

Abstract
Tryptophan (Trp) is a naturally occurring amino acid, which exhibits fluorescence emission properties that are dependent on the polarity of the local environment around the Trp side chain. However, this sensitivity also complicates interpretation of fluorescence emission data. A non-natural analogue of tryptophan, β-(1-azulenyl)-L-alanine, exhibits fluorescence insensitive to local solvent polarity and does not impact the structure or characteristics of several peptides examined. In this study we investigated the effect of replacing Trp with β-(1-azulenyl)-L-alanine in the well-known bee-venom peptide melittin. This peptide provides a model framework for investigating the impact of replacing Trp with β-(1-azulenyl)-L- alanine in a functional peptide system that undergoes significant shifts in Trp fluorescence emission upon binding to lipid bilayers. Microbiological methods including assessment of the antimicrobial activity by minimal inhibitory concentration (MIC) assays and bacterial membrane permeability assays indicated little difference between the Trp and the β-(1-azulenyl)-L-alanine-substituted versions of melittin. Circular dichroism spectroscopy showed both that peptides adopted the expected α-helical structures when bound to phospholipid bilayers and electrophysiological analysis indicated that both created membrane disruptions leading to significant conductance increases across model membranes. Both peptides exhibited a marked protection of the respective fluorophores when bound to bilayers indicating a similar membrane-bound topology. As expected, while fluorescence quenching and CD indicate the peptides are stably bound to lipid vesicles, the peptide containing β-(1-azulenyl)-L-alanine exhibited no fluorescence emission shift upon binding while the natural Trp exhibited >10 nm shift in emission spectrum barycenter. Taken together, the β-(1-azulenyl)-L-alanine can serve as a solvent insensitive alternative to Trp that does not have significant impacts on structure or function of membrane interacting peptides.
Keywords: fluorescence, tryptophan analogs, antimicrobial peptides, azulene
Introduction
Despite their seeming simplicity, peptides are involved in all processes in living organisms. Incredible diversity of structure and function and the relative ease of preparation of peptides make them an attractive therapeutic target(1-4). Moreover, peptides are often used as models to answer fundamental questions in an attempt to gain better understanding of more complex biomolecular systems(5-11). One area to which peptides have contributed significantly is the field of membrane protein biophysics, where they have been used as model systems to elucidate fundamental principles underlying protein-lipid interactions, membrane binding, stability of transmembrane structures, and protein-protein interactions in the bilayer5-11). These fundamental studies are often complementary to biophysical investigations of naturally occurring membrane interacting peptides such as antimicrobial peptides (AMPs), host defense peptides (HDPs), and peptide toxins.
The interactions of peptides or proteins with lipid bilayers can be investigated through a number of physical and spectroscopic methods. Surface plasmon resonance (SPR)(12-14) and isothermal titration calorimetry (ITC)(15-17) have been successfully applied to determine kinetic and thermodynamic components of the peptide-lipid interaction, however these methods are costly and not always amenable to interactions between proteins within the bilayer or for structural characterizations. Attenuated total reflectance infrared (ATR-IR) and solid-state nuclear magnetic resonance (SSNMR) spectroscopies offer valuable structural and topographic insights into membrane interacting peptides and proteins,(18-20) but they do require large amounts of material and the data analysis is often complicated due to dynamic rearrangements. Fluorescence spectroscopy has evolved as a premier tool in the study of protein- membrane interactions. Intrinsic fluorophores, such as tryptophan (Trp) and tyrosine (Tyr) allow for studies of the intricate details of protein/peptide-lipid interactions at a minimal cost and without disturbing the native interactions. The inherent environmental sensitivity of Trp fluorescence has been applied to binding and topography studies of peptides and proteins alike.(5, 21-23) While Trp’s intrinsic fluorescence is an invaluable tool for elucidating topographic changes and binding state of model peptides, the associated changes in its spectral properties (fluorescence lifetime, emission maximum, quantum yield) can often complicate the analysis of quenching, FRET and other assays dependent on spectral properties. Moreover, contributions of multiple tryptophan residues are difficult to deconvolute in complex membrane systems, where multiple protein species are interacting in the bilayer or at the bilayer surface. To address some of these issues, a wide variety of exogenous fluorophores have been utilized in the studies of membrane interactions as well as in the studies of protein-protein interactions in lipid bilayers.(24-26) These fluorophores often have absorption and emission spectral characteristics that differ significantly from those of the intrinsic fluorophores simplifying fluorescence data analysis. Unfortunately, these exogenous fluorophores are often very bulky and thereby may exert significant influence on short model peptides.
Recently, we reported that β-(1-azulenyl)-L-alanine (AzAla), a minimally disruptive mimic of tryptophan (Figure 1), can be successfully incorporated into calmodulin-binding peptides without loss of function.(7) While being a structurally conservative replacement, AzAla has a number of unique spectral and photophysical characteristics compared to Trp. AzAla’s additional absorbance peak centered at 342 nm is significantly shifted from the 280 nm absorbance maximum of Trp and it can be selectively excited in the presence of multiple Trp residues to produce an unambiguous fluorescent readout. Moreover, the fluorescence of AzAla is insensitive to the polarity of the local environment, unlike that of Trp. This allows for more straightforward analysis of quenching and FRET data regardless of the peptide binding state, orientation, or dynamic rearrangements when in the membrane. Despite obvious structural similarities of AzAla to Trp, the two amino acids have very different hydrophobicities,(7) thus it is important to establish whether Trp to AzAla substitution is conservative enough not to perturb therapeutically relevant interactions in vivo. In this paper we explore the effects of AzAla introduction on anti-microbial properties of melittin
Figure 1.
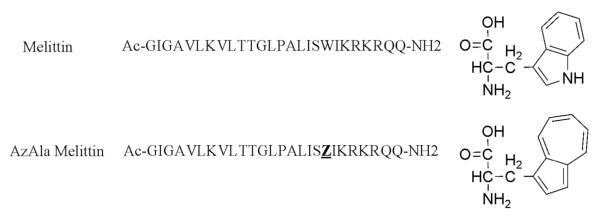
the sequences used in this study and the structures of Tryptophan (top) and β-(1-azulenyl)-L- alanine (AzAla). The single letter abbreviation Z represents the AzAla moiety in the lower sequence.
Melittin, derived from the toxin of the honeybee Apis mellifera, is one of the most well characterized membrane interacting peptides.(27-32) This 26 amino acid residue peptide binds to lipid bilayers resulting in membrane permeabilization through the formation of transmembrane pores or lesions. Melittin’s amphiphilic sequence has five cationic and a number of hydrophobic residues, including a single Trp at position 19. Melittin’s interaction with lipid bilayers is relatively non-specific, so changes in headgroup composition and bilayer thickness do not prevent pore formation, but rather modulate the degree of permeabilization.(33, 34) The permeabilization is a concentration dependent phenomenon that requires four to six melittin peptides to homooligomerize into a lesion competent structure, most likely a stable, “barrel-stave” model pore.(35) However, more recent data have indicated that the melittin pore may be more transient.(36) The high levels of activity against a broad spectrum of target membranes have made melittin a commonly used control molecule for membrane permeabilization of bacteria, fungi, and mammalian cells. The membrane permeabilizing activity of melittin has also made it an attractive system for use in pharmaceutical and biotechnology applications.(30, 32, 37, 38) In addition to the membrane lytic activity, melittin exhibits a number of other well-characterized interactions such as inhibition of various ion channels via calmodulin-mediated pathways.(39)
This study presents the characterization of a melittin variant in which the Trp19 is replaced with AzAla. Considering the well characterized behavior of melittin, it is an ideal model system to begin investigating the impacts of AzAla on peptide and protein function. The native Trp in melittin undergoes significant shifts in the fluorescence emission spectrum when binding to lipid membranes, providing an ideal test case for the impact of AzAla on a system which exhibits Trp solvent sensitivity. While on a structural level this is a conservative replacement, the impacts on protein-lipid interactions, protein- protein interactions, and functional activity have not been explored in great depth. This study presents the a thorough functional comparison of peptides containing Trp vs. AzAla as a demonstration that the AzAla moiety does not cause significant structural or functional disruptions, even in a relatively small 26-aa peptide. Herein, we focus on the structural and functional characterization of the AzAla-substituted melittin using a combination of spectroscopic and microbiological approaches. In the absence and presence of model membranes, circular dichroism (CD) spectroscopy indicates very similar secondary structure in the natural peptide sequence and the AzAla-substituted version while fluorescence quenching assays showed an identical trend of accessibility to aqueous and membrane bound quenchers. As expected, spectroscopic assays confirmed that the AzAla-substituted melittin did not undergo any significant spectral shifts when interacting with lipid bilayers. Microbiological assays of bacterial membrane permeabilization as well as electrophysiological measurements of current transmission across the bilayer showed similar or identical patterns of permeabilization induced by the native and AzAla- substituted melittin. These results confirm the utility of the AzAla amino acid as a chemical probe for membrane interacting peptides with seemingly little effect on the functionality of the host peptide.
Materials and Methods
Peptide Synthesis and Purification
The peptide sequences and structures of Trp and AzAla are shown in Figure 1
AzAla Melittin was synthesized by manual fluorenylmethylcarbonyl (Fmoc) solid phase peptide synthesis at elevated temperatures using our previously optimized protocols for hydrophobic peptides.(40-42) H-Rink Amide ChemMatrix resin was used as a solid support. Deprotection of resin and Fmoc group was carried out in 5% piperazine for 10 min. 2-(6-Chloro-1H-benzotriazole-1-yl)- 1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) /N,N-diisopropylethylamine (DIEA) was used for activation of amino acids. The coupling reaction was carried out for 10 min for all amino acids except for Arginine (20 min) and Azulenylalanine (15 min). The N-terminal of the peptide was acylated using a mixture of acetic anhydride and DIEA. A similar approach was used for synthesis of melittin with N,N,N’,N’-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) replacing HCTU. Deprotection of the side chains and cleavage of the peptide from the resin was achieved by subjecting it to a mixture of trifluoroacetic acid (TFA)/H2O/triisopropyl silane (TIS) (95:2.5:2.5, v/v) for 2 hours at room temperature. The crude peptide was then precipitated and washed with cold t-butyl methyl ether (MTBE). The peptide was then purified on a Varian ProStar 210 preparative reverse phase High Performance Liquid Chromatography system with a C4 column using a linear gradient of solvent A (0.1% TFA in Millipore H2O) and solvent B (90% CH3CN, 10% H2O, 0.1% TFA). The identity of the peptide was confirmed by Bruker Autoflex III Smartbeam MALDI-TOF mass spectrometer. The purity of the peptide was determined on a Shimadzu Prominence UFLC instrument with an analytical Zorbax Eclipse XDB-C18 column (4.6 mm x 150 mm) using a linear gradient of solvent A and solvent B. Peptide concentrations were determined using an Agilent 8453 UV-Vis spectrometer by measuring the absorbance at 280 nm for melittin and 342 nm for AzAla melittin. Peptides’s extinction coefficients at these wavelengths were 5500 M-1 cm-1 and 4212 M-1 cm-1, respectively.
Lipid Vesicle Preparation
Lipids were purchased from Avanti Polar Lipids and used without further purification. The lipids used for vesicle studies were 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) while 1,2-diphytanoyl- sn-glycero-3-phosphocholine (DPhyPC) was used for electrophysiological measurements. Vesicle compositions were either 100% POPC (PC), 100% DPhyPC, or 75:25 POPC:POPG (PC:PG). Appropriate volumes of lipids dissolved in chloroform were dried under a gentle stream of N2 and then subjected to high vacuum for 1 hour to complete drying. For most experiments, small unilamellar vesicles (SUVs) were formed by resuspending the lipid film in PBS buffer (50 mM sodium phosphate, 100 mM NaCl pH 7.1) before sonicating in a high power bath sonicator for 20 minutes. For CD and dual quencher analysis (DQA) experiments, individual samples were created by the ethanol dilution method. Briefly, the lipids or lipids with 10-doxyl nonadecane (10-DN) quencher were mixed and co-dried under a stream of N2 followed by complete solvent removal in vacuo, then the film was dissolved in 10 RL ethanol to which an appropriate volume of buffer was added while vortexing. Vesicles used for CD spectroscopy were created using HBS (4 mM HEPES, 30 mM NaCl, pH 7.2) and further sonicated before addition of peptide.
Fluorescence Spectroscopy
Measurements were performed on a JY-Horiba fluoromax2 instrument with emission and excitation slits set to 2.5 nm. Samples of wild type melittin were excited at 280 nm for binding and iodide quenching experiments, and 295 nm for acrylamide quenching experiments with emission measured over the range of 300-400 nm. Samples of AzAla-containing melittin were excited at 342 nm and emission was monitored over the range 350-450 nm. Spectral barycenter was calculated as an intensity-weighted average over the range wavelengths collected.(43)
Stern-Volmer quenching assays were performed by titrating a sample of peptide and buffer or lipid vesicles with either potassium iodide (KI) or acrylamide. Samples for quenching analysis were created by mixing peptide and lipid vesicles to final concentrations of 250 mM total lipid and 2 mM peptide and were allowed to incubate for 30 minutes before analysis. Fluorescence measurements were taken prior to addition of quencher as the F0. Aliquots of 4 M acrylamide or 3 M KI supplemented with 100 mM sodium thiosulfate (to prevent I3− formation) were added to the samples, allowed to incubate for 5 minutes, and then remeasured. Control samples for iodide quenching using KCl instead of KI were monitored to correct for potential effects from ionic strength changes, however these effects were negligible. Acrylamide samples were corrected for inner filter effects in the excitation path for the WT Trp-containing melittin samples. Fluorescence from background samples containing only lipid vesicles, buffer and quenchers were subtracted.
Samples for DQA were prepared by incubating the peptides with lipid vesicles lacking or containing the non-polar quencher 10-DN as previously described(44). Wavelength regimes for Trp- containing peptides were λex = 280 nm, λem = 340 nm (10-DN samples) or λex = 295 nm, λem = 340 nm for acrylamide; λex = 342 nm, λem = 382 nm (both quencher samples). Quenching ratio was calculated as previously reported(44).
Circular Dichroism Spectroscopy
Jasco J-715 CD spectrometer was used to collect CD spectra using a quartz cuvette with a 1 cm path length. The CD spectra were collected using a step scan mode averaging over three runs with an averaging time of 4 s. Samples were prepared in a buffer containing 4 mM HEPES, 30 mM NaCl, pH 7.2. The peptide concentrations were maintained at 35-40 μM unless specified otherwise. In order to obtain reliable mean residue ellipticity (MRE) values, care was taken that the absorbance of the sample never exceeded 1.5.
Minimal Inhibitory Concentration assay
Antibacterial activity of the melittin peptides was determined in a standard broth dilution assay according to the Clinical and Laboratory Standards Institute guidelines.(45, 46) Peptides were dissolved in a mixture of ethanol and PBS buffer, and stock solutions were prepared by 2-fold serial dilutions with 0.01% acetic acid in PBS to yield 7 different concentrations while 0.01% in PBS was used as a negative control. The bacterial strains Escherichia coli D31(47) and Staphylococcus aureus ATCC® 25923™, were aerobically cultured in Mueller Hinton broth (MHB). An overnight culture of the bacterial strains was diluted 1:250 in fresh MHB and grown at 37oC with shaking until the culture reached log phase (OD600 of 0.5-0.6) and diluted in fresh MHB to a final concentration of ~5 × 105 CFU/mL. The final plate was made by adding 10RL of the peptide stock solutions to the wells of a sterile 96-well plate to which 90RL of diluted bacterial culture was added. The assay plate was incubated at 37 °C for 18 h. Bacterial growth was assayed by absorbance at OD600 using Multiskan microplate reader (Thermo Fisher). The minimum inhibitory concentration (MIC) was defined as the lowest peptide concentration to completely inhibit growth as judged by OD600. All MIC values are averages of at least triplicate samples.
Membrane permeabilization of E. coli
Membrane permeabilization assays were performed as previously reported.(48, 49) Briefly, for inner membrane (IM) permeability, a colony of Escherichia coli D31 was inoculated in a solution of LB Broth (Difco). The culture was incubated at 37° C with shaking overnight. The culture was then diluted 1:100 in LB supplemented with 2 mM isopropyl β-D-1- thiogalactopyranoside (IPTG) and incubated at 37° C with shaking until the OD600 reached 0.2-0.5. Using a 96-well polystyrene plate, peptides and the control (cetyl trimethyl ammonium bromide, CTAB) were serially diluted from stocks. Next, Z-buffer (100mM Na2HPO4, 10mM KCl, 1mM MgSO4, 40mM β–mercaptoethanol, pH 7.1) and an aliquot of the bacterial culture were added to each well. Just prior to reading, ortho-Nitrophenyl-β-galactoside (ONPG) dissolved in Z-buffer (15 μL, 4mg /ml) was added to the wells. The absorbance at 420 nm was then monitored using a ThermoSkan plate reader for 90 minutes with shaking between individual readings to prevent cell settling. All assays were performed at least in triplicate.
For outer membrane (OM) permeability, a colony of Escherichia coli D31 was inoculated in a solution of LB Broth (Difco) supplemented with ampicillin (100 Rg/mL). The culture was incubated at 37° C with shaking overnight. The culture was then diluted 1:100 in LB and incubated at 37° C with shaking until the OD600 was approximately 0.2. The culture was then centrifuged at 2500 rpm for 15 min, and the pellet was resuspended in an equal volume of PBS buffer (10 mM phosphate, 200 mM NaCl, pH 7.0). The resuspended bacterial cells were added to the wells of a 96-well plate containing serially diluted peptides or polymyxin B as a control. Nitrocefin was added to the solution to a concentration of 50 Rg/mL. The plate was then in the same manner as the IM permeability procedure, except the sample absorbance was measured at 486 nm. All assays were performed at least in triplicate.
Membrane permeabilization of S. aureus
Permeabilization of S. aureus membranes was performed as previously described. Briefly, DAPI (from a 600 nM stock) was added to 100 RL of bacterial suspension in each well for a final concentration of 1×108 cfu/ml and 85.7 nM DAPI. Stock solutions of peptides were prepared and subsequently serially diluted in a reservoir and from which 20RL of each of the peptide dilutions were added to appropriate wells in the assay plate. As a control, only buffer with no peptide was added to control wells. The fluorescence was recorded immediately before (time 0) and after addition (time 1), 10, 30, and 60 minutes using a SpectraMax M5 microplate reader using λex = 358 and λem = 461.
Electrophysiology
A 1 cm2 PTFE (Teflon) sheet with a 100 μm hole in the center was attached with PDMS (Kwik-Cast, WPI) to a previously fabricated large holder. The Teflon sheet was positioned ca. 500 micrometer above a microscope cover slip mounted onto a homemade holder that sits on an inverted microscope (Axio Observer D, Zeiss, Germany). Two μL of pre-paint mixture (0.5 mg/ml DPhyPC in pentane) was ejected around the hole and allowed to evaporate over a period of several minutes. Unsupported horizontal lipid bilayer membranes are formed using a painting technique similar to the one developed by Mueller et al.(50). Briefly, electrolyte solution (1 mol/L pH 7.2 buffered with TRIS and pH adjusted with citric acid) wets both sides of the teflon sheet. Matching Ag/AgCl electrodes are inserted into patch pipettes formed from a laser-based pipette puller (P-2000 Sutter Instruments). The pipettes are preloaded with the electrolyte solution. The electrodes are then inserted into the electrolyte. Using video microscopy, a glass microcapillary (femtotip II, Eppendorf, NY) is positioned with a motorized manipulator (MPC-275, Sutter Instruments) ca. 100 micron above the hole in the teflon partition. Several picoliters of 10 mg/ml DPhyPC:hexadecane solution is ejected from the tip and adhered to the teflon surface. A glass rod with a glass ball formed at the end is placed in a mechanical manipulation stage and paints lipid across the hole. The lipid/solvent mixture thins over a period of several seconds to form a lipid bilayer membrane.
After the formation of a lipid bilayer membrane, a new tip containing 10 μmol/L of either wild type or azulene modified peptide is positioned ca. 50 μm directly above the membrane. A transmembrane voltage is applied with ground referenced to the TRANS side of the membrane. Backing pressure is then applied (Femtojet, Eppendorff) to the peptide tip and the resulting transmembrane current is recorded. The current is measured with an amplifier headstage (Axopatch 200B, Molecular Devices, CA), digitized (Digidata 1440A, Molecular Devices) and filtered with a 3-pole low-pass Bessel filter. After some time the current begins to increase as peptides partition across the membrane and the backing pressure is then reduced to zero.
When a significant number of peptides are present in the membrane as measured by the transmembrane current, the Ag/AgCl electrode tip on the CIS side of the membrane is patched down onto the membrane. If the current is zero or extremely large, the tip is removed from the surface and a backing pressure is applied to remove the patch membrane from the end of the electrode. The process is repeated until a single or a few peptides are localized within the patch area and the patch electrode remains in contact with the membrane for the duration of the measurement.
Results and Discussion
Effects of AzAla Introduction on Peptide Function – Antibacterial Activity
Melittin is widely known to be a membrane permeabilizing agent(28, 36, 37, 51) and as such has been used extensively as a positive control in antimicrobial assays investigating the activity of antimicrobial peptides, polymers and peptidomimetics. The antimicrobial activity of the AzAla substituted melittin was compared side by side with native melittin against E. coli and S. aureus. Minimal inhibitory concentration (MIC) assays were performed using the standard broth dilution method where a fixed concentration of bacteria is exposed to serial dilutions of peptide and allowed to incubate overnight. The MIC is defined as the lowest concentration of peptide that prevents growth of the bacterial inoculum as measured by optical turbidity of the sample. The native melittin and the AzAla substituted melittin exhibited identical MIC values against E. coli or S. aureus over the range of concentrations tested, indicating that the abililty to disrupt the bacterial membrane resulting in cell death or cellular stasis was not affected by the AzAla moiety (Table 1).
Table 1.
Comparison of Peptide Characteristics
| Melittin | AzAla Melittin | |
|---|---|---|
| MIC (μM) | ||
| E.coli | 7.5 | 7.5 |
| S.aureus | 7.5 | 7.5 |
| λmax (nm) | ||
| Buffer | 355 ± 1 | 380 ± 1 |
| PC Vesicles | 346 | 380 ± 1 |
| PCPG Vesicles | 335 ± 1 | 380 ± 1 |
| Acrylamide Ksv (M−1) | ||
| Buffer | 20.2 | 6.57 |
| PC Vesicles | 5.35 | 1.34 |
| PCPG Vesicles | 1.59 | 0.43 |
| Iodide Ksv (M−1) | ||
| Buffer | 13.07 | 20.06 |
| PC Vesicles | 2.16 | 2.62 |
| PCPG Vesicles | 1.89 | 2.76 |
The membrane permeabilizing activity of melittin and the AzAla substituted melittin was further investigated against intact E. coli. The assays involve the use of chromogenic enzyme substrates, which are minimally permeable across the E. coli outer or inner membrane under normal conditions. If the membrane integrity is compromised, in this case by exposure to melittin, the rate of transit across the membrane increases allowing for substrate breakdown to increase as measured by an increase in the absorbance of the product. The effects on outer membrane permeability are assayed using the β-lactamase and nitrocefin enzyme-substrate pair while inner membrane permeability is assayed using the β- galactosidase and ONPG pair. Figure 2A shows the outer membrane permeabilization (nitrocefin) while Figure 2B shows the inner membrane permeabilization (ONPG). In both cases, melittin and AzAla substituted melittin exhibited nearly identical dose response curves in which low concentrations of peptide did not induce leakage while the highest concentrations tested resulted in similar maximal levels of permeabilization (Figure 2B and S4). Interestingly, neither melittin nor AzAla substituted melittin completely permeabilized the inner membrane of E. coli compared to detergent control. This is not surprising considering the peptide is required to first bind and traverse the outer membrane before gaining access to the inner membrane. This is in contrast to melittin induced outer membrane permeabilization, which was similar to the known membrane disrupting peptide polymyxin.
Figure 2.
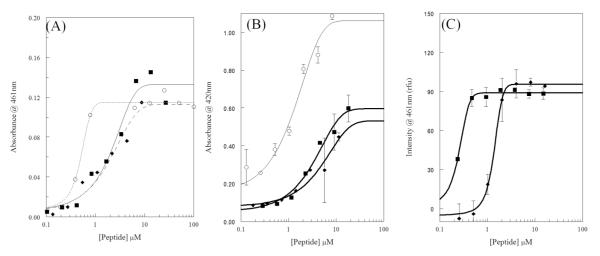
Bacterial membrane permeabilization. For all graphs melittin is shown as squares (■), AzAla-Melittin as diamonds (◆), and control compounds as open circles (○). (A) Permeabilization of E.coli outer membrane (OM) assayed by enzymatic breakdown of nitrocefin. Control is polymyxinB. Curve scheme is: solid – melittin, dashes – AzAla Melittin, dots – polymyxinB. (B) Permeabilization of E.coli inner membrane (OM) assayed by enzymatic breakdown of ONPG. Control is CTAB. (C) Permeabilization of S.aureus membrane assayed by binding of DAPI to bacterial DNA. All data were fit to a dose-response functions and are averages of at least 3 replicates. Error bars are omitted for graphical clarity in (A).
A similar approach was used to assay membrane permeabilization in S. aureus. In this case, the DNA binding fluorophore DAPI was used as the reporter. DAPI has a low intrinsic fluorescence and is relatively impermeant across the gram positive bacterial membrane. Once membrane integrity is compromised, DAPI can more easily cross the membrane and bind to bacterial DNA resulting in an increase in fluorescence intensity. The DAPI leakage results are shown in Figure 2C which again display a similar dose-response pattern for both melittin and AzAla substituted melittin. While the overall profile is similar, the EC50 value for AzAla substituted melittin is ~5 fold larger than that of the natural melittin sequence (0.25 mM vs. 1.38 mM) although both sequences result in the same level of maximal permeabilization. This could be indicative of slightly altered pore size, pore properties, or pore forming kinetics between the two peptides (see below).
Effects of AzAla Introduction on Peptide Function – Membrane interactions & structure
The MIC and bacterial membrane permeabilization assays indicate that the AzAla substitution did not significantly impact the function of the peptide on a macroscopic level. However, as the AzAla moiety was developed to be a probe of biophysical interactions, the effects on membrane interactions and peptide structure were investigated using spectroscopy and electrophysiology. Circular dichroism (CD) spectroscopy was used to characterize the secondary structure of the peptides in the presence and absence of model lipid bilayers. Due to the short length of melittin, no higher order structures can form, thus CD spectroscopy can yield an almost complete understanding of structural conformations of the peptide. The CD spectra of native and AzAla modified melittin are identical in the absence (Figure 3A) and presence (Figure 3B) of lipid vesicles. This indicates that the presence of the non-natural amino acid does not disrupt the membrane bound, active conformation of the peptide nor does it impact the structural rearrangement from random coil in solution to α-helix when bound to the bilayer on the timescales tested.
Figure 3.
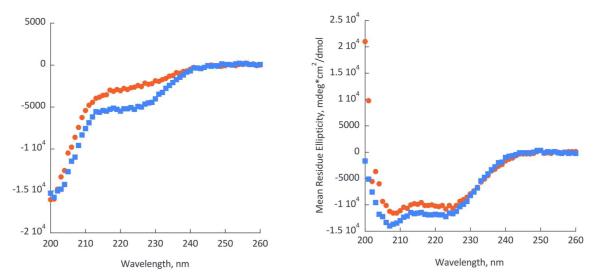
Circular Dichroism spectra of mellitin (red circles) and AzAla-labeled mellitin (blue squares) in the absence (left) and in the presence of small unilamellar vesicles (POPC:POPG = 75:25). Buffer conditions: 4 mM HEPES, pH 7.2, 30 mM NaCl.
The ability to bind and interact with lipid bilayers is a key component of the membrane permeabilizing activity of melittin. The naturally occurring Trp in the melittin sequence serves as a good probe for membrane binding as it exhibits a blue shift in the fluorescence emission spectrum upon partitioning into the lipid bilayer. While useful for studying the native peptide sequence, due to the environmental insensitivity of the AzAla moiety the AzAla melittin did not exhibit a similar shift when binding to lipid vesicles, as expected (Table 1, Figures 4, S1, and S2). The Trp from the native sequence exhibited a more than 12 nm shift in emission spectrum barycenter when bound to lipid vesicles containing a mixture of PC and PG headgroups, and a more than 7 nm shift when bound to vesicles composed of 100% PC headgroups (Figure 4). In comparison, the AzAla peptide exhibited at most 3 nm shifting when bound to PC:PG vesicles and no shift when bound to PC vesicles. Monitoring the maximum emission wavelength (λmax) showed shifts in the range of 10-20 nm for the native Trp and no λmax shift was detected for the AzAla peptide (Figures S1 and S2). Taken together, the spectral data indicate that the environment around the Trp is different in the PC vs PC:PG vesicle bound states (due to different spectral barycenters and λmax in the bound state) and, more importantly, the AzAla moiety is exhibiting the expected stability in fluorescence emission spectra regardless of local polarity..,
Figure 4.

Binding of Melittin and AzAla Melittin to lipid vesicles by change in spectral barycenter as a function of lipid concentration. Melittin was titrated with PC vesicles is shown as diamonds(◆), melittin titrated with PC:PG vesicles shown as squares(■), AzAla-melittin titrated with PC vesicles is shown as triangles(▲), and AzAla-melittin titrated with PC:PG vesicles is shown as circles (•). Data are averages of at least 3 replicate samples.
While the melittin binding curves indicate that the peptide was fully bound to the PCPG bilayers at 50 RM lipid, the binding of the melittin and AzAla substituted melittin was confirmed using fluorescence quenching. Peptide fluorescence was quenched by titrating acrylamide (Figures 5A and 5B) or iodide (Figures 5C and 5D) into solutions of peptide with and without lipid vesicles of varied composition. Both membrane permeant acrylamide and membrane impermeant iodide quenching data clearly show that both the native peptide and the AzAla mutant yield linear Stern-Volmer quenching profiles. As previously reported, the magnitudes of the Stern-Volmer quenching constants vary from Trp to AzAla, but the trends of Ksv for the peptides in the absence of lipid or in the presence of 100% PC, or 75:25 PC:PG vesicles are consistent (Table 1)(7). In both cases, the samples lacking lipid vesicles exhibited the greatest degree of quenching and resultant highest Ksv values, indicative of significant exposure to the quencher. The samples that had vesicles composed of 100% PC lipids resulted in an intermediate Ksv value, indicating that the peptide-lipid interactions shielded the fluorophores to some degree. Finally, the samples with 75:25 PC:PG vesicles exhibited the lowest Ksv values indicating the highest degree of fluorophore shielding, presumably through the interaction with the bilayer. The significant decrease in Ksv from aqueous to membrane bound in both peptides indicates that both peptides undergo topographical orientations in the membrane to prevent quencher access to the fluorophore. This was further confirmed using 10-doxyl nonadecane (10-DN), a membrane-imbedded quencher, which exhibited strong quenching of both peptides (Figure S3). The hydrophobic 10-DN molecule is restricted to the nonpolar core of the lipid bilayer such that quenching by 10-DN requires the fluorophore to be in close proximity to the acyl chain region of the lipid bilayer. The strong degree of quenching exhibited by 10-DN indicates that both peptides are interacting with the core of the lipid bilayer when bound to PC:PG vesicles and to a lesser degree in PC bilayers. This is consistent with the pattern of acrylamide quenching in Figure 5C and D which shows enhanced protection from acrylamide in PC:PG vesicles, presumably due to a conformation that allows the fluorophore to become more deeply imbedded in the nonpolar core of the bilayer. Importantly, this trend is consistent in both the native and AzAla substituted peptides. It should also be noted that the interpretation of DQA quenching results are relative as the exact fluorescence lifetime values for AzAla at various positions in the lipid bilayer have not yet been determined. Additionally, slight changes in peptide orientation when in a pore-forming conformation may alter accessibility to one or both quenchers, influencing the final Q-ratio.
Figure 5.

Fluorescence quenching of Melittin (A&C) or AzAla Melittin (B&D) in the presence or absence of lipid vesicles. Fluorescence quenchers used are KI (A&B) or acrylamide (C&D). Quenching of 2RM peptide in pH 7 phosphate buffer is shown by diamonds (◆), in the presence of DOPC vesicles (250RM total lipid concentration) is shown by squares (■), or in the presence of DOPC:DOPG (3:1) vesicles (250RM total lipid concentration) is shown by triangles (▲). Data are averages of at least 3 replicate samples.
The final step in characterization is examining the pore forming activity of the peptides. Melittin and AzAla substituted melittin were delivered to an unsupported bilayer membrane composed of DPhyPC and voltage induced currents were recorded. Peptides were delivered from an aqueous stock directly above the membrane surface with a micropipette tip (Femtotip, Eppendorf) and currents were recorded for approximately 10 minutes. First, we demonstrated the peptides partition across the membrane and enable transmembrane current by positioning an electrode pipette tip above the membrane (Figs. 6A and 6B). It can be seen that for both the melittin and AzAla substituted melittin, current flows across the membrane. We observe significant differences between both growth and decay rates between the two peptides. The difference in the signal growth rates most likely results from differences in local peptide concentration at the membrane surface. This is expected given the uncertainties associated with the tip positions with respect to the membrane and the uncertainties in the applied pressure at the low pressures used (~ 10hPa). A greater difference is seen in the decay rate where the current decayed approximately ten times faster for the azulene modified peptide. To investigate any spurious causes for this difference such as differences in the interaction between the peptides and the lipid/solvent annulus we patched the electrode pipette tip down onto the membrane surface to study current fluctuations at or near the single peptide limit. It can be seen from Figs. 6C and 6D that at the single peptide level, the magnitude and duration of the current fluctuations are slightly less for the azulene modified peptides, but do not differ by an order of magnitude. This suggests differences in the measurement protocol (whole membrane vs. patch) are mostly responsible for the differences seen in Figs. 6A and 6B while any small remaining differences (also seen in our membrane permeabilization assays in S.aureus), will require further investigation of azulene modified pore forming kinetics. Nevertheless, for the purposes outlined herein, the overall similarity of the pore kinetics between the two peptides supports the assumption that the azulene modified pore could serve as a viable functional alternative to the wild-type melittin.
Figure 6.

Electrophysiological characterization of pore formation – Pore formation kinetics and properties for azulene modified and wild type peptides in the bulk under a 60 mV applied transmembrane potential. (A) Bulk insertion of azulene modified peptide. At t = 5 s a 10 hPa backing pressure was applied for a period of ca. 30 seconds. In response to this pulse the transmembrane current increases exponentially with a time constant of (7.25 ± 0.01) s. At approximately t = 50 s, the current decreases with an exponential decay time of (2.38 ± 0.01) s. (B) Bulk insertion of wild type peptide. At t = 5 s a 50 hPa pressure pulse was applied for a period of 10 s. In response to this pulse the current increases with an exponential time constant of (0.90 ± 0.01) s and decreases with an exponential time constant of (27.8 ± 0.1) s. Both traces were digitally filtered with a low-pass 10 Hz Hanning filter for presentation purposes. (C,D) Typical current traces for individual azulene modified (C) and wild type (D) pores show discrete current steps that are similar in both cases. Both current traces were measured under a 60 mV applied transmembrane potential. Both traces were digitally filtered with a low-pass 10 Hz Hanning filter for presentation purposes.
Conclusion
We have recently reported that the AzAla moiety is a versatile probe for protein-protein interactions in aqueous or membrane environments and could be extended to analyze protein-lipid interactions(7). The results presented here extend the original characterization of the AzAla moiety to the well-characterized model system of melittin to support the non-disruptive substitution of AzAla for Trp in protein sequences. Although the Trp residue in the native melittin sequence does not impede many modes of characterization, the data shows that the replacement by AzAla provides alternative methods of characterization for this and other sequences if the Trp fluorescence is convoluted without introducing major structural or functional impacts. While some subtle differences were noted in specific assays, the overall trends of structure and function were retained when the native Trp was substituted with the AzAla group. Of specific interest is that the AzAla substituted melittin showed nearly identical structural conformations by CD indicating the non-proteinogenic amino acid is not impacting the structural propensities of the molecule. One of the major strengths of the AzAla moiety is the insensitivity to environmental polarity compared to the naturally occurring Trp. Assays of the topographical/exposure patterns by fluorescence quenching show that the two variants of the peptide behave similarly, but the AzAla substituted melittin did not undergo any significant shifting of λmax or barycenter from aqueous to membrane bound conformations. This confirms the utility of this non-proteinogenic amino acid as a useful addition to the molecular toolkit available for studying detailed structure-function relationships in proteins or peptides that interact in a lipid bilayer environment. With the advent of an efficient way for translational incorporation of AzAla into proteins(52) we expect is to find a wide variety of chemical biology applications.
Supplementary Material
Acknowledgements
G.A.C. and I.V.K would like to thank Bill DeGrado for his mentorship, guidance, support and inspiration over the years. We would also like to acknowledge Bill for his early work on melittin and making this fascinating model system accessible to us. This work was funded by in part by Virginia Commonwealth University (start-up funds to J.E.R.), NSF-EFRI (grant 1332349 to I.V.K), ORAU Ralph E. Powe Junior Faculty Enhancement award and a Humboldt Research Fellowship to I.V.K., and the NIH (grant 1R15GM094330 to G.A.C.).
References
- 1.Caputo GA, Litvinov RI, Li W, Bennett JS, Degrado WF, Yin H. Computationally designed peptide inhibitors of protein-protein interactions in membranes. Biochemistry. 2008;47:8600–8606. doi: 10.1021/bi800687h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, DeGrado WF. Computational design of peptides that target transmembrane helices. Science. 2007;315(5820):1817–1822. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 3.Hancock RE, Chapple DS. Peptide antibiotics. Antimicrobial agents and chemotherapy. 1999;43(6):1317–1323. doi: 10.1128/aac.43.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hancock RE. Cationic peptides: effectors in innate immunity and novel antimicrobials. The Lancet infectious diseases. 2001;1(3):156–164. doi: 10.1016/S1473-3099(01)00092-5. [DOI] [PubMed] [Google Scholar]
- 5.Caputo GA, London E. Position and ionization state of Asp in the core of membrane-inserted alpha helices control both the equilibrium between transmembrane and nontransmembrane helix topography and transmembrane helix positioning. Biochemistry. 2004;43:8794–8806. doi: 10.1021/bi049696p. [DOI] [PubMed] [Google Scholar]
- 6.Caputo GA, London E. Analyzing transmembrane protein and hydrophobic helix topography by dual fluorescence quenching. Methods in molecular biology. 2013;974:279–295. doi: 10.1007/978-1-62703-275-9_13. [DOI] [PubMed] [Google Scholar]
- 7.Moroz YS, Binder W, Nygren P, Caputo GA, Korendovych IV. Painting proteins blue: beta-(1-azulenyl)-L-alanine as a probe for studying protein-protein interactions. Chemical communications. 2013;49:490–492. doi: 10.1039/c2cc37550h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gratkowski H, Lear JD, DeGrado WF. Polar side chains drive the association of model transmembrane peptides. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(3):880–885. doi: 10.1073/pnas.98.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lear JD, Gratkowski H, DeGrado WF. De novo design, synthesis and characterization of membrane-active peptides. Biochemical Society transactions. 2001;29(Pt):4, 559–564. doi: 10.1042/bst0290559. [DOI] [PubMed] [Google Scholar]
- 10.DeGrado WF, Gratkowski H, Lear JD. How do helix-helix interactions help determine the folds of membrane proteins? Perspectives from the study of homo-oligomeric helical bundles. Protein science : a publication of the Protein Society. 2003;12(4):647–665. doi: 10.1110/ps.0236503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson JP, Weinger JS, Engelman DM. Motifs of serine and threonine can drive association of transmembrane helices. Journal of molecular biology. 2002;316:799–805. doi: 10.1006/jmbi.2001.5353. [DOI] [PubMed] [Google Scholar]
- 12.Papo N, Shai Y. Effect of drastic sequence alteration and D-amino acid incorporation on the membrane binding behavior of lytic peptides. Biochemistry. 2004;43:6393–6403. doi: 10.1021/bi049944h. [DOI] [PubMed] [Google Scholar]
- 13.Papo N, Shai Y. Can we predict biological activity of antimicrobial peptides from their interactions with model phospholipid membranes? Peptides. 2003;24:1693–1703. doi: 10.1016/j.peptides.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Gehman JD, Luc F, Hall K, Lee TH, Boland MP, Pukala TL, Bowie JH, Aguilar MI, Separovic F. Effect of antimicrobial peptides from Australian tree frogs on anionic phospholipid membranes. Biochemistry. 2008;47:8557–8565. doi: 10.1021/bi800320v. [DOI] [PubMed] [Google Scholar]
- 15.Rezansoff AJ, Hunter HN, Jing W, Park IY, Kim SC, Vogel HJ. Interactions of the antimicrobial peptide Ac-FRWWHR-NH(2) with model membrane systems and bacterial cells. The journal of peptide research : official journal of the American Peptide Society. 2005;65:491–501. doi: 10.1111/j.1399-3011.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- 16.Wieprecht T, Apostolov O, Beyermann M, Seelig J. Membrane binding and pore formation of the antibacterial peptide PGLa: thermodynamic and mechanistic aspects. Biochemistry. 2000;39:442–452. doi: 10.1021/bi992146k. [DOI] [PubMed] [Google Scholar]
- 17.Wieprecht T, Apostolov O, Seelig J. Binding of the antibacterial peptide magainin 2 amide to small and large unilamellar vesicles. Biophysical chemistry. 2000;85:187–198. doi: 10.1016/s0301-4622(00)00120-4. [DOI] [PubMed] [Google Scholar]
- 18.Cady S, Wang T, Hong M. Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein. J Am Chem Soc. 2011;133:11572–11579. doi: 10.1021/ja202051n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, DeGrado WF. Computational design of peptides that target transmembrane helices. Science (New York, N.Y.) 2007;315:1817–1822. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 21.Heuck AP, Hotze EM, Tweten RK, Johnson AE. Mechanism of membrane insertion of a multimeric beta-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Molecular cell. 2000;6(5):1233–1242. doi: 10.1016/s1097-2765(00)00119-2. [DOI] [PubMed] [Google Scholar]
- 22.Malenbaum SE, Collier RJ, London E. Membrane topography of the T domain of diphtheria toxin probed with single tryptophan mutants. Biochemistry. 1998;37(51):17915–17922. doi: 10.1021/bi981230h. [DOI] [PubMed] [Google Scholar]
- 23.Tang J, Signarvic RS, DeGrado WF, Gai F. Role of helix nucleation in the kinetics of binding of mastoparan X to phospholipid bilayers. Biochemistry. 2007;46:13856–13863. doi: 10.1021/bi7018404. [DOI] [PubMed] [Google Scholar]
- 24.Saar-Dover R, Ashkenazi A, Shai Y. Peptide interaction with and insertion into membranes. Methods in molecular biology (Clifton, N.J.) 2013;1033:173–183. doi: 10.1007/978-1-62703-487-6_12. [DOI] [PubMed] [Google Scholar]
- 25.Matsuda M, Hatanaka W, Takeo M, Kim CW, Niidome T, Yamamoto T, Kishimura A, Mori T, Katayama Y. Short Peptide Motifs for Long-Lasting Anchoring to the Cell Surface. Bioconjugate chemistry. 2014 doi: 10.1021/bc500465j. [DOI] [PubMed] [Google Scholar]
- 26.Kuroda K, Caputo GA, DeGrado WF. The role of hydrophobicity in the antimicrobial and hemolytic activities of polymethacrylate derivatives. Chemistry (Weinheim an der Bergstrasse, Germany) 2009;15:1123–1133. doi: 10.1002/chem.200801523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Argiolas A, Pisano JJ. Bombolitins, a new class of mast cell degranulating peptides from the venom of the bumblebee Megabombus pennsylvanicus. The Journal of biological chemistry. 1985;260:1437–1444. [PubMed] [Google Scholar]
- 28.DeGrado WF, Musso GF, Lieber M, Kaiser ET, Kezdy FJ. Kinetics and mechanism of hemolysis induced by melittin and by a synthetic melittin analogue. Biophysical journal. 1982;37:329–338. doi: 10.1016/S0006-3495(82)84681-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee MT, Chen FY, Huang HW, Department of Physics, N. C. U. C.-L. T. Energetics of pore formation induced by membrane active peptides. Biochemistry. 2004;43(12):3590–3599. doi: 10.1021/bi036153r. [DOI] [PubMed] [Google Scholar]
- 30.Rayahin JE, Buhrman JS, Gemeinhart RA. Melittin-glutathione S-transferase fusion protein exhibits anti-inflammatory properties and minimal toxicity. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2014;65C:112–121. doi: 10.1016/j.ejps.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yevtushenko DP, Romero R, Forward BS, Hancock RE, Kay WW, Misra S. Pathogen-induced expression of a cecropin A-melittin antimicrobial peptide gene confers antifungal resistance in transgenic tobacco. Journal of experimental botany. 2005;56(416):1685–1695. doi: 10.1093/jxb/eri165. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Zhao B, Huang C, Meng XM, Bian EB, Li J. Melittin restores PTEN expression by down-regulating HDAC2 in human hepatocelluar carcinoma HepG2 cells. PloS one. 2014;9:e95520. doi: 10.1371/journal.pone.0095520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradrick TD, Philippetis A, Georghiou S. Stopped-flow fluorometric study of the interaction of melittin with phospholipid bilayers: importance of the physical state of the bilayer and the acyl chain length. Biophysical journal. 1995;69:1999–2010. doi: 10.1016/S0006-3495(95)80070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stromstedt AA, Wessman P, Ringstad L, Edwards K, Malmsten M. Effect of lipid headgroup composition on the interaction between melittin and lipid bilayers. Journal of colloid and interface science. 2007;311:59–69. doi: 10.1016/j.jcis.2007.02.070. [DOI] [PubMed] [Google Scholar]
- 35.Runnels LW, Scarlata SF. Theory and application of fluorescence homotransfer to melittin oligomerization. Biophysical journal. 1995;69:1569–1583. doi: 10.1016/S0006-3495(95)80030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiedman G, Herman K, Searson P, Wimley WC, Hristova K. The electrical response of bilayers to the bee venom toxin melittin: evidence for transient bilayer permeabilization. Biochimica et biophysica acta. 2013;1828:1357–1364. doi: 10.1016/j.bbamem.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jamasbi E, Batinovic S, Sharples RA, Sani MA, Robins-Browne RM, Wade JD, Separovic F, Hossain MA. Melittin peptides exhibit different activity on different cells and model membranes. Amino acids. 2014;46:2759–2766. doi: 10.1007/s00726-014-1833-9. [DOI] [PubMed] [Google Scholar]
- 38.Hood JL, Jallouk AP, Campbell N, Ratner L, Wickline SA. Cytolytic nanoparticles attenuate HIV-1 infectivity. Antiviral therapy. 2013;18:95–103. doi: 10.3851/IMP2346. [DOI] [PubMed] [Google Scholar]
- 39.Saimi Y, Kung C. Calmodulin as an ion channel subunit. Annual review of physiology. 2002;64:289–311. doi: 10.1146/annurev.physiol.64.100301.111649. [DOI] [PubMed] [Google Scholar]
- 40.Korendovych IV, Kim YH, Ryan AH, Lear JD, Degrado WF, Shandler SJ. Computational design of a self-assembling beta-peptide oligomer. Org Lett. 2010;12:5142–5145. doi: 10.1021/ol102092r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korendovych IV, Shandler SJ, Montalvo GL, DeGrado WF. Environment-and sequence-dependence of helical type in membrane-spanning peptides composed of beta3-amino acids. Organic letters. 2011;13:3474–3477. doi: 10.1021/ol201218y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shandler SJ, Korendovych IV, Moore DT, Smith-Dupont KB, Streu CN, Litvinov RI, Billings PC, Gai F, Bennett JS, DeGrado WF. Computational design of a beta-peptide that targets transmembrane helices. Journal of the American Chemical Society. 2011;133:12378–12381. doi: 10.1021/ja204215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nanda V, Cristian L, Toptygin D, Brand L, Degrado WF. Nanosecond Dynamics of InfluenzaA/M2TM and an Amantadine Resistant Mutant Probed by Time-Dependent Red Shifts of a Native Tryptophan. Chemical physics. 2013;422 doi: 10.1016/j.chemphys.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caputo GA, London E. Using a novel dual fluorescence quenching assay for measurement of tryptophan depth within lipid bilayers to determine hydrophobic alpha-helix locations within membranes. Biochemistry. 2003;42:3265–3274. doi: 10.1021/bi026696l. [DOI] [PubMed] [Google Scholar]
- 45.NCCLS Approved standards M7-A3. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. 2003.
- 46.Wiegand I, Hilpert K, Hancock RE. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nature protocols. 2008;3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 47.Burman LG, Nordstrom K, Boman HG. Resistance of Escherichia coli to penicillins. V. Physiological comparison of two isogenic strains, one with chromosomally and one with episomally mediated ampicillin resistance. Journal of bacteriology. 1968;96:438–446. doi: 10.1128/jb.96.2.438-446.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gibney KA, Sovadinova I, Lopez AI, Urban M, Ridgway Z, Caputo GA, Kuroda K. Poly(ethylene imine)s as antimicrobial agents with selective activity. Macromolecular bioscience. 2012;12:1279–1289. doi: 10.1002/mabi.201200052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi H, Palermo EF, Yasuhara K, Caputo GA, Kuroda K. Molecular design, structures, and activity of antimicrobial peptide-mimetic polymers. Macromolecular bioscience. 2013;13:1285–1299. doi: 10.1002/mabi.201300126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mueller P, Rudin DO, Tien HT, Wescott WC. METHODS FOR THE FORMATION OF SINGLE BIMOLECULAR LIPID MEMBRANES IN AQUEOUS SOLUTION. J. Phys. Chem. 1963;67:534–535. [Google Scholar]
- 51.Piers KL, Hancock RE. The interaction of a recombinant cecropin/melittin hybrid peptide with the outer membrane of Pseudomonas aeruginosa. Molecular microbiology. 1994;12(6):951–958. doi: 10.1111/j.1365-2958.1994.tb01083.x. [DOI] [PubMed] [Google Scholar]
- 52.Shao J, Korendovych IV, Broos J. Biosynthetic incorporation of the azulene moiety in proteins with high efficiency. Amino acids. 2015;47:213–216. doi: 10.1007/s00726-014-1870-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.