

Abstract
Mitochondrial defects are the cause of numerous disorders affecting the oxidative phosphorylation system (OXPHOS) in humans leading predominantly to neurological and muscular degeneration. The molecular origin, manifestations, and progression of mitochondrial diseases have a broad spectrum, which makes very challenging to find a globally effective therapy. The study of the molecular mechanisms underlying the mitochondrial dysfunction indicates that there is a wide range of pathways, enzymes and molecules that could be potentially targeted for therapeutic purpose. Therefore, focusing on the pathology of the disease is essential to design new treatments. In this review, we will summarize and discuss the different therapeutic interventions tested in some mouse models of mitochondrial diseases laying emphasis on the molecular mechanisms of action and their potential applications.
1. Introduction
Mitochondrial diseases include a wide group of human disorders affecting the oxidative phosphorylation system (OXPHOS). Common OXPHOS alterations in mitochondrial disorders are associated with mutations in multiples genes encoded by both the mitochondrial (mtDNA) and the nuclear (nDNA) genome (Schon, 2012; Wallace, 2010). Mitochondrial diseases are no longer considered orphan diseases. Epidemiological studies predict 1 in 5,000 children to be affected by them (Schaefer, 2004). During the last decade, many compounds have been tested to ameliorate or delay the symptoms of these devastating disorders. However with a few remarkable exceptions (Hirano, 2012), to date no effective treatments are available to cure mitochondrial diseases. The efforts of the scientific community to find cures include different approaches, such as preventing the disease transmission from mother to child, gene therapy, exercise training, correction of metabolic alterations, specialty diets and antioxidant treatments [for review of current treatments in humans see (Pfeffer, 2013; Schon, 2010)]. Reliable clinical trials are hampered by the inability to collect large study groups due to the extremely heterogeneous nature of mitochondrial diseases (Pfeffer, 2013). For this reason, a “personalized medicine” is considered nowadays a prospect for treatment. In this chapter (Part III of review miniseries) we discuss the different therapeutic strategies that have been tested in some of the mouse models described in Parts I and II, highlighting principles, limitations and potential applications of the tested interventions. We have grouped the different treatments according to their mechanism of action in the following categories: (i) heteroplasmic shift, (ii) replacement of defective genes, (iii) activation of mitochondrial biogenesis, (iv) nutritional intervention, and (v) other alternative treatments (Figure 1).
Figure 1.
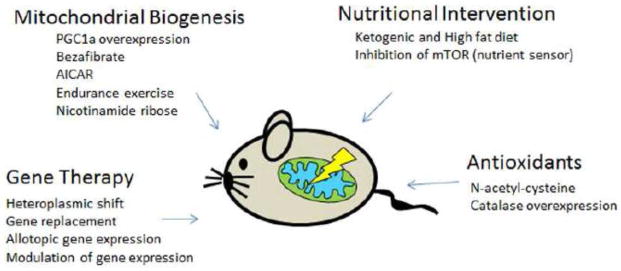
General therapeutic approaches tested in mouse models of mitochondrial diseases.
2. Therapeutic interventions tested in mitochondrial deficient mouse models
2.1. Heteroplasmic shift
Different copy numbers of wild type (wt) and mutated mtDNA can coexist into the mitochondria without being detrimental. The ratio of the levels of the two molecules defines the heteroplasmy level of the mutation and the mtDNA mutations must reach a certain load to exert their biochemical, cellular and clinical phenotype (threshold effect). Hence, a reasonable therapeutic approach to prevent mitochondrial dysfunction is based on the reduction of the mtDNA mutant load. Since the pathological threshold levels of heteroplasmy tend to be very high, a small reduction in the % of heteroplasmy is expected to be beneficial (Thorburn and Dahl, 2001; Zeviani and Di Donato, 2004). One of the approaches to change the heteroplasmic levels involves the use of restriction endonucleases that will recognize specific restriction sites only present in the mutant mtDNA. The restriction endonuclease can be expressed and targeted to the mitochondria to digest the unwanted population of mtDNA. To test the feasibility of this approach, Moraes’ group took advantage of an existing mouse model carrying two different non pathogenic haplotypes of murine mtDNA, NZB and BALB (Jenuth, 1997). They used ApaLI (Mito-ApaLI) that recognizes a site only in BALB mtDNA causing a shift towards the NZB haplotype (Bayona-Bafaluy, 2005). A viral transduction of Mito-ApaLI in skeletal muscle and brain of NZB/BALB mice by local injection produced a rapid heteroplasmic shift in both tissues (Bayona-Bafaluy, 2005). The same approach has given promising results when delivered systemically to heart and liver tissue using adeno-associated and adenovirus in the NZB/BALB mice (Bacman, 2010). The heteroplasmic shift towards NZB mtDNA observed in these tissues was not followed by depletion or deletion of mtDNA, most likely because the NZB replicated faster avoiding mtDNA depletion caused by the rapid digestion of the BALB genome (Bacman, 2007). Systemic delivery of Mito-ApaLI in newborn NZB/BALB mice decreased the NZB haplotype specifically in skeletal muscle and heart. The expression of the transgene was stable over time and no collateral effects were observed (Bacman, 2012). However, the success of this type of approach in patients is somehow limited because it requires the presence of a unique restriction site in the mutant mtDNA to avoid mtDNA depletion, which is a potential problem that has to be taken in consideration when using this innovative strategy.
A refinement of this approach involves the use of re-engineered transcription activator-like effector nucleases (TALENs) targeted to the mitochondria (mito-TALEN) (Bacman, 2013). A heteroplasmy shift towards wild type mtDNA was detected in cells carrying the common deletion and in cybrids carrying the Leber’s hereditary optic neuropathy (LHON) point mutation 14459G>A in the ND6 gene using the mito-TALENs approach. Since not all the pathogenic mutations in the mtDNA described in humans give rise to sites recognized by restriction endonucleases, the use of mito-TALENs opens the possibility for a more effective custom design intervention. However, mito-TALENs technology remains to be tested in vivo in animal models.
A different strategy to enrich wild type mtDNA content in heteroplasmic cells has been achieved by using ketone bodies in culture media deprived of glucose (Santra, 2004). The ketone bodies mimic the ability of galactose-containing media to shift heteroplasmy levels due to the inability of the deficient cells to metabolize this carbohydrate. The selective pressure forces the cells to rely on OXPHOS and not on glycolysis to produce ATP (Santra, 2004). Based on this principle, the effect of a ketogenic diet has been tested in vivo using the Deletor mouse, which carries a dominant mutation in the twinkle-helicase that led to accumulation of multiple mtDNA deletions during aging and a late-onset progressive respiratory chain deficiency (Tyynismaa, 2005). Deletor mouse fed with a ketogenic diet showed an increase in the respiratory chain activity and a general improvement on the course of the disease. Unlike what happens in cell culture, no heteroplasmic shift was detected in the Deletor mouse, as the percentage of deleted mtDNA molecules remained unchanged (Ahola-Erkkilä, 2010). Instead, the ketogenic diet induced mitochondrial biogenesis, diminished the amount of COX (cytochrome c oxidase) negative fibers in muscle and restored the metabolic and lipidomic changes observed in the Deletor mice to wild type levels. The rationale of using ketogenic diet to induce mitochondrial biogenesis is discussed later in the nutritional modulation section 2.4.1.
2.2. Gene replacement
Attempts to correct the genetic defects on mitochondrial diseases using gene therapy have been tested in animal models. However, this proves challenging when the cause of the disease relays on defective mtDNA. To date, there is no in vivo technology available that allows the direct manipulation of the mtDNA to replace a defective gene as it can be done for the nuclear DNA. However, in recent years clever approaches have been developed to circumvent this limitation (Table I).
Table I.
Genetic approaches tested in mouse models of mitochondrial diseases.
| Therapeutic approach | Mouse model | Effects of the therapeutic intervention | References | ||
|---|---|---|---|---|---|
| mtDNA heteroplasmic shift | Gene therapy | Local injection of Ad5- and AAV1,2-mito-ApaLI restriction endonuclease | NZB/BALB adult mice | mtDNA shift towards NZB haplotype in skeletal muscle | (Bayona-Bafaluy, 2005) |
| Systemic injection of AAV6 and Ad5-mito-ApaLI restriction endonuclease to heart and liver | NZB/BALB adult mice | mtDNA shift towards NZB haplotype in heart and liver | (Bacman, 2010) | ||
| Systemic injection of AVV9-mito-ApaLI restriction endonuclease to striated muscle | NZB/BALB newborn mice | mtDNA shift towards the NZB haplotype in all striated muscles | (Bacman, 2012) | ||
| Gene Replacement Replacement of the defective gene |
Nuclear gene expression | Local injection of AAV2-Ant1construct in skeletal muscle | Ant1−/− mice | Increased export of ATP and restored the morphology of the affected muscles | (Flierl, 2005) |
| Intravitreal injection of AVV2-Aif1 construct | Harlequin-CI deficient mice | Protection of retinal ganglion cells and optic nerve integrity, preservation of CI function in optic nerves, as well as the prevention of glial response | (Bouaita, 2012) | ||
| Local injection of AAV1-Ndufs4 construct to the vestibular nucleus | Vestibular nucleus specific Ndufs4 KO mice | Delay of disease onset, increased lifespan and improved breathing response | (Quintana, 2012) | ||
| Intravenous injection of AVV2/8-human TYMP | Tymp−/− Upp1−/− | Reduced accumulation of dThd and dUrd in serum, liver, muscle and brain but not small intestine | (Torres-Torronteras, 2014) | ||
| AVV2/8-Mpv17 | Mpv17 −/− | Prevented liver failure induced by the ketogenic diet and restored liver mtDNA copy number and OXPHOS ability | (Bottani, 2014) | ||
| Intravenous injection of AAVrh10-human Fxn construct | Heart-skeletal muscle specific Fxn KO | Prevented the onset of cardiac failure and reversed the cardiac contractile dysfunction | (Perdomini, 2014) | ||
| Allotopic expression | AAV2-mediated delivery of human ND4 to inner retinal neurons | Mice injected with the human mutated form of ND4 (G11778A) | Suppression of the visual loss and optic atrophy induced by the mutant ND4 homolog | (Yu, 2012) | |
| Modulation of gene expression | Histone deacetylase inhibitors (pimelic o aminoben zamides) | Friedreich’s ataxia | Increased Frataxin protein levels and improvement in the phenotype. | (Sandi, 2011) | |
2.2.1. Viral delivery of nuclear genes
One of the first pioneering experiments to replenish the defective gene was performed on the Ant1−/− mice using adenovirus-mediated expression of the wild type gene (Agostino, 2003; Kawamata, 2011). ANT1 encodes for the isoform mainly expressed in skeletal muscle and heart of the adenine nucleotide translocator, which is essential for the exchange between cytosolic ADP and mitochondrial ATP. Local injection of adenovirus associated vector (AAV), carrying the wt Ant1, formed a functional ADP/ATP carrier, increased the export of ATP and restored the morphology of the affected muscles (Flierl, 2005). Interestingly, this approach was more effective in newborn mice rather than in adults, probably due to the large presence of muscle precursor cells in the injected locus.
A similar approach has been applied to two different mouse models of complex I deficiency. Intravitreal administration of an AAV2 vector containing the wt version of the Aif1 gene prevented the severe optic atrophy exhibited in the Harlequin mice (Bouaita, 2012). The results clearly showed that Aif1 mRNA levels and CI function in optic nerves were restored. Moreover, the treatment prevented the activation of glial and microglial response (Bouaita, 2012). In the brain specific Ndufs4−/− KO mouse the expression of the wild type NDUFS4 in the vestibular nucleus of the dorsal medulla delay the onset of the disease, improved some phenotypes and increased lifespan (Quintana, 2012).
Virus delivery therapy has also been successfully used for the correction of defects on enzymes involved in mitochondrial depletion syndrome (MDS). AVV was used to restore thymidine phosphorylase (TP) enzyme activity in a previously described double KO mouse model (Tymp−/−, Upp1−/−) of mitochondrial neurogastrointestinal encephalopathy (MNGIE) syndrome (Lopez, 2009). TP is involved in the catabolism of nucleotides and defects of this enzyme induced the accumulation of deoxythymidine (dThd) and deoxyuridine (dUrd) in serum with repercussions on mtDNA replication. MNGIE patients suffer of progressive external ophthalmoplegia or PEO, severe gastrointestinal dysmotility, peripheral neuropathy and leukoencephalopathy (Hirano, 2004). An ex vivo hematopoietic gene therapy was tested in partially myeloablated Tymp−/−, Upp1−/− mice by cell infusion of transduced hematopoietic linage negative cells derived from the double KO mouse. The hematopoietic cells were previously transduced with a lentivirus carrying the human TYMP cDNA before transfusion into a syngenic myeloablated double KO. This approach resulted in an increase of TP activity and reduced the nucleoside overload in peripheral blood of the KO mice to wild type levels all without apparent toxic effects (Torres-Torronteras, 2011). AAV transduction was also tested in vivo in the Tymp−/−, Upp1−/− mice. In this occasion, the TYMP expression was targeted to the liver with the AVV2/8, which has high tropism for liver, also contained the thyroxine-binding globulin promoter to express the human TYMP cDNA specifically in this tissue. Single intravenous injection on 8–12 week old Tymp−/−, Upp1−/− mice corrected the biochemical TP defect and lowered the accumulation of dThd and dUrd in serum and several tissues (liver, muscle and brain) with the exception of the small intestine (Torres-Torronteras, 2014). Although the authors underline the feasibility of this kind of therapy in a MNGIE mouse model, there are some limitations of these studies, such as the lack of a disease phenotype in MNGIE mice (except for the nucleoside chemical imbalance) and the difference in dThd and dUrd catabolism between mouse and humans. These differences may limit the effectiveness in the use of this mouse model in testing therapeutic interventions.
In the recombinant Ethe1−/− mouse model for ethylmalonic encephalopathy (EE), AVV2/8-mediated liver gene therapy resulted in full restoration of the metabolic and clinical hallmarks of the disease (Di Meo, 2012). In this case, the mouse model nicely reproduced the pathogenic mechanism of EE such as the accumulation of hydrogen sulfide (H2S) in the bloodstream and tissues due to the lack of function of the Ethe1 enzyme. Expression of the human ETHE1 protein, mainly in liver, successfully restored its enzymatic activity and correct thiosulfate levels in the plasma, a biomarker reflecting the accumulation of H2S. The treated animals showed restored locomotor activity and spectacular prolongation in the lifespan, living until 6–8 months after birth, whereas untreated mice live around 5 weeks (Di Meo, 2012). Therefore, restoring the levels of ETHE1 enzyme in a filtering organ such as the liver can decrease the levels of H2S in blood, thus clearing the toxic compound from the blood stream. The results obtained in this mouse model clearly indicated AVV2/8-mediated liver ETHE1 therapy is an effective approach that can be applied for EE patients.
AVV transduction has also been used to correct defects due to MPV17 mutations causing hepatocerebral forms of mitochondrial depletion syndrome. When fed for two months with ketogenic diet (KD), Mpv17 KO mice (Mpv17−/−) displayed liver cirrhosis and hepatomegaly accompanied by a progressively weight loss. Moreover, Mpv17 KO mouse showed decreased mtDNA content and mitochondrial transcripts (Bottani, 2014). Liver specific transduction of human wt MPV17 (hMPV17) injected 3 weeks before starting the ketogenic diet with the recombinant virus prevented liver failure, weight loss and changes in the amount of mtDNA and transcripts. In contrast, AVV-hMpv17 injection after 1 month of KD administration did not prevented but just delayed the liver damage progression in the KO mice, highlighting the need for a very early treatment, at least for certain pathologies.
AVV-based gene therapy has also been successfully achieved in a mouse model of Friedreich’s ataxia, a disease characterized by neurodegeneration, hypertrophic cardiomyopathy and diabetes (Perdomini, 2014). This disease is due to reduced levels of FRATAXIN (FXN), a mitochondrial protein implicated in the biosynthesis of the Fe/S clusters. Early administration via intravenous injection of AVV-expressing human FRATAXIN fully prevented the onset of the cardiac disease in the heart and skeletal muscle specific Fxn KO model. More importantly and contrary to what it was observed in other models, the administration of the AVV-frataxin vector after the onset of the cardiac phenotype was able to completely reverse the cardiomyopathy of these mice (Perdomini, 2014).
2.2.2. Allotopic gene expression
Allotopic expression refers to the expression of recoded mtDNA genes by the nuclear genome or by a transfected plasmid/vector. This approach could be used to expresses wild type copies of a mutated mtDNA gene to restore the functional defect. This strategy resulted more complicated than expected due to the different genetic code of the mitochondrial genome and the necessity to import the newly produced proteins into the mitochondria from the cytoplasm. AVV mediated gene delivery of mtDNA-encoded genes has been performed in cultured cell models (Guy, 2002; Perales-Clemente, 2011) and recently this promising strategy has been tested in a murine model (Yu, 2012). Although allotopic protein expression raises lots of questions, including the real import and assembly into its respective complex (Perales-Clemente, 2011), these attempts demonstrate that the efficient expression and import of re-encoded mitochondrial proteins is a feasible alternative of treatment. In fact, viral injection of wild type version of the mitochondrial encoded gene ND4 prevented the visual loss induced by a previous viral vector delivery of a mutant human ND4 gene carrying the pathogenic mutation R340V, which has been proven to cause blindness in rodents (Ellouze, 2008; Yu, 2012).
A cleaver and innovative approach to bypass defects on CIII or CIV of the respiratory chain, which has been under development for the last several years, involves the allotopic expression of the alternative oxidase (AOX) (El-Khoury, 2014). AOX is an enzyme found in plants and few other organisms that can oxidize ubiquinol transfering electrons to oxygen and produce water. Expression of AOX will also prevent the formation of superoxide in those mitochondrial defects that might produce a over-reduced pool of ubiquinone. After proof of principle studies performed in Drosophila and human cells (Fernandez-Ayala, 2009; Kakizaki, 2010), the group of Rustin and Jacobs created a mouse that expresses the recoded version of the AOX gene from Ciona intestinalis in mouse (MitAOX mouse) (El-Khoury, 2013). The expression of AOX in the mouse did not caused any deleterious or counter productive effect. AOX did not interfer with the normal function of the respiratory chain. Moreover, AOX functionally interacted with the respiratory chain to confer prolonged protection of the whole animal against cyanide gas exposure bypassing the respiratory inhibition. AOX also reduced oxidative stress caused by the blockage of the respiratory chain (El-Khoury, 2013). These results are very encouraging and have a great potential. Future studies to test if the MitAOX mouse or AOX-viral vectors can rescue or ameliorate the pathological phenotypes in mouse models with CIII or CIV deficiency are required.
2.3. Activation of mitochondrial biogenesis
Mitochondrial biogenesis is frequently observed in patients with mitochondrial diseases and the common believe is that it is an adaptive response triggered by the OXPHOS defect (Rossmanith, 2008). Similarly, mitochondrial biogenesis has also been observed in multiple mouse models of mitochondrial diseases. Therefore, bolstering this natural adaptive response of mitochondrial proliferation could be exploited as a therapeutic intervention. In the last few years, our group and others have demonstrated that increased mitochondrial biogenesis can boost residual OXPHOS capacity, thus preventing the bioenergetic crisis. Activation of mitochondrial biogenesis in cells and animal models with respiratory chain defects restored bioenergetic capacity by improving ATP synthesis (Bastin, 2008; Srivastava, 2009; Wenz, 2008; Wenz, 2009). Key regulators of mitochondrial biogenesis are the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) (Scarpulla, 2008), and the nuclear hormone receptor peroxisome proliferator-activated receptor (PPAR) family (Bonnefont, 2009). PGC-1α is physiologically induced by conditions of shortage of energy and/or increased demand for ATP through the activation of certain transcription factors (Handschin and Spiegelman, 2006). Beside PGC-1α, Sirtuins (SIRT1-SIRT7) are important metabolic players that can boost mitochondrial function through the deacetylation of targets proteins (Houtkooper, 2012). Among others, targets of SIRT1, the most studied sirtuin, are PGC-1α and the Forkhead box O (FOXO) transcription factors, both of which regulate transcription programs related to increase mitochondrial function (Andreux, 2013). Increments in the mitochondrial biogenesis as a therapeutic intervention (Table II) have been achieved by the following strategies:
Table II.
Interventions to enhance mitochondrial biogenesis in mouse models of mitochondrial diseases.
| Therapeutic approach | Mouse model | Effect of the therapeutic intervention | Reference | ||
|---|---|---|---|---|---|
| Activation of mitochondrial biogenesis | Transgenic overexpression of PGC1-α | Mating mice overexpressing PGC1-α specifically in muscle | Skeletal muscle specific Cox10 KO | Delayed mitochondrial myopathy; increased expression of OXPHOS subunits and mtDNA content; increased lifespan | (Wenz, 2008) |
| Skeletal muscle specific Surf1 KO | Increased mtDNA content, increased citrate synthase activity, increased steady-state levels of CIV specific subunits and partial but significant recovery of CIV activity | (Viscomi, 2011) | |||
| Mutator mouse | Activation of mitochondrial biogenesis in skeletal muscle and improvement of oxidative capacity both in muscle and heart; no changes in the physical appearance | (Dillon, 2012b) | |||
| Pharmacological treatment | Bezafibrate (0.5% w/w) added to standard diet | Skeletal muscle specific KO | Increased CIV activity; higher expression of some subunits of OXPHOS system and citrate synthase activity; improved performance on a treadmill | (Wenz, 2008) | |
| Neuron specific Cox10 KO | Induction of mitochondrial biogenesis; recovering of COX activity; reduced neuronal loss, inflammation and glia activation. Marked delay of the encephalopathy phenotype | (Noe, 2013) | |||
| Surf1 KO and muscle specific Cox15 KO | Toxic effect with hepatomegaly and loss of weight. Increased levels of PPARα and δ and several fatty acid oxidation (FAO) related target genes | (Viscomi, 2011) | |||
| Mutator mouse | Improvement of skin and spleen phenotype; no change markers of mitochondrial content/function in skeletal muscle; increased expression of FAO enzymes; Hepatomegaly | (Dillon, 2012a) | |||
| Deletor mouse | Treatment delayed the accumulation of COX deficient muscle fibers and multiple mtDNA deletions. Mitochondrial biogenesis was not induced. | (Yatsuga and Suomalain en, 2012) | |||
| AICAR (I.P. injected) | Surf1KO, muscle specific Cox15 KO, Sco2KO/KI | Partial recovery of the CIV activity; no increase of mtDNA levels or citrate synthase activity; no activation in mitochondrial biogenesis but rather an increase in the OXPHOS transcription/translation | (Viscomi, 2011) | ||
| NAD+ dependent mitochondrial biogenesis (Sirt1 activation) | Nicotinamide riboside (400mg/Kg in the diet) | Deletor mouse | Robust induction of mitochondrial biogenesis Decreased mtDNA deletions | (Khan, 2014) | |
| Sco2KO/KI | Improved motor performance, increased mitochondrial respiratory chain activities in muscle | (Cerutti, 2014) | |||
| Pan-PARP inhibitor (MRLB-45696 and PJ34 50mg/Kg in the diet) | Sco2KO/KI | Improved motor performance, increased mitochondrial respiratory chain activities in muscle and brain | (Cerutti, 2014) | ||
| Genetic ablation of Parp1 gene | Sco2KO/KI | ||||
| Endurance exercise training | Muscle specific Cox10 KO mice | Improvement on the phenotype and survival Activation of mitochondrial biogenesis | (Wenz, 2008) | ||
| Mutator mouse | Delayed of the aging and increased lifespan | (Safdar, 2011) | |||
2.3.1. Transgenic overexpression of PGC-1α
The transgenic overexpression of PGC-1α in skeletal muscle on the Cox10 KO mouse model of mitochondrial myopathy was tested. PGC-1α overexpression driven by the muscle creatine kinase promoter (MCK) induced a successful mitochondrial proliferation, accompanied by an increased expression of many OXPHOS subunits and mtDNA copy number, leading to a significant delay in the onset of the mitochondrial myopathy displayed by the COX10 KO mouse (Wenz, 2008). The rescued mice lived longer than Cox10 KO mice and showed improved performance on a treadmill. Consistently, the beneficial effect of the transgenic overexpression of PGC-1α was also confirmed in the Surf1 KO, another model of COX deficiency. The boost of mitochondrial biogenesis included increased mtDNA content, increased citrate synthase activity and increased levels of COX subunits leading to a partial but significant recovery of COX activity (Viscomi, 2011).
Transgenic overexpression of PGC-1α in the skeletal muscle, under the same promoter, has been shown to also improve some of the aging phenotypes of the Mutator mice (Dillon, 2012b). The Mutator mouse model is a knock-in carrying a mutant version of the mitochondrial polymerase gamma with defective proofreading 3′–5′ exonuclease activity (Trifunovic, 2004). The defective proofreading activity caused the accumulation of mtDNA point mutation and subsequent mitochondrial dysfunction progressing with age (Kujoth, 2005; Williams, 2010). The overexpression of PGC-1α in the Mutator mice increased mitochondrial biogenesis and function in skeletal muscle (Dillon, 2012b). However, the mitochondrial biogenesis did not reduce the accumulation of mtDNA mutations but rather caused a small increase. These results indicate that increased mitochondrial biogenesis in the muscle was able to improve some premature aging phenotypes by mechanisms other than reverting the accumulation of mtDNA mutations. In fact, the PGC-1α overexpressing mouse have decreased aging phenotypes such as sarcopenia and chronic inflammation even though the overexpression was limited to muscle tissue (Wenz, 2009).
2.3.2. Pharmacological induction of mitochondrial biogenesis
An induction of mitochondrial biogenesis can also be achieved by pharmacological means although this does not tend to be as powerful as the transgenic expression of PGC-1α. Drug treatments also require special considerations on dose, time and frequency of administration and could result on undesired side effects.
2.3.2.1. Bezafibrate treatment
Increases in the levels of PGC-1α have also been achieved by treating different mouse models with bezafibrate, a PPAR pan-agonist that acts on the PPARs/PGC-1α axis, enhancing lipid metabolism and oxidative capacity (Bastin, 2008; Tenenbaum, 2005). Bezafibrate is been used in humans for many years to treat dyslipidemia. In contrast to the unanimous beneficial effects induced by transgenic overexpression of PGC-1α, the bezafibrate treatment remained controversial in mouse models. In some instances bezafibrate had beneficial effects (Dillon, 2012a; Noe, 2013; Wenz, 2008; Yatsuga and Suomalainen, 2012), in others the benefit was absent or even toxic (Viscomi, 2011).
Muscle specific Cox10 KO mice fed with bezafibrate showed increased COX activity, higher expression of some subunits of the OXPHOS system and 2.5 fold increase in citrate synthase activity. Thus, bezafibrate treatment increased mitochondrial biogenesis in this model. The higher energetic capacity of bezafibrate-fed mice resulted in a better performance on the treadmill (Wenz, 2008).
Bezafibrate had also a beneficial effect in the Deletor mouse model, which under the diet, showed reduced percent of COX negative fibers and reduced mtDNA deletion load in skeletal muscle (Yatsuga and Suomalainen, 2012). Although there was an improvement of the phenotype exhibited in the Deletor mouse with bezafibrate this was not due to an increased mitochondrial biogenesis. Surprisingly, mitochondrial function and protein expression were reduced in the bezafibrate treated Deletor mouse, in fact the drug did not affect the expression levels of PGC-1α or the PPARs receptors. Thus the beneficial mechanism of action of bezafibrate in this mouse model remains to be clarified. When comparing the different models, the bezafibrate diet regimen in the Deletor mouse was the same as the one tested in the Cox10 KO, however there was a difference on the age of the mouse when treatment was initiated. The bezafibrate treatment in the Deletor mice was started at 12 months of age, in agreement with the late disease onset, whereas in Cox10 KO the drug was provided at 5 weeks of age when mice still without major phenotypic abnormalities. These differences in age may influence the effectiveness of the treatment. It is important to note that the Deletor mice treated with bezafibrate showed increased lipid oxidation and hepatomegaly as side effects of the treatment (Yatsuga and Suomalainen, 2012).
Similarly to the results obtained in Cox10 KO myopathy mice, bezafibrate conferred neuroprotection in the brain specific Cox10 KO mouse model (Noe, 2013). Cox10 KO mice fed with bezafibrate diet showed a marked delay of the encephalopathy compared with the KO fed with standard diet. Bezafibrate reduced neuronal loss, inflammation and glia activation. At a molecular level, this improvement was paralleled by an increase of PGC1-α expression in the cortex and hippocampus, an increase of COX specific activity and higher amount of some OXPHOS subunits. Bioenergetic improvement determined also a drastic reduction of the oxidative damage in the brain of bezafibrate-fed Cox10 KO mice and an increased response in the antioxidant defenses (Noe, 2013).
Contrasting results were obtained on other myopathy models of COX deficiency caused by the ablation of Surf1 and Cox15. In this case, Bezafibrate did not have any beneficial effect or was detrimental to the Cox15 KO. The diet regimen was the same as the one tested in the Cox10 KO animals; however the drug seemed to have toxic effects, leading to weight loss and hepatomegaly (Viscomi, 2011). Despite this hepatotoxic effect, a significant increase of the expression of PPARα, PPARβ/δ and several fatty acid oxidation (FAO) related target genes was observed in the Surf1 KO bezafibrate treated animals, confirming the effectiveness of the drug on target genes in vivo. On the other hand, no significant changes in PGC-1α mRNA levels, mtDNA content, citrate synthase, and OXPHOS complexes activity were detected in bezafibrate treated versus untreated Surf1 KO mice. There is no obvious explanation between these discrepancies in the different myopathy mouse models. The responsiveness to the drug may depend on a number of still unknown variables. A clear difference in the response is that in the Cox10 KO mouse bezafibrate increased the expression of PGC-1α, whereas that increased was not detected in the other models.
The premature aging Mutator mouse was also fed with the bezafibrate diet for 8 months. The treatment resulted in improved phenotypes but also with a clear side effect, hepatomegaly (Dillon, 2012a). Mice treated had delayed hair loss and improved skin and spleen aging-like phenotypes, but no improvements were detected in other tissues. This data indicated specific differences in the tissues response to the drug. Besides the beneficial effect, no increases in mitochondrial proliferation were detected in skin and spleen, however increases in markers of fatty acid oxidation were observed (Dillon, 2012a).
There is no evidence of liver damage by the use of bezafibrate in humans, thus the hepatomegaly observed in some mouse models suggests a rodent-specific side effect, and most likely is dose-dependent one. The dose used in the animal models was about 80 times higher than the dose used for the treatment of dyslipidemia in humans (10 mg/Kg/day) (Djouadi and Bastin, 2011). For this reason, further studies reducing the bezafibrate dose in mouse are needed to determine if beneficial effects can be maintained without the problematic side effects.
2.3.2.2. AICAR treatment
Since PGC-1α is one of the dowstream targets of AMPK (5′-adenosine monophosphate-activated protein kinase), another strategy to increase mitochondrial biogenesis is by activating this kinase using its specific agonist AICAR (5-aminoimidazole-4-carboxiamide) (Jager, 2007). AICAR induces the phosphorylation of AMPK in its major regulatory phosphorylation site (AMPK-P172) in vivo (Fogarty, 2010; Narkar, 2008). Intraperitoneal injections of AICAR in the muscle specific, Cox15 KO, Surf1 KO and Sco2KO/KI mouse models partially rescued the myopathy phenotype (Viscomi, 2011). Interestingly, the levels of AMPK-P172 were already higher in skeletal muscle of the untreated Surf1 KO, Sco2KO/KI and Cox15 KO mice when compared to control animals and further increased upon AICAR treatment. After 1 month of treatment, Surf1 KO, Cox15 KO, and Sco2KO/KI mice showed partial recovery of the COX defect and other OXPHOS complexes in muscle. However, despite the increased levels of COX subunits and transcripts, AICAR treatment induced a mild increase of PGC-1α levels only in the Sco2KO/KI mice, whereas the mtDNA content and citrate synthase activity were not higher in the other AICAR treated groups. These results suggest that the recovery in the biochemical phenotype observed in the Surf1 KO animals after AICAR treatment was determined by an increase in the transcription and translation of the OXPHOS components rather than to an effect on mitochondrial proliferation. AICAR treatment significantly restored the motor activity in the Sco2KO/KI mouse but not in the muscle specific Cox15 KO model. This difference in effectiveness is most likely due to the severe phenotype of the COX15 KO compared to the milder phenotype on the Sco2KO/KI. AICAR might represent a promising agent for therapeutic intervention at least for mild mitochondrial myopathies. Long-term administration of AICAR needs to be addressed to rule out any possible toxic.
2.3.2.3. NAD+ dependent mitochondrial biogenesis
Sirtuin proteins are deacetylase enzymes that are able to activate the mitochondrial transcriptional program and therefore increase mitochondrial function (Houtkooper, 2012). Since NAD+ is the rate limiting co-substrate for the sirtuins to catalyze the deacetylation of target proteins, strategies to increase the NAD+ pool and hence activate sirtuins have been studied in vivo. Recently, two approaches have been tested: (i) dietary supplementation with the NAD+ precursor nicotinamide ribose and (ii) the inhibition of alternative NAD+-consuming enzymes such us poly(ADP-ribose)polymerase-1 (PARP1).
2.3.2.3.1. Nicotinamide riboside supplementation (NR)
Nicotinamide riboside (NR) is a vitamin B3-analogue and NAD+ precursor able to increase NAD+ content, sirtuin activity and expression of mitochondrial genes in muscle, liver and brown adipose tissues but not in brain and white adipose tissue in control mice (Canto, 2012). Nicotinamide riboside supplementation was tested in two mouse models of mitochondrial disease: the Deletor mice, a late onset mitochondrial myopathy model (Khan, 2014), and the Sco2KOKI mice, a muscle specific COX deficient mouse (Cerutti, 2014). Nicotinamide ribose increased mitochondrial function and delayed the onset of disease in both mouse models. However, the mechanism underlying the delay of the pathological phenotype seems to be different in the two models. In the Deletor mouse, NR administration induced a robust mitochondrial biogenesis in skeletal muscle (Khan, 2014), whereas in the Sco2KOKI mouse, the restored mitochondrial function has been associated with increased transcription of genes related to OXPHOS and fatty acid oxidation (FAO) (Cerutti, 2014). The different response observed beetween models was attributed to the length of the treatment, that was shorter in the Sco2 KOKI model (1 month) and much longer in the Deletor model (4 months). These results suggest a time-dependent activation of different mitochondriogenic programs, where the induction of mitochondrial biogenesis takes longer that the induction of OXPHOS and FAO related genes. Interestingly, activation of the mitochondrial unfolded protein response (mtUPR) was found in both mouse models after NR-supplementation. mtUPR is a stress-response protective mechanism, that can contribute to improve mitochondrial function. Moreover, in the Deletor model, NR supplementation after the manifestation of the disease (17 months at initiation of treatment) was shown to induce mitochondrial biogenesis and succesfully delay disease progression (Khan, 2014). This finding is of special relevance for the medical treatment because it is most likely that treatment of affected patients will be initiated after onset of disease as it may take long time to obtain the exact diagnosis or cause of the clinical condition. However, NR supplementation in the diet did not induce mitochondrial gene expression or mitochondrial function in the brain, neither in the Deletor, nor Sco2 KOKI mouse model (Cerutti, 2014; Khan, 2014).
2.3.2.3.2. Inhibition of NAD+-dependent enzyme PARP1
Another strategy to increase NAD+ levels consist in the inhibition of the enzyme poly(ADP-ribose)polymerase-1 (PARP1), which is involved in DNA damage detection and repair. PARP1 is one of the highest consumer of NAD+ in the nucleus. Genetic ablation of Parp-1 was shown to increased Sirt 1 activity in skeletal muscle, as well as enhanced mitochondrial oxidative capacity and endurance in control animals (Bai, 2011). Accordingly, ablation of Parp1 gene or administration of a dual PARP1 and PARP2 inhibitor (pan-PARP inhibitor) MRL-45696 as a food mix in the Sco2KO/KI mouse model improved motor performance and restored COX activity in skeletal muscle (Cerutti, 2014). As seen with the NR treatment in Sco2KO/KI model, mitochondrial biogenesis was not activated in animals treated with the pan-PARP inhibitor. The increased mitochondrial function detected was due to increase of the mRNA expression levels of OXPHOS and FAO related genes (Cerutti, 2014).
In contrast to the NR treatment, genetic or pharmacologic inactivation of Parp1 gene increased mitochondrial function not only in skeletal muscle but also in brain (Cerutti, 2014). Therefore, treatment with pan-PARP inhibitors represents good prospect therapy for neuropathies and myopathies of mitochondrial origin. Long-term treatment (16 weeks) of control mice with MRL-45696 did not reveal any evidence of toxicity in this time frame (Pirinen, 2014). Pan-PARPs inhibitors are currently been used in the treatment of certain cancers with few side effects (Audeh, 2010; Bundred, 2013).
2.3.3. Exercise regimen
Metabolic adaptations to increase mitochondrial biogenesis by stimulating the AMPK/PGC-1α pathway can be achieved by endurance exercise (Benton, 2008; Ojuka, 2004; Wu, 2002). In fact, Cox10 KO mice subjected to endurance exercise improved significantly their phenotypic outcome and survival. The beneficial effects were due to activation of mitochondrial biogenesis, increase in ATP content and switch of muscle fiber type from glycolytic to oxidative (Wenz, 2008). Likewise, a 5 months exercise endurance regime in the Mutator mice delayed the aging course and expanded the lifespan of the animals (Safdar, 2011). Moreover, the endurance exercise reduced the frequency of mtDNA mutations and depletion, restored mitochondrial morphology and oxidative capacity to normal levels in the Mutator mice. The amelioration of the pathology in the Mutator mouse was attributed to a stimulation of mitochondrial biogenesis by restoring PGC-1α to control values; to a reduction of apoptosis to basal levels in different tissues; and possibly to a regulation of other systemic factors (Safdar, 2011). There are several studies indicating that exercise also could result beneficial for patients suffering from mitochondrial myopathies although the exercise intolerance common in this type of disorders can raise concerns. Exercise have been showed to be beneficial, in terms on increased oxidative capacity, muscle strength and improved quality of life in patients with mtDNA mutations, however the mechanisms underlying the improvement are not known. Exercise can alter the proportion of mutant and wild type mtDNA in regenerated muscle fibers (Murphy, 2008; Taivassalo, 2006; Taivassalo, 2001). However, giving the heterogeneous nature of mitochondrial disorders, it is still unclear whether moderate exercise will result beneficial in all cases. More studies addressing this issue are needed.
2.4. Nutritional intervention
Similar to the pharmacological approaches, changes in diet (alternative fuel sources), caloric restriction or administration of chemicals that can target or stimulate metabolic pathways involved in cellular nutrient sensing have the potential to result beneficial for the treatment of mitochondrial diseases. This type of strategy has been tried in few mouse models (Table III) and in humans. Depending on the metabolic defect, just simple changes on diet content could result beneficial.
Table III.
Interventions to regulate metabolism and oxidative stress tested in mouse models of mitochondrial diseases.
| Therapeutic approach | Mouse model | Effects of the therapeutic intervention | Reference | ||
|---|---|---|---|---|---|
| Nutritional Modulation | Administration of Ketogenic Diet | Deletor mouse | Slowed and improved progression of phenotype; decreased number of CIV negative fibers; restored to normal levels lipids, aminoacids and ketone bodies in plasma; no changes on mt DNA | (Ahola-Erkkilä, 2010) | |
| Administration of a diet with high fat content | Harlequin (CI deficient mice) | Neuroprotective effects, better performance of rotarod and tail suspension test | (Schiff, 2011) | ||
| Inhibition of mTOR pathway | Rapamycin treatment (injected or administered in the diet) | Ndufs4−/− mouse | Increased lifespan, improvement of neurological signs, better performance on rotarod, disappearance of dispnea | (Johnson, 2013b) | |
| Other treatments | Control of mPTP opening | Cyclosporin A treatment | “Mutator mouse” | Reduced apoptosis and amelioration of the heart phenotype | (Mott, 2004) |
| Antioxidant treatments | Overexpression of catalase in heart | Partial rescue of the aging phenotype | (Dai, 2010) | ||
| N-acetyl-L-cysteine treatment | (Ahlqvist, 2012) | ||||
| Metabolic bypass | Dietary supplementation with dCMP and dTMP | TK2 knock-in mouse | Delayed disease onset, increased mtDNA levels Reduced severity of encephalopathy and prolonged lifespan | (Garone, 2014) | |
2.4.1. Ketogenic and High Fat diet
The ketogenic (KD), and high fat (HFD) diets consist in high fat content and a low carbohydrate content that has been shown to have beneficial effect on mitochondrial diseases in a dual manner. On one hand, these diets have the ability of increase fatty acids oxidation to feed electrons to the electron transfer flavoprotein (ETF) that subsequently will feed into ubiquinone in the electron transport chain with the potential to bypass complex I. In the other hand, fatty acids are known to act as natural ligands of the Peroxisomal Proliferator Activator Receptors (PPAR), inducing their activation and as described in section 2.3, and stimulate mitochondrial biogenesis (Bastin, 2008). HFD consisted of fat 30 kcal%, carbohydrate 52 kcal% and protein 18 kcal% whereas a control diet consisted of fat 8 kcal%, carbohydrate 72 kcal% and protein 20 kcal% (Schiff, 2011). Compared to the HFD, the fat content in the KD diet is much higher, 89.5 kcal%, and the levels of carbohydrate are severely reduced to 0.1 kcal%. In the KD diet, also he levels of protein are reduced close to half respect to the HFD (10.4 kcal%). Administration of a high fat diet significantly slowed the disease progression and the neurodegenerative phenotype of the Harlequin complex I-deficient mice (Schiff, 2011). However, the mechanisms involved in the neuroprotective effects of the HFD in vivo, remained unclear. HFD-fed mice, both control and Harlequin mice showed increased levels of β-hydroxybutyrate in blood, suggesting increased production of ketone bodies by the liver under this diet. Increased β-hydroxybutyrate could constitute an additional source of metabolic fuel for the brain. Therefore, it is believed that the beneficial effects are due to an increased in the energy metabolism of the brain. Unfortunately mitochondrial mass was not assayed in this study to determine if activation of mitochondrial biogenesis played any role in the beneficial effects obtained. As described in section 2.1, ketogenic diet significantly slowed down disease progression in the Deletor mice without inducing a heteroplasmic shift. However, the ketogenic diet was harmful for the control mice, as they became obese and displayed liver steatosis and inflammation (Ahola-Erkkilä, 2010). In the Mpv17 KO mouse, the KD diet failed to increased mitochondrial biogenesis and instead induced severe liver toxicity in both controls and Mpv17 KO animals (Bottani, 2014). Hence, further clinical evaluation of the high content fat diet in OXPHOS-deficient patients is required as the beneficial results observed in some OXPHOS defective mice are encouraging.
2.4.2. Rapamycin administration
Cells are able to recognize and respond to the nutrient availability in their environment through complex signaling pathways, which integrate the sensing and response, making these pathways good candidates for therapeutic intervention of OXPHOS defects. One of the key factors in the coordination of nutrient sensing, cell growth, cell proliferation and cell survival is the kinase mTOR (mammalian target of rapamycin). Indeed, mTOR regulates metabolism by integrating several cellular interconnected pathways: levels of amino acids in the lysosome, energetic sensing by AMPK, and extracellular signals through insulin/IGF (Johnson, 2013a). Reductions in the nutrient signaling, achieved by glucose restriction and by genetic inhibition of mTOR, have been shown to rescue short replicative lifespan in several yeast mutants defective on mitochondrial function (Schleit, 2013). Based on these findings, Johnson and colleagues studied the effect of the inhibition of mTOR with rapamycin in the mammalian model of Leigh syndrome, the Ndufs4 KO mouse (Johnson, 2013a). Ndufs4 gene encodes for a catalytic subunit of CI. This mouse showed a progressive neurodegenerative phenotype characterized by weight loss, lethargy, ataxia and early dead, associated with CI deficiency (Quintana, 2010). Daily intraperitoneal injections of rapamycin, the Ndufs4 KO mice induced a significant improvement on lifespan (up to 25% in males and 38% in females) and a delay in the onset of the pathological features (Johnson, 2013b). Treatment with encapsulated rapamycin diet was even more effective, increasing the lifespan up to 4 months and inducing a striking improvement of neurological and myopathic signs, and disappearance of dyspnea. Moreover, the peculiar glia activation observed in the Ndufs4 KO mice, was only evident in mice treated with vehicle whereas they were totally absent in age matched rapamycin treated KO mice (Johnson, 2013b). While the precise mechanism of this recue effect remains unclear, rapamycin did not have any effect on the OXPHOS complexes activity and/or stability but induced a metabolic shift to an increase on the amino acid catabolism and a decrease on the glycolytic metabolism. These results strongly suggest that boosting alternative metabolic pathways to produce energy should be considered as therapeutic approach for mitochondrial diseases. Although rapamycin treatment showed many of beneficial effects, the authors underline the occurrence of adverse effects such as immunosuppression, hyperlipidemia and decreased wound healing ability, which might limit the use of this drug use in the clinics, particularly in young patients. Nonetheless, other mTOR inhibitors or rapamycin-like drugs exerting mild side effects could be specifically designed to circumvent undesired side effects.
2.5. Other treatments
Since many of mitochondrial dysfunctions are attributed to increased levels of ROS, the use of antioxidants or inhibitors of the mitochondrial permeability transition pore (mPTP) have been shown to ameliorate the phenotype of certain defects. For example, in the Mutator mouse model, a subtle contribution of reactive oxygen species (ROS) in the generation of the aged phenotype has been revealed by the observation that the overexpression of catalase in heart or the treatment with N-acetyl-L-cysteine partially rescued the aging symptoms (Ahlqvist, 2012; Dai, 2010) (Table III). The severity of these phenotypes was also mitigated and even reverted by the inhibition of the mPTP opening with cyclosporin A treatment (Mott, 2004).
Another strategy to improve phenotypic alterations due to abnormal chromatin structure has been proposed. Since chromatin structure is regulated by histone acetylation/deacetylation, a treatment with different histone deacetylase (HDAC) inhibitors was tested in a mouse model of Friedreich’s ataxia with successful results. The trinucleotide expansion found in Fxn gene impairs normal transcription due to the altered chromatin structure. An improvement of the neurological phenotype and a partial restoration of the levels of the frataxin in the brain was observed in the mouse treated with HDAC inhibitors (Sandi, 2011) (Table I). These results support further assessment of this class of compounds for treatment of Friedreich’s ataxia and other defects involving epigenomic alterations.
Metabolic bypass therapy is another approach where it is possible to supplement with compounds that are downstream to go around the enzymatic defect of a particular pathway. This approach was tested in the TK2 knock-in mouse. TK2 is a mitochondrial thymidine kinase 2 that phosphorylates thymidine and deoxycitidine pyrimidine nucleosides to produce their respective monophosphate forms dTMP and dCMP, which are later converted to deoxynucleotide triphosphates (dNTPs). The dNTPs are used for the mtDNA synthesis, therefore abnormalities in the TK2 gene cause severe mtDNA depletion and multiple deletions. The TK2 knock-in mouse recapitulates the infantile encephalopathy and prematurely dies at P16 (Akman, 2008). The TK2 defect causes a dNTP imbalance with low dTTP and dCTP in tissues. When this two dNTPs precursors were given orally to the TK2 knock-in mouse, starting at postnatal day 4 there was an amelioration of the encephalopathy phenotype, increase in mtDNA levels and significant extension on the life span (Garone, 2014) (Table III). These results indicate that the metabolic bypass therapy has a great potential to be translated to patients with block in the dNTP synthesis and be used in other disorders that have an unbalanced nucleotide pool.
3. Concluding remarks
The aim of this review was to summarize the therapeutic approaches that have been tested in mouse models of mitochondrial diseases. These approaches are diverse and mainly include gene therapy, induction of mitochondrial biogenesis and nutritional intervention. Beside the positive effects observed in some of the cases, very little has been translated in the clinical practice. Unfortunately, we still lack of a cure for mitochondrial diseases. Recent in vitro studies have shown many compounds potentially beneficial for the treatment of mitochondrial diseases (Saada, 2011), but their use in vivo remains scarce. We foresee that in the next few years this will be a very fruitful field in medical research now that the field counts with numerous mouse models that recapitulate human disease phenotype. The mouse models will be instrumental to test in vivo new therapeutic strategies and will facilitate the translation into the clinics.
Highlights.
There is no cure for mitochondrial diseases.
Mouse models have been created to understand the mechanism of disease.
Mouse models are instrumental to test in vivo therapeutic strategies.
Therapeutic interventions include gene therapy, heteroplasmic shift, mitochondrial biogenesis, nutritional intervention and exercise regime.
Acknowledgments
We would like to express our gratitude to Jose Oca-Cossio for critical comments of this manuscript. FD research has been funded by the James and Esther King Research Program Florida Department of Health (grant number 08KN-01), by the National Institute of Health (grant number GM101225) and by the United Mitochondrial Disease Foundation (grant number 14050R).
Footnotes
Conflict of interest statement: Authors have nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Susana Peralta, Email: SPeralta@med.miami.edu.
Alessandra Torraco, Email: alessandra.torraco@opbg.net.
Luisa Iommarini, Email: luisa.iommarini2@unibo.it.
Francisca Diaz, Email: FDiaz1@med.miami.edu.
References
- Agostino A, Valletta L, Chinnery PF, Ferrari G, Carrara F, Taylor RW, Schaefer AM, Turnbull DM, Tiranti V, Zeviani M. Mutations of ANT1, Twinkle, and POLG1 in sporadic progressive external ophthalmoplegia (PEO) Neurology. 2003;60:1354–1356. doi: 10.1212/01.wnl.0000056088.09408.3c. [DOI] [PubMed] [Google Scholar]
- Ahlqvist KJ, Hämäläinen RH, Yatsuga S, Uutela M, Terzioglu M, Götz A, Forsström S, Salven P, Angers-Loustau A, Kopra OH, Tyynismaa H, Larsson NG, Wartiovaara K, Prolla T, Trifunovic A, Suomalainen A. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012;15:100–109. doi: 10.1016/j.cmet.2011.11.012. [DOI] [PubMed] [Google Scholar]
- Ahola-Erkkilä S, Carroll CJ, Peltola-Mjösund K, Tulkki V, Mattila I, Seppänen-Laakso T, Oresic M, Tyynismaa H, Suomalainen A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet. 2010;19:1974–1984. doi: 10.1093/hmg/ddq076. [DOI] [PubMed] [Google Scholar]
- Akman HO, Dorado B, Lopez LC, Garcia-Cazorla A, Vila MR, Tanabe LM, Dauer WT, Bonilla E, Tanji K, Hirano M. Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum Mol Genet. 2008;17:2433–2440. doi: 10.1093/hmg/ddn143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov. 2013;12:465–483. doi: 10.1038/nrd4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Duan D, Moraes CT. Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2012;19:1101–1106. doi: 10.1038/gt.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Garcia S, Moraes CT. Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2010;17:713–720. doi: 10.1038/gt.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Hernandez D, Moraes CT. Modulating mtDNA heteroplasmy by mitochondria-targeted restriction endonucleases in a ‘differential multiple cleavage-site’ model. Gene Ther. 2007;14:1309–1318. doi: 10.1038/sj.gt.3302981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med. 2013;19:1111–1113. doi: 10.1038/nm.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-de Murcia J, Auwerx J. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin J, Aubey F, Rötig A, Munnich A, Djouadi F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients’ cells lacking its components. J Clin Endocrinol Metab. 2008;93:1433–1441. doi: 10.1210/jc.2007-1701. [DOI] [PubMed] [Google Scholar]
- Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci U S A. 2005;102:14392–14397. doi: 10.1073/pnas.0502896102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton CR, Wright DC, Bonen A. PGC-1alpha-mediated regulation of gene expression and metabolism: implications for nutrition and exercise prescriptions. Appl Physiol Nutr Metab. 2008;33:843–862. doi: 10.1139/H08-074. [DOI] [PubMed] [Google Scholar]
- Bonnefont JP, Bastin J, Behin A, Djouadi F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. 2009;360:838–840. doi: 10.1056/NEJMc0806334. [DOI] [PubMed] [Google Scholar]
- Bottani E, Giordano C, Civiletto G, Di Meo I, Auricchio A, Ciusani E, Marchet S, Lamperti C, d’Amati G, Viscomi C, Zeviani M. AAV-mediated liver-specific MPV17 expression restores mtDNA levels and prevents diet-induced liver failure. Molecular therapy: the journal of the American Society of Gene Therapy. 2014;22:10–17. doi: 10.1038/mt.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaita A, Augustin S, Lechauve C, Cwerman-Thibault H, Benit P, Simonutti M, Paques M, Rustin P, Sahel JA, Corral-Debrinski M. Downregulation of apoptosis-inducing factor in Harlequin mice induces progressive and severe optic atrophy which is durably prevented by AAV2-AIF1 gene therapy. Brain. 2012;135:35–52. doi: 10.1093/brain/awr290. [DOI] [PubMed] [Google Scholar]
- Bundred N, Gardovskis J, Jaskiewicz J, Eglitis J, Paramonov V, McCormack P, Swaisland H, Cavallin M, Parry T, Carmichael J, Dixon JM. Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: a phase I multicentre trial in patients scheduled for elective breast cancer surgery. Invest New Drugs. 2013;31:949–958. doi: 10.1007/s10637-012-9922-7. [DOI] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, Viscomi C, Zeviani M. NAD(+)-Dependent Activation of Sirt1 Corrects the Phenotype in a Mouse Model of Mitochondrial Disease. Cell Metab. 2014;19:1042–1049. doi: 10.1016/j.cmet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–544. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo I, Auricchio A, Lamperti C, Burlina A, Viscomi C, Zeviani M. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol Med. 2012;4:1008–1014. doi: 10.1002/emmm.201201433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon LM, Hida A, Garcia S, Prolla TA, Moraes CT. Long-term bezafibrate treatment improves skin and spleen phenotypes of the mtDNA mutator mouse. PLoS One. 2012a;7:e44335. doi: 10.1371/journal.pone.0044335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon LM, Williams SL, Hida A, Peacock JD, Prolla TA, Lincoln J, Moraes CT. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum Mol Genet. 2012b;21:2288–2297. doi: 10.1093/hmg/dds049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouadi F, Bastin J. Species differences in the effects of bezafibrate as a potential treatment of mitochondrial disorders. Cell Metab. 2011;14:715–716. doi: 10.1016/j.cmet.2011.11.003. author reply 717. [DOI] [PubMed] [Google Scholar]
- El-Khoury R, Dufour E, Rak M, Ramanantsoa N, Grandchamp N, Csaba Z, Duvillie B, Benit P, Gallego J, Gressens P, Sarkis C, Jacobs HT, Rustin P. Alternative oxidase expression in the mouse enables bypassing cytochrome c oxidase blockade and limits mitochondrial ROS overproduction. PLoS genetics. 2013;9:e1003182. doi: 10.1371/journal.pgen.1003182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khoury R, Kemppainen KK, Dufour E, Szibor M, Jacobs HT, Rustin P. Engineering the alternative oxidase gene to better understand and counteract mitochondrial defects: state of the art and perspectives. British journal of pharmacology. 2014;171:2243–2249. doi: 10.1111/bph.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellouze S, Augustin S, Bouaita A, Bonnet C, Simonutti M, Forster V, Picaud S, Sahel JA, Corral-Debrinski M. Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet. 2008;83:373–387. doi: 10.1016/j.ajhg.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Ayala DJ, Sanz A, Vartiainen S, Kemppainen KK, Babusiak M, Mustalahti E, Costa R, Tuomela T, Zeviani M, Chung J, O’Dell KM, Rustin P, Jacobs HT. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. 2009;9:449–460. doi: 10.1016/j.cmet.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Flierl A, Chen Y, Coskun PE, Samulski RJ, Wallace DC. Adeno-associated virus-mediated gene transfer of the heart/muscle adenine nucleotide translocator (ANT) in mouse. Gene Ther. 2005;12:570–578. doi: 10.1038/sj.gt.3302443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty S, Hawley SA, Green KA, Saner N, Mustard KJ, Hardie DG. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem J. 2010;426:109–118. doi: 10.1042/BJ20091372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garone C, Garcia-Diaz B, Emmanuele V, Lopez LC, Tadesse S, Akman HO, Tanji K, Quinzii CM, Hirano M. Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol Med. 2014;6:1016–1027. doi: 10.15252/emmm.201404092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Qi X, Pallotti F, Schon EA, Manfredi G, Carelli V, Martinuzzi A, Hauswirth WW, Lewin AS. Rescue of a mitochondrial deficiency causing Leber Hereditary Optic Neuropathy. Ann Neurol. 2002;52:534–542. doi: 10.1002/ana.10354. [DOI] [PubMed] [Google Scholar]
- Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- Hirano M, Garone C, Quinzii CM. CoQ(10) deficiencies and MNGIE: two treatable mitochondrial disorders. Biochim Biophys Acta. 2012;1820:625–631. doi: 10.1016/j.bbagen.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, Marti R, Spinazzola A, Nishino I, Nishigaki Y. Thymidine phosphorylase deficiency causes MNGIE: an autosomal recessive mitochondrial disorder. Nucleosides, nucleotides & nucleic acids. 2004;23:1217–1225. doi: 10.1081/NCN-200027485. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Shoubridge EA. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013a;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, Sedensky MM, Kaeberlein M. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013b;342:1524–1528. doi: 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizaki Y, Seymour RS, Ito K. A novel functional element in the N-terminal region of Arum concinnatum alternative oxidase is indispensable for catalytic activity of the enzyme in HeLa cells. Biochim Biophys Acta. 2010;1797:20–28. doi: 10.1016/j.bbabio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Kawamata H, Tiranti V, Magrané J, Chinopoulos C, Manfredi G. adPEO mutations in ANT1 impair ADP-ATP translocation in muscle mitochondria. Hum Mol Genet. 2011;20:2964–2974. doi: 10.1093/hmg/ddr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsstrom S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, Suomalainen A. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014;6:721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lopez LC, Akman HO, Garcia-Cazorla A, Dorado B, Marti R, Nishino I, Tadesse S, Pizzorno G, Shungu D, Bonilla E, Tanji K, Hirano M. Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Hum Mol Genet. 2009;18:714–722. doi: 10.1093/hmg/ddn401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott JL, Zhang D, Freeman JC, Mikolajczak P, Chang SW, Zassenhaus HP. Cardiac disease due to random mitochondrial DNA mutations is prevented by cyclosporin A. Biochem Biophys Res Commun. 2004;319:1210–1215. doi: 10.1016/j.bbrc.2004.05.104. [DOI] [PubMed] [Google Scholar]
- Murphy JL, Blakely EL, Schaefer AM, He L, Wyrick P, Haller RG, Taylor RW, Turnbull DM, Taivassalo T. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain. 2008;131:2832–2840. doi: 10.1093/brain/awn252. [DOI] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe N, Dillon L, Lellek V, Diaz F, Hida A, Moraes CT, Wenz T. Bezafibrate improves mitochondrial function in the CNS of a mouse model of mitochondrial encephalopathy. Mitochondrion. 2013;13:417–426. doi: 10.1016/j.mito.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojuka EO. Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle. Proc Nutr Soc. 2004;63:275–278. doi: 10.1079/PNS2004339. [DOI] [PubMed] [Google Scholar]
- Perales-Clemente E, Fernández-Silva P, Acín-Pérez R, Pérez-Martos A, Enríquez JA. Allotopic expression of mitochondrial-encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res. 2011;39:225–234. doi: 10.1093/nar/gkq769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomini M, Belbellaa B, Monassier L, Reutenauer L, Messaddeq N, Cartier N, Crystal RG, Aubourg P, Puccio H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat Med. 2014;20:542–547. doi: 10.1038/nm.3510. [DOI] [PubMed] [Google Scholar]
- Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, Hirano M, Zeviani M, Bindoff LA, Yu-Wai-Man P, Hanna M, Carelli V, McFarland R, Majamaa K, Turnbull DM, Smeitink J, Chinnery PF. New treatments for mitochondrial disease-no time to drop our standards. Nature reviews Neurology. 2013;9:474–481. doi: 10.1038/nrneurol.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirinen E, Canto C, Jo YS, Morato L, Zhang H, Menzies KJ, Williams EG, Mouchiroud L, Moullan N, Hagberg C, Li W, Timmers S, Imhof R, Verbeek J, Pujol A, van Loon B, Viscomi C, Zeviani M, Schrauwen P, Sauve AA, Schoonjans K, Auwerx J. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014;19:1034–1041. doi: 10.1016/j.cmet.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Kruse SE, Kapur RP, Sanz E, Palmiter RD. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci U S A. 2010;107:10996–11001. doi: 10.1073/pnas.1006214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Zanella S, Koch H, Kruse SE, Lee D, Ramirez JM, Palmiter RD. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J Clin Invest. 2012;122:2359–2368. doi: 10.1172/JCI62923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmanith W, Freilinger M, Roka J, Raffelsberger T, Moser-Thier K, Prayer D, Bernert G, Bittner RE. Isolated cytochrome c oxidase deficiency as a cause of MELAS. Journal of medical genetics. 2008;45:117–121. doi: 10.1136/jmg.2007.052076. [DOI] [PubMed] [Google Scholar]
- Saada A. The use of individual patient’s fibroblasts in the search for personalized treatment of nuclear encoded OXPHOS diseases. Mol Genet Metab. 2011;104:39–47. doi: 10.1016/j.ymgme.2011.07.016. [DOI] [PubMed] [Google Scholar]
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sandi C, Pinto RM, Al-Mahdawi S, Ezzatizadeh V, Barnes G, Jones S, Rusche JR, Gottesfeld JM, Pook MA. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol Dis. 2011;42:496–505. doi: 10.1016/j.nbd.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra S, Gilkerson RW, Davidson M, Schon EA. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann Neurol. 2004;56:662–669. doi: 10.1002/ana.20240. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci. 2008;1147:321–334. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders--past, present and future. Biochim Biophys Acta. 2004;1659:115–120. doi: 10.1016/j.bbabio.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Schiff M, Benit P, El-Khoury R, Schlemmer D, Benoist JF, Rustin P. Mouse studies to shape clinical trials for mitochondrial diseases: high fat diet in Harlequin mice. PLoS One. 2011;6:e28823. doi: 10.1371/journal.pone.0028823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleit J, Johnson SC, Bennett CF, Simko M, Trongtham N, Castanza A, Hsieh EJ, Moller RM, Wasko BM, Delaney JR, Sutphin GL, Carr D, Murakami CJ, Tocchi A, Xian B, Chen W, Yu T, Goswami S, Higgins S, Holmberg M, Jeong KS, Kim JR, Klum S, Liao E, Lin MS, Lo W, Miller H, Olsen B, Peng ZJ, Pollard T, Pradeep P, Pruett D, Rai D, Ros V, Singh M, Spector BL, Vander Wende H, An EH, Fletcher M, Jelic M, Rabinovitch PS, MacCoss MJ, Han JD, Kennedy BK, Kaeberlein M. Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell. 2013;12:1050–1061. doi: 10.1111/acel.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nature reviews Genetics. 2012;13:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, DiMauro S, Hirano M, Gilkerson RW. Therapeutic prospects for mitochondrial disease. Trends Mol Med. 2010;16:268–276. doi: 10.1016/j.molmed.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum Mol Genet. 2009;18:1805–1812. doi: 10.1093/hmg/ddp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taivassalo T, Gardner JL, Taylor RW, Schaefer AM, Newman J, Barron MJ, Haller RG, Turnbull DM. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. 2006;129:3391–3401. doi: 10.1093/brain/awl282. [DOI] [PubMed] [Google Scholar]
- Taivassalo T, Shoubridge EA, Chen J, Kennaway NG, DiMauro S, Arnold DL, Haller RG. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann Neurol. 2001;50:133–141. doi: 10.1002/ana.1050. [DOI] [PubMed] [Google Scholar]
- Tenenbaum A, Motro M, Fisman EZ, Tanne D, Boyko V, Behar S. Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch Intern Med. 2005;165:1154–1160. doi: 10.1001/archinte.165.10.1154. [DOI] [PubMed] [Google Scholar]
- Thorburn DR, Dahl HH. Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. American journal of medical genetics. 2001;106:102–114. doi: 10.1002/ajmg.1380. [DOI] [PubMed] [Google Scholar]
- Torres-Torronteras J, Gomez A, Eixarch H, Palenzuela L, Pizzorno G, Hirano M, Andreu AL, Barquinero J, Marti R. Hematopoietic gene therapy restores thymidine phosphorylase activity in a cell culture and a murine model of MNGIE. Gene Ther. 2011;18:795–806. doi: 10.1038/gt.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Torronteras J, Viscomi C, Cabrera-Perez R, Camara Y, Di Meo I, Barquinero J, Auricchio A, Pizzorno G, Hirano M, Zeviani M, Marti R. Gene Therapy Using a Liver-targeted AAV Vector Restores Nucleoside and Nucleotide Homeostasis in a Murine Model of MNGIE. Molecular therapy. 2014;22:901–907. doi: 10.1038/mt.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C, Zeviani M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A. 2009;106:20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Williams SL, Huang J, Edwards YJ, Ulloa RH, Dillon LM, Prolla TA, Vance JM, Moraes CT, Zuchner S. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 2010;12:675–682. doi: 10.1016/j.cmet.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams RS. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- Yatsuga S, Suomalainen A. Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum Mol Genet. 2012;21:526–535. doi: 10.1093/hmg/ddr482. [DOI] [PubMed] [Google Scholar]
- Yu H, Koilkonda RD, Chou TH, Porciatti V, Ozdemir SS, Chiodo V, Boye SL, Boye SE, Hauswirth WW, Lewin AS, Guy J. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci U S A. 2012;109:E1238–1247. doi: 10.1073/pnas.1119577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127:2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]