

Abstract
Trapped ion mobility spectrometry coupled to mass spectrometry (TIMS-MS) was utilized for the separation and identification of familiar explosives in complex mixtures. For the first time, molecular adduct complex lifetimes, relative stability, binding energies and candidate structures are reported for familiar explosives. Experimental and theoretical results showed that the adduct size and reactivity, complex binding energy and the explosive structure tailors the stability of the molecular adduct complex. TIMS flexibility to adapt the mobility separation as a function of the molecular adduct complex stability (i.e., short or long IMS experiments / low or high IMS resolution) permits targeted measurements of explosives in complex mixtures with higher confidence levels.
Keywords: explosives, trapped ion mobility spectrometry, ion neutral collision cross section, dissociation rates, ion stability
INTRODUCTION
Methods for the determination of trace levels of explosives and explosive related materials were developed rapidly and placed into service following several incidents in the 1980s involving catastrophic attacks with bombs on large civilian aircraft.1, 2 The method chosen and distributed widely was ion mobility spectrometry (IMS) which was still in nascent stages of discovery concerning principles of ionization chemistry and best practices for measurements of ion mobility.3–7 Nonetheless, embodiments of IMS were able to operate economically for on-site screening of hand-luggage at security check points of passengers and were distributed in airports world-wide. Measurements by the Explosive Trace Detectors (ETDs) with IMS depend upon the collection and vaporization of explosive residue, formation of molecular ions through chemical reactions in the gas phase, and their separation in a weak electric field as they drift in a bath gas.8 A necessary requirement for an IMS measurement is that molecular ions formed from a substance should be distinctive and should have lifetimes sufficient to pass through the drift region with a characteristic mobility. This can be challenging with explosive molecular ions which may exhibit brief lifetimes and undergo reactions or decompositions in either the reaction region or in the drift region.9, 10 While sufficient understanding existed on the ionization chemistry and stability of ions in air at ambient pressure to justify the development of ETDs based on IMS, precise knowledge of the kinetics of ion decompositions and even the means to measure ion lifetimes in air at ambient pressure were not developed until recently.
Explosive ions are formed in IMS based ETDs through chemical reactions where an explosive molecule, M, is electrostatically associated with a reactant or reagent ion, commonly Cl−, through ion-dipole or ion-induced dipole interaction.6, 11, 12 The ions have thermal energies in the ion source of an IMS analyzer and ion and molecule associations are favorable with an energy barrier. Excess energy from the association can be lost by collisions, by reactions, and by dissociation of the explosive from the ion by the high collision frequency and abundance of small polar neutrals in the purified air of the IMS drift tube. Common reactions with explosives include hydrogen abstraction of protons that are acidic enough to be lost as HCl from an adduct [M+Cl]− and loss of NO3− which appears to arise as a Cl− displacement reaction with fracture at a weak carbon-oxygen bond.8 In other instances, the original adduct [M+Cl]− has lifetime sufficient to pass through the drift region and reach the detector as an intact ion. In other instances, the ion may survive in the reaction region (~ 3 ms) and undergo reactions or dissociation in the drift region, appearing as a distortion in the baseline of the mobility spectrum.13 Methods were described to extract kinetic information from baseline distortions and refined methods developed recently as a kinetic IMS instrument to obtain rate data for specific ions over a range of temperatures without interferences from unwanted ion neutral interactions.14 Reactions including loss of NO3− and Cl− from thermalized ions require energy which has been measured with the kinetic IMS method as 60 to 89 kJ/mol and match favorably with ab initio calculations.9, 10 These reactions are dependent not only on temperature and moisture but also on the precursor ion. While commercial ETDs produce Cl− by dissociative electron capture in a beta emitter source, electrospray ionization (ESI) sources affords flexibility and convenience to form adducts from other anions by spiking the ESI starting solution with various salts.15, 16 For example, measurement of multiple adduct forms of a targeted compound increases the identification confidence while reduces the probability of having interferences from the sample matrix.
With the recent development of trapped ion mobility spectrometry (TIMS), higher mobility resolution and the capability to interrogate and simultaneously measure molecular ion-neutral collision cross section (CCS) as a function of the time after the molecular ion formation has permitted kinetic studies of molecular ion-neutral bath gas interactions in the millisecond to second time scale.17–22 In the current study, the unique potential of TIMS to hold ions while interacting with bath gas molecules (“TIMS” thermostat) is utilized to study at the single molecular level the stability and dissociation kinetics of familiar explosives with different adduct forms. In particular, ion-neutral collision cross sections (CCS) are measured using TIMS for a series of familiar explosive standards in nitrogen as a bath gas and compared with traditional drift tube IMS measurements and theoretical calculations. TIMS-MS capability to separate and identify explosives from complex samples is also demonstrated. In addition, for the first time, molecular ion stability and lifetimes are reported for a series of familiar explosive molecular adduct forms.
EXPERIMENTAL SECTION
Chemicals
Individual standards of 2-methyl-1,3,5-trinitrobenzene (TNT), 1,3,5-trinitroperhydro-1,3,5-triazine (RDX), 3-nitrooxy-2,2-bis(nitrooxymethyl)propyl nitrate (PETN) and octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) were obtained from AccuStandard (New Haven, CT) and used as received. Ammonium chloride, ammonium formate, ammonium acetate and ammonium nitrate salts and chromatography grade water, methanol and acetonitrile solvents were obtained from Fisher Scientific (Suwanee, GA) and used as received. TNT, RDX and HMX were dissolved in 1:1 water:methanol v:v ratio, and PETN was dissolved in 1:1:1 water:methanol:acetonitrile v:v ratio to a final concentration of 1 μM. Each ammonium salt containing solution was prepared separately and added to each explosive solution to a final concentration of 10 mM of ammonium salt. An electrospray ionization source (ESI, Bruker Daltonics Inc., MA) was used for all analyses in negative ion mode. Sample purity was confirmed with sub ppm mass accuracy for each standard using ultra-high resolution mass spectrometry in Solarix 7T FT-ICR MS mass spectrometer (Bruker Daltonics Inc., Billerica, MA). A complex mixture of TNT + cappuccino was prepared by doping a standard cappuccino coffee solution with the TNT standard (1 μM) to 100:1 v:v ratio ; the complex mixture sample was diluted in 1:1:1 water:methanol:acetonitrile v:v ratio to a final concentration of 10 nM of TNT standard.
TIMS-MS operation
Details regarding the TIMS operation and specifics compared to traditional IMS can be found elsewhere.17, 19, 21, 23, 24 Briefly, in TIMS mobility separation is based on holding the ions stationary using an electric field against a moving gas. The separation in a TIMS device can be described by the center of the mass frame using the same principles as in a conventional IMS drift tube.25 In traditional drift tube cells, mobility separation is related to the number of ion-neutral collision (or drift time); analogously, the mobility separation in a TIMS device is related to the bath gas drift velocity, ion confinement and ion elution parameters. The mobility, K, of an ion in a TIMS cell is described by:
| (1) |
where νg, E, Velution and Vbase are the velocity of the gas, applied electric field, elution and base voltages, respectively. The constant A was determined using reported mobilities of explosives.8, 26 In TIMS operation, multiple geometric isomers/conformers can be trapped simultaneously at different E values resulting from a voltage gradient applied across the IMS tunnel. After thermalization, trapped species are eluted by decreasing the electric field in stepwise decrements (referred to as the “ramp”). Each mobility-separated isomer/conformer eluting from the TIMS cell can be described by a characteristic voltage difference (i.e., Velution − Vbase). Eluted ions are then mass analyzed and detected by a maXis impact Q-ToF mass spectrometer (Bruker Daltonics Inc, Billerica, MA).
In a TIMS device, the total analysis time can be described as:
| (2) |
where, Ttrap is the thermalization/trapping time, ToF is the time after the mobility separation, and Vramp and Tramp are the voltage range and time required to vary the electric field, respectively. The elution voltage can be experimentally determined by varying the ramp time for a constant ramp voltage. This procedure also determines the time ions spend outside the separation region To (e.g., ion trapping and time-of-flight).
The TIMS funnel is controlled using in-house software, written in National Instruments Lab VIEW, and synchronized with the maXis Impact Q-ToF acquisition program.17, 23 TIMS separation was performed using nitrogen as a bath gas at ca. 300 K and typical pressures at the entrance and back regions of the TIMS analyzer were P1 = 2.6 and P2 = 1.0 mbar, respectively (see more details in ref19). The same RF (2040 kHz and 200–350 Vpp) was applied to all electrodes including the entrance funnel, the mobility separating section, and the exit funnel. At all times, the axial electric field was kept under the low field limit (E/p < 10 V cm1 torr−1) throughout the TIMS and no significant ion heating is produced by the RF confinement.
Mobility values (K) were correlated with CCS (Ω) using the equation:
| (3) |
where ze is the charge of the ion, kB is the Boltzmann constant, N* is the number density at standard temperature and pressure conditions, and mI and mb refer to the masses of the ion and bath gas, respectively.25
The analysis of the molecular adducts decomposition was considered as a first order reaction. The molecular adduct abundance at a given time is defined by the equation:
| (4) |
where k is the decomposition rate (k= 1/td), td is the lifetime of the molecular adduct complex, and Io is the initial abundance.
Theoretical calculations
Geometries and binding energies of candidate structures were optimized at the DFT/B3LYP/6–31+g(d) level using Gaussian 09 software.27 Vibrational frequencies were calculated to guarantee that the optimized structures correspond to a real minima in the energy space, and zero-point energy corrections were applied to calculate the relative stability. Partial atomic charges were calculated using the Merz-Singh-Kollman scheme constrained to the molecular dipole moment.28, 29 Theoretical ion-neutral collision cross sections were calculated using the trajectory method (TM) in MOBCAL version for nitrogen30, 31 with a bath gas at ca. 300K. It should be noted that the MOBCAL version for nitrogen was used assuming the similarity of the molecules to those used to develop the Lennard-Jones potential at 300 K in refs30, 31; for other molecules, alternatives methods may be more accurate (see reference32). All optimized geometries and MOBCAL input files can be found in the supporting information.
RESULTS AND DISCUSSION
A prerequisite for a good analytical IMS performance is the ability to separate and identify molecular species with high reproducibility. The IMS resolution of hand held IMS instruments (e.g., ETDs) is commonly RIMS=20 or below; however, laboratory research IMS instruments using drift tube IMS designs can routinely reach RIMS=80–100.33–37 Recently, we have reported the advantages of TIMS technology to achieve higher mobility resolution (RIMS=150–250).19, 20 Different from other IMS forms (e.g., field asymmetric IMS,38 differential mobility spectrometer39–41, segmented quadrupole drift cell,42 cylindrical drift tubes,43 and traveling wave ion guide44), TIMS mobility resolution varies with the size, mass and charge of the molecule of interest; that is, different trapping conditions are required to compensate for molecular ion diffusion and for coulombic repulsion of molecular ions during the trapping and elution steps. In practice, this translates into a lower mobility resolution for high mobility and low mass-to-charge ratio species when compared to previously reported values during fast TIMS mobility scans (see Figure 1 for common explosives). One alternative to increase the TIMS mobility resolution is to reduce the ramp speed which results in higher IMS resolution. For example, a high mobility resolution of RTIMS>120 can be achieved for the analysis of explosives which results on a 3–5 fold increase in resolution when compared to commercially available ETD instruments.
Figure 1.
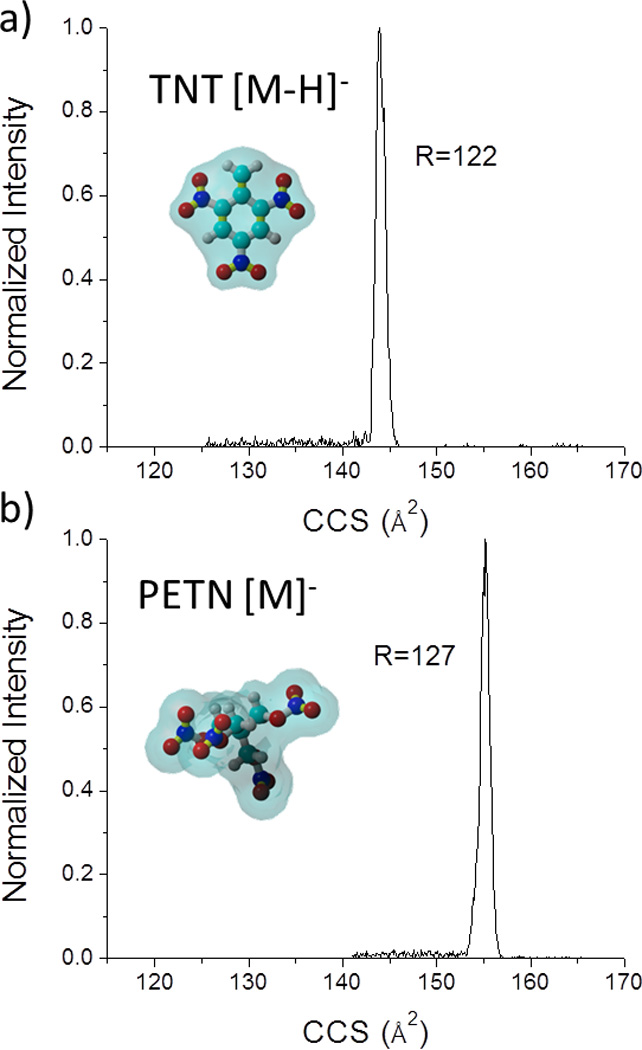
Typical IMS projection spectra for a) TNT and b) PETN using ESI-TIMS-MS.
The high mobility resolution of a TIMS device provides great potential for the analysis of explosives in complex mixtures when coupled to mass spectrometry (see Figure 2). That is, the ability to separate common interferences, to increase peak capacity, and to reduce chemical noise using orthogonal separations permits better identification of explosives using accurate CCS (<5% accuracy using external calibration) and m/z measurements (in the example presented, mass resolution was RTOF=30–40k). Nevertheless, when internal calibrants are used for CCS determination in a TIMS device over a narrower CCS range the accuracy is better than a few percent. When compared to other hyphenated MS techniques for the analysis of familiar explosives,15, 28, 45–51 TIMS-MS provides higher throughput, dynamic range and reduced analysis time. While and increase in peak capacity is observed during TIMS-MS analysis, the most challenging part involves the identification of compounds from the 2D IMS-MS plots. If standards are available for the a priori selected target (see Figure 2c), the identification can be achieved by direct correlation of the IMS and MS data. It should be noted, that additional IMS-MS/MS can further increase the identification capabilities. Another alternative is the coupling of TIMS to ultrahigh resolution MS analyzers (see example in ref52); however, it should be note that TIMS-TOF-MS performs at much shorter acquisition times.
Figure 2.
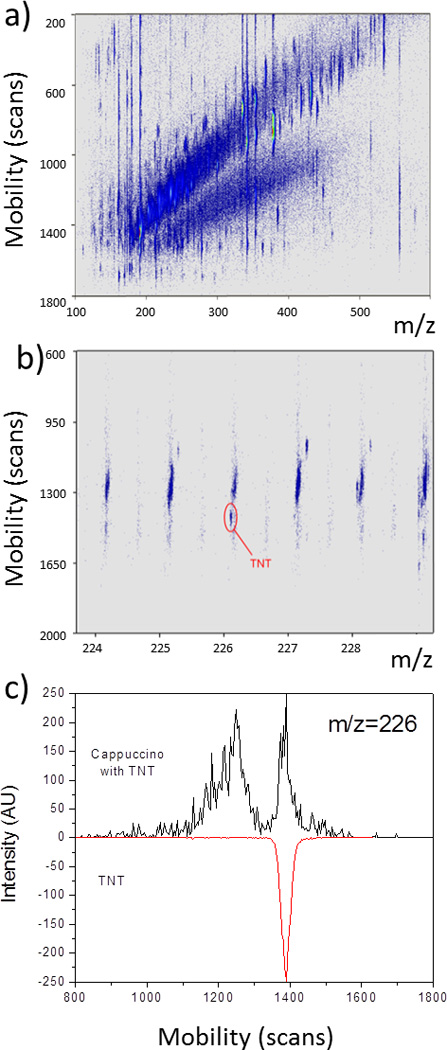
a) 2D IMS-MS contour plot of a complex mixture (cappuccino + TNT); b) inset in the m/z=224–229 range, and c) IMS projection plots of m/z=226 for the complex mixture and a TNT standard.
While TIMS-MS provides high confidence for the analysis of common explosives, one way to further improve the confidence level is to simultaneously measure different molecular adducts.15, 16 That is, each measured molecular adduct form provides a two point identification (i.e., CCS and m/z). Multiple molecular adducts can be formed during ESI of explosives by spiking the ESI starting solution with various salts (see example in Figure 3). In practice, this translates in a CCS and m/z shift for each adduct form, thus increasing the confidence level (see more details in Table 1). Compound identification from complex mixtures is typically challenging by the existence of molecular interferences in the IMS or MS domain. The use of multiple IMS and MS identification points from multiple adduct forms of a targeted compound increases the identification confidence while reduces the probability of having interferences from the sample matrix. In addition, since TIMS permits the measurement of CCS using first principles, the identification can be complemented with theoretical calculations; this approach can be very useful for the case of molecular adduct complexes that can exist as multiple conformations in the gas phase (see example in ref52). Table 1 summarizes theoretical and experimental CCS of all the molecular adduct complexes observed (all structures are provided in the supplemental information, see Figure S1). A Ko error of less than 0.5% was observed in TIMS replica measurements. Close inspection shows that a good agreement is observed between the theoretical and TIMS experimental values (<5% difference). The largest difference between Ko values measured by TIMS and literature values can be attributed to the sample introduction (see ref 8). For example, Ko values of 1.45, 1.48 and 1.54 has been reported for TNT [M−H]− for sample introduction by desorption, ESI, and vapor (membrane), respectively.
Figure 3.
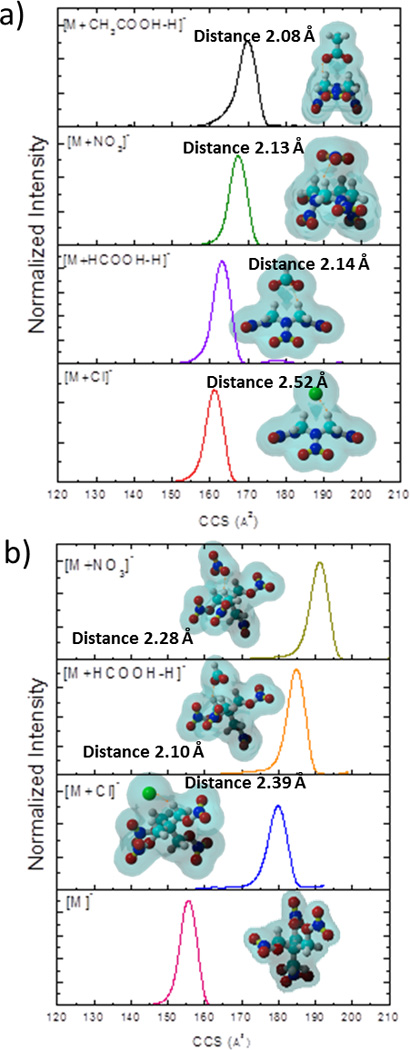
Typical TIMS spectra for a) HMX and b) PETN as a function of the adduct form. Distances between the molecules and the adducts are shown.
Table 1.
Experimental (TIMS), literature,8, 26 and theoretical mobility values of molecular adduct complexes from familiar explosives.
| Compound | Ionic Form | m/z | TIMS Experimental | Reported Ko (cm2/V·s) |
Theoretical CCS (Å2) |
|
|---|---|---|---|---|---|---|
| Ko (cm2/V·s) |
CCS (Å2) |
|||||
| TNT | [M−H]− | 226.010 | 1.48 | 143 | 1.48 | 136 |
| RDX+NH4Cl | [M+Cl]− | 257.003 | 1.44 | 147 | 1.44* | 149 |
| RDX+NH4NO3 | [M+NO3]− | 284.022 | 1.36 | 154 | 1.35* | 152 |
| HMX+NH4Cl | [M+Cl]− | 331.015 | 1.29 | 161 | 1.25 | 162 |
| HMX+HCO2 | [M+HCOOH-H]− | 341.044 | 1.28 | 162 | - | 161 |
| HMX+NH4C2H3O2 | [M+CH3COOH-H]− | 355.059 | 1.23 | 169 | - | 169 |
| HMX+NH4NO3 | [M+NO3]− | 358.034 | 1.23 | 167 | - | 165 |
| PETN | [M*]− | 316.013 | 1.37 | 152 | - | 151 |
| PETN+NH4Cl | [M+Cl]− | 350.982 | 1.17 | 178 | 1.20 | 182 |
| PETN+HCO2 | [M+HCOOH-H]− | 361.011 | 1.14 | 182 | - | 179 |
| PETN+NH4NO3 | [M+NO3]− | 378.001 | 1.11 | 187 | 1.14 | 188 |
Literature values used in the TIMS calibration are denoted with *. A Ko error of less than 0.5% was observed in the TIMS replicate measurements.
The measurement of multiple adduct forms of familiar explosives depends on the probability of forming the molecular adduct complex and their relative stability. During ESI ion formation, changes in the relative salt content can be used to preferentially target the formation of an adduct form as a way to avoid potential CCS and/or m/z interferences. In addition, the relative stability of the molecular ion complex during the TIMS-MS measurements will provide the best adduct candidate for effective detection. Explosives present different affinities to form a molecular adduct complex. For example, TNT presents very low affinity to form a molecular adduct; however, HMX, RDX and PETN form a variety of complexes (e.g., [M+Cl]−, [M+HCOOH-H]−, [M+CH3COOH-H]− and [M+NO3]−). Inspection of the molecular adduct lifetimes shows that the larger the adduct size the lower the complex stability (see Figure 4 and Table 2). For example, PETN [M]− shows the largest lifetime (400 ms) when compared to the other molecular adducts [M+Cl]− (85 ms), [M+HCOOC-H]− (92 ms), and [M+NO3]− (85 ms). Moreover, the explosive structure influences the probability of forming molecular adducts. For example, HMX presents larger binding energy and longer lifetimes (~3–4×) for the molecular adduct forms when compared with RDX and PETN (see Table 2). Inspection of the HMX complexes optimized geometries shows that the multiple coordination between the HMX molecule and the adduct favors the stability of the complex. That is, if the charge is protected, TIMS-MS experiments shows no ion loss in up to two seconds of trapping (e.g., m/z= 301 C3N3(CF3)3 [M]− from Agilent tuning mix53, Figure 4a). Moreover, if the charge is exposed (e.g., TNT [M−H]−), ions can undergo charge neutralization via charge transfer with the bath gas molecules (e.g., proton transfer). In the case of the molecular adduct, the reactive nature of the adduct ion and the probability to collide with a bath gas molecule increases the chances for decomposition of the molecular adduct complex by transferring the charge carrying adduct to a bath gas molecule (e.g., decomposition by adduct transfer). That is, TIMS-MS experiments suggest that the collision rate and bath gas composition (or impurities) can be a the defining factors for the observation of the molecular adduct complex. Although we cannot establish the mechanism for the molecular adduct complex decomposition, preliminary results suggest that the electrostatic nature of the complex can be lost by the interaction with a third partner (bath gas molecule), a short life complex formations, followed by the detachment of the adduct from the molecular complex.
Figure 4.
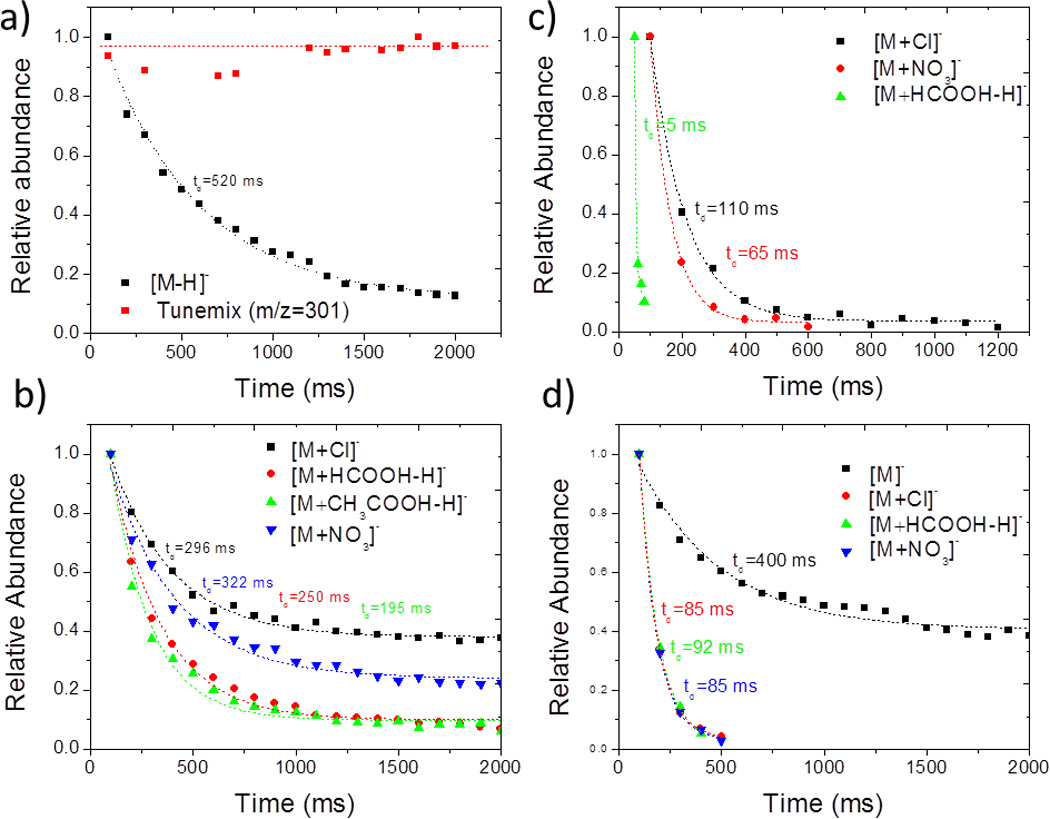
Relative abundance of familiar explosive molecular ions as a function of the trapping time: a) TNT, b) HMX, c) RDX and d) PETN. Notice that for m/z= 301 C3N3(CF3)3 [M]− no ion loss in up to 2 seconds of trapping is observed (Figure 4a).
Table 2.
Lifetime (td), decomposition constant (k), absolute binding energy and molecular-adduct distances of molecular adduct complexes from familiar explosives.
| Compound | Ionic Form | td (ms) |
k (s−1) | Binding Energy (kcal/mol) |
Distance molecule- adduct (Å) |
|---|---|---|---|---|---|
| TNT | [M−H]− | 520 | 1.92 | - | |
| RDX+NH4Cl | [M+Cl]− | 110 | 9.09 | 36.93 | 2.52 |
| RDX+NH4NO3 | [M+NO3]− | 65 | 15.38 | 33.73 | 1.97 |
| HMX+NH4Cl | [M+Cl]− | 296 | 3.38 | 47.47 | 2.52 |
| HMX+HCO2 | [M+HCOOH-H]− | 250 | 4.00 | 50.25 | 2.14 |
| HMX+NH4C2H3O2 | [M+CH3COOH-H]− | 195 | 5.13 | 52.61 | 2.08 |
| HMX+NH4NO3 | [M+NO3]− | 322 | 3.11 | 41.06 | 2.13 |
| PETN | [M]− | 400 | 2.50 | - | |
| PETN+NH4Cl | [M+Cl]− | 85 | 11.76 | 30.95 | 2.39 |
| PETN+HCO2 | [M+HCOOH-H]− | 92 | 10.87 | 33.57 | 2.10 |
| PETN+NH4NO3 | [M+NO3]− | 85 | 11.76 | 28.50 | 2.28 |
During TIMS analysis, short analysis time will increase the probability to observe a molecular adduct complex; however, slower electric field ramp speed will provide higher mobility separations but longer measurement times. That is, high resolution TIMS separation can be limited by the molecular adduct complex lifetime and initial population (or abundance). Moreover, this observation can be extrapolated to the case of traditional drift tube IMS measurements in that long drift times will reduce the probability to observe a molecular complex ion form. In any IMS separation, since the number of collision defines the mobility resolution, the probability to observe a molecular adduct complex at high IMS resolution is limited by their stability and the composition of the bath gas.
CONCLUSIONS
The analytical capabilities of TIMS-MS for the separation and identification of familiar explosives has been demonstrated. In particular, a three to five fold increase in mobility resolution was observed for TIMS analyzer when compared with commercial ETD IMS devices. The use of molecular adducts complexes increases the confidence level and permits the identification of familiar explosives using first principle CCS and m/z measurements. For the first time, lifetimes, relative stability, binding energies and candidate structures are reported for molecular adducts of familiar explosives. Inspection of the molecular adduct interaction with the residual bath gas showed three major trends: i) molecular ion (e.g., [M−H]−) are more stable than their molecular adduct counterparts (e.g., [M+Cl]−, [M+HCOOH-H]−, [M+CH3COOH-H]− and [M+NO3]−), ii) the stability of the chloride and nitrate adducts is higher than the formate and acetate adduct, and iii) HMX forms the most stable molecular adduct complexes when compared with RDX and PETN. We interpret this relative stability as a consequence of the probability of decomposition and of charge exchange with the bath gas of the molecular adduct complexes. That is, the adduct size and reactivity, complex binding energy and the explosive structure define the stability of the molecular adduct complex. TIMS flexibility to modify the mobility separation as a function of the molecular adduct stability (i.e., short or long IMS experiments / low or high IMS resolution) permits targeted measurements of explosives in complex mixtures.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institute of Health (Grant No. R00GM106414). The authors will like to thank Dr. Alexander Mebel and the Instructional & Research Computing Center (Florida International University) for helpful discussions during the theoretical calculations. We will like to acknowledge the technical support provided by the Advance Mass Spectrometry Facility at Florida International University.
Footnotes
ASSOCIATED CONTENT
Supporting Information">Supporting Information
Electronic supplementary information (ESI) available: Detailed information (e.g., geometry files and charges) of the explosives molecular adduct complexes considered here.
The authors declare no competing financial interest.
REFERENCES
- 1.Lucero DP. J Test. Eval. 1985;13:12. [Google Scholar]
- 2.Fetterolf DD, Clark TD. J Forens. Sci. 1993;38:28–39. [Google Scholar]
- 3.Kilpatrick WD. Plasma Chromatography (trademark) and Dynamite Vapor Detection. Defense Technical Information Center; 1971. [Google Scholar]
- 4.Karasek FW. Research/Development. 1974;25:3. [Google Scholar]
- 5.Karasek FW, Denney DW. Journal of Chromatography A. 1974;93:141–147. doi: 10.1016/s0021-9673(00)83025-3. [DOI] [PubMed] [Google Scholar]
- 6.Spangler GE, Lawless PA. Anal. Chem. 1978;50:884–892. [Google Scholar]
- 7.Spangler GE, Carrico JP, Kim SH. presented in part at the Proceedings of the International Symposium on Analysis and Detection of Explosive; Quantico, VA. 1983. [Google Scholar]
- 8.Ewing RG, A AD, Eiceman GA, Ewing GJ. Talanta. 2001;54:515–529. doi: 10.1016/s0039-9140(00)00565-8. [DOI] [PubMed] [Google Scholar]
- 9.Rajapakse MY, Stone JA, Eiceman GA. J Phys. Chem. A. 2014;118:2683–2692. doi: 10.1021/jp412444b. [DOI] [PubMed] [Google Scholar]
- 10.Rajapakse RMMY, Stone JA, Eiceman GA. Int. J. Mass Spectrom. 2014;371:28–35. [Google Scholar]
- 11.Karasek FW, Tatone OS, Kane DM. Anal. Chem. 1973;45:1210–1214. [Google Scholar]
- 12.Lawrence AH, Neudorfl P. Anal. Chem. 1988;60:104–109. [Google Scholar]
- 13.Ewing RG, Eiceman GA, Harden CS, Stone JA. Int. J. Mass Spectrom. 2006;255–256:76–85. [Google Scholar]
- 14.Valadbeigi Y, Farrokhpour H, Tabrizchi M. J Phys Chem A. 2014;118:7663–7671. doi: 10.1021/jp506140m. [DOI] [PubMed] [Google Scholar]
- 15.Gapeev A, Sigman M, Yinon J. Rapid Commun. Mass Spectrom. 2003;17:943–948. doi: 10.1002/rcm.1006. [DOI] [PubMed] [Google Scholar]
- 16.Mathis JA, McCord BR. Rapid Commun. Mass Spectrom. 2005;19:99–104. doi: 10.1002/rcm.1752. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez-Lima FA, Kaplan DA, Park MA. Rev. Sci. Instr. 2011;82:126106. doi: 10.1063/1.3665933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez-Lima F, Kaplan D, Suetering J, Park M. Int. J. Ion Mobility Spectrom. 2011;14:93–98. doi: 10.1007/s12127-011-0067-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez DR, DeBord JD, Ridgeway ME, Kaplan DA, Park MA, Fernandez-Lima F. Analyst. 2014;139:1913–1921. doi: 10.1039/c3an02174b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silveira JA, Ridgeway ME, Park MA. Anal. Chem. 2014;86:5624–5627. doi: 10.1021/ac501261h. [DOI] [PubMed] [Google Scholar]
- 21.Schenk ER, Ridgeway ME, Park MA, Leng F, Fernandez-Lima F. Anal. Chem. 2014;86:1210–1214. doi: 10.1021/ac403386q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molano-Arevalo JC, Hernandez DR, Gonzalez WG, Miksovska J, Ridgeway ME, Park MA, Fernandez-Lima F. Anal Chem. 2014;86:10223–10230. doi: 10.1021/ac5023666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez-Lima FA, Kaplan DA, Suetering J, Park MA. Int. J. Ion Mobility Spectrom. 2011;14:93–98. doi: 10.1007/s12127-011-0067-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schenk ER, Mendez V, Landrum JT, Ridgeway ME, Park MA, Fernandez-Lima F. Anal. Chem. 2014;86:2019–2024. doi: 10.1021/ac403153m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDaniel EW, Mason EA. Mobility and diffusion of ions in gases. New York, New York: John Wiley and Sons, Inc.; 1973. [Google Scholar]
- 26.Tam M, Hill HH. Anal. Chem. 2004;76:2741–2747. doi: 10.1021/ac0354591. [DOI] [PubMed] [Google Scholar]
- 27.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery J, J A, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, revision C.02. Wallingford CT: Gaussian, Inc.; 2004. [Google Scholar]
- 28.Singh UC, Kollman PA. J Comput. Chem. 1984;5:129–145. [Google Scholar]
- 29.Besler BH, Merz KM, Kollman PA. J Comput. Chem. 1990;11:431–439. [Google Scholar]
- 30.Campuzano I, Bush MF, Robinson CV, Beaumont C, Richardson K, Kim H, Kim HI. Anal. Chem. 2011;84:1026–1033. doi: 10.1021/ac202625t. [DOI] [PubMed] [Google Scholar]
- 31.Kim HI, Kim H, Pang ES, Ryu EK, Beegle LW, Loo JA, Goddard WA, Kanik I. Anal. Chem. 2009;81:8289. doi: 10.1021/ac900672a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larriba C, Hogan CJ. J Phys. Chem. A. 2013;117:3887–3901. doi: 10.1021/jp312432z. [DOI] [PubMed] [Google Scholar]
- 33.Kanu AB, Dwivedi P, Tam M, Matz L, Hill HH. J Mass Spectrom. 2008;43:1–22. doi: 10.1002/jms.1383. [DOI] [PubMed] [Google Scholar]
- 34.Dugourd P, Hudgins RR, Clemmer DE, Jarrold MF. Rev. Sci. Instrum. 1997;68:1122–1129. [Google Scholar]
- 35.Merenbloom SI, Glaskin RS, Henson ZB, Clemmer DE. Anal. Chem. 2009;81:1482–1487. doi: 10.1021/ac801880a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kemper PR, Dupuis NF, Bowers MT. Int. J. Mass Spectrom. 2009;287:46–57. [Google Scholar]
- 37.Blase RC, Silveira JA, Gillig KJ, Gamage CM, Russell DH. Int. J. Mass Spectrom. 2011;301:166–173. [Google Scholar]
- 38.Kolakowski BM, Mester Z. Analyst. 2007;132:842–864. doi: 10.1039/b706039d. [DOI] [PubMed] [Google Scholar]
- 39.Eiceman GA, Krylov EV, Krylova NS, Nazarov EG, Miller RA. Anal. Chem. 2004;76:4937–4944. doi: 10.1021/ac035502k. [DOI] [PubMed] [Google Scholar]
- 40.Krylov EV, Coy SL, Vandermey J, Schneider BB, Covey TR, Nazarov EG. Rev Sci Instrum. 2010;81:024101. doi: 10.1063/1.3284507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nazarov EG, Miller RA, Eiceman GA, Stone JA. Anal Chem. 2006;78:4553–4563. doi: 10.1021/ac052213i. [DOI] [PubMed] [Google Scholar]
- 42.Guo Y, Wang J, Javahery G, Thomson BA, Siu KWM. Anal. Chem. 2004;77:266–275. doi: 10.1021/ac048974n. [DOI] [PubMed] [Google Scholar]
- 43.Glaskin RS, Ewing MA, Clemmer DE. Anal. Chem. 2013;85:7003–7008. doi: 10.1021/ac4015066. [DOI] [PubMed] [Google Scholar]
- 44.Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, Thalassinos K, Bateman RH, Bowers MT, Scrivens JH. Int. J. Mass Spectrom. 2007;261:1–12. [Google Scholar]
- 45.Casetta B, Garofolo F. Org. Mass Spectrom. 1994;29:517–525. [Google Scholar]
- 46.Garofolo F, Longo A, Migliozzi V, Tallarico C. Rapid Commun. Mass Spectrom. 1996;10:1273–1277. [Google Scholar]
- 47.Yinon J, McClellan JE, Yost RA. Rapid Commun. Mass Spectrom. 1997;11:1961–1970. [Google Scholar]
- 48.Zhao X, Yinon J. J Chromatogr. A. 2002;977:59–68. doi: 10.1016/s0021-9673(02)01349-3. [DOI] [PubMed] [Google Scholar]
- 49.Wu Z, Hendrickson CL, Rodgers RP, Marshall AG. Anal. Chem. 2002;74:1879–1883. doi: 10.1021/ac011071z. [DOI] [PubMed] [Google Scholar]
- 50.Zhao X, Yinon J. J Chromatogr. A. 2002;946:125–132. doi: 10.1016/s0021-9673(01)01533-3. [DOI] [PubMed] [Google Scholar]
- 51.Sánchez C, Carlsson H, Colmsjö A, Crescenzi C, Batlle R. Anal. Chem. 2003;75:4639–4645. doi: 10.1021/ac034278w. [DOI] [PubMed] [Google Scholar]
- 52.Benigni P, Thompson CJ, Ridgeway ME, Park MA, Fernandez-Lima F. Anal. Chem. 2015;87:4321–4325. doi: 10.1021/ac504866v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flanagan LA. Hewlett-Packard Company>. 5872357 U.S. Patent. 1999
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.