

Abstract
We investigated Ca2+ handling in isolated brain synaptic and nonsynaptic mitochondria and in cultured striatal neurons from the YAC128 mouse model of Huntington’s disease (HD). Both synaptic and nonsynaptic mitochondria from 2- and 12-month-old YAC128 mice had larger Ca2+ uptake capacity than mitochondria from YAC18 and wild-type FVB/NJ mice. Synaptic mitochondria from 12-month-old YAC128 mice had further augmented Ca2+ capacity compared with mitochondria from 2-month-old YAC128 mice and age-matched YAC18 and FVB/NJ mice. This increase in Ca2+ uptake capacity correlated with an increase in the amount of mutant huntingtin protein (mHtt) associated with mitochondria from 12-month-old YAC128 mice. We speculate that this may happen due to mHtt-mediated sequestration of free fatty acids thereby increasing resistance of mitochondria to Ca2+-induced damage. In experiments with striatal neurons from YAC128 and FVB/NJ mice, brief exposure to 25 or 100μM glutamate produced transient elevations in cytosolic Ca2+ followed by recovery to near resting levels. Following recovery of cytosolic Ca2+, mitochondrial depolarization with FCCP produced comparable elevations in cytosolic Ca2+, suggesting similar Ca2+ release and, consequently, Ca2+ loads in neuronal mitochondria from YAC128 and FVB/NJ mice. Together, our data argue against a detrimental effect of mHtt on Ca2+ handling in brain mitochondria of YAC128 mice.
Keywords: Huntington’s disease, striatum, mitochondria, neuron, calcium, permeability transition pore
Introduction
Huntington’s disease (HD) is a hereditary neurodegenerative disorder linked to a mutation in a single gene encoding huntingtin protein (Htt) (MacDonald et al. 1993). Although a causative link between this mutation and HD pathogenesis is well documented, the precise molecular mechanisms of the detrimental effect of mutant huntingtin (mHtt) remain obscure. One of the major hypotheses posits that mHtt leads to abnormalities in Ca2+ signaling in affected neurons (Bezprozvanny and Hayden 2004) probably due to augmented activity of the NMDA-subtype of glutamate receptors (Zhang et al. 2008), aberrations in inositol 1,4,5-trisphosphate (IP3) receptor function (Tang et al. 2003), and defects in mitochondrial Ca2+ handling (Panov et al. 2002).
Mitochondria possess Ca2+ channels in the inner membrane (Baughman et al. 2011; De et al. 2011) that allow Ca2+ influx into mitochondria and Ca2+ accumulation in the mitochondrial matrix (Bernardi 1999). In early studies, investigators found decreased Ca2+ uptake capacity in brain mitochondria from YAC72 mice and a rat HD model (Panov et al. 2002; Gellerich et al. 2008) and in mitochondria of a conditionally immortalized striatal progenitor cell line STHQ111/Q111 expressing mHtt with 111 glutamines (Milakovic et al. 2006; Lim et al. 2008). It is well established that the magnitude of mitochondrial Ca2+ uptake capacity is limited by sensitivity of mitochondria to the detrimental effect of Ca2+ (Chalmers and Nicholls 2003). A large Ca2+ load in mitochondria induces mitochondrial damage manifested in induction of the permeability transition pore (PTP) that causes mitochondrial swelling and depolarization (Bernardi 1999). This limits the ability of mitochondria to accumulate Ca2+, produce ATP, and retain pro-apoptotic proteins like cytochrome c (Rasola and Bernardi 2011). Consequently, facilitated PTP induction in mitochondria associated with mHtt was proposed to explain Ca2+ handling defects in mitochondria from HD animal and cell models (Milakovic et al. 2006; Gellerich et al. 2008). Indeed, increased propensity to Ca2+-stimulated PTP induction was found in mitochondria in neurons from HD mice and immortalized striatal cells (Choo et al. 2004; Fernandes et al. 2007; Lim et al. 2008; Quintanilla et al. 2013). On the other hand, in our previous study, we did not find an increased likelihood of PTP induction in striatal and cortical mitochondria isolated from HD mice compared to mitochondria from wild-type mice (Brustovetsky et al. 2005). Later, Oliveira et al. (2007) demonstrated increased Ca2+ uptake capacity in brain nonsynaptic mitochondria isolated from R6/2 and YAC128 mice compared to mitochondria from wild-type littermates (Oliveira et al. 2007). Moreover, in experiments with cultured cortical neurons expressing N-terminal or full-length mHtt, investigators failed to find a significant effect of mHtt on mitochondrial Ca2+ accumulation following exposure of neurons to excitotoxic glutamate (Chang et al. 2006). Thus, the question of whether mHtt increases sensitivity of brain mitochondria and, particularly, neuronal mitochondria to Ca2+-induced damage remains open.
In the present study, we investigated whether mHtt facilitates PTP induction and examined the effect of mHtt on Ca2+ uptake capacity in synaptic and nonsynaptic mitochondria isolated from YAC128, YAC18, and wild-type FVB/NJ mice. Consistent with our previous data (Brustovetsky et al. 2005), our current results do not support facilitated PTP induction in brain mitochondria from YAC128 mice. Both synaptic and nonsynaptic mitochondria isolated from early symptomatic 2-month-old YAC128 mice had larger Ca2+ uptake capacity than mitochondria from age-matched YAC18 and FVB/NJ mice consistent with the previous report by Oliveira et al. (2007) (Oliveira et al. 2007). Moreover, synaptic mitochondria from late-stage symptomatic 12-month-old YAC128 mice had even greater Ca2+ uptake capacity compared with mitochondria from 2-month-old YAC128 mice and age-matched YAC18 and FVB/NJ mice. The augmented Ca2+ uptake capacity of synaptic mitochondria from 12-month-old YAC128 mice correlated with an increased amount of mHtt associated with mitochondria. Finally, in experiments with glutamate-exposed striatal neurons from YAC128 and FVB/NJ mice, we found no evidence for mHtt-induced impairment of mitochondrial Ca2+ accumulation.
Materials and Methods
Materials
Pyruvate, malate, succinate, glutamate, EGTA, ADP, oligomycin, rotenone, antimycin A, 2,4-dinitrophenol, N-methyl-D-glucamine and carbonylcyanide-p-trifluorometh-oxyphenylhydrazone (FCCP) were purchased from Sigma (St. Louis, MO, USA). Tetraphenylphosphonium chloride was from Fluka (Buchs, Switzerland). Percoll was from GE Healthcare Bio-Sciences (Pittsburgh, PA, USA). Bovine serum albumin (BSA), free from free fatty acids, was from MP Biomedicals (Irvine, CA, USA). Alamethicin was from Enzo (Farmingdale, NY, USA).
Animals
All procedures with animals were performed in accordance with the Institutional Animal Care and Use Committee approved protocol. Transgenic YAC18 and YAC128 mice as well as wild-type FVB/NJ mice were purchased from Jackson Laboratories (Bar Harbor, ME) and breeding colonies were established in Laboratory Animal Resource Center at Indiana University School of Medicine, Indianapolis, IN. YAC128 mice express full-length human mHtt containing 128 glutamines in the polyglutamine (polyQ) stretch in addition to wild-type mouse Htt whereas YAC18 mice express full-length human Htt containing 18 glutamines (Hodgson et al. 1996; Hodgson et al. 1999). Male YAC18 and YAC128 mice were bred with female FVB/NJ mice (background strain). The mice were housed under standard conditions with free access to water and food. Previously, it was reported that YAC128 mice at 2 months of age begin to show behavioral abnormalities and are considered to be early symptomatic (Van Raamsdonk et al. 2005a; Van Raamsdonk et al. 2005b). A recent study using electron microscopy demonstrated a significant reduction in excitatory synapses of striatal neurons in 12-month-old YAC128 mice (Singaraja et al. 2011). At 12 months of age, YAC128 mice show significant striatal loss and are considered to be at an advanced stage of HD pathology (Slow et al. 2003). Therefore, we were interested in assessing mitochondrial Ca2+ handling in mice at both ages. Consequently, in our experiments, we used early symptomatic 2-month-old and mature 12-month-old YAC128 mice, control age-matched YAC18 mice, and their age-matched wild-type FVB/NJ littermates of both sexes.
Genotyping
All offspring were genotyped with a PCR assay on tail DNA. Briefly, PCR of tail DNA was performed according to the protocol provided by Jackson Laboratory using oligonucleotide primers oIMR6533 (GGCTGA GGAAGCTGAGGAG) and TmoIMR1594 (CCGCTCAGGTTCTGCTTTTA) purchased from Invitrogen. The PCR reaction mixture contained 1μl DNA template and 23μl Platinum PCR SuperMix (Invitrogen) supplemented with 0.39μM of each primer (Invitrogen), total volume 25μl. Cycling conditions were 5 min at 95°C, 35 cycles at 30 sec at 95°C, 30 sec at 56°C, 60 sec at 72°C, and 10 min at 72°C. Reaction products were analyzed on 1.2% agarose gel run at 100V for 60 min with Tris-acetate-EDTA running buffer containing 1X GelRed™ Nucleic Acid Gel Stain (Biotium, CA).
Isolation of brain nonsynaptic and synaptic mitochondria
Percoll gradient-purified brain nonsynaptic and synaptic mitochondria from YAC128, YAC18 and wild-type FVB/NJ mice were isolated as we described previously (Brustovetsky et al. 2002; Shalbuyeva et al. 2007). The scheme illustrating the procedure for isolation and purification of synaptic and nonsynaptic mitochondria is shown in Supplemental Figure 1, and a brief description is in the Figure legend. Supplemental Figure 2 illustrates the distinct separation of nonsynaptic mitochondria and synaptosomes. Synaptosomes are further used to release synaptic mitochondria.
Nonsynaptic mitochondria are from neuronal somata and glial cells, whereas synaptic mitochondria are of pure neuronal origin and derived from nerve terminals (synaptosomes). The use of both types of mitochondria allows a complete assessment of brain mitochondrial functions. While Ca2+ uptake capacity of nonsynaptic mitochondria from YAC128 mice has been previously assessed (Oliveira et al. 2007), Ca2+ uptake capacity of synaptic mitochondria was evaluated for the first time in our study. Earlier, it was hypothesized that BSA can displace mHtt from its binding sites on mitochondria (Panov et al. 2003). Therefore, BSA was omitted from all solutions used in our experiments with isolated mitochondria unless stated otherwise.
Mitochondrial Ca2+ uptake capacity
Mitochondrial Ca2+ uptake was measured with a miniature Ca2+-selective electrode in a 0.3 ml chamber at 37°C under continuous stirring. A decrease in the external Ca2+ concentration indicated mitochondrial Ca2+ uptake. The standard incubation medium contained 125 mM KCl, 0.5 mM MgCl2, 3 mM KH2PO4, 10 mM Hepes, pH 7.4, 10μM EGTA, and was supplemented either with 3 mM pyruvate plus 1 mM malate or 3 mM succinate plus 3 mM glutamate. Here and in other experiments, succinate was used in combination with glutamate to prevent oxaloacetate inhibition of succinate dehydrogenase (Lehninger et al. 1993). Additionally, the incubation medium was supplemented with 0.1 mM ADP and 1μM oligomycin as described previously (Chalmers and Nicholls 2003). Ca2+ was added to mitochondria as 10μM CaCl2 pulses. Data were quantified as Ca2+ uptake capacity per mg of mitochondrial protein.
Mitochondrial swelling and membrane potential
Mitochondrial swelling was evaluated at 37°C and continuous stirring by following changes in light scattering of mitochondrial suspension at 525 nm with an incident light beam at 180° in a 0.3 ml chamber. The incubation medium for light scattering measurements contained 215 mM mannitol, 70 mM sucrose, 0.5 mM MgCl2, 3 mM KH2PO4, 10 mM Hepes, pH 7.4, 10μM EGTA, 3 mM succinate, 3 mM glutamate. A decrease in light scattering of mitochondrial suspension indicated mitochondrial swelling. Maximal mitochondrial swelling was induced by alamethicin (30μg/ml). Maximal swelling was taken as 100%. Ca2+-induced swelling was calculated as a percentage of maximal swelling (Li et al. 2010). Mitochondrial membrane potential was measured in a standard KCl-based incubation medium at 37°C with a tetraphenylphosphonium (TPP+) electrode by following TPP+ distribution between the incubation medium and mitochondria (Kamo et al. 1979). A decrease in external TPP+ concentration corresponds to mitochondrial polarization, while an increase of TPP+ in the incubation medium corresponds to depolarization.
Mitochondrial reactive oxygen species production
Reactive oxygen species (ROS) production by mitochondria was monitored at 37°C in the standard KCl-based incubation medium with Amplex Red assay for H2O2 (Molecular Probes, Eugene, OR) using a Perkin-Elmer LS 55 luminescence spectrometer and excitation/emission wavelengths 550/590 nm as described previously (Votyakova and Reynolds 2001).
Immunoblotting
Isolated mitochondria pretreated with Protease Inhibitor Cocktail (Roche) were solubilized by incubation in NuPAGE LDS sample buffer (Invitrogen, Carlsbad, CA) supplemented with a reducing agent at 70°C for 15 minutes. Bis-Tris gels (4–12%, Invitrogen) and Tris-Acetate gels (3–8%, Invitrogen) were used for electrophoresis (40μg protein per lane). After SDS-PAGE, proteins were transferred to Hybond-ECL nitrocellulose membrane (Amersham Biosciences). Blots were incubated for 1 hour at room temperature in blocking solution of 5% dry milk, phosphate-buffered saline, pH 7.2, and 0.15% Triton X-100. Then, where indicated, blots were incubated with one of the following primary antibodies: mouse monoclonal anti-polyQ 1C2 (mAb1574, Millipore, 1:1000), specific for mHtt; mouse monoclonal anti-cyclophilin D (EMD, San Diego, CA, 1:1000); rabbit monoclonal anti-MEK1/2 (Pierce, Rockford, IL, 1:1000); rabbit polyclonal anti-VDAC1 (Calbiochem, 1:1000); rabbit polyclonal anti-calnexin (Abcam, Cambridge, MA, 1:1000); and mouse monoclonal anti-COX IV (Life Technologies, Grand Island, NY, 1:2000). To assess neuronal and glial populations in cell cultures, cells were lysed in Tris Buffer Saline (TBS) supplemented with 1% Nonidet P-40 and Protease Inhibitor Cocktail (Roche) by incubating for 15 minutes on ice. Then, cell lysates were centrifuged at 35,000 rpm (100,000×g) for 30 minutes. Aliquots of supernate were supplemented with NuPAGE LDS sample buffer (Invitrogen, Carlsbad, CA), containing a reducing agent, incubated at 70°C for 15 minutes, and then tested by western blotting with mouse monoclonal anti-MAP2 antibody, rabbit polyclonal anti-GFAP antibody (Millipore, Temecula, CA, 1:1000), and mouse monoclonal anti-GAPDH antibody (Abcam, Cambridge, MA, 1:2000). Blots were incubated with goat anti-mouse or goat anti-rabbit IgG (1:20000) coupled with horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and developed with Supersignal West Pico chemiluminescent reagents (Pierce, Rockford, IL). Molecular mass markers See Blue Plus 2 Standards (5μl) and HiMark Pre-stained High Molecular Weight Protein Standards (10μl) (Invitrogen) were used to determine molecular masses of the bands. NIH ImageJ 1.48v software (http://rsb.info.nih.gov//ij) was used to quantify band densities
Cell culturing
Striatal neuronal cultures were prepared from individual striata of postnatal day 1 YAC128 mice and their wild-type littermates as previously described (Dubinsky 1993), but without pooling cells from different animals together. We used neuronal-glial co-culture derived from postnatal day 1 mouse pups because it is more physiologically relevant and allows for the study of more mature, better developed cells than pure neuronal culture derived from embryonic animals. For all platings, 35 mg·ml−1 uridine plus 15 mg·ml−1 5-fluoro-2′-deoxyuridine were added 24 hours after plating to inhibit proliferation of non-neuronal cells. Neurons were cultured in a 5% CO2 atmosphere at 37°C in Neurobasal medium with B27 supplement (Life Technologies).
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 15 minutes. Then, cells were incubated with Protein-Free Blocking Buffer (Pierce, Rockford, IL) for an hour at room temperature. Cells were incubated overnight with primary antibodies anti-MAP2 (neuronal marker, mouse monoclonal, Millipore, Temecula, CA, 1:500), anti-GFAP (astroglial marker, rabbit polyclonal, Millipore, Temecula, CA, 1:1000), anti-DARPP-32 (marker of medium spiny neurons, rabbit monoclonal, Pierce, Rockford, IL, 1:200), and anti-GABA (rabbit polyclonal, Pierce, Rockford, IL, 1:20000). Nuclei were stained with DAPI (nuclear marker, Invitrogen, Carlsbad, CA). Then, cells were incubated with secondary donkey anti-mouse antibody conjugated with AlexaFluor 488 and donkey anti-rabbit AlexaFluor 568 (Invitrogen, Carlsbad, CA, 1:1000). Bright field and fluorescence images were acquired using a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon CFI Super Fluoro 20× 0.75 NA objective and CCD camera Cool SNAPHQ (Roper Scientific, Tucson, AZ) controlled by MetaMorph 6.3 software (Molecular Devices, Downingtown, PA).
Calcium imaging
To follow changes in cytosolic Ca2+, striatal neurons (10–12 days in vitro, DIV) were loaded at 37°C with 2.6μM Fura-2FF-AM (Molecular Probes, Eugene, OR) in the standard bath solution containing 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM NaHEPES, pH 7.4, and 5 mM glucose. The osmolarity of the bath solutions was similar to that in the growth medium (280 mosm). Osmolarity of the bath solution was measured with an osmometer, Osmette II™ (Precision Systems Inc., Natick, MA). Fluorescence imaging was performed with an inverted microscope, Nikon Eclipse TE2000-S, using a Nikon CFI Plan Fluor 20× 0.45 NA objective and a back-thinned EM-CCD camera Hamamatsu C9100-12 (Hamamatsu Photonic Systems, Bridgewater, NJ) controlled by Simple PCI software 6.1 (Compix Inc., Sewickley, PA). The excitation light was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA). The excitation filters (340±5 and 380±7) were controlled by a Lambda 10-2 optical filter changer (Sutter Instruments, Novato, CA). Fluorescence was recorded through a 505 nm dichroic mirror at 535±25 nm. The images were taken every 15 seconds during the time-course of the experiment using the minimal exposure time that provided acceptable image quality. The changes in cytosolic Ca2+ concentration ([Ca2+]c) were monitored by following a ratio of F340/F380, calculated after subtracting the background from both channels. After 1.5 minutes of fluorescence recording in the standard bath solution, glutamate (25 or 100μM) plus 10μM glycine was applied to the neurons. At the end of the experiment, the bath solution with glutamate and Ca2+ was replaced by a glutamate- and Ca2+-free solution, Na+ was replaced by equimolar N-methyl-D-glucamine (NMDG) and 1μM FCCP was applied to the neurons to depolarize neuronal mitochondria and to release Ca2+ accumulated in mitochondria. Quantification of Fura-2FF signals was carried out per manufacturer instructions. [Ca2+]c was calculated using the Grynkiewicz method (Grynkiewicz et al. 1985), assuming Kd for Fura-2FF is 5.5μM. In all experiments, the fluorescence background was subtracted from the signals. Since Ca2+ binding and spectroscopic properties of fluorescent dyes can differ significantly in the intracellular milieu, the cytosolic Ca2+ concentrations presented in this paper should be deemed estimates as stated previously by other investigators (Dietz et al. 2007; Stanika et al. 2009).
Statistics
Data are shown as mean ± SEM of several independent experiments. Statistical analysis of the experimental results consisted of unpaired t-test or one-way ANOVA followed by Bonferroni’s post hoc test (GraphPad Prism® 4.0, GraphPad Software Inc., SanDiego, CA, USA). Every experiment was performed using several different preparations of isolated mitochondria or several separate neuronal-glial platings.
RESULTS
In the majority of our experiments, we used both synaptic and nonsynaptic mitochondria isolated from YAC128 mice and their wild-type littermates, FVB/NJ mice. Previously, it was reported that in vivo exposure to 3-nitropropionic acid, a mitochondrial toxin that inhibits succinate dehydrogenase (Alston et al. 1977) and simulates some aspects of HD in animal models (Palfi et al. 1996), alters corticostriatal synaptic activity (Mendoza et al. 2014). Synaptic deficiency in HD is well established (Sepers and Raymond 2014). Corticostriatal synaptic abnormalities in YAC128 mice were seen at 1 month of age (Joshi et al. 2009). Synaptic dysfunction was observed in 1.5- to 2-month-old YAC128 mice (Milnerwood and Raymond 2007). On the other hand, synaptic activity in medium spiny neurons from 12-month-old YAC128 mice was significantly reduced (Joshi et al. 2009). Consistent with this, a recent study using electron microscopy demonstrated a significant reduction in excitatory synapses onto striatal neurons in 12-month-old YAC128 mice (Singaraja et al. 2011). Therefore, to determine the mechanisms of synaptic deficiency in YAC128 mice, it is essential to establish whether mHtt impairs synaptic mitochondria in these animals.
Following isolation and discontinuous Percoll-gradient purification, both synaptic and nonsynaptic mitochondria from YAC128 mice were tested for purity. The purity of isolated mitochondria was assessed by western blotting with antibodies against calnexin (an endoplasmic reticulum marker), MEK1/2 (a cytosolic marker), and COX IV (a mitochondrial marker). The purity control data are shown in Figure 1A. These data show the lack of endoplasmic reticulum and cytosolic contaminations in synaptic and nonsynaptic mitochondria isolated and purified from FVB/NJ and YAC128 mice. Since mHtt is predominantly localized in the cytosol, our data confirm that if mHtt is detected in mitochondrial fractions, it is not due to cytosolic contamination.
Figure 1. Purity of synaptic and nonsynaptic mitochondria isolated from YAC128 and wild-type FVB/NJ mice (A). Detection of human mutant huntingtin (mHtt) in brain nonsynaptic (A) and synaptic (B) mitochondria isolated from YAC128 mice (B–E).

In A, homogenates, cytosolic and mitochondrial fractions were analyzed using western blotting with antibodies against calnexin (ER marker), MEK1/2 (cytosolic marker), and COXIV (mitochondrial marker). Fractions were solubilized and proteins were resolved by SDS-PAGE using 4–12% Bis-Tris gels as described in Materials and Methods. In B and D, mitochondria (Mtc) were analyzed immediately after isolation (Mtc from ice), after 30 minutes of incubation in the standard incubation medium supplemented with 3 mM succinate and 3 mM glutamate without (washing) and with 0.1% BSA, free from fatty acids, (washing with BSA), and after 30 minutes of incubation at pH 11.5 (alkali treatment). Mitochondrial porin (voltage-dependent anion channel, VDAC) was used as a loading control. In C and E, the western blotting data were quantified using NIH ImageJ software and presented as a mHtt/VDAC ratio. Data are mean±SEM, *p<0.01 compared with mitochondria from ice and after washing, N=7.
Both synaptic and nonsynaptic mitochondria from YAC128 mice retained attached mHtt that could not be washed out after 30 minutes of incubation at 37°C in the standard KCl-based incubation medium with or without 0.1% BSA (Fig. 1B–E). On the other hand, alkali treatment for 30 minutes at pH 11.5, previously used to remove mHtt from mitochondrial membranes (Choo et al. 2004), decreased mHtt level below the detection limit of western blotting (Fig. 1B–E). The western blotting data were quantified using NIH ImageJ software and presented as a mHtt/VDAC ratio. These data are consistent with the previous findings obtained in an early study, demonstrating mHtt association with mitochondria (Choo et al. 2004).
Earlier, some investigators reported an increased propensity to PTP induction in mitochondria exposed to mHtt (Choo et al. 2004; Fernandes et al. 2007; Lim et al. 2008; Quintanilla et al. 2013). However, in our previous study we did not find evidence for increased susceptibility to PTP induction in striatal and cortical mitochondria from HD mice compared with mitochondria from wild-type mice (Brustovetsky et al. 2005). In this earlier study, we investigated PTP induction in nonsynaptic mitochondria. In the present study, we for the first time tested synaptic (neuronal) mitochondria from YAC128 and wild-type FVB/NJ mice (Fig. 2). Mitochondrial swelling and depolarization are the major manifestations of PTP induction (Bernardi 1999). Consequently, we investigated an induction of the PTP by simultaneously monitoring a decrease in light scattering of the mitochondrial suspension, indicative of swelling of the organelles, and a release of TPP+ from mitochondria, indicative of mitochondrial depolarization. Ca2+, in a concentration-dependent manner, induced swelling and depolarization of synaptic mitochondria (Fig. 2). To quantify swelling of mitochondria, at the end of the experiment we applied alamethicin (AL), an antibiotic that causes maximal mitochondrial swelling (Brustovetsky and Dubinsky 2000b). Maximal swelling was taken as 100% and mitochondrial swelling following 15 minutes incubation with Ca2+ was calculated as a percentage of maximal swelling. Both mitochondrial swelling and depolarization in response to Ca2+ were comparable in mitochondria isolated from YAC128 and wild-type mice. Thus, in our experiments mHtt failed to increase propensity to PTP induction in synaptic (neuronal) mitochondria from YAC128 mice.
Figure 2. Ca2+-induced mitochondrial swelling and depolarization in synaptic mitochondria from wild-type FVB/NJ (A, C, E) and YAC128 (B, D, F) mice.

Mitochondrial swelling was evaluated by following a decline in light scattering of mitochondrial suspension (red traces). A decrease in light scattering indicates mitochondrial swelling. Changes in mitochondrial membrane potential were evaluated by following distribution of tetraphenylphosphonium (TPP+) between incubation medium and mitochondrial matrix (black traces). The decrease in TPP+ concentration in the incubation medium ([TPP+]ext) indicates TPP+ accumulation in mitochondria and high membrane potential. The release of TPP+ from mitochondria indicates a decrease in membrane potential. Mitochondrial swelling and membrane potential were evaluated simultaneously at 37°C. Where indicated 25, 50, or 100μM Ca2+ was applied to mitochondria (Mtc). At the end of the experiments, 60μM 2,4-dinitrophenol (2,4-DNP) was applied to completely depolarize mitochondria and, then, 30μg/ml alamethicin (AL) was added to induce maximal mitochondrial swelling, taken as 100%. The amount of Ca2+-induced swelling was determined as percentage of maximal swelling (shown in A as X%). In G, percentage of Ca2+-induced mitochondrial swelling. Data are mean±SEM, N=8.
Next, we tested the effect of mHtt on Ca2+ uptake capacity of mitochondria isolated from YAC128 mice and compared it with Ca2+ uptake capacity of mitochondria from YAC18 and wild-type FVB/NJ mice. A possible decrease in mitochondrial Ca2+ uptake capacity could serve as an indicator of mitochondrial impairment in HD. Conversely, unchanged or augmented Ca2+ uptake capacity suggests a lack of defects in mitochondrial Ca2+ handling. In our hands, Ca2+ uptake capacity of nonsynaptic mitochondria from 2- and 12-month-old YAC128 mice was slightly but statistically significantly increased compared with mitochondria from YAC18 and FVB/NJ mice (Fig. 3). This result is consistent with data previously reported by Oliveira et al (2007) (Oliveira et al. 2007). The slight increase in Ca2+ uptake capacity was observed with both Complex I substrates pyruvate/malate and the Complex II substrate succinate used in combination with glutamate to prevent accumulation of oxaloacetate and inhibition of succinate dehydrogenase (Brustovetsky and Dubinsky 2000a). Synaptic mitochondria from 2-month-old YAC128 mice also had a slight but statistically significant increase in Ca2+ uptake capacity compared with mitochondria from age-matched YAC18 and FVB/NJ mice regardless of oxidative substrates (Fig. 4A, D). BSA (Fig. 4B) and cyclosporin A (not shown) considerably increased Ca2+ uptake capacity in synaptic mitochondria from all mouse strains tested. Interestingly, Ca2+ uptake capacity of synaptic mitochondria from 12-month-old YAC128 mice was noticeably increased compared with mitochondria from 2-month-old YAC128 mice and age-matched 12-month-old YAC18 and FVB/NJ mice (Fig. 4C). These experiments also showed that Ca2+ uptake capacity of mitochondria from YAC18 mice was indistinguishable from Ca2+ capacity of mitochondria derived from FVB/NJ mice. Therefore, in the following experiments we used only mitochondria from YAC128 and wild-type FVB/NJ mice.
Figure 3. Ca2+ uptake capacity of brain nonsynaptic mitochondria isolated from YAC128, YAC18, and wild-type FVB/NJ mice.

Mitochondria were incubated at 37°C in the standard incubation medium supplemented either with 3 mM succinate plus 3 mM glutamate or with 3 mM pyruvate plus 1 mM malate as indicated in the Figure. In all Ca2+ uptake experiments, 100μM ADP and 1μM oligomycin were present in the incubation medium. In A and B, mitochondria were isolated from 2-month-old mice. In C, mitochondria were isolated from 12-month-old mice. In A–C, where indicated 10μM Ca2+ pulses were applied to mitochondria until mitochondria fail to uptake additional Ca2+. In D, statistical analysis of Ca2+ uptake capacity of mitochondria from YAC128, YAC18, and wild-type FVB/NJ mice. Data are mean±SEM, *p<0.05 compared with mitochondria from FVB/NJ and YAC18 mitochondria, N=9.
Figure 4. Ca2+ uptake capacity of brain synaptic mitochondria isolated from YAC128, YAC18, and wild-type FVB/NJ mice.

Mitochondria were incubated at 37°C in the standard incubation medium supplemented either with 3 mM succinate plus 3 mM glutamate or with 3 mM pyruvate plus 1 mM malate as indicated in the Figure. In all Ca2+ uptake experiments, 100μM ADP and 1μM oligomycin were present in the incubation medium. In B, incubation medium was additionally supplemented with 0.1% BSA (free from fatty acids). In A, B, and D, mitochondria were isolated from 2-month-old mice. In C, mitochondria were isolated from 12-month-old mice. In A–D, where indicated 10μM Ca2+ pulses were applied to mitochondria until mitochondria fail to uptake additional Ca2+. In E, statistical analysis of Ca2+ uptake capacity of mitochondria from YAC128, YAC18, and wild-type FVB/NJ mice. Data are mean±SEM, *p<0.05 comparing with FVB/NJ and YAC18 mitochondria; **p<0.01 compared with mitochondria from 12-month-old FVB/NJ and YAC18 mice, N=9.
Previously, it was reported that the level of cyclophilin D (CyD), a key regulatory component of the PTP that sensitizes the pore to Ca2+ (Baines et al. 2005; Schinzel et al. 2005), was reduced in mitochondria from older animals (Eliseev et al. 2007). Therefore, we tested whether the increased Ca2+ uptake capacity in mitochondria from YAC128 mice, particularly, in synaptic mitochondria from 12-month-old transgenic animals, correlates with a decrease in CyD. Our experiments revealed a lack of difference in the amount of CyD in synaptic and nonsynaptic mitochondria from 2- and 12-month-old YAC128 and FVB/NJ mice (Suppl. Fig. 3). Consequently, this could not explain increased Ca2+ uptake capacity in mitochondria from YAC128 mice compared with mitochondria from wild-type animals.
ROS are major activators of the PTP (Bernardi 1999) and PTP induction is the key factor that restricts Ca2+ uptake capacity (Chalmers and Nicholls 2003). It seemed conceivable that different levels of ROS production could underlie the difference in Ca2+ uptake capacity. Therefore, in the following experiments we examined mitochondrial H2O2 generation by synaptic mitochondria from YAC128 and FVB/NJ mice using the Amplex Red assay (Brustovetsky et al. 2003). With succinate plus glutamate, ROS generation was much higher than with pyruvate plus malate. BSA further augmented ROS generation (Fig. 5A, B), presumably due to an increase in membrane potential in both YAC128 and wild-type mitochondria that may lead to an increase in ROS generation (Korshunov et al. 1997). Ca2+, on the other hand, depolarizes mitochondria (Bernardi 1999) and, consequently, inhibited ROS generation with succinate plus glutamate (Fig. 5C), but did not significantly change ROS generation in the presence of pyruvate plus malate (Fig. 5D). Importantly, there was no difference in ROS generation between mitochondria from YAC128 and FVB/NJ mice. Figure 5E shows a statistical analysis of ROS measurements. Thus, ROS production could not account for the difference in Ca2+ uptake capacity between mitochondria from transgenic and wild-type animals.
Figure 5. H2O2 production by synaptic mitochondria from 2-month-old YAC128 and wild-type FVB/NJ mice.

Mitochondrial H2O2 production was measured under continuous stirring in 0.4 ml cuvette at 37°C in the standard KCl-based incubation medium. In A–C, the standard incubation medium was supplemented with 3 mM succinate plus 3 mM glutamate. In D, the incubation medium was supplemented with 3 mM pyruvate plus 1 mM malate. In B, 0.1% BSA (free from fatty acids) was present in the medium. In C and D, 12.5 or 200μM Ca2+ was applied to mitochondria, respectively, as indicated. In E, statistical analysis of H2O2 production under different experimental conditions. Data are mean±SEM, N=11. *p<0.01 compared with H2O2 generation by wild-type mitochondria oxidizing succinate plus glutamate, but without BSA and Ca2+; #p<0.01 compared with H2O2 generation by YAC128 mitochondria oxidizing succinate plus glutamate, but without BSA and Ca2+.
Next, we examined the level of mHtt associated with mitochondria and found a 2-fold increase in the amount of mHtt bound to synaptic mitochondria from 12-month-old YAC128 mice compared with mitochondria from 2-month-old YAC128 mice (Fig. 6A, B). This increase was not due to an increase in cytosolic mHtt because it remained at the same level (Fig. 6C, D). Thus, augmented Ca2+ uptake capacity of synaptic mitochondria from 12-month-old YAC128 mice correlated with the increased amount of mHtt associated with these mitochondria.
Figure 6. The level of mutant huntingtin (mHtt) associated with brain mitochondria isolated from 2- and 12-month-old YAC128 and wild-type FVB/NJ mice.
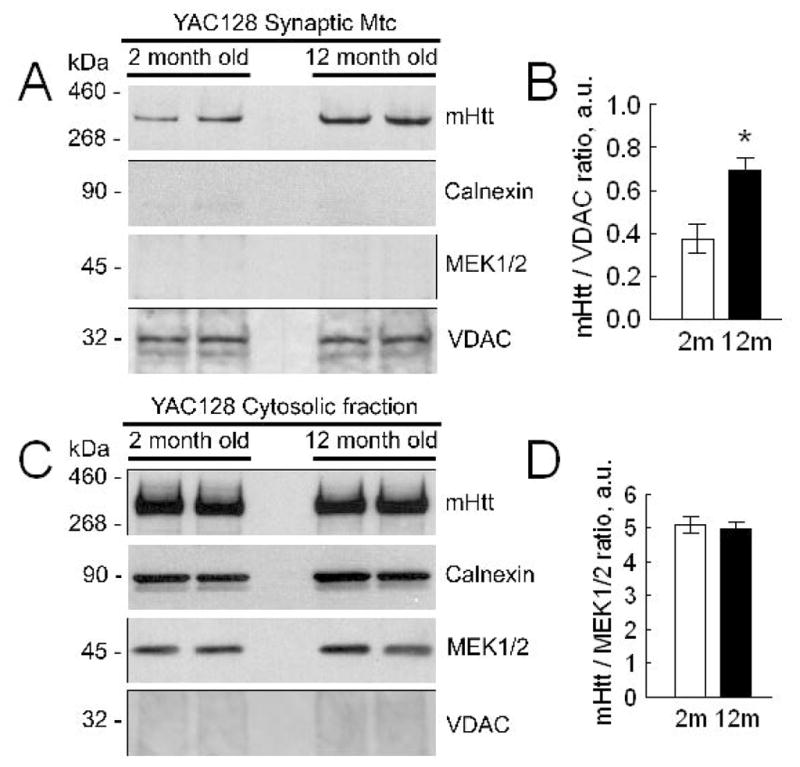
In A, western blots of brain synaptic mitochondria from 2- and 12-month-old YAC128 and wild-type FVB/NJ mice with anti-polyQ 1C2 antibody, specific for mHtt. Isolated mitochondria were solubilized and proteins were resolved by SDS-PAGE using 3–8% Tris-acetate gels. Calnexin was used as a marker of endoplasmic reticulum. MEK1/2 was used as a cytosolic marker. Mitochondrial porin (voltage-dependent anion channel, VDAC) was used as a mitochondrial marker and loading control. In B, statistical analysis of western blot densitometry with data expressed as a ratio of mHtt band intensity to band intensity of VDAC. Data are mean±SEM, *p<0.05 compared with mitochondria from 2-month-old YAC128 mice, N=8. In C, western blots of brain cytosolic fractions from 2- and 12-month-old FVB/NJ and YAC128 mice with anti-polyQ 1C2 antibody, specific for mHtt. Cytosolic fractions were resolved by SDS-PAGE using 3–8% Tris-acetate gels. MEK1/2 was used as a cytosolic marker and loading control. In D, statistical analysis of western blot densitometry with data expressed as a ratio of mHtt band intensity to band intensity of MEK1/2. Data are mean±SEM, N=8.
Finally, we evaluated accumulation of Ca2+ in mitochondria of striatal neurons exposed to excitotoxic glutamate. Previously, this approach was used in studies with cortical neurons, expressing mHtt (Chang et al. 2006). In our experiments, we used striatal neurons in culture derived from individual postnatal day 1 (PN1) YAC128 and FVB/NJ mouse pups. The viability of cultured striatal neurons derived from these animals was tested using propidium iodide staining for dead neurons and calcein-AM staining for live neurons and found to be similar: at 10–12 DIV 91±3% of neurons from FVB/NJ mice and 93±4% of neurons from YAC128 mice were alive. Suppl. Figure 4A shows the representative bright-field image of cell culture used in our experiments. All pups used for harvesting neurons were genotyped and cultured cells were tested for the presence of mHtt using western blotting (Suppl. Fig. 4B). The cell culture contained both neurons and astrocytes stained by neuronal marker MAP2 (green) and glial marker GFAP (red) (Mao and Wang 2001), respectively (Suppl. Fig. 4D). To determine consistency in glia/neuron ratio, we performed western blotting with antibodies against MAP2 and GFAP. GAPDH was used as a loading control. Then, we performed densitometry and calculated MAP2/GFAP ratio as a characteristic of our cell cultures (Suppl. Fig. 5). These experiments showed very small variability (mean±SEM: 1.10±0.08 for FVB/NJ mice, N=6, and 1.12±0.07 for YAC128 mice, N=6) between different platings as well as the lack of difference between cell cultures prepared from YAC128 and wild-type littermates.
Judging by staining with anti-GABA antibody the majority of striatal neurons (85%) were GABA positive, typical for striatal neurons (Ivkovic and Ehrlich 1999; Mao and Wang 2001), (Fig. Suppl. 4F) and 50% of all cells were positive for DARPP32, a marker for medium spiny neurons (Ivkovic and Ehrlich 1999) (Suppl. Fig. 4H). Supplemental Figures 4C, E, and G show bright-field images for the corresponding immunofluorescence images. In calcium imaging experiments, we used short applications of 25 and 100μM glutamate (plus 10μM glycine) and evaluated subsequent release of mitochondrial Ca2+ into the cytosol following depolarization of the organelles with 1μM FCCP (Fig. 7). The magnitude of elevation in cytosolic Ca2+ due to Ca2+ release from mitochondria was used to evaluate mitochondrial Ca2+ uptake capacity in situ (Chang et al. 2006). Simultaneously with FCCP application, we replaced external Na+ for N-methyl-D-glucamine (NMDG), a bulk organic cation that cannot be transported by Na+/Ca2+ exchanger and hence prevents Ca2+ extrusion from the cell by this mechanism. In these experiments, glutamate applications produced transient increases in cytosolic Ca2+ followed by recovery of cytosolic Ca2+ to near resting level after removal of glutamate (Fig. 7). Neurons from YAC128 mice had a more robust response to glutamate. Zhang et al (2008) also reported elevated cytosolic Ca2+ in response to glutamate in YAC128 medium spiny neurons (Zhang et al. 2008). Depolarization of mitochondria with 1μM FCCP in Ca2+-free bath solution (to prevent influx of external Ca2+) triggered a massive increase in cytosolic Ca2+ that was similar in neurons from both YAC128 and wild-type mice (Fig. 7). This result suggests that mHtt does not decrease the ability of neuronal mitochondria to accumulate Ca2+ and, therefore, most likely does not affect mitochondrial contribution to maintenance of Ca2+ homeostasis in striatal neurons exposed to excitotoxic glutamate. Overall, the presented results demonstrate the lack of difference in mitochondrial Ca2+ accumulation in isolated mitochondria and cultured neurons from YAC128 and FVB/NJ mice, suggesting similar sensitivity to deleterious Ca2+ and comparable propensity to PTP induction.
Figure 7. triatal neurons from YAC128 and wild-type FVB/NJ mice have comparable mitochondrial Ca2+ accumulation following transient glutamate-induced elevations in cytosolic Ca2+.

In A, B, E, and F, the averaged fluorescence signals (mean±SEM) from the representative experiments are shown. Cytosolic Ca2+ was followed by monitoring Fura-2FF F340/F380 fluorescence ratio at 37°C. In these experiments, striatal neurons (10–12 DIV) were exposed to 25μM or 100μM glutamate (in both cases, with 10μM glycine) for 2 minutes as indicated. Then, glutamate and glycine were removed to let [Ca2+]c recover. After [Ca2+]c reached near resting level (7 minutes after glutamate removal) neurons were treated with 1μM FCCP to depolarize mitochondria and release accumulated Ca2+. To avoid ambiguity concerning possible Ca2+ influx from the outside of the cell, external Ca2+ was removed simultaneously with glutamate and glycine. In addition, to prevent Ca2+ extrusion from the cell by Na+/Ca2+ exchanger, the external Na+ was replaced by equimolar N-methyl-D-glucamine (NMDG) as indicated. In C, D, G, and H, the areas under the curve (AUC) for the averaged fluorescence signals are shown. The AUC for glutamate-induced increase in [Ca2+]c (Glu AUC) was calculated for the 120 second period beginning with glutamate application. The AUC for FCCP-induced increase in [Ca2+]c (FCCP Glu) was calculated for the 300 second period following FCCP application. Data are mean± SEM. In C, *p<0.05 compared with wild-type cells, N=8–9 separate experiments with 20–25 individual neurons in each experiment.
DISCUSSION
In earlier studies, mitochondrial dysfunction and a decreased ability of mitochondria to accumulate Ca2+ have been proposed to contribute to HD pathogenesis (Panov et al. 2002; Choo et al. 2004; Milakovic et al. 2006). On the other hand, in experiments with nonsynaptic mitochondria, we and other investigators failed to find an increase in sensitivity to deleterious Ca2+ and instead observed an increase in resistance to Ca2+ that was manifested in reduced propensity to PTP induction and augmented Ca2+ uptake capacity (Brustovetsky et al. 2005; Oliveira et al. 2007). In the present study, we demonstrated that both synaptic and nonsynaptic mitochondria isolated from YAC128 mice retain their ability to accumulate Ca2+ compared with mitochondria from wild-type animals. Moreover, mitochondria from YAC128 mice, especially synaptic mitochondria from 12-month-old animals, had larger Ca2+ uptake capacity compared with mitochondria from wild-type littermates. The augmented Ca2+ uptake capacity of synaptic mitochondria from 12-month-old YAC128 mice correlated with an increased amount of mHtt associated with these mitochondria. Notably, an increased amount of full-length mHtt was also found to be associated with brain nonsynaptic mitochondria isolated from 12-month-old knock-in 150Q/150Q mice compared with younger knock-in animals (Orr et al. 2008). Finally, following application of excitotoxic glutamate to cultured striatal neurons from YAC128 mice, neuronal mitochondria accumulated similar amounts of Ca2+ compared with mitochondria in striatal neurons from wild-type animals. Thus, mHtt failed to reduce Ca2+ uptake capacity in brain mitochondria from YAC128 mice. On the contrary, the level of mHtt binding to mitochondria positively correlates with mitochondrial resistance to deleterious Ca2+.
In our experiments we found that BSA increased Ca2+ uptake capacity of brain mitochondria isolated from both YAC128 mice and their wild-type littermates. It is known that ROS production by mitochondria, oxidizing succinate, depends on the reverse electron flow in the mitochondrial electron transport chain that brings electrons back to Complex I. The reverse electron flow, in turn, depends on mitochondrial membrane potential: hyperpolarization leads to an increase in reverse electron flow and, subsequently, to elevated ROS production (Korshunov et al. 1997; Korshunov et al. 1998). It is well established that ROS increase propensity to PTP induction in mitochondria (Zoratti and Szabo 1995). However, it is also known that free fatty acids (FFA) increase propensity of PTP induction as well (Zoratti and Szabo 1995). BSA binds FFA (Spector 1975) and exerts a strong protective effect against PTP (see Fig. 5 and (Wieckowski et al. 2000)). Based on these observations, our data concerning BSA effects suggest that the protective action of BSA prevails over deleterious effect of augmented ROS generation.
Over the last 15 years, studies from several laboratories implicated mHtt in facilitating PTP induction and impairment of mitochondrial Ca2+ accumulation (Panov et al. 2002; Choo et al. 2004; Milakovic et al. 2006; Gizatullina et al. 2006; Fernandes et al. 2007; Lim et al. 2008; Gellerich et al. 2008; Quintanilla et al. 2013). An early report by Panov et al. presented data that suggested bioenergetic abnormalities and a decrease in Ca2+ accumulation by mitochondria exposed to mHtt (Panov et al. 2002). Because inhibition of the PTP with cyclosporin A and ADP failed to eliminate the difference between mitochondria from wild-type and HD mice, PTP involvement was ruled out. The effect of mHtt on mitochondrial Ca2+ uptake capacity appeared to be elusive and in a subsequent paper Panov et al. reported that “the defect in Ca2+ handling in brain mitochondria was consistently observed only if brain mitochondria were isolated without BSA” (Panov et al. 2003). The authors presumed that BSA could displace mHtt from its binding sites on mitochondria. Our data do not support this hypothesis. We found that mHtt is associated with mitochondria, consistent with previous reports (Choo et al. 2004), but BSA does not displace mHtt from mitochondria.
In our earlier study, we did not observe an increased predisposition to Ca2+-induced PTP induction in striatal and cortical nonsynaptic mitochondria from HD mice compared with mitochondria from wild-type littermates (Brustovetsky et al. 2005). On the contrary, we found increased resistance to Ca2+ in striatal mitochondria exposed to mHtt. Consistent with our findings, Oliveira et al. reported that nonsynaptic mitochondria from R6/2 and YAC128 mice had increased Ca2+ uptake capacity compared with mitochondria from wild-type animals (Oliveira et al. 2007). Both our study (Brustovetsky et al. 2005) and a study by Oliveira et al. (Oliveira et al. 2007) suggested the lack of mHtt-induced impairment of mitochondrial Ca2+ handling, argued against facilitation of PTP induction by mHtt, and did not support involvement of PTP in HD pathogenesis. In line with this, Perry et al. found that R6/2 mice crossed with CyD-knockout mice and, thus, expressing reduced or no CyD, had increased neuronal mitochondrial Ca2+ uptake capacity without a sign of improvement in either behavioral or neuropathological features of HD (Perry et al. 2010). The authors concluded that increased Ca2+ capacity of neuronal mitochondria is not beneficial in R6/2 mice. Chang et al. exposed neurons expressing N-terminal mHtt to glutamate and used FCCP-induced depolarization of mitochondria to release accumulated Ca2+ (Chang et al. 2006). The authors failed to find a significant effect of mHtt on the ability of neuronal mitochondria to accumulate Ca2+ following exposure of neurons to excitotoxic glutamate. Recently, Wang et al. stimulated Ca2+ mobilization in medium spiny neurons by activating group I metabotropic glutamate receptors and triggering IP3 production and found significantly higher Ca2+ load in mitochondria in neurons from YAC128 mice compared with neurons from wild-type mice (Wang et al. 2013). Overall, these data strongly argue against mHtt-induced impairment of mitochondrial Ca2+ uptake and facilitated PTP induction, and their role in HD pathogenesis. Thus, there are two distinct views on the possible mHtt-induced impairment of mitochondrial Ca2+ handling. One group of investigators postulates mHtt-induced facilitation of PTP induction, leading to defects in mitochondrial Ca2+ uptake, whereas the other group does not find evidence for these detrimental mHtt effects. The reason for this discrepancy is not clear, but it might be related to the use of different HD models and variations in methodological approaches.
In our present study, we did not find evidence of impaired Ca2+ uptake capacity in either synaptic or nonsynaptic mitochondria from YAC128 mice. On the contrary, both types of mitochondria had increased Ca2+ capacity, especially, synaptic mitochondria from 12-month-old YAC128 mice. The mechanism underlying augmented Ca2+ uptake capacity of mitochondria that are associated with increased levels of mHtt is unknown. At the moment, we can only speculate about a possible scenario. Mitochondrial Ca2+ uptake capacity mainly depends on the ability of mitochondria to resist an induction of the PTP (Chalmers and Nicholls 2003). Induction of the PTP causes mitochondrial depolarization that precludes further Ca2+ uptake and results in a release of previously accumulated Ca2+ (Rasola and Bernardi 2011). It is known that FFA are effective activators of PTP (Zoratti and Szabo 1995) and FFA binding by BSA (Spector 1975) most likely underlies BSA-mediated protection against PTP induction (Wieckowski et al. 2000). In our experiments, BSA significantly increased Ca2+ uptake capacity of synaptic (Fig. 5B) and nonsynaptic (not shown) mitochondria from 2-month-old FVB/NJ, YAC18, and YAC128 mice. Furthermore, in experiments with 12-month-old mice, we found that BSA significantly increased Ca2+ uptake capacity of mitochondria from FVB/NJ mice, but was ineffective in mitochondria from YAC128 mice (Fig. 5C). Keeping in mind the increased amount of mHtt associated with mitochondria from 12-month-old YAC128 mice and elevated Ca2+ uptake capacity in these mitochondria, the lack of further augmentation with BSA suggests that mHtt and BSA effects may have a similar mechanism, e.g. binding of FFA. Although additional experiments are necessary to test this hypothesis and untangle the potential mechanism of FFA binding to mHtt, existing literature lends support to this notion. It was reported that in Fatty-Acid-Binding Proteins (FABPs), a family of intracellular proteins that bind FFA, arginine and tryptophan form H-bonds directly with fatty acids, whereas glutamine interacts with fatty acids through two intervening water molecules (Hamilton 2002). Because mHtt has a large number of glutamines, it is conceivable that mHtt may interact with FFA more strongly than wild-type Htt and potentially may sequester FFA. Alternatively, the elongated polyQ stretch in mHtt might result in a change in mHtt conformation, leading to increased ability of mHtt to bind FFA. However, these hypotheses require further experimental testing.
The increased Ca2+ uptake capacity of mitochondria from YAC128 mice may reflect compensatory adaptation to augmented Ca2+ influx via overactivated NMDA receptors and/or increased Ca2+ release from endoplasmic reticulum via abnormally activated IP3 receptors (Bezprozvanny and Hayden 2004). Similar to findings by Chang et al. and Wang et al. (Chang et al. 2006; Wang et al. 2013), in experiments with cultured striatal neurons from YAC128 mice and wild-type littermates, we did not find evidence of the decreased ability of neuronal mitochondria to accumulate Ca2+. Consequently, comparable susceptibility to PTP induction and the lack of mHtt-induced deficiency in mitochondrial Ca2+ uptake capacity in mitochondria from YAC128 and wild-type mice argue against possible involvement of these mechanisms in HD pathogenesis.
Supplementary Material
Acknowledgments
This work was supported by NIH/NINDS grant R01 NS078008 to N.B.
The abbreviations used are
- HD
Huntington’s disease
- Htt
wild-type mouse huntingtin protein
- mHtt
human mutant huntingtin
- ROS
reactive oxygen species
- BSA
bovine serum albumin
- TPP+
tetraphenylphosphonium cation
- NMDG
N-methyl-D-glucamine
- CyD
cyclophilin D
- PN1
postnatal day 1
- VDAC
voltage-dependent anion channel
- 2,4-DNP
2,4-dinitrophenol
- DIV
days in vitro
- PTP
the permeability transition pore
- IP3
inositol 1,4,5-trisphosphate
- FFA
free fatty acids
Footnotes
The authors have no conflicts of interest to declare.
References
- Alston TA, Mela L, Bright HJ. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc Natl Acad Sci U S A. 1977;74:3767–3771. doi: 10.1073/pnas.74.9.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem. 2002;80:207–218. doi: 10.1046/j.0022-3042.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Purl KJ, Capano M, Crompton M, Dubinsky JM. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J Neurosci. 2003;23:4858–4867. doi: 10.1523/JNEUROSCI.23-12-04858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, Dubinsky JM. Dual responses of CNS mitochondria to elevated calcium. J Neurosci. 2000a;20:103–113. doi: 10.1523/JNEUROSCI.20-01-00103.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, Dubinsky JM. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J Neurosci. 2000b;20:8229–8237. doi: 10.1523/JNEUROSCI.20-22-08229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, LaFrance R, Purl KJ, Brustovetsky T, Keene CD, Low WC, Dubinsky JM. Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington’s Disease. J Neurochem. 2005;93:1361–1370. doi: 10.1111/j.1471-4159.2005.03036.x. [DOI] [PubMed] [Google Scholar]
- Chalmers S, Nicholls DG. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem. 2003;278:19062–19070. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- De SD, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz RM, Kiedrowski L, Shuttleworth CW. Contribution of Na(+)/Ca(2+) exchange to excessive Ca(2+) loading in dendrites and somata of CA1 neurons in acute slice. Hippocampus. 2007;17:1049–1059. doi: 10.1002/hipo.20336. [DOI] [PubMed] [Google Scholar]
- Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. Journal of Neuroscience. 1993;13:623–631. doi: 10.1523/JNEUROSCI.13-02-00623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliseev RA, Filippov G, Velos J, VanWinkle B, Goldman A, Rosier RN, Gunter TE. Role of cyclophilin D in the resistance of brain mitochondria to the permeability transition. Neurobiol Aging. 2007;28:1532–1542. doi: 10.1016/j.neurobiolaging.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Fernandes HB, Baimbridge KG, Church J, Hayden MR, Raymond LA. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington’s disease. J Neurosci. 2007;27:13614–13623. doi: 10.1523/JNEUROSCI.3455-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina ZZ, Nguyen HP, Trumbeckaite S, Vielhaber S, Seppet E, Zierz S, Landwehrmeyer B, Ries O, von HS, Striggow F. Impaired regulation of brain mitochondria by extramitochondrial Ca2+ in transgenic Huntington disease rats. J Biol Chem. 2008;283:30715–30724. doi: 10.1074/jbc.M709555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizatullina ZZ, Lindenberg KS, Harjes P, Chen Y, Kosinski CM, Landwehrmeyer BG, Ludolph AC, Striggow F, Zierz S, Gellerich FN. Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann Neurol. 2006;59:407–411. doi: 10.1002/ana.20754. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hamilton JA. How fatty acids bind to proteins: the inside story from protein structures. Prostaglandins Leukot Essent Fatty Acids. 2002;67:65–72. doi: 10.1054/plef.2002.0400. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Smith DJ, McCutcheon K, Koide HB, Nishiyama K, Dinulos MB, Stevens ME, Bissada N, Nasir J, Kanazawa I, Disteche CM, Rubin EM, Hayden MR. Human huntingtin derived from YAC transgenes compensates for loss of murine huntingtin by rescue of the embryonic lethal phenotype. Hum Mol Genet. 1996;5:1875–1885. doi: 10.1093/hmg/5.12.1875. [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Ehrlich ME. Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J Neurosci. 1999;19:5409–5419. doi: 10.1523/JNEUROSCI.19-13-05409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PR, Wu NP, Andre VM, Cummings DM, Cepeda C, Joyce JA, Carroll JB, Leavitt BR, Hayden MR, Levine MS, Bamford NS. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci. 2009;29:2414–2427. doi: 10.1523/JNEUROSCI.5687-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamo N, Muratsugu M, Hongoh R, Kobatake Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J Membr Biol. 1979;49:105–121. doi: 10.1007/BF01868720. [DOI] [PubMed] [Google Scholar]
- Korshunov SS, Korkina OV, Ruuge EK, Skulachev VP, Starkov AA. Fatty acids as natural uncouplers preventing generation of O2- and H2O2 by mitochondria in the resting state. FEBS Lett. 1998;435:215–218. doi: 10.1016/s0014-5793(98)01073-4. [DOI] [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- Lehninger AL, Nelson DL, Cox MM. Principles of Biochemistry. Worth Publishers; New York: 1993. [Google Scholar]
- Li T, Brustovetsky T, Antonsson B, Brustovetsky N. Dissimilar mechanisms of cytochrome c release induced by octyl glucoside-activated BAX and by BAX activated with truncated BID. Biochim Biophys Acta. 2010;1797:52–62. doi: 10.1016/j.bbabio.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Fedrizzi L, Tartari M, Zuccato C, Cattaneo E, Brini M, Carafoli E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J Biol Chem. 2008;283:5780–5789. doi: 10.1074/jbc.M704704200. [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf AM, Lehrach H, Buckler AJ, Church D, Doucette-Stamm L, O’Donovan MC, Riba-Ramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Mao L, Wang JQ. Upregulation of preprodynorphin and preproenkephalin mRNA expression by selective activation of group I metabotropic glutamate receptors in characterized primary cultures of rat striatal neurons. Brain Res Mol Brain Res. 2001;86:125–137. doi: 10.1016/s0169-328x(00)00276-x. [DOI] [PubMed] [Google Scholar]
- Mendoza E, Miranda-Barrientos JA, Vazquez-Roque RA, Morales-Herrera E, Ruelas A, De la Rosa G, Flores G, Hernandez-Echeagaray E. In vivo mitochondrial inhibition alters corticostriatal synaptic function and the modulatory effects of neurotrophins. Neuroscience. 2014;280:156–170. doi: 10.1016/j.neuroscience.2014.09.018. [DOI] [PubMed] [Google Scholar]
- Milakovic T, Quintanilla RA, Johnson GV. Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: functional consequences. J Biol Chem. 2006;281:34785–34795. doi: 10.1074/jbc.M603845200. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Corticostriatal synaptic function in mouse models of Huntington’s disease: early effects of huntingtin repeat length and protein load. J Physiol. 2007;585:817–831. doi: 10.1113/jphysiol.2007.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira JM, Jekabsons MB, Chen S, Lin A, Rego AC, Goncalves J, Ellerby LM, Nicholls DG. Mitochondrial dysfunction in Huntington’s disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyot MC, Peschanski M, Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J Neurosci. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov AV, Burke JR, Strittmatter WJ, Greenamyre JT. In vitro effects of polyglutamine tracts on Ca2+-dependent depolarization of rat and human mitochondria: relevance to Huntington’s disease. Arch Biochem Biophys. 2003;410:1–6. doi: 10.1016/s0003-9861(02)00585-4. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Perry GM, Tallaksen-Greene S, Kumar A, Heng MY, Kneynsberg A, van GT, Detloff PJ, Albin RL, Lesort M. Mitochondrial calcium uptake capacity as a therapeutic target in the R6/2 mouse model of Huntington’s disease. Hum Mol Genet. 2010;19:3354–3371. doi: 10.1093/hmg/ddq247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Jin YN, von BR, Johnson GV. Mitochondrial permeability transition pore induces mitochondria injury in Huntington disease. Mol Neurodegener. 2013;8:45. doi: 10.1186/1750-1326-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011;50:222–233. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepers MD, Raymond LA. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov Today. 2014;19:990–996. doi: 10.1016/j.drudis.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Shalbuyeva N, Brustovetsky T, Brustovetsky N. Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J Biol Chem. 2007;282:18057–18068. doi: 10.1074/jbc.M702134200. [DOI] [PubMed] [Google Scholar]
- Singaraja RR, Huang K, Sanders SS, Milnerwood AJ, Hines R, Lerch JP, Franciosi S, Drisdel RC, Vaid K, Young FB, Doty C, Wan J, Bissada N, Henkelman RM, Green WN, Davis NG, Raymond LA, Hayden MR. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum Mol Genet. 2011;20:3899–3909. doi: 10.1093/hmg/ddr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, van RJ, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16:165–179. [PubMed] [Google Scholar]
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A. 2009;106:9854–9859. doi: 10.1073/pnas.0903546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005a;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Slow EJ, Hossain SM, Leavitt BR, Hayden MR. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J Neurosci. 2005b;25:4169–4180. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Chen Q, Wang X, Wang QC, Wang Y, Cheng HP, Guo C, Sun Q, Chen Q, Tang TS. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J Biol Chem. 2013;288:3070–3084. doi: 10.1074/jbc.M112.407726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieckowski MR, Brdiczka D, Wojtczak L. Long-chain fatty acids promote opening of the reconstituted mitochondrial permeability transition pore. FEBS Lett. 2000;484:61–64. doi: 10.1016/s0014-5793(00)02127-x. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li Q, Graham RK, Slow E, Hayden MR, Bezprozvanny I. Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington’s disease. Neurobiol Dis. 2008;31:80–88. doi: 10.1016/j.nbd.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.