were infected with CPXV V5-219-HA, collected at sequential times and lysed directly in buffer containing SDS and β-mercaptoethanol. The proteins were resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose filters and probed with antibodies to the epitope tags followed by infrared dyes, which allowed the two tags to be visualized in the same gel. The major CPXV219 bands were prominent at 4 h, increased at 6 h and unchanged at 8 h suggesting an early promoter (Fig. 3B), which was consistent with the putative promoter sequence preceding the ORF (not shown). Expression of CPXV219 in the presence of cytosine arabinoside (AraC), an inhibitor of DNA replication that prevents intermediate and late gene expression, confirmed regulation by an early promoter (Fig. 3B). Antibodies to both N- and C-terminal tags recognized the slowest migrating band with an estimated mass of >260-kDa (which appeared yellow due to the overlap of red and green), indicating that it was the full-length protein. This protein was present in increased amounts, relative to the smaller ones, at 24 h. Antibody to the N-terminal tag recognized a 110-kDa band in addition to the full-length protein, whereas antibody to the C-terminal tag recognized a prominent 160-kDa band as well as smaller bands ranging from 80 to 40 kDa in mass (Fig. 3B).
Association of the N- and C-terminal fragments of CPXV219
Western blotting indicated that most of the CPXV219 is present as N- and C-terminal fragments. Although the fragments separated under denaturing and reducing conditions, there was a possibility that they remain associated in infected cells. To investigate whether the fragments associate with each other, we carried out immunoprecipitation (IP) experiments. HeLa cells were infected with the CPXV V5-219-HA and solubilized with 1% NP-40. The lysates were incubated with antibody to the V5 or HA tags in the presence or absence of 0.4% SDS to allow separation of loosely bound proteins, followed by the addition of protein G dynabeads. After washing, the proteins were eluted from the beads and analyzed by SDS-PAGE and Western blotting (Fig. 3C). When anti-V5 MAb was used to capture the CPXV219 protein, irrespective of SDS, the N-terminal V5 and C-terminal HA fragments were detected by Western blotting indicating their association. Similarly, when anti-HA MAb was used to capture the CPXV219 protein, both fragments were detected by Western blotting. Control experiments showed absence of pull-down with irrelevant anti-FLAG MAb. Intracellular co-localization shown below by confocal microscopy also supported the association of the N- and C-terminal fragments.
Proteolytic processing of CPXV219
We considered several possibilities for the appearance of multiple forms of CPXV219: proteolytic cleavage, internal transcription initiation and internal translation initiation. Additional CPXV mutants were constructed to rule out a possibility that the 160-kDa and smaller C-terminal fragments arise from internal transcription or translation start sites. Starting with the virus containing CPXV219 that had GFP replacing the N-terminus and HA at the C-terminus (used for making CPXV V5-219-HA), we swapped GFP with either (i) the N-terminus of CPXV219 in which the first methionine was mutated from ATG to AGT (CPXV 219M1mut-HA) or (ii) in which the codons at position 5 and 10 were mutated to stop sequences (CPXV 219Stop-HA) or (iii) the original N-terminus was restored (CPXV 219Rev-HA). The recombinant viruses with HA-tags at the C-termini were clonally purified and characterized by PCR and DNA sequencing of the relevant portion of the genome. HeLa cells were infected with the recombinant viruses and the lysates were analyzed by SDS-PAGE and Western blotting with antibody to the C-terminal HA tag (Fig. 3D). No C-terminally labeled bands were detected from cells infected with CPXV 219Stop-HA or CPXV 219-M1mut providing evidence against internal initiation and supporting the model in which CPXV219 is produced as a long polypeptide and subsequently cleaved.
Cleavage of CPXV219 could be carried out by viral or cellular proteinases. The two predicted viral proteinases are homologs of VACV I7 (Ansarah-Sobrinho and Moss, 2004a; Byrd and Hruby, 2005) and G1 (Ansarah-Sobrinho and Moss, 2004b; Hedengren-Olcott et al., 2004), which are expressed post-replicatively. Although not characterized in CPXV, the putative promoter sequences suggested that they are also expressed post-replicatively. Since cleavage of CPXV219 occurred in the presence of a DNA replication inhibitor (Fig. 3B), it seemed unlikely that either the I7 or G1 homologs were involved. In view of the predicted signal peptide and C-terminal TM domain, we considered that proteinase cleavage might occur by a cellular enzyme during transit through the secretory system. The drug brefeldin A (BFA) inhibits protein transport from the endoplasmic reticulum to the Golgi apparatus indirectly by preventing formation of COPI-mediated transport vesicles (Doms et al., 1989; Lippincott-Schwartz et al., 1989). When CPXV V5-219-HA infected cells were treated with BFA, there was an accumulation of the 260-kDa band and reductions of the smaller 160- and 110-kDa bands, suggesting that cleavage occurs in the Golgi or a post-Golgi compartment during transport of CPXV219 through the secretory pathway (Fig. 3B).
CPXV219 undergoes SP cleavage
In a prior construct, we placed the V5 tag after the putative SP to ensure that it would be retained in the mature protein. To confirm SP cleavage actually occurs, we followed a previously described strategy (Husain et al., 2006) that takes advantage of two MAbs that react with the 19 amino acid FLAG tag: one (M1) reacts with the FLAG epitope only when it is located N-terminally, whereas the other (M2) reacts with internal as well as N-terminal FLAG epitope. Thus, reactivity with M1 MAb could occur only after SP cleavage. Three recombinant viruses were constructed with FLAG tags at the N-terminus (Fig. 4A). One construct (CPXV SP-FLAG-219) contained the predicted SP sequence of CPXV219 immediately preceding the FLAG epitope tag; a positive control (CPXV VGSP-FLAG-219) was created by replacing the predicted CPXV219 SP sequence with the known SP sequence of the G protein of vesicular stomatitis virus (VSV) followed by the FLAG epitope tag; a negative control (CPXV VGSP*-FLAG-219) was generated using a VSV-G SP sequence with two amino acids mutated that impair cleavage (Shaw et al., 1988). Cells were infected with the three viruses and the lysates were analyzed by SDS-PAGE and Western blotting. Using the M2 antibody, we showed that the three proteins were expressed and contained the FLAG epitope (Fig. 4A). However, only the SP-FLAG-219 and VGSP-FLAG-219 were detected by the M1 MAb that is specific for N-terminal FLAG, which could only arise by SP cleavage at the ER (Fig. 4A). The negative control VGSP*-FLAG-219 was not detected with M1 MAb (Fig. 4A). These results confirmed the prediction of signal peptide cleavage at the N-terminus of CPXV219.
Fig. 4.

Signal peptide cleavage and glycosylation of CPXV219. (A) Western blots. HeLa cells were infected with 10 PFU per cell of viruses expressing CPXV219 proteins that contain a single FLAG epitope tag following the putative CPXV219 SP (SP-219), the VSV-G SP (VG-SP) replacing the CPXV 219 SP, or non-cleavable VSV G SP (VG*-SP) replacing the CPX V219 SP. After 18 h, whole cell lysates were treated with β–mercaptoethanol and SDS and analyzed by SDS-PAGE and Western blotting. Separate membranes were blotted with anti-FLAG clone M2 or anti-FLAG antibody clone M1 and visualized by infrared fluorescence (upper) and chemiluminescence (lower). (B) Glycosylation. HeLa cells were infected with 3 PFU per cell of CPXV V5-219-HA or parental CPXV-BR. After 18 h, the cells were harvested, lysed with 50 mM Tris pH 7.0, 150 mM NaCl, and 0.5% NP-40 detergent and nuclei removed by centrifugation. The clarified lysates were incubated with PNGaseF, EndoH, or left untreated and analyzed by SDS-PAGE and Western blotting. Antibodies to V5 (green) and HA (red) were visualized by infrared fluorescence. Yellow results from coincidence of red and green. The positions of marker proteins are shown on the left of each panel.
CPXV219 is N-glycosylated in the ER and traffics to the Golgi apparatus
The finding of SP cleavage as well as the effect of BFA on cleavage of CPXV219 suggested that the protein traffics through the secretory pathway. Glycosylation could account for apparent mass of >260 kDa determined by SDS-PAGE, whereas the ORF sequence predicts a mass of 215 kDa. Bioinformatics predicted numerous N-glycosylation sites clustered mainly between codons 69 to 364 and additional sites between codons 997 and 1776 (Fig. 1A). To test these prediction, lysates of cells infected with CPXV V5-219-HA were digested with peptide N-glycosidase F (PNGaseF), which cleaves between the innermost GlcNAc and asparagine residues of N-linked glycoproteins and endoglycosidase H (EndoH), which also cleaves asparagine-linked oligosaccharides but not after further processing in the Golgi apparatus. Thus, EndoH-resistance can occur after transit to the Golgi apparatus. Undigested and digested proteins were analyzed by SDS-PAGE and Western blotting with antibodies to V5 and HA. As predicted, the N-terminal segment of CPXV219 exhibited a large mobility shift, equivalent to 45-kDa, upon PNGase digestion whereas the C-terminal segment was reduced by only about 10-kDa (Fig. 4B). The combined mass of the PNGase-digested N- and C-terminal fragments was similar to that predicted for the full-length CPXV219, indicating that there is one or a few closely spaced cleavage sites at about 600 amino acids from the start codon. Mobility shifts of full-length, N-terminal and C-terminal fragments also occurred after EndoH digestion (Fig. 4B). However, EndoH digestion did not decrease the mobility of the N-terminal fragment as much as PNGase suggesting partial resistance due to carbohydrate processing in the Golgi apparatus.
Cellular localization of CPXV219
Fluorescent probes conjugated to anti-V5 and anti-HA antibodies were used to localize V5-CPXV219-HA and its cleavage products by confocal microscopy. Extensive co-localization of the probes within the cell and at the plasma membrane supported the association of the N- and C-terminal fragments (Fig. 5A). Another recombinant CPXV with C-terminal triple-FLAG (219-3xFLAG), which allowed sensitive detection with anti-FLAG M2 MAb, was used for intracellular localization. Polyclonal antibodies to cell proteins were also used for co-localization. Cells in which CPXV219 co-localized with cellular calnexin, ERGIC-53 and GM130 targeting the ER, ER-Golgi intermediate compartment and cis-Golgi apparatus, respectively, are shown in Fig. 5B. The plasma membrane also appeared to be stained with FLAG antibody (Fig. 5B). Taken together these results suggested that CPXV219 traverses the entire secretory pathway rather than halting in one compartment.
Fig. 5.

Cellular localization of CPXV219. (A) HeLa cells were infected with 5 PFU per cell of CPXV V5-219-HA for 8 h. The cells were fixed and permeabilized and stained with monoclonal fluorescent conjugated antibodies to the V5 and HA epitope tags. The cells were also stained with DAPI to visualize nuclei and virus factories (arrows). (B) HeLa cells were infected with 3 PFU per cell of CPXV 219-3XFLAG for 8 h and then fixed and permeabilized. Cells were stained with antibodies against the C-terminal FLAG of CPXV219 (red) and either calnexin (green), ERGIC 53/p53 (green) or GM130 (green), which localize in the ER, ERGIC and Golgi network, respectively. DNA in nuclei and virus factories were stained with DAPI (blue). Yellow represents the colocalization of green and red. Representative images are shown.
Additional experiments were carried out to confirm the plasma membrane localization of the CPXV219 and determine whether the protein is accessible from outside the cell. In order to gain high sensitivity, the cells were infected with CPXV 219-3xFLAG virus and stained with anti-FLAG M2 antibody with and without Triton X-100 permeabilization. As a positive control, infected cells were stained with a MAb that binds to the VACV B5 homolog on the surface of unpermeabilized cells. Antibody to L1 a mostly intracellular virion component was used as a negative control. Staining of CPXV219-FLAG in unpermeabilized cells produced a distinct ring that delineated the outside of the cell with the same pattern as B5, but not seen with L1 (Fig. 6A). After permeabilization, CPXV219-FLAG, B5, and L1 were all stained. The extracellular staining of the C-terminal tag of CPXV219-FLAG demonstrated that the 40 amino acids following the TM domain are extracellular. The relatively weak signal obtained with the N-terminal V5 tag precluded attempts to demonstrate extracellular localization of the N-terminal V5 tag (data not shown).
Fig. 6.

Localization of CPXV219 on the cell surface. (A) HeLa cells were infected with 3 PFU per cell of CPXV 219-3XFLAG and stained with FLAG M2 MAb (red) and polyclonal anti-B5 (green) or anti-L1 (green) antibody either prior to treatment (UnPerm) or following fixation in 4% paraformaldehyde and permeabilization with Triton X-100 (Perm). DNA was stained with DAPI (blue). (B) Biotinylation. HeLa cells were infected with 10 PFU/cell of CPXV 219-3XFLAG for 18 h and then incubated with (+) or without (−) a non-membrane permeable biotin (NHS-SS-Biotin), washed to remove excess biotin, and harvested. Lysates were incubated with neutravidin beads and the bound proteins were eluted and analyzed by SDS-PAGE and Western blotting with anti-V5 (α-V5) or anti-HA (α-HA) MAb. The positions of marker proteins are shown on the left.
To support the immunofluorescence data, we used a non-membrane permeable biotin (NHS-SS-Biotin) to label extracellular proteins on live cells infected with CPXV V5-219-HA. Lysates were prepared and incubated with beads attached to NeutrAvidin to purify biotinylated proteins. After washing the beads, the bound proteins were eluted and resolved by SDS-PAGE. The C-terminal fragment of the biotinylated protein was detected using anti-HA and the N-terminal fragment with anti-V5 (Fig. 6B), whereas the proteins were not detected in the control without biotinylation. These results demonstrate that the N- and C-terminal fragments of CPXV219 remain associated at the cell surface.
CPXV219 does not contribute to virulence in mice
Although CPXV mutants with total or partial deletion of CPXV219 replicated well in cell culture, we considered the possibility that the protein might be more important during animal infection. CPXV 219Stop, without epitope tags or reporter genes, and CPXV 219Rev were used to infect BALB/c mice via the intranasal (IN) route with doses of 104 to 106 PFU. At the low dose of 104 PFU, there was minor weight loss and 100% survival of mice infected with the mutant and revertant viruses (Fig. 7A, B). At 105 PFU, there was slightly greater weight loss and lower survival with the revertant virus compared to the mutant virus, whereas at 106 PFU the weight loss was similar for both and few mice survived (Fig. 7A, B). The calculated LD50 values of 1.0 × 105 and 2.1 × 105 for CPXV 219Rev and CPXV 219Stop, respectively, suggested at most a minor effect of CPXV219 expression on virulence in BALB/c mice.
Fig. 7.

Comparison of CPXV 219rev and CPXV 219Stop in mouse infection models. (A) Infection of BALB/c mice. CPXV 219rev and CPXV 219Stop were administered at doses of 104, 105 and 106 PFU intranasally to groups of 5 BALB/c mice and the animals were weighed daily. (B) Percent survival of BALB/c mice from panel A. (C) Survival of CAST/EiJ mice. CPXV 219Rev and CPXV 219Stop were administered at doses of 101, 102 and 103 PFU intranasally to groups of 5 CAST/EiJ mice and the animals were weighed daily. Percent survival was plotted. (D) Organ titers of mice from panel C. At time of death organs of CAST/EiJ mice were collected, weighed, and frozen. Organs were then homogenized and titered on BS-C-1 cells. The PFU/g was calculated for spleen, liver, lungs, and brain and the total PFU/organ for the ovaries and nasal turbinates.
We also compared the virulence of CPXV 219Stop and CPXV 219Rev in CAST/Eij mice, which are more susceptible to CPXV and other orthopoxvirus infections than BALB/c mice (Americo et al., 2010; Americo et al., 2014). Because of their sensitivity to CPXV, the CAST/Eij mice were infected IN with CPXV 219Rev and CPXV 219Stop at 1, 10, or 100 PFU. Weight loss and time to death were monitored. All of the low dose 1 PFU animals survived. At 10 PFU, 60% of the mice infected with CPXV 219Rev survived and 20% of those infected with CPXV 219Stop survived. At the high dose of 100 PFU, none of the mice infected with CPXV 219Rev survived, and 20% of those infected with CPXV 219Stop survived (Fig. 7C). Weight loss for all groups was minimal even up to death and none reached the 70% level that would trigger termination. LD50 values of 15 and 5 PFU were calculated for CPXV 219Rev and CPXV 219Stop, respectively. Similar results were obtained in a second study (not shown). At the times of death, organs were harvested and titered by plaque asssy on BS-C-1 cells (Fig. 7D). The organs contained similar titers of CPXV 219Rev and CPXV 219Stop and differences were not significant. We concluded that CPXV219 does not appear to contribute greatly to virulence during an IN infection of either BALB/c or more sensitive CAST/Eij mice.
Discussion
Our study of the CPXV219 protein was motivated by the very large size of the ORF and its conservation in most but not all chordopoxvirues. Indeed, members of some genera have multiple copies suggesting an important role. CPXV219 was synthesized within the first 4 h of infection and in the presence of an inhibitor of DNA replication, indicating an early stage promoter. By constructing a recombinant virus with epitope tags at both ends, we detected the full-length protein, which migrated with an apparent mass of >260 kDa, and more abundant 110 kDa N-terminal and 160 kDa C-terminal fragments by Western blotting under denaturing conditions. Immunoprecipitation experiments, however, indicated that the fragments remained associated prior to denaturation in vitro. The possibility that the C-terminal fragment arose from internal initiation was ruled out by its failure to form either when stop codons were introduced near the N-terminus or by removing the start codon. An increase in amount of full-size protein occurred when BFA was added, suggesting that proteolytic cleavage occurred during transit through the Golgi apparatus. Further studies demonstrated cleavage of a signal peptide and transit through the secretory pathway. Extensive N-glycosylation, also occurred during trafficking; approximately 45- and 10-kDa reductions in the masses of the N- and C-terminal fragments occurred following PNGase digestion. If we estimate that each N-glycosylation contributes 2.2 kDa to protein mass, then approximately 20 of the 21 predicted sites in the N-terminal fragment were glycosylated and 4 or 5 of the 10 predicted sites in the C-terminus were glycosylated. The acquisition of endoH-resistance, particularly of the N-terminal fragment, was further evidence of Golgi or post-Golgi transport.
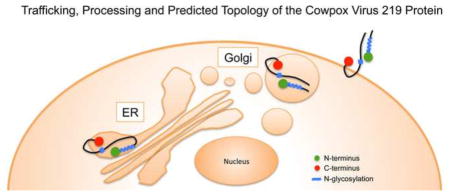
The CPXV219 protein was visualized throughout the secretory pathway including the plasma membrane and was detected in unpermeabilized cells by labeling with fluorescent antibody to a C-terminal epitope tag and confocal microscopy. Based on the available data, we suggest the topological model in Fig. 8. In step I, CPXV219 is co-translationally inserted into the ER with cleavage of the signal peptide and glycosylation. In step II, the protein is transported to the Golgi network where modification of carbohydrate and proteolytic cleavage occur. In step III, the two fragments while still associated are transported to the plasma membrane. The C-terminus was considered to be outside of the plasma membrane because the epitope tag was accessible to antibody in unpermeabilized cells. The site of CPXV219 cleavage at approximately amino acid 600 of the full-length protein must have been in the ER based on the inhibition by BFA and at least part of the region between amino acid 1,000 and 1,800 must also have been in the ER since that is where the potential N-glycosylation sites are located. Assuming that the predicted TM near the C-terminus is utilized, then CPXV219 must traverse the membrane twice. There are two additional hydrophobic regions estimated between amino acids 925 to 938 and 1317 to 1325, one of which must traverse the membrane. Based on the location of the potential N-glycosylation sites, we predict that the more distal hydrophobic site was used. This would mean that a loop between amino acids 1325 and 1856 is cytoplasmic. The entire N-terminal fragment must have been in the ER based on the cleaved signal peptide, the extent of N-glycosylation and the absence of additional hydrophobic domains. Since fluorescence microscopy showed the N-terminal fragment colocalizing with the C-terminal fragment within the cytoplasm and at the plasma membrane and co-immunoprecipitating with the C-terminal fragment, we also placed it on the cell surface in our model. However, further experimentation is needed to directly confirm the predicted topology.
Fig. 8.

Topological model of CPXV219. I, the CPXV protein is co-translationally synthesized at the ER, with removal of the signal peptide, and glycosylated. II, the protein is transported to the Golgi network where the carbohydrate is modified and cleavage occurs to form two major fragments. III, the two fragments while still associated are transported to the plasma membrane where the segments that were in the ER are now extracellular. The linear model of CPXV219 shows placement of the signal peptide (SP), epitope tags (V5 and HA), cleavage site, N-glycosylated region, C-terminal transmembrane domain (TM) and internal hydrophobic domain (HD) that may serve as a second TM.
In a recent study Alzhanova and coworkers (Alzhanova et al., 2014) analyzed the monkeypox virus (MPXV) homolog of CPXV219. Their study and ours were consistent in providing evidence for proteolytic cleavage of the full-length protein and cell surface expression of the C-terminal fragment, despite methodological differences. In contrast to our approach, which involved construction of recombinant CPXV containing epitope tags at both the N- and C-termini, Alzhanova and co-workers (Alzhanova et al., 2014) relied on transient expression by plasmids in uninfected cells or by adenovirus vectors. Because the majority of their studies were carried out with only a C-terminal epitope tag, they did not demonstrate the formation of a stable N-terminal fragment.
The inhibition of cleavage by BFA suggested the action of a Golgi or post-Golgi proteinase. The approximately 65- and 150-kDa sizes of the deglycosylated CPXV219 fragments suggested that the major cleavage site or sites are about 600 amino acids from the N-terminus. The Arg-X-X-Arg and Arg-Arg sequences between amino acids 589 and 596 are candidate furin cleavage sites. A conserved run of Ser residues is just downstream of the putative cleavage sites and additional Ser residues are about 30 amino acids further downstream. A region rich in prolines (35%) and arginines (15%) with 14 PXXP sequences occurs between amino acids 706 and 778 of CPXV219 and is conserved in other orthopoxvirus homologs. Such proline-rich regions are found in SH3 domains and may be involved in protein-protein interactions (Kay et al., 2000).
Despite the conservation of CPXV219 homologs in most poxviruses, we found that introduction of stop codons near the N-terminus or deletion of the entire ORF had no effect on replication in tissue culture cells. This suggested that CPXV219 likely had a role in countering host defenses. However, the CPXV219 null mutant was not attenuated in either BALB/c mice or the more sensitive CAST mice (Americo et al., 2014). We considered that other CPXV genes might make the role of CPXV219 partially redundant or that the activity of CPXV219 is fine-tuned for its natural rodent host and is nonfunctional in mice. Both ideas are consistent with the recent report by Alzhanova and coworkers (Alzhanova et al., 2014). While searching for a MPXV gene product that renders T-cells non-responsive they identified MPX197, a homolog of CPXV219. In a rhesus macaque model, deletion of the MPX197 resulted in increased survival, reduced viral load, and increased levels of both CD4+ and CD8+ pox-specific T cells. The MPXV, VARV and CPXV homologs each inhibit human and monkey T cells but are less active on mouse T cells, providing a possible explanation for our finding that a CPXV219 null mutant did not affect the disease course of CPXV in mice. In addition, CPXV encodes two inhibitors of MHC class I presentation (Alzhanova et al., 2009; Byun et al., 2009; Byun et al., 2007) that could compensate for the absence of CPXV219. In the future, we plan to investigate the role of the ectromelia homolog in the mouse, its natural host.
Materials and Methods
Cells
BS-C-1 cells were propagated in minimum essential medium with Earle’s salts supplemented with 10% fetal bovine serum, 100 U/mL of penicillin, and 100 μg/ml of streptomycin (Quality Biologicals, Gaithersburg, MD). HeLa cells were propagated in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and antibiotics as described above.
Recombinant CPXV219 viruses
DNA sequences were generated using overlapping PCR for the purpose of homologous recombination in the generation of mutant viruses. The flanking regions of the C-terminus of CPXV219 were amplified from CPXV-BR genomic DNA using SR42 (5′-AAG ATG CTA AAA ATA AGA AAA GGA CAT ATA -3′), SR96* (5′-CCA TTT ATA GCA TAG AAA AAA ACA AAA TGA AAT ATT CAC GTA TTC ATT AGA ATC GTC ATG-3′), SR3* (5′-ATG CAC GAG CTG TAC AAG TAA TAA TAA AAA AAT AGT TTA ACT CTT TTT AGA ACC AGT TTG -3′) and SR4 (5′-GTT TAT ATC ACA GCA TTC TAC AAA CAG TCT AAA C -3′). In a second PCR the overlapping tails of the above primers (underlined) were used to flank GFP under the control of a VACV p11 promoter amplified using SR94 (5′-TTT CAT TTT GTT TTT TTC TAT GCT ATA AAT GGT G -3′) and SR20 (5′-TTA CTT GTA CAG CTC GTC CAT GCC GAG AGT GAT CCC -3′). The resulting PCR product containing an independently regulated GFP flanked by the regions up- and downstream of the 219 C-terminus was gel purified and transfected (lipofectamine 2000, Invitrogen) into cells infected with CPXV-BR. Recombinant viruses that express GFP were detected by fluorescence microscopy and isolated by three rounds of plaque purification beneath a 5% methylcellulose overlay. The correct site and sequence of the CPXV219-CtdelGFP recombinant was confirmed by PCR.
C-terminally tagged CPXV219 was generated using the same homologous recombination method described above but replacing GFP with the wild-type C-terminal CPXV219 sequence and an HA tag (N- YPYDVPDYA -C) immediately prior to the stop codon. The primers SR105 (5′-AGC GTA GTC TGG GAC GTC GTA TGG GTA GTA CCG ATT ATC CAT AAT TTC CAT AGA TAC TG -3′) and SR106 (5′-CGA CGT CCC AGA CTA CGC TTA ATA AAA AAA TAG TTT AAC TCT TTT TAG AAC CAG TTT GG -3′) were used in overlapping PCR to generate the recombination fragment. Non-fluorescent plaques were purified in tissue culture and generation of CPXV 219-CtHA was confirmed by PCR and sequencing.
The same technique was also used to generate the N-terminal V5 tag (N- GKPIPNPLLGLDST –C) of the doubly tagged mutants of CPXV219. The primers SR1 (5′-AAC ATG AGT ATT CTA GGT GTC TCT ATT GAA TG -3′), SR93 (5′-CAC CAT TTA TAG CAT AGA AAA AAA CAA AAT GAA AGG TTG CCG GTC ATA AAC AAA CC -3′), SR95 (5′-CAT GCA CGA GCT GTA CAA GTA ATA TAA TTG TAA ACT AAC TAC AAA TTC TGC ATG TGA TG -3′) and SR55 (5′-GCA TAA GTT ACC TCT TTA ACA CTA GAA AGC TTT TGG -3′) were used, along with the previously described GFP amplification, to create the GFP deletion construct CPXV219-NtdelGFP. Primers SR138 (5′-GGT CGA GTC GAG GCC GAG GAG GGG GTT GGG GAT GGG CTT GCC ACA CCA CGA ACA TGT CGC -3′) and SR139 (5′-CGG CCT CGA CTC GAC CTA TGA AAC ATG TGT AAG AAA ATC TGC ATT G -3′) were used to generate the N-terminal recombination product in which the V5 tag was inserted directly following the signal peptide cleavage site. Plaques were purified and confirmed by PCR and sequencing.
Mutants used to confirm the signal peptide were similarly generated from the CPXV 219-NtdelGFP in which a single FLAG tag (N- DYKDDDDK -C) was inserted immediately following the wild-type CPXV219 signal peptide (N-MNLQRLSLAIYLTATCSWC-C), a known signal peptide (SP) sequence from the VSV-G protein (VG-SP: N-MKCLLYLAFLFIGVNC-C), or the known SP with a single mutation (bold) disrupting cleavage (VG-SP*: N- MKCLLYLAFLFIGVNR -C) as shown in (Husain et al., 2006).
Viruses were propagated in BS-C-1 cells and released through 3x repeated freeze/thaw cycles. Samples were sonicated prior to each use and viral titers were determined in duplicate using BS-C-1 cells.
Antibodies
The HA epitope tag was detected with anti-HA clone HA-7 Mab (Sigma), anti-HA rabbit Pab (Invitrogen) or horseradish peroxidase (HRP)-conjugated rabbit anti-HA polyclonal antibody (PAb) (Thermo). The V5 epitope tag was detected with anti-V5 SV5-Pk1 MAb (Thermo) or with HRP conjugated anti-V5 rabbit PAb (Bethyl). Directly conjugated anti-HA mouse IgG1, clone 16B12 (Life Technologies) and anti-V5 FITC clone SV5-Pk1 (Thermo) were used for confocal microscopy. The FLAG epitope tag was detected with either anti-FLAG M2 MAb or anti-FLAG M1 MAb (Sigma) specific for FLAG at the free N-terminus. Anti-calnexin, anti-ERGIC-53/p53 and GM130 (C-terminal) rabbit PAbs were obtained form Sigma. The anti-B5 rat MAb 19C2 (Schmelz et al., 1994) and anti-L1 rabbit PAb (Lustig et al., 2005) were described previously.
Western blotting
Cells were harvested and lysed in 2x SDS sample buffer added directly to the cell monolayer. Cell lysates were disrupted in a cup sonicator and heated to 95°C for 10 min. Equal volumes of lysates were analyzed by SDS-PAGE using a 4–12% Tris-glycine gel and 1x Tris-glycine SDS running buffer (Invitrogen) at 125V for 2 h. Samples were transferred via a semi-dry transfer (Invitrogen) and nitrocellulose membranes were blocked in 5% milk in Tris-buffered saline with 0.1% Tween-20. Antibodies were incubated at a 1:1000 dilution in 5% milk overnight. Proteins used for Western blots were detected using anti-mouse and/or anti-rabbit secondary antibodies conjugated to IRdye 680 or 800 and visualized using a LI-COR Odessey infared imager (LI-COR Biosciences, Lincoln, NE). For visualization of the anti-FLAG M1 antibody, goat anti-mouse HRP secondary (KCL, diluted 1:10,000) was visualized using chemiluminescence (ECL, Thermo).
Glycosidase treatment of cell lysate
HeLa cells (2.5×105) were washed in phosphate-buffered saline (PBS) and lysed in 75 μl of 50 mM Tris pH 7.0, 150 mM NaCl, and 0.5% NP-40 detergent on ice for 20 min. Nuclei were removed by low-speed centrifugation (400xg, 5 min, 4°C). The supernatants were adjusted to contain 0.5% SDS and 1% β-mercaptoethanol and heated at 95°C for 10 min. PNGaseF (1500 U) or EndoH (1500 U; New England Biolabs) was added and incubated overnight at 37°C. Samples were analyzed by SDS-PAGE and Western blotting.
Confocal microscopy
HeLa cells were grown on coverslips (12 Circle #1, Thermo) to approximately 80% confluency in 24-well plates and infected with CPXV219 mutants at multiplicity of 3 PFU/cell for the indicated amount of time at 37°C. Cells were washed briefly in PBS and fixed in 4% paraformaldehyde in PBS. Cells were permeabilized in 0.1% Triton X-100 for 10 min and blocked in 10% fetal bovine serum in PBS for at least 1 h at room temperature. Antibody was incubated overnight at a 1:250 dilution in blocking buffer at 4°C, washed in PBS, and incubated with secondary (Life Technologies, Alexa-fluor mouse-488, rabbit-594) at a 1:250 dilution in blocking buffer for 4–18 h. Cell nuclei were stained with 4′, 6-diamidino-2-phenylindole, dihydrochloride (DAPI, Invitrogen) 1:1000 in H2O for 30 min, washed in PBS, mounted to glass slides with a drop of prolong gold anti-fade mounting media (Invitrogen) and viewed with a confocal microscope.
IP analysis
HeLa cells were infected at a multiplicity of 5 PFU per cell for 24 h and harvested in lysis buffer containing 1% NP-40, 50 mM Tris-HCl, 150 mM NaCl, 100 μl EDTA and complete protease inhibitor (Roche) and allowed to proceed at 4°C for 30 min. The lysates were cleared by centrifugation at 1600 rpm for 15 min at 4°C. IP was carried out overnight using 2–4 μg of either anti-V5 or anti-HA MAb in the presence or absence of 0.4% SDS using 15–25 μl of magnetic protein G Dynabeads (Invitrogen). After incubation, the beads were washed in PBS containing 0.5% Tween-20 and resuspended in 2x SDS sample buffer (0.125 M Tris-HCl, pH 6.8, 4% SDS, 0.005% Bromophenol Blue, and 20% glycerol) with 5% β-mercaptoethanol (Sigma), heated to 95°C for 10 min, and analyzed by Tris-glycine SDS-PAGE.
Biotinylation of extracellular proteins
HeLa cells were infected with CPXV 219-CtFLAG at a multiplicity of 10 PFU per cell for 18 h, washed in PBS, labeled with 0.5mg/ml of a non-membrane permeable biotin (NHS-SS-Biotin, Thermo), washed to remove excess biotin, quenched by 10 min wash in 2.5% fetal bovine serum in EMEM, and incubated in lysis buffer containing 10 mM Tris-HCl, 10 mM NaCl, 5 mM MgCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.5% NP-40 at 4°C for 30 min. Lysates were incubated with 15–25 μl of Neutravidin agarose beads (Thermo) and washed. The beads were treated with 2x SDS sample buffer with 5% β-mercaptoethanol (Sigma), heated to 95°C for 10 min, and analyzed by Tris-glycine SDS-PAGE and Western blotting.
Intranasal infection model
All experimental procedures involving mice were approved by the National Institutes of Allergy and Infectious Disease Animal Care and Use Committee and carried out in a humane manner. Female BALB/c and CAST/EiJ mice were purchased from Taconic Biosciences and Jackson Laboratory, respectively. Animals were kept in a clean environment in sterile microisolator cages. Groups of 5 BALB/c or CAST/Eij mice were anesthetized with isofluorane and inoculated in a single nostril with a 10 μl suspension of sucrose-cushion purified virus. The infection inoculum was titered by plaque assay to confirm dosages. Mice were weighed daily following infection and euthanized if reached 70% of their initial body weight in accordance with NIAID Animal Care and Use protocols. All experiments were performed in an animal biosafety level 2 (ABSL2) suite with approval of the NIAID Animal Care and Use Committee.
Highlights.
Homologs of the cowpox virus 219 protein are encoded by most chordopoxviruses
The cowpox virus 219 homologs are the largest poxvirus proteins
The protein traffics the secretory pathway where glycosylation and cleavage occur
N- and C- terminal fragments remain associated and inserted into the plasma membrane
The 219 protein is not required for cowpox virus replication and virulence in mice
Acknowledgments
We thank Catherine Cotter for her assistance with tissue culture and training in laboratory techniques, and Jeffrey Americo for discussions. Patricia Earl provided assistance with animal experiment planning and execution. The Comparative Medicine Branch provided daily weights, monitoring, and animal maintenance. The Biological Imaging Section assisted with confocal microscopy. The research was supported by the Division of Intramural Research, NIAID, NIH. S.R. is a Ph.D. candidate in the Department of Cell Biology and Molecular Genetics at the University of Maryland, College Park, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alzhanova D, Edwards DM, Hammarlund E, Scholz IG, Horst D, Wagner MJ, Upton C, Wiertz EJ, Slifka MK, Fruh K. Cowpox virus inhibits the transporter associated with antigen processing to evade T cell recognition. Cell Host Microbe. 2009;6:433–445. doi: 10.1016/j.chom.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzhanova D, Hammarlund E, Reed J, Meermeier E, Rawlings S, Ray CA, Edwards DM, Bimber B, Legasse A, Planer S, Sprague J, Axthelm MK, Pickup DJ, Lewinsohn DM, Gold MC, Wong SW, Sacha JB, Slifka MK, Fruh K. T Cell Inactivation by Poxviral B22 Family Proteins Increases Viral Virulence. Plos Path. 2014;10 doi: 10.1371/journal.ppat.1004123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Americo JL, Moss B, Earl PL. Identification of wild-derived inbred mouse strains highly susceptible to monkeypox virus infection for use as small animal models. J Virol. 2010;84:8172–8180. doi: 10.1128/JVI.00621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Americo JL, Sood CL, Cotter CA, Vogel JL, Kristie TM, Moss B, Earl PL. Susceptibility of the wild-derived inbred CAST/Ei mouse to infection by orthopoxviruses analyzed by live bioluminescence imaging. Virology. 2014;449:120–132. doi: 10.1016/j.virol.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansarah-Sobrinho C, Moss B. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J Virol. 2004a;78:6335–6343. doi: 10.1128/JVI.78.12.6335-6343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansarah-Sobrinho C, Moss B. Vaccinia virus G1 protein, a predicted metalloprotease, is essential for morphogenesis of infectious virions but not for cleavage of major core proteins. J Virol. 2004b;78:6855–6863. doi: 10.1128/JVI.78.13.6855-6863.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd CM, Hruby DE. A conditional-lethal vaccinia virus mutant demonstrates that the I7L gene product is required for virion morphogenesis. Virol J. 2005;2:4. doi: 10.1186/1743-422X-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun M, Verweij MC, Pickup DJ, Wiertz EJ, Hansen TH, Yokoyama WM. Two mechanistically distinct immune evasion proteins of cowpox virus combine to avoid antiviral CD8 T cells. Cell Host Microbe. 2009;6:422–432. doi: 10.1016/j.chom.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun M, Wang X, Pak M, Hansen TH, Yokoyama WM. Cowpox virus exploits the endoplasmic reticulum retention pathway to inhibit MHC class I transport to the cell surface. Cell Host Microbe. 2007;2:306–315. doi: 10.1016/j.chom.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Dabrowski PW, Radonic A, Kurth A, Nitsche A. Genome-Wide Comparison of Cowpox Viruses Reveals a New Clade Related to Variola Virus. Plos One. 2013;8 doi: 10.1371/journal.pone.0079953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damon I. Poxviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6. Wolters Kluwer/Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 2160–2184. [Google Scholar]
- Doms RW, Russ G, Yewdell JW. Brefeldin A redistributes resident and itinerant Golgi proteins to the enodplasmic reticulum. J Cell Biol. 1989;109:61–72. doi: 10.1083/jcb.109.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubser C, Hue S, Kellam P, Smith GL. Poxvirus genomes: a phylogenetic analysis. J Gen Virol. 2004;85:105–117. doi: 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- Haller SL, Peng C, McFadden G, Rothenburg S. Poxviruses and the evolution of host range and virulence. Infect Genet Evol. 2014;21:15–40. doi: 10.1016/j.meegid.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedengren-Olcott M, Byrd CM, Watson J, Hruby DE. The vaccinia virus G1L putative metalloproteinase is essential for viral replication in vivo. J Virol. 2004;78:9947–9953. doi: 10.1128/JVI.78.18.9947-9953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain M, Weisberg AS, Moss B. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc Natl Acad Sci USA. 2006;103:19506–19511. doi: 10.1073/pnas.0609406103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Bonafacino JS, Yuan LC, Klausner RD. Rapid redistribution of Golgi proteins in the ER in cells treated with brefeldin A, evidence for membrane cycling from Golgi to ER. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig S, Fogg C, Whitbeck JC, Eisenberg RJ, Cohen GH, Moss B. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J Virol. 2005;79:13454–13462. doi: 10.1128/JVI.79.21.13454-13462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Poxviridae. In: Knipe DM, Howley PM, editors. Fields Virology. 6. Wolters Kluwer/Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 2129–2159. [Google Scholar]
- Schmelz M, Sodeik B, Ericsson M, Wolffe EJ, Shida H, Hiller G, Griffiths G. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J Virol. 1994;68:130–147. doi: 10.1128/jvi.68.1.130-147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AS, Rottier PJ, Rose JK. Evidence for the loop model of signal-sequence insertion into the endoplasmic reticulum. Proc Natl Acad Sci USA. 1988;85:7592–7596. doi: 10.1073/pnas.85.20.7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol. 2003;77:7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZY, Zikos D, Osterrieder N, Tischer BK. Generation of a complete single-gene knockout bacterial artificial chromosome library of cowpoxv irus and identification of its essential genes. J Virol. 2014;88:490–502. doi: 10.1128/JVI.02385-13. [DOI] [PMC free article] [PubMed] [Google Scholar]