

Abstract
Tamoxifen, a triphenylethylene antiestrogen and one of the first-line endocrine therapies used to treat estrogen receptor-positive breast cancer, has a number of interesting, off-target effects, and among these is the inhibition of sphingolipid metabolism. More specifically, tamoxifen inhibits ceramide glycosylation, and enzymatic step that can adventitiously support the influential tumor-suppressor properties of ceramide, the aliphatic backbone of sphingolipids. Additionally, tamoxifen and metabolites N-desmethyltamoxifen and 4-hydroxytamoxifen, have been shown to inhibit ceramide hydrolysis by the enzyme acid ceramidase. This particular intervention slows ceramide destruction and thereby depresses formation of sphingosine 1-phosphate, a mitogenic sphingolipid with cancer growth-promoting properties. As ceramide-centric therapies are becoming appealing clinical interventions in the treatment of cancer, agents like tamoxifen that can retard the generation of mitogenic sphingolipids and buffer ceramide clearance via inhibition of glycosylation, take on new importance. In this review, we present an abridged, lay introduction to sphingolipid metabolism, briefly chronicle tamoxifen’s history in the clinic, examine studies that demonstrate the impact of triphenylethylenes on sphingolipid metabolism in cancer cells, and canvass works relevant to the use of tamoxifen as adjuvant to drive ceramide-centric therapies in cancer treatment. The objective is to inform the readership of what could be a novel, off-label indication of tamoxifen and structurally-related triphenylethylenes, an indication divorced from estrogen receptor status and one with application in drug resistance.
Keywords: Tamoxifen, P-glycoprotein, Sphingolipid, Glucosylceramide synthase, Acid ceramidase, Multidrug resistance
1. Introduction
The antiestrogen tamoxifen demonstrates a myriad of non-genomic activities; curiously among these is inhibition of sphingolipid (SL) metabolism at ceramide glycosylation and at ceramide hydrolysis. This is principally noteworthy because sphingolipids are remarkably active lipids that orchestrate events that regulate cancer cell death as well as proliferation, ceramide more specifically, a pro-death player, and sphingosine 1-phosphate (S1P), a pro-life manager (Fig. 1). In survival mode, cancer cells have or acquire enzymatic operatives to suppress ceramide potency, glycosylation by glucosylceramide synthase (GCS) producing glucosylceramide (GC), and hydrolysis by acid ceramidase (AC), ceramide destruction. Destruction however is a double-edged sword that boosts mitogenic potential through phosphorylation of sphingosine by sphingosine kinase (SphK). Tamoxifen, tamoxifen metabolites, and related triphenylethylenes (TPE’s), as inhibitors of ceramide glycosylation and hydrolysis, can thus govern ceramide-regulated apoptotic cell death by effectively undoing ceramide resistance mechanisms and thwarting downstream mitogenicity. In the scheme of cancer therapeutics, whether ceramide-generating agents are administered or whether cell-deliverable ceramides are used as anti-cancer agents, use of TPE’s in ceramide-centric drug regimens presents an interesting direction we believe is worthy of exploitation. This review will focus on the role of TPE’s as regulators of SL metabolism and as adjuvants to ceramide-based cancer therapies.
Fig. 1.

Anabolic and catabolic routes of ceramide metabolism. Ceramide, shown in blue, the aliphatic backbone of sphingolipids, can be converted to sphingomyelin, ceramide 1-phosphate, and glucosylceramide. Ceramide, a tumor suppressor, induces apoptosis. Glycosylation (conversion to GC) is a prominent metabolic pathway in multidrug resistant cancer cells; glycosylation as well leads to ceramide resistance. If ceramide is hydrolyzed by ceramidase, sphingosine and free fatty acid are generated. The sphingosine can be metabolized to sphingosine 1-phosphate, a mitogenic entity, by sphingosine kinase. Various points in ceramide metabolism can be activated or inhibited, providing a useful strategy for studying ceramide-related events. GC, glucosylceramide. Note: arrows to designate several of the back-reactions are not included in this figure; for example, beta-glucocerebrosidase, also known as D-glucosyl-N-acylsphingosine glucohydrolase, which catalyzes cleavage by hydrolysis of the beta-glucosidic linkiage of glucosylceramide.
2. Sphingolipid metabolism in a nutshell
Sphingolipids are a class of nitrogen-containing lipids that harbor ceramide, a tumor suppressor lipid [1-3], as the aliphatic backbone (Fig. 1). If SL’s contain sugars, they are termed glycosphingolipids. SL’s serve structural roles in cellular membranes, and ceramide in particular has wide impact on cell function via mosaic signaling cascades [2]. By and large, tumor cells are protected from ceramide’s apoptosis-inducing, deleterious effects by constructive metabolic steps that produce a wide variety of noteworthy lipids such as sphingomyelin (SM), GC, precursor of higher cerebrosides, globosides, and gangliosides, and galactosylceramides, precursors of sulfatides. Of particular interest here is ceramide glycosylation, the major metabolic pathway utilized by multidrug resistant cancer cells to facilitate ceramide clearance [4-6]. Whereas ceramide is a powerful tumor censor, the glycosylated product, GC, formed by the action of GCS (Ceramide: UDP-Glc Glucosyltransferase), is ineffectual in this realm. Upregulated ceramide glycosylation is a mechanism of ceramide resistance in cancer cells [7-11]. Also relevant is ceramide hydrolysis, specifically by acid ceramidase , another sentinel enzyme regulator of cancer cell growth [12-14]. Similar with GCS, AC thwarts the tumor-killing properties of ceramide via hydrolysis, producing fatty acid and sphingosine, the latter a substrate for Sphk, the enzyme catalyzing production of S1P, a cancer cell mitogen [15,16] .
Ceramide can be synthesized de novo via enzymatic reactions that open with condensation of serine and palmitoyl-CoA to form sphinganine (Fig. 2); this reaction is catalyzed by serine palmitoyltransferase (SPT). The process concludes with an acylation step conducted by dihydroceramide synthase to generate dihydroceramide; this is followed by insertion of a 4,5-trans double in the sphingoid base moiety. A myriad of anticancer agents stimulate the de novo pathway and as well provoke degradation of SM to produce ceramide [17]. Both the de novo and the sphingomyelinase pathways are crucial therapeutic targets for the generation of ceramide in cancer cells, and whereas ceramide produced by these pathways can elicit apoptotic responses, anticancer efficacy can be limited by ceramide metabolic clearance (Fig. 1).
Fig. 2.
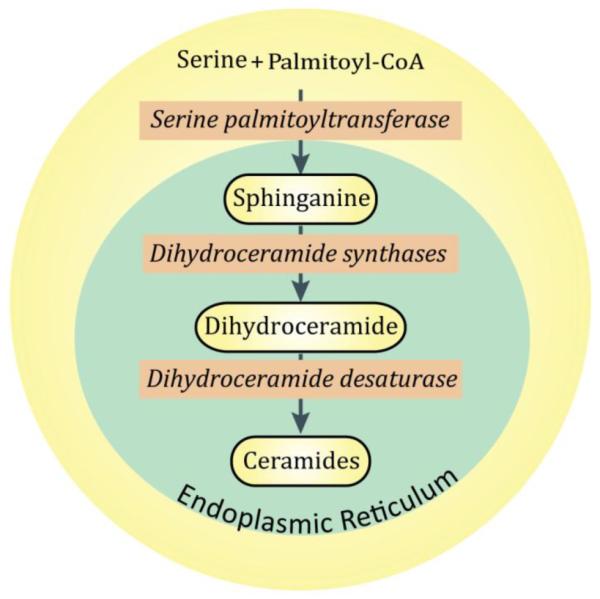
De novo pathway enzymes for production of ceramide. Ceramide synthesis is initiated by serine palmitoyltransferase and culminates with addition of a 4,5-trans double bond in dihydroceramide, catalyzed by dihydroceramide desaturase. Ceramide synthases, which number 6, are the subject of engaged investigations due to their selectivity for an array of fatty acyl-CoAs, giving rise to a myriad of molecular species of ceramides with distinct biological properties. Enzymes of the de novo pathway are shown in italics.
A number of agents have been designed to block ceramide clearance via the glycosylation pathway, chief among these are the “P-drugs” such as D-threo-1-phenyl-2-hexadecanoylamino-3-morpholino-1-propanol (PPMP) and D-threo-1-phenyl-2-decanoylamino-3-morphomolino-1-propanol (PDMP), competitive inhibitors of GCS [18]. Tamoxifen shares GCS-inhibitory activity in common with the P-drugs [19], albeit inhibition is not of a competitive nature but instead linked to the intracellular trafficking of glycolipids [20], a topic addressed later, in detail. It is this non-genomic, off-target activity of tamoxifen and related TPE’s that presents interesting prospective in the area of ceramide and cancer therapy.
3. A brief history of tamoxifen, from “morning after pill” to front-line breast cancer drug
When it was first discovered that estrogen was involved in the growth and development of breast cancer, scientists in an effort to treat this disease began to experiment with a variety of laboratory-synthesized, non-steroidal, estrogen-like chemicals, one being stilboestrol, first synthesized in 1938 and termed an endocrine disruptor. Because side effects were often severe, much of this work was abandoned in the early 1960’s. During the same period, scientists at Imperial Chemical Industries Pharmaceuticals (ICI) were charged with the task of designing new contraceptives in the form of a type of “morning after” pill. Several promising compounds were discovered, tamoxifen among them. Although tamoxifen never proved useful in human contraception, renewed interest in developing estrogen blockers led to its use in the treatment of breast cancer in the United Kingdom, in 1972. Tamoxifen went on to become the endocrine treatment of choice for all stages of breast cancer and a gold standard antiestrogen.
Tamoxifen is really a pro-drug that has little affinity for the estrogen receptor. It is metabolized in the liver by cytochrome P450 to active metabolites, 4-hydroxytamoxifen and N-desmethyl-4-hydroxytamoxifen (Fig. 3). The most prominent metabolite in humans is N-desmethyltamoxifen (DMT) [21,22]. Binding studies reveal that 4-hydroxytamoxifen binds to the estrogen receptor with an affinity equal to estradiol, which is 25-50 times higher than tamoxifen’ s affinity, whereas DMT binds the receptor with < 1 percent the affinity of tamoxifen [23]. The poor affinity of DMT for the estrogen receptor coupled with its activity as a potent inhibitor of SL metabolism (Sections 6, 8), makes for a compelling argument regarding therapeutic application for this weakly estrogenic metabolite.
Fig. 3.
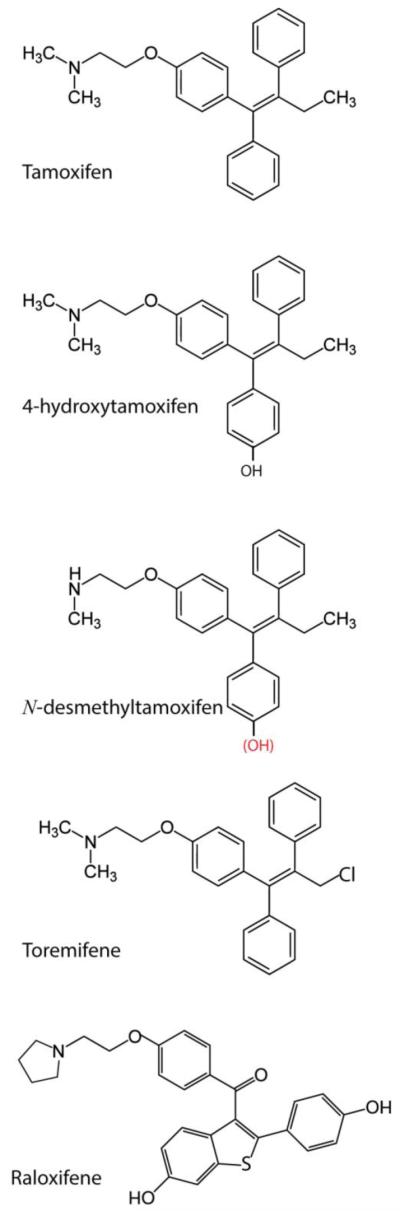
Chemical structure of triphenylethylenes and related SERM, raloxifene. Tamoxifen is composed of an aromatic triphenylethylene nucleus and an aminoethoxy side chain. Tamoxifen is metabolized in humans mainly to N-desmethyltamoxifen, 4-hydroxytamoxifen, 4-hydroxy-N-desmethyltamoxifen (endoxifen, structure shown with red hydroxyl group), and N-didesmethyltamoxifen (not shown). Metabolism is directed by the cytochrome P450 enzyme system. 4-hydroxytamoxifen is a potent anti-estrogen and is considered the most active metabolite; however, endoxifen is also a potent anti-estrogen. Raloxifene (Evista) belongs to the benzothiophene class of compounds and shares inhibition of ceramide glycosylation properties with tamoxifen. Toremifene is a chlorinated derivative of tamoxifen.
The aromatic nucleus of tamoxifen is composed of three phenyl groups bridged by ethylene, the simplest alkene. A prominent moiety, the aminoethoxy side chain, made up of a dimethylamine function and an ethyl part, is necessary for high-affinity binding to the estrogen receptor [24]. Tamoxifen, with nanomolar affinity for the estrogen receptor, is also referred to as a Selective Estrogen Receptor Modulator (SERM), meaning that in some tissues tamoxifen imparts beneficial estrogenic effects [25-29], as discussed below. In the United States, tamoxifen is approved for prophylactic use in prevention of breast cancer [30].
4. Non-sphingolipid-associated activities of TPE’s
4.1 Cardiovascular and bone health
Although designed to be a specific estrogen receptor antagonist, tamoxifen demonstrates a number of impressive, off-target activities, a subject that has been earlier reviewed by Kellen [31] . Increasingly, researchers are finding that a number of these off-target effects provide health benefits, and capital among these is the cardio-protective asset of tamoxifen and related agents such as raloxifene and toremifene (Fig. 3) [32-36]. It has been postulated that tamoxifen exerts cardio-protection via membrane antioxidant-mediated reactions that promote protection of LDL and cardiac membranes against oxidative damage [32,37], a key shift that contributes to a decrescendo of cholesterol, homocysteine, and lipoprotein (a) levels. Although it’s no secret that SERMs are useful in lowering cardiovascular risk factors as well as in treating bone disorders [38-40], this is a complex area in which the majority of studies were conducted in breast cancer patients, thus data need be carefully scrutinized. To fill in the gap, the reader is directed to reviews on SERMs and associated clinical findings [41,42].
Also, much in line with cardio-protection are the effects of tamoxifen on cholesterol metabolism by Acyl-CoA:cholesterol acyl transferase (ACAT), the enzyme catalyzing biosynthesis of cholesterol esters. Tamoxifen is a potent inhibitor of cholesterol esterification, and it is this activity that is in part purported to underlie the athero-protective action [43,44], this because over-accumulation of cholesterol esters in arteries contributes to development of atherosclerotic lesions [45]. A curious aside in relation to neoplasms, avasimibe, an ACAT-1 blocker, inhibits cholesterol ester synthesis in human glioma cell lines and induces cell cycle arrest and apoptosis by way of caspase-8 and -3 activation [46]. In a breast cancer model, ACAT inhibition reduced MDA-MB-231 cell migration [47], demonstrating a role for cholesterol esterification in metastasis. These observations highlight the array of biology that can be affected by off-target responses to tamoxifen and related TPE’s.
4.2 TPE’s in cancer and other diseases
Several additional activities that are part of the TPE repertoire merit mention. For example, work in estrogen receptor-negative T-lymphoblastic leukemia cells demonstrates that tamoxifen, clomiphene, and nafoxidine (in the low micromorar range) are cytotoxic and induce apoptosis [48] . Whether perturbations in SL metabolism underlie these responses remains a question. Studies in nude mice have shown that tamoxifen reverses multidrug resistance in colorectal cancer, independent of estrogen receptor status [49]. Additional works also illustrate the benefits of tamoxifen in colorectal cancer [50-52] perhaps demonstrating the need for an in depth evaluation in this area. Other, more recent findings suggest that tamoxifen might be of utility in treating Duchenne muscular dystrophy [53]. Toremifene, a chlorinated derivative of tamoxifen (Fig. 3), has been positioned as a promising agent for prevention of prostate cancer [54] and shown to avert prostate cancer in a transgenic mouse model [55].
Tamoxifen has been shown active in combination with cisplatin in non-small cell lung cancer [56] and recently demonstrated to enhance erlotinib-induced cytotoxicity in non-small cell lung cancer [57]; interestingly, compared to single agent erlotinib, the combination reduced activation of phospho-AKT and phospho-ERK 1/2 and reduced thymidine phosphorylase levels, underscoring the implications of tamoxifen in rational design of novel drug regimens. Moreover, favorable responses in glioma are believed to be linked to the interaction of tamoxifen with protein kinase C (PKC) [58]. In MCF-7 human breast cancer cells, tamoxifen was shown to induce selective membrane association of PKC-epsilon [59], and in estrogen receptor-negative MDA-MB-231 cells, phosphatidic acid, a lipid second messenger, was generated in response to tamoxifen exposure [31,60]. In a human mammary fibroblast cell line, tamoxifen was shown to activate phospholipase C and D and elicit PKC translocation [61].
Last but not least, tamoxifen has been extensively studied since the late 1970’s as a possible chemotherapeutic agent for treatment of malignant melanoma [62]. In an attempt to enhance outcome in patients, a number of combination therapy regimens have been investigated. The “Dartmouth Regimen” the most extensively used, is comprised of dacarbazine (DTIC), cisplatin, carmustine (BCNU), and tamoxifen [63,64], Even though early studies related favorable overall response rates [65], the benefit of tamoxifen in the Dartmouth has been questioned [66]. A salient point in this instance regards molecular mechanisms of tamoxifen in melanomas, which to this day remain elusive. Some ideas on the subject include inhibition of tumor cell invasion and migration [67,68] and inactivation of insulin-like growth factor-1 receptor [69,70], which is important in tumor cell survival. The mechanisms underlying tamoxifen-induced programmed cell death in cancer, in general, have been reviewed by Mandlekar and Kong [71] .
5. Tamoxifen as modulator of multidrug resistance in cancer
This area of discussion speaks volumes and deserves a separate heading due to the influential role tamoxifen has played in attempts to remedy multidrug resistance. This area is also key to tamoxifen’s impact on SL metabolism, a mystery that will be disclosed in Section 7.
Multidrug resistance, inherent or acquired and an all too frequent characteristic of cancer cells, represents a serious barrier to successful treatment. Although many resistance mechanisms have been described, those that involve proteins belonging to the ABC (ATP binding cassette) transporter superfamily are of particular interest. Drug transport proteins such as P-glycoprotein (P-gp) and multidrug resistance protein (MRP), unwanted plasma membrane occupants in tumor cells, continue to be a major focus of laboratory and clinical studies in drug resistance [72,73]. By and large, research in this area has been directed towards overcoming the chemotherapy efflux capacity of the transporters with the goal of attaining lethal drug concentrations within tumor cells [74-78]. One way of limiting chemotherapy efflux is through the employ of antagonists to the drug pumps [79]. Whereas this goal remains promising, clinical use of a myriad of antagonists has provided little progress in treating drug-resistant forms of cancer [80].
The tantalizing question is, Why the big interest in tamoxifen in multidrug resistance? It’s no secret anymore that tamoxifen interacts directly with P-gp but itself is not a substrate for efflux [81,82]. In binding to P-gp, tamoxifen inhibits chemotherapy efflux, a good thing and reason for excitement in the clinical setting. Of the many P-gp antagonists that have been investigated, verapamil and cyclosporin A for example (Fig. 4), tamoxifen has had a long, enduring run in the clinic, having been paired with etoposide,vinblastine,CHOP(cyclophosphamide/doxorubicin/vincristine/prednisone/etoposide), daunorubicin, epirubicin, doxorubicin and dexverapamil, and paclitaxel, for treatment of advanced epithelial tumors, relapsed or refractory acute leukemia, hepatocellular carcinoma, colorectal cancer, non-Hodgkin’s lymphoma, metastatic melanoma, and advanced renal cell carcinoma [83-92]. Needless-to-say, such versatility is impressive, especially in estrogen receptor-negative circumstances. Although effective in vitro in reversing multidrug resistance, the combination regimens have been poorly beneficial, in vivo. However, divorcing tamoxifen from actions as modulator of chemotherapy efflux and employing it as a regulator of SL metabolism could be key to newfound utility.
Fig. 4.

Chemical structure of cyclosporin A and verapamil, prominent clinical agents employed as drug resistance modulators. Verapamil is a phenylalkylamine, L-type calcium channel blocker used in the treatment of hypertension, angina, and arrhythmia, and on the docket of the World Health Organization’s list of essential medicines. Cyclosporin A, a cyclic, 11-amino acid peptide, is an immunosuppressant used in organ transplantation to prevent rejection. Both verapamil and cyclosporin A inhibit ceramide glycosylation in cancer cells.
6. Tamoxifen effects on ceramide glycosylation
As noted above, keeping levels of ceramide “in check” is one way cancer cells manage life and death directions. In the mid-1990’s, we were the first to demonstrate that multidrug resistant cancer cells contained elevated levels of glycosylated ceramide, GC, compared to wild-type cancer cell counterparts [4,93], thus establishing a connection between chemotherapy resistance and alterations in ceramide metabolism. These findings marked a new push to examine the role of SL’s in chemotherapy response. In a unique model of acquired resistance developed by Gottesman and colleagues [94] , it was subsequently shown in cervical carcinoma cell lines, representing sequentially increased resistance to vinblastine, that GC levels increased proportionally; similar results were obtained in doxorubicin-resistant sublines [95]. Interestingly, a crescendo in P-gp expression in these cell lines mimicked the increases in GC levels, highlighting a possible correlation between GC production and P-gp expression. Until that time, relatively few works had delved into the area of glycolipids in drug-resistant cancer. Around the same time, groups from the United States and France demonstrated that daunorubicin induced apoptosis in leukemia cells via activation of de novo ceramide synthesis and activation of sphingomyelin hydrolysis [96], hence a landmark finding implicating ceramide in the cytotoxic action of heterocyclic anticancer agents. Thus, in terms of “protection”, it made sense for cancer cells to upregulate ceramide glycosylation, thereby capping cytotoxic threat.
The first report providing evidence that tamoxifen antagonized glycolipid synthesis surfaced in 1996 [19]. The study, conducted in KB-V-1 multidrug resistant cervical carcinoma cells and in a human melanoma cell line, showed in the former, that a 24 hr exposure to 5 μM tamoxifen reduced GC content by nearly 100%. In the melanoma model, tamoxifen exposure lowered endogenous levels of GC and higher metabolites, lactosylceramides and gangliosides; this was of particular interest not only because tamoxifen is a component of the Dartmouth regimen, but because melanoma is of neuroendocrine origin and thus rich in gangliosides that are posited to be associated with metastatic potential [97]. Using a crude enzyme preparation from melanoma cells, results of cell-free enzymatic assays revealed that tamoxifen was a poor GCS inhibitor, suggesting that inhibition of GC synthesis observed in intact cells was not due to direct GCS interaction.
In an effort to corroborate initial findings, work on the effects of tamoxifen on glycolipid metabolism was forwarded. In studies using radiolabeled palmitic acid, which enters the ceramide synthetic pathway de novo (Fig. 2) , Lavie et al [98] showed that tamoxifen induced a time-dependent, near-total depression of GC synthesis in multidrug resistant MCF-7/AdrR (NCI/ADR-NIH) cells, a work also revealing that triphenylbutene, which is “tamoxifen” devoid of the dimethylaminoethanol moiety, was a relatively ineffective inhibitor. Interestingly, when compared with the specific GCS inhibitor PPMP, tamoxifen potency was on a par [4] . In addition, this study showed that tamoxifen inhibited conversion of short-chain ceramide, C6-ceramide, to C6-GC in cells exposed to this apoptosis-inducing ceramide analog, a relevant finding in light of present day efforts using C6-ceramide in combinatorial anticancer regimens [99]. Toremifene (Fig. 3), a triphenylethylene with structure and pharmacology similar to tamoxifen, has likewise been shown effective in inhibiting GC synthesis in drug resistant cancer cells [100].
Limited information is available regarding the structure activity relationship of tamoxifen for inhibition of ceramide glycosylation, although DMT has been shown as effective as tamoxifen in halting C6-ceramide glycosylation [101]. Knowledge in this area would provide a useful guide for the rational engineering of potent ligands based on the chemical structure of tamoxifen. Poirot and colleagues [29] have reported in depth on the structural features of tamoxifen involved in multiple targeting with regard to not only genomic effects but to non-genomic targets such as the microsomal anti-estrogen binding site, protein kinase C, calmodulin-dependent enzymes, and cholesterol acyltransferase.
Several works on tamoxifen and regulation of ceramide glycosylation support the initial findings. For example, in a study on the accumulation of GC in cytoplasmic droplets in multidrug resistant cancer cells, Morjani et al [5] found that tamoxifen mimicked the action of PPMP in attenuating GC buildup and proposed that tamoxifen elicited influence via disruption of lipid metabolism. Scarlatti et al [102], investigating ceramide-mediated macroautophagy, showed that tamoxifen-dependent accumulation of autophagic vacuoles in HT-29 colon cancer cells could be mimicked by another GCS inhibitor, PDMP. In experiments involving anticancer peptides, Furlong et al [103] concluded that the manipulation of cellular ceramide levels by tamoxifen might be essential to enhance the apoptosis-inducing potential of these peptides, and in a study on the anti-apoptotic role of GCS, Baran and colleagues [104] showed in a breast cancer cell model, that tamoxifen downregulated GCS expression, and suggested a link between this downregulation and the accumulation of apoptotic ceramides.
7. Tamoxifen effects on ceramide glycosylation—proposed mechanism
Shortly after the discovery demonstrating the inhibitory effects of tamoxifen on glycolipid synthesis, it was revealed that this action was shared with verapamil and cyclosporin A (Fig. 4) [98] , classical drug resistance reversing agents [105]. Further, the potencies of these agents were similar, as tested in NCI/ADR-RES cells. As it was construed unlikely that tamoxifen, the phenylalkylamine, L-type calcium channel blocker, verapamil, and a cyclic, 11-amino acid peptide from Tolypocladium inflatum, cyclosporin A, agents with vastly dissimilar structures (Fig. 3,4), could all inhibit GCS, questions about mechanism of action flourished.
Work implicating GC in the biochemistry of multidrug resistance [4,93,100], studies revealing that elevated levels of GC in cancer cells correlate with overexpression of P-gp [4,95], the finding that commonly employed P-gp antagonists suppressed GC synthesis in drug resistant cancer cells [98], and novel research from The Netherlands, showing that Golgi-resident P-gp can function as a SM and GC transmemebrane flippase [20,106], gave life to the idea that P-gp played a role in ceramide metabolism and may even be involved in regulating ceramide-orchestrated cancer cell death. The experiments of De Rosa et al [107] also demonstrated that drug resistance proteins assist in neutral glycosphingolipid biosynthesis. To this end, in very enlightening works, Smyth et al [107] showed that P-gp protected drug-resistant leukemia cells from caspase-dependent apoptosis mediated by cytotoxic drugs and by ligation of Fas (a ceramide-linked death cascade), implying that P-gp exerts a role in regulating some caspase-dependent apoptotic pathways. In furtherance, Shabbits and Mayer [108] demonstrated that P-gp modulated ceramide-mediated sensitivity to paclitaxel, a ceramide generating taxane [109], suggesting that ceramide metabolism and apoptosis are regulated not only by GCS but by P-gp. Even more enlightening, a subsequent study in leukemia demonstrated that resistance to short-chain ceramide-induced apoptosis in P-gp-positive cell lines could be alleviated by exposure to P-gp antagonists GF120918 and cyclosporin A, wherein expression of P-gp was identified in the Golgi [110]. Thus, P-gp appears to be a secret ingredient in the ceramide glycosylation pathway. And lastly, the reader should consider work by Sietsma et al [111], on the role of SL’s in drug resistance mechanisms. This insightful review, although focusing on neuroblastoma, discusses refined ensemble between sphingolipids and ATP-binding cassette transporters that may stanchion the drug resistant phenotype.
The idea that P-gp protects cells from ceramide-governed cytotoxicity has been supported by an informative set of studies in HeLa cells that have conditional expression of P-gp [112], a clean system with which to dissect the role of the drug pump in ceramide metabolism and cytotoxic response. That study revealed that P-gp-expressing HeLa cells converted ceramide to GC at > 4-times the rate of P-gp-void cells. P-gp-rich cells were also resistant to ceramide whilst cells devoid of P-gp were sensitive. Further and most interesting, ceramide resistance inherent in P-gp-rich cells was reversed by tamoxifen, and it turns out, this reversal was accompanied by a block in GC production. In cell-free assays, tamoxifen did not inhibit GC synthesis, indicating that machinery of the intact cell is requisite for the tamoxifen effects. Thus, it appears that P-gp is an essential guardian of ceramide-associated cytotoxicity.
Follow-up work in wild-type and multidrug resistant counterparts cemented the role of P-gp as ceramide sentry. For example, consider the study by Chapman et al [112], demonstrating that multidrug resistant ovarian cancer cells possessed an enhanced capacity for conversion of short-chain ceramides to short-chain GC, compared to wild-type counterpart, albeit the levels of cell-free GCS activity were similar. Curiously, whereas low-dose GCS inhibitor, ethylenedioxy-P4 (0.2 μM), blocked C6-GC formation nearly totally, it did not magnify C6-ceramide cytotoxicity, indicating that inhibition of GCS failed to sensitize cells to C6-ceramide. However, increasing the dose of ethylenedioxy-P4 (5.0 μM) produced C6-ceramide sensitization, suggesting that at higher doses, GCS inhibitors interact with P-gp in much the same fashion as tamoxifen. This idea is championed by the work of Sietsma et al [113], who demonstrated that the GCS inhibitor and chemical cousin of ethylenedioxy-P4, PDMP, decreased paclitaxel and vincristine efflux in neuroblastoma cells, and thus acts as a P-gp antagonist. In a culminating, informative work, Chai et al [114] demonstrated that the chemosensitizing properties of the P-drug GCS inhibitors was mediated primarily through modulation of P-gp function, bringing us, as in sonata allegro form, to the recapitulation of the original theme: that P-gp potentiates ceramide glycosylation, and if antagonized, augments ceramide sensitivity, both features previous ascribed to GCS. Bolstering this theme, Norris-Cervetto et al [115] published work showing that inhibition of GCS is not sufficient to reverse drug resistance in cancer cells. In this instance, the GCS inhibitor was NB-DNJ (N-butyl-deoxynojirimycin, miglustat), a water-soluble imido sugar with little affection for P-gp.
Although it may be an over-simplification, the upshot from these studies is that P-gp confers resistance to ceramide-regulated cell death by hastening it’s conversion to GC (Fig. 5). GC, synthesized at the cytosolic surface of the Golgi, can be flipped into the Golgi by Golgi-resident P-gp, where it is converted to lactosylceramide by lactosylceramide synthase. We believe the efficiency of this “hand-off” contributes to ceramide resistance by providing a driving force to perpetuate ceramide → GC conversion that limits negative feedback inhibition of GCS. But, when this conveyance is interrupted, for example by P-gp antagonists (Fig. 5), product inhibition occurs and contributes to a reduction in GC levels and in some instances, increases in ceramide. We are now seeing that this type of SL trafficking can be harnessed for enhancing sensitivity to ceramide-generating anticancer agents as well as sensitivity to short-chain ceramides [101,116-118], a topic covered in Section 9.
Fig. 5.

Schematic illustrating effect of tamoxifen and classical P-gp antagonists on GC synthesis. Shown here using short-chain, C6-ceramide as starting substrate, conversion to GC is catalyzed by GCS (shown in blue) at the ER-Golgi interface. Resultant GC can be transported into the Golgi lumen by the flippase actions (shown in green) of P-gp where it is further metabolized to LC by LCS (shown in blue). Tamoxifen and other P-gp antagonists inhibit transit of newly synthesized GC into the Golgi lumen, interference that effectively halts ceramide glycosylation, resulting in ceramide buildup. P-gp, P-glycoprotein; GC, glucosylceramide; GCS, glucosylceramide synthase; ER, endoplasmic reticulum; LC, lactosylceramide; LCS, lactosylceramide synthase.
8. Effects of tamoxifen on acid ceramidase activity
Acid ceramidase, also known as N-acylsphingosine deacylase, was first described in 1963, by Shimon Gatt [119], in studies being conducted at Hadassah Medical School, The Hebrew University, Jerusalem. Due to its command over ceramide fate, this lysosomal enzyme now holds a lofty position in cancer biology; a vast body of literature has been generated revealing that AC, in addition to enhancing cell survival and resistance to programmed cell death, stands as a critical regulator of cell progression, migration, and invasion [117,120-124] .
By catalyzing ceramide hydrolysis (see Fig. 1), AC functions as a gatekeeper removing ceramide and thereby blunting a host of ceramide-directed cell death cascades [1-3] . In addition, ceramide hydrolysis frees sphingosine, a substrate for Sphk, a rising star in the arena of cancer therapy [125]. Sphingosine is used in the generation of S1P, a mitogenic, pro-survival virtuoso [126]. Thus AC, a “Janus-type” enzyme on one hand powers-down apoptotic responses, and on the other, contributes to promotion of cancer cell growth.
Of neutral, alkaline, and acid ceramidases, presently AC holds the dominant position, having been designated an important target in cancer therapy [127]. Highlighting this designation are studies in prostate cancer, where AC expression relates to poor outcome [128] and resistance to radiation [129]. In melanoma, AC expression modulates sensitivity to dacarbazine [130]. These authors showed that DTIC decreased AC activity and increased intracellular ceramide levels, actions driven by reactive oxygen species-dependent activation of cathepsin B-mediated digestion of AC. Because tamoxifen has long been used for treatment of melanoma, and DTIC and tamoxifen share AC as a common target, these intriguing connections are interesting food-for-thought. And worthy of mention, a genetic study showed that AC is among the most important candidate genes in melanoma diagnostics [131]. With this, we believe that design and evaluation of inhibitors of AC are important priorities in cancer medicine, but perhaps more specifically in melanoma and in prostate cancer.
The first demonstration of AC inhibition by tamoxifen was conducted in mutant p53, triple-negative MDA-MB-468 human breast cancer cells [116] . This report showed that tamoxifen, formulations of nanoliposomal tamoxifen, DMT, and 4-hydroxytamoxifen inhibited AC activity in intact cells, whereas triphenylbutene, the aromatic tamoxifen nucleus, was devoid of activity. Pre-exposure of cells to tamoxifen also led to AC inhibition, as documented in cell-free assays conducted with lysates. Tamoxifen did not inhibit AC activity when added directly to the enzyme incubation, suggesting that the mechanism underlying enzyme inhibition required intact cells to proceed. Subsequent work demonstrated that tamoxifen inhibited AC in a wide variety of cancer cells, prostate, breast, pancreatic, colorectal, and leukemia [124,132].
Clues regarding tamoxifen mechanism of action were presented in and interesting work by Elojeimy et al [133] using desipramine, the tricyclic anti-depressant, which is an amphiphilic, lysomotropic agent, similar to chlorpromazine and chloroquine. This group showed that desipramine’s inhibition of AC in prostate cancer cells was accompanied by downregulation of AC protein levels elicited by cathepsin B-driven proteolytic degradation. Using this knowledge as a spring board, Morad et al [124] were able to establish that tamoxifen affected rapid, marked lysosomal membrane permeability that was accompanied by dose- and time-dependent diminution of AC protein expression. Lysosomes contain numerous hydrolytic enzymes with acidic pH optima, including proteases, and lysosomal membrane permeability can lead to release of these enzymes and their destructive element. As lysosomes appear to be an early, primary tamoxifen target, Morad et al [124] assessed the impact of various protease inhibitors and were able to demonstrate that cathepsin B was responsible for tamoxifen-induced AC diminuendo. In summary, these studies reveal another off-target, SL-associated effect of tamoxifen and tamoxifen metabolites, inhibition of AC. However, as the experts may see it, inhibition of this type is non-specific, but never-the-less, depressing the action of AC has relevance in the realm of SL signaling and cancer biology [134,135] .
As mentioned Section 2, “Sphingolipid Metabolism”, enzymatic degradation of ceramide frees sphingosine that can be converted to S1P by Sphk (see Fig. 1). Halting this transaction can be beneficial in terms of cancer therapy, because cancers love Sphk [136-140]. In a recent study in leukemia, both tamoxifen and DMT were shown to effectively block the formation of S1P and its precursor [132]. In concert, GC levels were greatly diminished through tamoxifen-orchestrated inhibition of ceramide glycosylation, resulting in increased levels of ceramide. Thus, tamoxifen blocks two important junctures in sphingolipid metabolism, ceramide glycosylation and hydrolysis. The study also showed, for the first time, that tamoxifen exposure can elicit downregulated expression of SphK1 resulting in greatly diminished Sphk activity. Interestingly, although downregulation of SphK1 alone would limit formation of S1P, this action is a redundant step in light of inhibition of AC by tamoxifen (see Fig. 1). Complementary and with clinical implications is the work of Bonhoure et al [141] showing that inhibition of Sphk1 elicits production of apoptotic markers and downregulates expression of anti-apoptotic XIAP in leukemia, effects that were accompanied by release of cytochrome c and SMAC/Diablo.
9. Tamoxifen as adjuvant with ceramide-centric therapies
In most instances when GCS inhibitors such as PPMP are employed to enhance efficacy of ceramide centric therapies, tamoxifen can substitute and yield similar results. This is likely because both agents interact with P-gp, which halts GC transit resulting in a shut-down of ceramide glycosylation and buildup of ceramides. The question is, Why is it more efficacious to target P-gp as opposed to GCS? This puzzle is curiously thought-provoking. Presented are the following cases in point: i) the water-soluble imido sugar inhibitors of GCS are ineffective in reversing drug resistance in multidrug resistant cell lines, whereas PDMP is effective [115], suggesting that inhibition of GCS does not reverse drug resistance. Thus, Norris-Cervetto et al [115] posed that chemosensitization achieved by PDMP cannot be caused by inhibition of GCS; ii) the observations of Shabbits et al [108] imply that ceramide metabolism and apoptosis effects are regulated not only by GCS but by P-gp; iii) definitive work by Chai et al [114] indicates that the sensitizing properties of the P-drug GCS inhibitors like PPMP are primarily mediated via modulation of P-gp function; iv) Chapman et al showed that although sub-micromolar ethylenedioxy-P4, a P-drug, inhibited GCS it failed to sensitize multidrug resistant ovarian cancer cells to C6-ceramide [142]. The take-home message in these studies, from our standpoint, is that in order to magnify the effects of ceramide-type therapies it is more advantageous to employ P-gp antagonists as opposed to GCS inhibitors, in other words, you are better off targeting SL transit. Hence, P-gp antagonists should be effective enhancers of ceramide-centric therapies, and indeed they are, as has been shown for not only tamoxifen, but as well for VX-710, cyclosporin A, and verapamil [101,142]. This raises the interesting idea of a “rebirth” or new indication for classical drug sensitizers, which by-and-large have failed as inhibitors of heterocyclic chemotherapy drug “efflux”.
Because ceramide is a versatile inducer of programmed cell death, its use as a drug is certainly attractive. However, the development of ceramide-based therapies has been to a degree limited by resistance of cancer cells that is in part driven by the upregulated metabolic clearance of ceramide. This predicament can be circumvented by targeting ceramide metabolism at glycosylation and hydrolysis. Discussion in this section will be limited to approaches for intensifying the effects of ceramide-based therapies through the employ of tamoxifen, its metabolites, or related triphenylethylenes. Noteworthy in this instance is the use of short-chain ceramides in nanoliposomal formulations, a strategy that has shown promise in both single-agent and combination approaches in systemic treatment of breast cancer, hepatocellular carcinoma, large granular lymphocytic leukemia, and melanoma [99]. It merits mention, however, that the capacity of different types of cancer cells to metabolize ceramide varies greatly [117]; therefore, inclusion of agents like tamoxifen or allied SL inhibitors can have significant application in cancer therapy, as often alluded to in this review.
For the most part, the majority of studies employing tamoxifen as adjuvant to ceramide-based therapies have been conducted with N-(4-hydroxylphenyl)retinamide (4-HPR, fenretinide) and with C6-ceramide (Table 1). Although 4-HPR produces a myriad of effects culminating in cytotoxicity, ceramide production, if not first violin, is considered a principal player in the cytotoxic response [143-145]. In this regard, an interesting side-action of 4-HPR is worthy of mention. 4-HPR can act as an inhibitor of dihydroceramide desaturase [146-148] therefore, dihydroceramides rather than ceramides can accumulate [149]. Although there is great discussion regarding the cytotoxic responses elicited by dihydroceramides versus ceramides [150], this point will not be illuminated here. Earlier studies from 2000, clearly showed the utility of adjuvant tamoxifen with 4-HPR in neuroblastoma, a benefit that was on a par with combination PPMP-4-HPR [151] . In prostate cancer cells, 4-HPR activates SPT resulting in ceramide generation; however, the ceramide produced is converted to GC in prostate cancer (Fig. 6), [118], a pro-survival mechanism. In that study the inclusion of tamoxifen synergized with 4-HPR to enhance the production of ceramide, block production on GC, and intensify cytotoxic endpoint. Such results are in line with a Commentary by Norman Radin in his article on chemotherapy by slowing glycolipid synthesis [152]. Because tamoxifen also limits AC activity (Fig. 6), we venture that 4-HPR-tamoxifen would be an especially effective regimen in prostate cancer, where AC portends poor prognosis [14]. Moreover, in leukemia, Morad et al [132] recently showed that tamoxifen and DMT are effective enhancers of C6-ceramide- and 4-HPR-induced therapeutic activity, supporting the utility of TPE’s as adjuvants with ceramide-centric therapies.
Table 1.
Adjuvant triphenylethyenes enhance therapeutic potential of ceramide-centric therapies
| Adjuvant | Ceramide-centric therapy | Cancer model | Reference |
|---|---|---|---|
| Tamoxifen | 4-HPR | Neuroblastoma, Colon |
[151] |
| 4-HPR | Prostate | [118] | |
| C6-ceramide | Melanoma | [154] | |
| C6-ceramide | Colon, Breast, Leukemia |
[117] | |
| C6-ceramide | Ovarian | [142] | |
| C6-ceramide | Cervical | [112] | |
| lactoferricin | Breast | [103] | |
| lactoferricin | Leukemia | [122] | |
| beta-Sitosterol | Breast | [155] | |
| Doxorubicin ± PSC 833 | Ovarian | [161] | |
| Gefitinib ,Etoposide, | Prostate | [162] | |
| C6-ceramide, doxorubicin | Ovarian | [163] | |
| Tamoxifen, DMT | 4-HPR, C6-ceramide | Leukemia | [132] |
| C6-ceramide | Colon | [101] | |
|
Tamoxifen, DMT,
4-hydroxytamoxifen |
C6-ceramide | Breast | [116] |
| 4-hydroxytamoxifen | Persin | Breast | [156] |
| Tamoxifen nanoparticles | Paclitaxel | Ovarian | [157] |
DMT, N-desmethyltamoxifen; 4-HPR, fenretinide; AML, acute myeloid leukemia
Fig. 6.

A combinatorial approach to suppression of cancer growth that employs 4-HPR and tamoxifen. Using prostate cancer cells (PC-3) as an example, 4-HPR activates SPT. This step forms sphinganine that is used for synthesis of ceramides, a process resulting in ceramide production. The addition of tamoxifen blocks cellular conversion of ceramide to GC, resulting in ceramide buildup (up arrows) with concurrent magnification of ceramide cancer suppressor effects. Tamoxifen also inhibits AC, a vital enzymatic junction. This inhibition leads to diminished levels of sphingosine that in turn limit production of mitogenic S1P. Not depicted here: the de novo pathway intermediates of ceramide biosynthesis and the impact of 4-HPR on dihydroceramide desaturase. SPT, serine palmitoyltransferase; GC, glucosylceramide; AC, acid ceramidase; P-gp, P-glycoprotein; S1P, sphingosine 1-phosphate.
The employ of a cell-deliverable ceramide, such as C6-ceramide, in combination with tamoxifen, has been shown to be an effective regimen in numerous studies in various cancer cell models (Table 1). For example, tamoxifen complements the therapeutic potential of C6-ceramide in prostate, breast, ovarian, and colorectal cancer, and in neuroblastoma and melanoma. In hormone-insensitive, triple-negative breast cancer cells, tamoxifen, DMT, and 4-hydroxytamoxifen were all shown of benefit when paired with C6-ceramide [153]; combinations reduced cell viability, elicited DNA fragmentation, induced cell cycle arrest, and promoted a robust increase in mitochondrial membrane potential. Interestingly and on point, in melanoma, where tamoxifen has been used in the Dartmouth regimen, paring with C6-ceramide inhibited cell proliferation, induced caspase-dependent apoptosis, promoted cell cycle arrest, diminished adhesion and migration, and downregulated survivin, a driving force of chemotherapy resistance in melanoma [154] .
Other ceramide-targeted agents that have been used in combination with tamoxifen include beta-sitosterol [155], a dietary phytosterol found in legumes, lactoferricin B [103,122], a cationic antimicrobial peptide, and the plant toxin, persin [156], derived from avocado leaves (Table 1). The magic between beta-sitosterol and tamoxifen in enhancing breast cancer cell kill lies is the potent activation of SPT by the former and blockade of ceramide glycosylation by the latter [155] and suggests that this natural product in combination with tamoxifen could be of benefit in management of breast cancer. Lactoferricin B activity has been examined in both breast cancer and in T-cell leukemia [103,122], where proapoptotic activity was enhanced by either C6-ceramide or tamoxifen. However, as lactoferricin B is not a ceramide-generating agent, the mechanisms underlying the potentiating effects remain unsolved. With persin, cytotoxic activity was magnified in human breast cancer cells by 4-hydroxytamoxifen, independent of estrogen receptor status and was associated with increased de novo ceramide synthesis. To modulate drug resistance in ovarian carcinoma via enhancement of intracellular ceramide levels, Amiji and colleagues [157] employed unique tamoxifen-loaded biodegradable polymeric nanoparticles in combination with paclitaxel and demonstrated a clinically translatable strategy.
10. Concluding remarks
As reviewed herein, tamoxifen, metabolites, and allied TPE’s have a unique influence on SL metabolism, an influence that can be harnessed to enhance the pro-apoptotic properties of ceramide, bearing relevance to clinical application. We and others have demonstrated that with a high-dose tamoxifen regimen, steady-state serum concentrations as high as 4.6 μM are attainable [158,159], and intra-tumoral concentrations can be 5-7 fold greater than serum [160]. Thus, use of tamoxifen in an adjuvant setting wherein high doses are needed, is within the realm of utility. However, we must address whether tamoxifen’s not insignificant side-effects might be changed by the proposed combination therapies. Firstly, there exist other TPE’s that could be employed, such as Raloxifene (Evista) and Fereston (toremifene). These TPE’s were developed as alternatives to tamoxifen in an effort to deliver agents with less side-effect. One of these agents, toremifene, has been demonstrated to block ceramide glycosylation in multidrug resistant human ovarian cancer cells [100] as well as inhibit AC activity in prostate cancer cells [124]. With specific regard to combination regimens, human peripheral blood mononuclear cells are refractory to C6-ceramide-tamoxifen at concentrations as high as 10 + 10 μM, over a 48 hr exposure period (unpublished data, investigator’s laboratory). But perhaps most appealing, however, is DMT, the tamoxifen metabolite with practically no affinity for the estrogen receptor [23], yet a potent inhibitor of SL metabolism, as related in Sections 6 and 8.
Finally, a most important message here regards the utility of P-gp antagonists, that here-to-for have been administered with heterocyclic, cytotoxic chemotherapy drugs, which may not have been the best partnering agents. Whether partnering classical P-gp antagonists such as tamoxifen, verapamil, or cyclosporin A, with ceramide-centric therapies “is the ticket” remains to be explored in relevant clinical models.
Highlights.
Tamoxifen is a potent regulator of sphingolipid metabolism
Tamoxifen inhibits ceramide glycosylation as well as ceramide hydrolysis
Tamoxifen can magnify ceramide-induced apoptosis in cancer cells
Tamoxifen and ceramide-centric therapies may have clinical relevance
Acknowledgements
This work was supported by the National Institutes of Health (NCI) grant number 5 P01 CA171983-02.
Abbreviations
- SL
sphingolipid
- S1P
sphingosine 1-phosphate
- GCS
glucosylceramide synthase
- GC
glucosylceramide
- AC
ceramidase
- SphK
sphingosine kinase
- PPMP
D-threo-1-phenyl-2-hexadecanoylamino-3-morpholino-1-propanol
- PDMP
D-threo-1-phenyl-2-decanoylamino-3-morphomolino-1-propanol
- TPE’s
triphenylethylenes
- DMT
N-desmethyltamoxifen
- SM
sphingomyelin
- SERM
selective estrogen receptor modulator
- ACAT
Acyl-CoA:cholesterol acyl transferase
- DTIC
dacarbazine
- BCNU
carmustine
- ABC
ATP binding cassette
- P-gp
P-glycoprotein
- MRP
multidrug resistance protein
- NB-DNJ
N-butyl-deoxynojirimycin
- 4-HPR
N-(4-hydroxylphenyl) retinamide
- ER
endoplasmic reticulum
- LC
lactosylceramide
- LCS
lactosylceramide synthase
- SPT
serine palmitoyltransferase
- AML
acute myeloid leukemia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Truman JP, Garcia-Barros M, Obeid LM, Hannun YA. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim Biophys Acta. 2014;1841:1174–1188. doi: 10.1016/j.bbalip.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer. 2013;13:51–65. doi: 10.1038/nrc3398. [DOI] [PubMed] [Google Scholar]
- [3].Saddoughi SA, Ogretmen B. Diverse functions of ceramide in cancer cell death and proliferation. Adv Cancer Res. 2013;117:37–58. doi: 10.1016/B978-0-12-394274-6.00002-9. [DOI] [PubMed] [Google Scholar]
- [4].Lavie Y, Cao H, Bursten SL, Giuliano AE, Cabot MC. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J Biol Chem. 1996;271:19530–19536. doi: 10.1074/jbc.271.32.19530. [DOI] [PubMed] [Google Scholar]
- [5].Morjani H, Aouali N, Belhoussine R, Veldman RJ, Levade T, Manfait M. Elevation of glucosylceramide in multidrug-resistant cancer cells and accumulation in cytoplasmic droplets. Int J Cancer. 2001;94:157–165. doi: 10.1002/ijc.1449. [DOI] [PubMed] [Google Scholar]
- [6].Gutierrez-Iglesias G, Hurtado Y, Palma-Lara I, Lopez-Marure R. Resistance to the antiproliferative effect induced by a short-chain ceramide is associated with an increase of glucosylceramide synthase, P-glycoprotein, and multidrug-resistance gene-1 in cervical cancer cells. Cancer Chemother Pharmacol. 2014;74:809–817. doi: 10.1007/s00280-014-2552-3. [DOI] [PubMed] [Google Scholar]
- [7].Xie P, Shen YF, Shi YP, Ge SM, Gu ZH, Wang J, Mu HJ, Zhang B, Qiao WZ, Xie KM. Overexpression of glucosylceramide synthase in associated with multidrug resistance of leukemia cells. Leuk Res. 2008;32:475–480. doi: 10.1016/j.leukres.2007.07.006. [DOI] [PubMed] [Google Scholar]
- [8].Liu YY, Patwardhan GA, Xie P, Gu X, Giuliano AE, Cabot MC. Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int J Oncol. 2011;39:425–431. doi: 10.3892/ijo.2011.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gouaze-Andersson V, Yu JY, Kreitenberg AJ, Bielawska A, Giuliano AE, Cabot MC. Ceramide and glucosylceramide upregulate expression of the multidrug resistance gene MDR1 in cancer cells. Biochim Biophys Acta. 2007;1771:1407–1417. doi: 10.1016/j.bbalip.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu YY, Han TY, Giuliano AE, Cabot MC. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001;15:719–730. doi: 10.1096/fj.00-0223com. [DOI] [PubMed] [Google Scholar]
- [11].Baran Y, Bielawski J, Gunduz U, Ogretmen B. Targeting glucosylceramide synthase sensitizes imatinib-resistant chronic myeloid leukemia cells via endogenous ceramide accumulation. J Cancer Res Clin Oncol. 2011;137:1535–1544. doi: 10.1007/s00432-011-1016-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zeidan YH, Jenkins RW, Korman JB, Liu X, Obeid LM, Norris JS, Hannun YA. Molecular targeting of acid ceramidase: implications to cancer therapy. Curr Drug Targets. 2008;9:653–661. doi: 10.2174/138945008785132358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Camacho L, Meca-Cortes O, Abad JL, Garcia S, Rubio N, Diaz A, Celia-Terrassa T, Cingolani F, Bermudo R, Fernandez PL, Blanco J, Delgado A, Casas J, Fabrias G, Thomson TM. Acid ceramidase as a therapeutic target in metastatic prostate cancer. J Lipid Res. 2013;54:1207–1220. doi: 10.1194/jlr.M032375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu X, Cheng JC, Turner LS, Elojeimy S, Beckham TH, Bielawska A, Keane TE, Hannun YA, Norris JS. Acid ceramidase upregulation in prostate cancer: role in tumor development and implications for therapy. Expert Opin Ther Targets. 2009;13:1449–1458. doi: 10.1517/14728220903357512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Orr Gandy KA, Obeid LM. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway in disease: review of sphingosine kinase inhibitors. Biochim Biophys Acta. 2013;1831:157–166. doi: 10.1016/j.bbalip.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1- phosphate, apoptosis and diseases. Biochim Biophys Acta. 2006;1758:2016–2026. doi: 10.1016/j.bbamem.2006.08.007. [DOI] [PubMed] [Google Scholar]
- [17].Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism--a strategy for overcoming drug resistance. J Natl Cancer Inst. 2001;93:347–357. doi: 10.1093/jnci/93.5.347. [DOI] [PubMed] [Google Scholar]
- [18].Abe A, Radin NS, Shayman JA, Wotring LL, Zipkin RE, Sivakumar R, Ruggieri JM, Carson KG, Ganem B. Structural and stereochemical studies of potent inhibitors of glucosylceramide synthase and tumor cell growth. J Lipid Res. 1995;36:611–621. [PubMed] [Google Scholar]
- [19].Cabot MC, Giuliano AE, Volner A, Han TY. Tamoxifen retards glycosphingolipid metabolism in human cancer cells. FEBS Lett. 1996;394:129–131. doi: 10.1016/0014-5793(96)00942-8. [DOI] [PubMed] [Google Scholar]
- [20].van Helvoort A, Smith AJ, Sprong H, Fritzsche I, Schinkel AH, Borst P, van Meer G. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell. 1996;87:507–517. doi: 10.1016/s0092-8674(00)81370-7. [DOI] [PubMed] [Google Scholar]
- [21].Lim YC, Desta Z, Flockhart DA, Skaar TC. Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother Pharmacol. 2005;55:471–478. doi: 10.1007/s00280-004-0926-7. [DOI] [PubMed] [Google Scholar]
- [22].Lien EA, Solheim E, Ueland PM. Distribution of tamoxifen and its metabolites in rat and human tissues during steady-state treatment. Cancer Res. 1991;51:4837–4844. [PubMed] [Google Scholar]
- [23].Fabian C, Tilzer L, Sternson L. Comparative binding affinities of tamoxifen, 4-hydroxytamoxifen, and desmethyltamoxifen for estrogen receptors isolated from human breast carcinoma: correlation with blood levels in patients with metastatic breast cancer. Biopharm Drug Dispos. 1981;2:381–390. doi: 10.1002/bdd.2510020407. [DOI] [PubMed] [Google Scholar]
- [24].Bignon E, Pons M, Dore JC, Gilbert J, Ojasoo T, Miquel JF, Raynaud JP, Crastes de Paulet A. Influence of di- and tri-phenylethylene estrogen/antiestrogen structure on the mechanisms of protein kinase C inhibition and activation as revealed by a multivariate analysis. Biochem Pharmacol. 1991;42:1373–1383. doi: 10.1016/0006-2952(91)90448-e. [DOI] [PubMed] [Google Scholar]
- [25].Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 2. Clinical considerations and new agents. J Med Chem. 2003;46:1081–1111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- [26].Jordan VC. Biochemical pharmacology of antiestrogen action. Pharmacol Rev. 1984;36:245–276. [PubMed] [Google Scholar]
- [27].Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J Med Chem. 2003;46:883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- [28].Meegan MJ, Lloyd DG. Advances in the science of estrogen receptor modulation. Curr Med Chem. 2003;10:181–210. doi: 10.2174/0929867033368501. [DOI] [PubMed] [Google Scholar]
- [29].de Medina P, Favre G, Poirot M. Multiple targeting by the antitumor drug tamoxifen: a structure-activity study. Curr Med Chem Anticancer Agents. 2004;4:491–508. doi: 10.2174/1568011043352696. [DOI] [PubMed] [Google Scholar]
- [30].Bentrem DJ, Jordan V. Craig. Tamoxifen, raloxifene and the prevention of breast cancer. Minerva Endocrinol. 2002;27:127–139. [PubMed] [Google Scholar]
- [31].Kellen JA. Tamoxifen: Beyond the Antiestrogen Birkhäuser. Boston: 1996. [Google Scholar]
- [32].Wiseman H. Tamoxifen as an antioxidant and cardioprotectant. Biochem Soc Symp. 1995;61:209–219. doi: 10.1042/bss0610209. [DOI] [PubMed] [Google Scholar]
- [33].Christodoulakos GE, Lambrinoudaki IV, Botsis DC. The cardiovascular effects of selective estrogen receptor modulators. Ann N Y Acad Sci. 2006;1092:374–384. doi: 10.1196/annals.1365.034. [DOI] [PubMed] [Google Scholar]
- [34].Hernandez E, Valera R, Alonzo E, Bajares-Lilue M, Carlini R, Capriles F, Martinis R, Bellorin-Font E, Weisinger JR. Effects of raloxifene on bone metabolism and serum lipids in postmenopausal women on chronic hemodialysis. Kidney Int. 2003;63:2269–2274. doi: 10.1046/j.1523-1755.2003.00005.x. [DOI] [PubMed] [Google Scholar]
- [35].Saarto T, Blomqvist C, Ehnholm C, Taskinen MR, Elomaa I. Antiatherogenic effects of adjuvant antiestrogens: a randomized trial comparing the effects of tamoxifen and toremifene on plasma lipid levels in postmenopausal women with node-positive breast cancer. J Clin Oncol. 1996;14:429–433. doi: 10.1200/JCO.1996.14.2.429. [DOI] [PubMed] [Google Scholar]
- [36].Morales M, Santana N, Soria A, Mosquera A, Ordovas J, Novoa J, Betancor P, Valeron PF, Diaz-Chico B, Chirino R. Effects of tamoxifen on serum lipid and apolipoprotein levels in postmenopausal patients with breast cancer. Breast Cancer Res Treat. 1996;40:265–270. doi: 10.1007/BF01806815. [DOI] [PubMed] [Google Scholar]
- [37].Wiseman H. Tamoxifen: new membrane-mediated mechanisms of action and therapeutic advances. Trends Pharmacol Sci. 1994;15:83–89. doi: 10.1016/0165-6147(94)90283-6. [DOI] [PubMed] [Google Scholar]
- [38].Jolly EE, Bjarnason NH, Neven P, Plouffe L, Jr., Johnston CC, Jr., Watts SD, Arnaud CD, Mason TM, Crans G, Akers R, Draper MW. Prevention of osteoporosis and uterine effects in postmenopausal women taking raloxifene for 5 years. Menopause. 2003;10:337–344. doi: 10.1097/01.GME.0000058772.59606.2A. [DOI] [PubMed] [Google Scholar]
- [39].Ko SS, Jordan VC. Treatment of osteoporosis and reduction in risk of invasive breast cancer in postmenopausal women with raloxifene. Expert Opin Pharmacother. 2011;12:657–674. doi: 10.1517/14656566.2011.557360. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [40].Lewiecki EM. Current and emerging pharmacologic therapies for the management of postmenopausal osteoporosis. J Womens Health (Larchmt) 2009;18:1615–1626. doi: 10.1089/jwh.2008.1086. [DOI] [PubMed] [Google Scholar]
- [41].Arun B, Anthony M, Dunn B. The search for the ideal SERM. Expert Opin Pharmacother. 2002;3:681–691. doi: 10.1517/14656566.3.6.681. [DOI] [PubMed] [Google Scholar]
- [42].Clarke BL, Khosla S. New selective estrogen and androgen receptor modulators. Curr Opin Rheumatol. 2009;21:374–379. doi: 10.1097/BOR.0b013e32832ca447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].de Medina P, Payre BL, Bernad J, Bosser I, Pipy B, Silvente-Poirot S, Favre G, Faye JC, Poirot M. Tamoxifen is a potent inhibitor of cholesterol esterification and prevents the formation of foam cells. J Pharmacol Exp Ther. 2004;308:1165–1173. doi: 10.1124/jpet.103.060426. [DOI] [PubMed] [Google Scholar]
- [44].Kusunoki J, Hansoty DK, Aragane K, Fallon JT, Badimon JJ, Fisher EA. Acyl-CoA:cholesterol acyltransferase inhibition reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;103:2604–2609. doi: 10.1161/01.cir.103.21.2604. [DOI] [PubMed] [Google Scholar]
- [45].Peng S, Guo W, Morrisett JD, Johnstone MT, Hamilton JA. Quantification of cholesteryl esters in human and rabbit atherosclerotic plaques by magic-angle spinning (13)C-NMR. Arterioscler Thromb Vasc Biol. 2000;20:2682–2688. doi: 10.1161/01.atv.20.12.2682. [DOI] [PubMed] [Google Scholar]
- [46].Bemlih S, Poirier MD, Andaloussi A. El. Acyl-coenzyme A: cholesterol acyltransferase inhibitor Avasimibe affect survival and proliferation of glioma tumor cell lines. Cancer Biol Ther. 2010;9:1025–1032. doi: 10.4161/cbt.9.12.11875. [DOI] [PubMed] [Google Scholar]
- [47].Antalis CJ, Uchida A, Buhman KK, Siddiqui RA. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin Exp Metastasis. 2011;28:733–741. doi: 10.1007/s10585-011-9405-9. [DOI] [PubMed] [Google Scholar]
- [48].Hayon T, Atlas L, Levy E, Dvilansky A, Shpilberg O, Nathan I. Multifactorial activities of nonsteroidal antiestrogens against leukemia. Cancer Detect Prev. 2003;27:389–396. doi: 10.1016/s0361-090x(03)00102-8. [DOI] [PubMed] [Google Scholar]
- [49].Shen LZ, Hua YB, Yu XM, Xu Q, Chen T, Wang JH, Wu WX. Tamoxifen can reverse multidrug resistance of colorectal carcinoma in vivo. World J Gastroenterol. 2005;11:1060–1064. doi: 10.3748/wjg.v11.i7.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nakayama Y, Sakamoto H, Satoh K, Yamamoto T. Tamoxifen and gonadal steroids inhibit colon cancer growth in association with inhibition of thymidylate synthase, survivin and telomerase expression through estrogen receptor beta mediated system. Cancer Lett. 2000;161:63–71. doi: 10.1016/s0304-3835(00)00600-5. [DOI] [PubMed] [Google Scholar]
- [51].Lindner DJ, Borden EC. Synergistic antitumor effects of a combination of interferon and tamoxifen on estrogen receptor-positive and receptor-negative human tumor cell lines in vivo and in vitro. J Interferon Cytokine Res. 1997;17:681–693. doi: 10.1089/jir.1997.17.681. [DOI] [PubMed] [Google Scholar]
- [52].Lointier P, Wildrick DM, Boman BM. Growth effects of tamoxifen on Lovo colon carcinoma cells and cultured cells from normal colonic mucosa. Anticancer Res. 1992;12:1523–1525. [PubMed] [Google Scholar]
- [53].Dorchies OM, Reutenauer-Patte J, Dahmane E, Ismail HM, Petermann O, Patthey-Vuadens O, Comyn SA, Gayi E, Piacenza T, Handa RJ, Decosterd LA, Ruegg UT. The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am J Pathol. 2013;182:485–504. doi: 10.1016/j.ajpath.2012.10.018. [DOI] [PubMed] [Google Scholar]
- [54].Taneja SS, Smith MR, Dalton JT, Raghow S, Barnette G, Steiner M, Veverka KA. Toremifene--a promising therapy for the prevention of prostate cancer and complications of androgen deprivation therapy. Expert Opin Investig Drugs. 2006;15:293–305. doi: 10.1517/13543784.15.3.293. [DOI] [PubMed] [Google Scholar]
- [55].Raghow S, Hooshdaran MZ, Katiyar S, Steiner MS. Toremifene prevents prostate cancer in the transgenic adenocarcinoma of mouse prostate model. Cancer Res. 2002;62:1370–1376. [PubMed] [Google Scholar]
- [56].Lara PN, Jr., Gandara DR, Longmate J, Gumerlock PH, Lau DH, Edelman MJ, Gandour-Edwards R, Mack PC, Israel V, Raschko J, Frankel P, Perez EA, Lenz HJ, Doroshow JH. Activity of high-dose toremifene plus cisplatin in platinum-treated non-small-cell lung cancer: a phase II California Cancer Consortium Trial. Cancer Chemother Pharmacol. 2001;48:22–28. doi: 10.1007/s002800100293. [DOI] [PubMed] [Google Scholar]
- [57].Ko JC, Chiu HC, Syu JJ, Jian YJ, Chen CY, Jian YT, Huang YJ, Wo TY, Lin YW. Tamoxifen enhances erlotinib-induced cytotoxicity through down-regulating AKT-mediated thymidine phosphorylase expression in human non-small-cell lung cancer cells. Biochem Pharmacol. 2014;88:119–127. doi: 10.1016/j.bcp.2014.01.010. [DOI] [PubMed] [Google Scholar]
- [58].Couldwell WT, Antel JP, Yong VW. Protein kinase C activity correlates with the growth rate of malignant gliomas: Part II. Effects of glioma mitogens and modulators of protein kinase C. Neurosurgery. 1992;31:717–724. doi: 10.1227/00006123-199210000-00015. discussion 724. [DOI] [PubMed] [Google Scholar]
- [59].Lavie Y, Zhang ZC, Cao HT, Han TY, Jones RC, Liu YY, Jarman M, Hardcastle IR, Giuliano AE, Cabot MC. Tamoxifen induces selective membrane association of protein kinase C epsilon in MCF-7 human breast cancer cells. Int J Cancer. 1998;77:928–932. doi: 10.1002/(sici)1097-0215(19980911)77:6<928::aid-ijc22>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- [60].Cabot MC, Zhang ZC, Giuliano AE. Tamoxifen elicits rapid transmembrane lipid signal responses in human breast cancer cells. Breast Cancer Res Treat. 1995;36:299–306. doi: 10.1007/BF00713401. [DOI] [PubMed] [Google Scholar]
- [61].Cabot MC, Zhang Z, Cao H, Lavie Y, Giuliano AE, Han TY, Jones RC. Tamoxifen activates cellular phospholipase C and D and elicits protein kinase C translocation. Int J Cancer. 1997;70:567–574. doi: 10.1002/(sici)1097-0215(19970304)70:5<567::aid-ijc13>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- [62].Nesbit RA, Woods RL, Tattersall MH, Fox RM, Forbes JF, MacKay IR, Goodyear M. Tamoxifen in malignant melanoma. N Engl J Med. 1979;301:1241–1242. doi: 10.1056/nejm197911293012218. [DOI] [PubMed] [Google Scholar]
- [63].Del Prete SA, Maurer LH, O’Donnell J, Forcier RJ, LeMarbre P. Combination chemotherapy with cisplatin, carmustine, dacarbazine, and tamoxifen in metastatic melanoma. Cancer Treat Rep. 1984;68:1403–1405. [PubMed] [Google Scholar]
- [64].McClay EF, Mastrangelo MJ, Bellet RE, Berd D. Combination chemotherapy and hormonal therapy in the treatment of malignant melanoma. Cancer Treat Rep. 1987;71:465–469. [PubMed] [Google Scholar]
- [65].McClay EF, McClay ME. Tamoxifen: is it useful in the treatment of patients with metastatic melanoma? J Clin Oncol. 1994;12:617–626. doi: 10.1200/JCO.1994.12.3.617. [DOI] [PubMed] [Google Scholar]
- [66].Rusthoven JJ, Quirt IC, Iscoe NA, McCulloch PB, James KW, Lohmann RC, Jensen J, Burdette-Radoux S, Bodurtha AJ, Silver HK, Verma S, Armitage GR, Zee B, Bennett K. Randomized, double-blind, placebo-controlled trial comparing the response rates of carmustine, dacarbazine, and cisplatin with and without tamoxifen in patients with metastatic melanoma. National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 1996;14:2083–2090. doi: 10.1200/JCO.1996.14.7.2083. [DOI] [PubMed] [Google Scholar]
- [67].Toma S, Ugolini D, Palumbo R. Tamoxifen in the treatment of metastatic malignant melanoma: still a controversy? (Review) Int J Oncol. 1999;15:321–337. [PubMed] [Google Scholar]
- [68].Matsuoka H, Tsubaki M, Yamazoe Y, Ogaki M, Satou T, Itoh T, Kusunoki T, Nishida S. Tamoxifen inhibits tumor cell invasion and metastasis in mouse melanoma through suppression of PKC/MEK/ERK and PKC/PI3K/Akt pathways. Exp Cell Res. 2009;315:2022–2032. doi: 10.1016/j.yexcr.2009.04.009. [DOI] [PubMed] [Google Scholar]
- [69].Hewish M, Chau I, Cunningham D. Insulin-like growth factor 1 receptor targeted therapeutics: novel compounds and novel treatment strategies for cancer medicine. Recent Pat Anticancer Drug Discov. 2009;4:54–72. doi: 10.2174/157489209787002515. [DOI] [PubMed] [Google Scholar]
- [70].Kanter-Lewensohn L, Girnita L, Girnita A, Dricu A, Olsson G, Leech L, Nilsson G, Hilding A, Wejde J, Brismar K, Larsson O. Tamoxifen-induced cell death in malignant melanoma cells: possible involvement of the insulin-like growth factor-1 (IGF-1) pathway. Mol Cell Endocrinol. 2000;165:131–137. doi: 10.1016/s0303-7207(00)00253-7. [DOI] [PubMed] [Google Scholar]
- [71].Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001;6:469–477. doi: 10.1023/a:1012437607881. [DOI] [PubMed] [Google Scholar]
- [72].Lavi O, Gottesman MM, Levy D. The dynamics of drug resistance: a mathematical perspective. Drug Resist Updat. 2012;15:90–97. doi: 10.1016/j.drup.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pluchino KM, Hall MD, Goldsborough AS, Callaghan R, Gottesman MM. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist Updat. 2012;15:98–105. doi: 10.1016/j.drup.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yang K, Wu J, Li X. Recent advances in the research of P-glycoprotein inhibitors. Biosci Trends. 2008;2:137–146. [PubMed] [Google Scholar]
- [75].Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. 2006;7:861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- [76].Baumert C, Hilgeroth A. Recent advances in the development of P-gp inhibitors. Anticancer Agents Med Chem. 2009;9:415–436. doi: 10.2174/1871520610909040415. [DOI] [PubMed] [Google Scholar]
- [77].Shepard RL, Cao J, Starling JJ, Dantzig AH. Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int J Cancer. 2003;103:121–125. doi: 10.1002/ijc.10792. [DOI] [PubMed] [Google Scholar]
- [78].O’Brien MM, Lacayo NJ, Lum BL, Kshirsagar S, Buck S, Ravindranath Y, Bernstein M, Weinstein H, Chang MN, Arceci RJ, Sikic BI, Dahl GV. Phase I study of valspodar (PSC-833) with mitoxantrone and etoposide in refractory and relapsed pediatric acute leukemia: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2010;54:694–702. doi: 10.1002/pbc.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist Updat. 2012;15:62–69. doi: 10.1016/j.drup.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol Sci. 2009;30:546–556. doi: 10.1016/j.tips.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Callaghan R, Higgins CF. Interaction of tamoxifen with the multidrug resistance P-glycoprotein. Br J Cancer. 1995;71:294–299. doi: 10.1038/bjc.1995.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bekaii-Saab TS, Perloff MD, Weemhoff JL, Greenblatt DJ, von Moltke LL. Interactions of tamoxifen, N-desmethyltamoxifen and 4-hydroxytamoxifen with P-glycoprotein and CYP3A. Biopharm Drug Dispos. 2004;25:283–289. doi: 10.1002/bdd.411. [DOI] [PubMed] [Google Scholar]
- [83].Stuart NS, Philip P, Harris AL, Tonkin K, Houlbrook S, Kirk J, Lien EA, Carmichael J. High-dose tamoxifen as an enhancer of etoposide cytotoxicity. Clinical effects and in vitro assessment in p glycoprotein expressing cell lines. Br J Cancer. 1992;66:833–839. doi: 10.1038/bjc.1992.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Cheng AL, Chen YC, Yeh KH, Chuang SE, Chen BR, Chen DS. Chronic oral etoposide and tamoxifen in the treatment of far-advanced hepatocellular carcinoma. Cancer. 1996;77:872–877. [PubMed] [Google Scholar]
- [85].Trump DL, Smith DC, Ellis PG, Rogers MP, Schold SC, Winer EP, Panella TJ, Jordan VC, Fine RL. High-dose oral tamoxifen, a potential multidrug-resistance-reversal agent: phase I trial in combination with vinblastine. J Natl Cancer Inst. 1992;84:1811–1816. doi: 10.1093/jnci/84.23.1811. [DOI] [PubMed] [Google Scholar]
- [86].Samuels BL, Hollis DR, Rosner GL, Trump DL, Shapiro CL, Vogelzang NJ, Schilsky RL. Modulation of vinblastine resistance in metastatic renal cell carcinoma with cyclosporine A or tamoxifen: a cancer and leukemia group B study. Clin Cancer Res. 1997;3:1977–1984. [PubMed] [Google Scholar]
- [87].Liu JH, Yang MH, Fan FS, Yen CC, Wang WS, Chang YH, Chen KK, Chen PM. Tamoxifen and colchicine-modulated vinblastine followed by 5-fluorouracil in advanced renal cell carcinoma: a phase II study. Urology. 2001;57:650–654. doi: 10.1016/s0090-4295(00)01096-7. [DOI] [PubMed] [Google Scholar]
- [88].Smith DC, Trump DL. A phase I trial of high-dose oral tamoxifen and CHOPE. Cancer Chemother Pharmacol. 1995;36:65–68. doi: 10.1007/BF00685734. [DOI] [PubMed] [Google Scholar]
- [89].Berman E, McBride M, Lin S, Menedez-Botet C, Tong W. Phase I trial of high-dose tamoxifen as a modulator of drug resistance in combination with daunorubicin in patients with relapsed or refractory acute leukemia. Leukemia. 1995;9:1631–1637. [PubMed] [Google Scholar]
- [90].Weinlander G, Kornek G, Raderer M, Hejna M, Tetzner C, Scheithauer W. Treatment of advanced colorectal cancer with doxorubicin combined with two potential multidrug-resistance-reversing agents: high-dose oral tamoxifen and dexverapamil. J Cancer Res Clin Oncol. 1997;123:452–455. doi: 10.1007/BF01372550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].El-Yazigi A, Berry J, Ezzat A, Wahab FA. Effect of tamoxifen on the pharmacokinetics of doxorubicin in patients with non-Hodgkin’s lymphoma. Ther Drug Monit. 1997;19:632–636. doi: 10.1097/00007691-199712000-00005. [DOI] [PubMed] [Google Scholar]
- [92].Nathan FE, Berd D, Sato T, Mastrangelo MJ. Paclitaxel and tamoxifen: An active regimen for patients with metastatic melanoma. Cancer. 2000;88:79–87. doi: 10.1002/(sici)1097-0142(20000101)88:1<79::aid-cncr12>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- [93].Lucci A, Cho WI, Han TY, Giuliano AE, Morton DL, Cabot MC. Glucosylceramide: a marker for multiple-drug resistant cancers. Anticancer Res. 1998;18:475–480. [PubMed] [Google Scholar]
- [94].Shen DW, Cardarelli C, Hwang J, Cornwell M, Richert N, Ishii S, Pastan I, Gottesman MM. Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J Biol Chem. 1986;261:7762–7770. [PubMed] [Google Scholar]
- [95].Gouaze V, Yu JY, Bleicher RJ, Han TY, Liu YY, Wang H, Gottesman MM, Bitterman A, Giuliano AE, Cabot MC. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol Cancer Ther. 2004;3:633–639. [PubMed] [Google Scholar]
- [96].Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- [97].Morton DL, Ravindranath MH, Irie RF. Tumor gangliosides as targets for active specific immunotherapy of melanoma in man. Prog Brain Res. 1994;101:251–275. doi: 10.1016/s0079-6123(08)61954-8. [DOI] [PubMed] [Google Scholar]
- [98].Lavie Y, Cao H, Volner A, Lucci A, Han TY, Geffen V, Giuliano AE, Cabot MC. Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. J Biol Chem. 1997;272:1682–1687. doi: 10.1074/jbc.272.3.1682. [DOI] [PubMed] [Google Scholar]
- [99].Jiang Y, DiVittore NA, Kaiser JM, Shanmugavelandy SS, Fritz JL, Heakal Y, Tagaram HR, Cheng H, Cabot MC, Staveley-O’Carroll KF, Tran MA, Fox TE, Barth BM, Kester M. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol Ther. 2011;12:574–585. doi: 10.4161/cbt.12.7.15971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lucci A, Giuliano AE, Han TY, Dinur T, Liu YY, Senchenkov A, Cabot MC. Ceramide toxicity and metabolism differ in wild-type and multidrug-resistant cancer cells. Int J Oncol. 1999;15:535–540. doi: 10.3892/ijo.15.3.535. [DOI] [PubMed] [Google Scholar]
- [101].Morad SA, Madigan JP, Levin JC, Abdelmageed N, Karimi R, Rosenberg DW, Kester M, Shanmugavelandy SS, Cabot MC. Tamoxifen magnifies therapeutic impact of ceramide in human colorectal cancer cells independent of p53. Biochem Pharmacol. 2013;85:1057–1065. doi: 10.1016/j.bcp.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–18391. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- [103].Furlong SJ, Mader JS, Hoskin DW. Lactoferricin-induced apoptosis in estrogen-nonresponsive MDA-MB-435 breast cancer cells is enhanced by C6 ceramide or tamoxifen. Oncol Rep. 2006;15:1385–1390. [PubMed] [Google Scholar]
- [104].Gucluler G, Piskin O, Baran Y. The roles of antiapoptotic sphingosine kinase-1 and glucosylceramide genes in drug induced cell death of MCF-7 breast cancer cells. J BUON. 2011;16:646–651. [PubMed] [Google Scholar]
- [105].Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–1876. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- [106].van Helvoort A, Giudici ML, Thielemans M, van Meer G. Transport of sphingomyelin to the cell surface is inhibited by brefeldin A and in mitosis, where C6-NBD-sphingomyelin is translocated across the plasma membrane by a multidrug transporter activity. J Cell Sci. 1997;110(Pt 1):75–83. doi: 10.1242/jcs.110.1.75. [DOI] [PubMed] [Google Scholar]
- [107].De Rosa MF, Sillence D, Ackerley C, Lingwood C. Role of multiple drug resistance protein 1 in neutral but not acidic glycosphingolipid biosynthesis. J Biol Chem. 2004;279:7867–7876. doi: 10.1074/jbc.M305645200. [DOI] [PubMed] [Google Scholar]
- [108].Shabbits JA, Mayer LD. P-glycoprotein modulates ceramide-mediated sensitivity of human breast cancer cells to tubulin-binding anticancer drugs. Mol Cancer Ther. 2002;1:205–213. [PubMed] [Google Scholar]
- [109].Charles AG, Han TY, Liu YY, Hansen N, Giuliano AE, Cabot MC. Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer Chemother Pharmacol. 2001;47:444–450. doi: 10.1007/s002800000265. [DOI] [PubMed] [Google Scholar]
- [110].Turzanski J, Grundy M, Shang S, Russell N, Pallis M. P-glycoprotein is implicated in the inhibition of ceramide-induced apoptosis in TF-1 acute myeloid leukemia cells by modulation of the glucosylceramide synthase pathway. Exp Hematol. 2005;33:62–72. doi: 10.1016/j.exphem.2004.10.005. [DOI] [PubMed] [Google Scholar]
- [111].Sietsma H, Dijkhuis AJ, Kamps W, Kok JW. Sphingolipids in neuroblastoma: their role in drug resistance mechanisms. Neurochem Res. 2002;27:665–674. doi: 10.1023/a:1020228117739. [DOI] [PubMed] [Google Scholar]
- [112].Chapman JV, Gouaze-Andersson V, Cabot MC. Expression of P-glycoprotein in HeLa cells confers resistance to ceramide cytotoxicity. Int J Oncol. 2010;37:1591–1597. doi: 10.3892/ijo_00000813. [DOI] [PubMed] [Google Scholar]
- [113].Sietsma H, Veldman RJ, Kolk D, Ausema B, Nijhof W, Kamps W, Vellenga E, Kok JW. 1-phenyl-2-decanoylamino-3-morpholino-1-propanol chemosensitizes neuroblastoma cells for taxol and vincristine. Clin Cancer Res. 2000;6:942–948. [PubMed] [Google Scholar]
- [114].Chai L, McLaren RP, Byrne A, Chuang WL, Huang Y, Dufault MR, Pacheco J, Madhiwalla S, Zhang X, Zhang M, Teicher BA, Carter K, Cheng SH, Leonard JP, Xiang Y, Vasconcelles M, Goldberg MA, Copeland DP, Klinger KW, Lillie J, Madden SL, Jiang YA. The chemosensitizing activity of inhibitors of glucosylceramide synthase is mediated primarily through modulation of P-gp function. Int J Oncol. 2011;38:701–711. doi: 10.3892/ijo.2010.888. [DOI] [PubMed] [Google Scholar]
- [115].Norris-Cervetto E, Callaghan R, Platt FM, Dwek RA, Butters TD. Inhibition of glucosylceramide synthase does not reverse drug resistance in cancer cells. J Biol Chem. 2004;279:40412–40418. doi: 10.1074/jbc.M404466200. [DOI] [PubMed] [Google Scholar]
- [116].Morad SA, Levin JC, Shanmugavelandy SS, Kester M, Fabrias G, Bedia C, Cabot MC. Ceramide--antiestrogen nanoliposomal combinations--novel impact of hormonal therapy in hormone-insensitive breast cancer. Mol Cancer Ther. 2012;11:2352–2361. doi: 10.1158/1535-7163.MCT-12-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chapman JV, Gouaze-Andersson V, Messner MC, Flowers M, Karimi R, Kester M, Barth BM, Liu X, Liu YY, Giuliano AE, Cabot MC. Metabolism of short-chain ceramide by human cancer cells--implications for therapeutic approaches. Biochem Pharmacol. 2010;80:308–315. doi: 10.1016/j.bcp.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Wang H, Charles AG, Frankel AJ, Cabot MC. Increasing intracellular ceramide: an approach that enhances the cytotoxic response in prostate cancer cells. Urology. 2003;61:1047–1052. doi: 10.1016/s0090-4295(02)02511-6. [DOI] [PubMed] [Google Scholar]
- [119].Gatt S. Enzymic Hydrolysis and Synthesis of Ceramides. J Biol Chem. 1963;238:3131–3133. [PubMed] [Google Scholar]
- [120].Dimanche-Boitrel MT, Rebillard A. Sphingolipids and response to chemotherapy. Handb Exp Pharmacol. 2013:73–91. doi: 10.1007/978-3-7091-1511-4_4. [DOI] [PubMed] [Google Scholar]
- [121].Fox TE, Bewley MC, Unrath KA, Pedersen MM, Anderson RE, Jung DY, Jefferson LS, Kim JK, Bronson SK, Flanagan JM, Kester M. Circulating sphingolipid biomarkers in models of type 1 diabetes. J Lipid Res. 2011;52:509–517. doi: 10.1194/jlr.M010595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Furlong SJ, Ridgway ND, Hoskin DW. Modulation of ceramide metabolism in T-leukemia cell lines potentiates apoptosis induced by the cationic antimicrobial peptide bovine lactoferricin. Int J Oncol. 2008;32:537–544. [PubMed] [Google Scholar]
- [123].Watters RJ, Fox TE, Tan SF, Shanmugavelandy S, Choby JE, Broeg K, Liao J, Kester M, Cabot MC, Loughran TP, Liu X. Targeting glucosylceramide synthase synergizes with C6-ceramide nanoliposomes to induce apoptosis in natural killer cell leukemia. Leuk Lymphoma. 2013;54:1288–1296. doi: 10.3109/10428194.2012.752485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Morad SA, Levin JC, Tan SF, Fox TE, Feith DJ, Cabot MC. Novel off-target effect of tamoxifen--inhibition of acid ceramidase activity in cancer cells. Biochim Biophys Acta. 2013;1831:1657–1664. doi: 10.1016/j.bbalip.2013.07.016. [DOI] [PubMed] [Google Scholar]
- [125].Bowles DE, Contreras-Ramos A, Sites RW. Gynomorphic mandible morphology in the dobsonfly, Corydalus cornutus. J Insect Sci. 2007;7:1–5. doi: 10.1673/031.007.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr Drug Targets. 2008;9:662–673. doi: 10.2174/138945008785132402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Liu X, Elojeimy S, Turner LS, Mahdy AE, Zeidan YH, Bielawska A, Bielawski J, Dong JY, El-Zawahry AM, Guo GW, Hannun YA, Holman DH, Rubinchik S, Szulc Z, Keane TE, Tavassoli M, Norris JS. Acid ceramidase inhibition: a novel target for cancer therapy. Front Biosci. 2008;13:2293–2298. doi: 10.2741/2843. [DOI] [PubMed] [Google Scholar]
- [128].Beckham TH, Lu P, Cheng JC, Zhao D, Turner LS, Zhang X, Hoffman S, Armeson KE, Liu A, Marrison T, Hannun YA, Liu X. Acid ceramidase-mediated production of sphingosine 1-phosphate promotes prostate cancer invasion through upregulation of cathepsin B. Int J Cancer. 2012;131:2034–2043. doi: 10.1002/ijc.27480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Mahdy AE, Cheng JC, Li J, Elojeimy S, Meacham WD, Turner LS, Bai A, Gault CR, McPherson AS, Garcia N, Beckham TH, Saad A, Bielawska A, Bielawski J, Hannun YA, Keane TE, Taha MI, Hammouda HM, Norris JS, Liu X. Acid ceramidase upregulation in prostate cancer cells confers resistance to radiation: AC inhibition, a potential radiosensitizer. Mol Ther. 2009;17:430–438. doi: 10.1038/mt.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Bedia C, Casas J, Andrieu-Abadie N, Fabrias G, Levade T. Acid ceramidase expression modulates the sensitivity of A375 melanoma cells to dacarbazine. J Biol Chem. 2011;286:28200–28209. doi: 10.1074/jbc.M110.216382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Musumarra G, Barresi V, Condorelli DF, Scire S. A bioinformatic approach to the identification of candidate genes for the development of new cancer diagnostics. Biol Chem. 2003;384:321–327. doi: 10.1515/BC.2003.037. [DOI] [PubMed] [Google Scholar]
- [132].Morad SA, Tan SF, Feith DJ, Kester M, Claxton DF, Loughran TP, Barth BM, Fox TE, Cabot MC. Modification of sphingolipid metabolism by tamoxifen and N-desmethyltamoxifen in acute myelogenous leukemia - Impact on enzyme activity and response to cytotoxics Biochim Biophys Acta. 2015;1851:919–928. doi: 10.1016/j.bbalip.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Elojeimy S, Holman DH, Liu X, El-Zawahry A, Villani M, Cheng JC, Mahdy A, Zeidan Y, Bielwaska A, Hannun YA, Norris JS. New insights on the use of desipramine as an inhibitor for acid ceramidase. FEBS Lett. 2006;580:4751–4756. doi: 10.1016/j.febslet.2006.07.071. [DOI] [PubMed] [Google Scholar]
- [134].Fabrias G, Bedia C, Casas J, Abad JL, Delgado A. Ceramidases in hematological malignancies: senseless or neglected target? Anticancer Agents Med Chem. 2011;11:830–843. doi: 10.2174/187152011797655104. [DOI] [PubMed] [Google Scholar]
- [135].Delgado A, Fabrias G, Bedia C, Casas J, Abad JL. Sphingolipid modulation: a strategy for cancer therapy. Anticancer Agents Med Chem. 2012;12:285–302. doi: 10.2174/187152012800228643. [DOI] [PubMed] [Google Scholar]
- [136].Dick TE, Hengst JA, Fox TE, Colledge AL, Kale VP, Sung SS, Sharma A, Amin S, Loughran TP, Jr., Kester M, Wang HG, Yun JK. The apoptotic mechanism of action of the sphingosine kinase 1 selective inhibitor SKI-178 in human acute myeloid leukemia cell lines. J Pharmacol Exp Ther. 2015;352:494–508. doi: 10.1124/jpet.114.219659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Plano D, Amin S, Sharma AK. Importance of sphingosine kinase (SphK) as a target in developing cancer therapeutics and recent developments in the synthesis of novel SphK inhibitors. J Med Chem. 2014;57:5509–5524. doi: 10.1021/jm4011687. [DOI] [PubMed] [Google Scholar]
- [138].Pyne NJ, Ohotski J, Bittman R, Pyne S. The role of sphingosine 1-phosphate in inflammation and cancer. Adv Biol Regul. 2014;54:121–129. doi: 10.1016/j.jbior.2013.08.005. [DOI] [PubMed] [Google Scholar]
- [139].Heffernan-Stroud LA, Obeid LM. Sphingosine kinase 1 in cancer. Adv Cancer Res. 2013;117:201–235. doi: 10.1016/B978-0-12-394274-6.00007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Selvam SP, Ogretmen B. Sphingosine kinase/sphingosine 1-phosphate signaling in cancer therapeutics and drug resistance. Handb Exp Pharmacol. 2013:3–27. doi: 10.1007/978-3-7091-1511-4_1. [DOI] [PubMed] [Google Scholar]
- [141].Bonhoure E, Pchejetski D, Aouali N, Morjani H, Levade T, Kohama T, Cuvillier O. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting sphingosine kinase-1. Leukemia. 2006;20:95–102. doi: 10.1038/sj.leu.2404023. [DOI] [PubMed] [Google Scholar]
- [142].Chapman JV, Gouaze-Andersson V, Karimi R, Messner MC, Cabot MC. P-glycoprotein antagonists confer synergistic sensitivity to short-chain ceramide in human multidrug-resistant cancer cells. Exp Cell Res. 2011;317:1736–1745. doi: 10.1016/j.yexcr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Wu JM, DiPietrantonio AM, Hsieh TC. Mechanism of fenretinide (4-HPR)-induced cell death. Apoptosis. 2001;6:377–388. doi: 10.1023/a:1011342220621. [DOI] [PubMed] [Google Scholar]
- [144].Morales MC, Perez-Yarza G, Rementeria NN, Boyano MD, Apraiz A, Gomez-Munoz A, Perez-Andres E, Asumendi A. 4-HPR-mediated leukemia cell cytotoxicity is triggered by ceramide-induced mitochondrial oxidative stress and is regulated downstream by Bcl-2. Free Radic Res. 2007;41:591–601. doi: 10.1080/10715760701218558. [DOI] [PubMed] [Google Scholar]
- [145].Maurer BJ, Metelitsa LS, Seeger RC, Cabot MC, Reynolds CP. Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)- retinamide in neuroblastoma cell lines. J Natl Cancer Inst. 1999;91:1138–1146. doi: 10.1093/jnci/91.13.1138. [DOI] [PubMed] [Google Scholar]
- [146].Zheng W, Kollmeyer J, Symolon H, Momin A, Munter E, Wang E, Kelly S, Allegood JC, Liu Y, Peng Q, Ramaraju H, Sullards MC, Cabot M, Merrill AH., Jr. Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim Biophys Acta. 2006;1758:1864–1884. doi: 10.1016/j.bbamem.2006.08.009. [DOI] [PubMed] [Google Scholar]
- [147].Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, Obeid LM, Bielawska A. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem. 2007;282:16718–16728. doi: 10.1074/jbc.M700647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Rahmaniyan M, Curley RW, Jr., Obeid LM, Hannun YA, Kraveka JM. Identification of dihydroceramide desaturase as a direct in vitro target for fenretinide. J Biol Chem. 2011;286:24754–24764. doi: 10.1074/jbc.M111.250779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Wang H, Maurer BJ, Liu YY, Wang E, Allegood JC, Kelly S, Symolon H, Liu Y, Merrill AH, Jr., Gouaze-Andersson V, Yu JY, Giuliano AE, Cabot MC. N-(4-Hydroxyphenyl)retinamide increases dihydroceramide and synergizes with dimethylsphingosine to enhance cancer cell killing. Mol Cancer Ther. 2008;7:2967–2976. doi: 10.1158/1535-7163.MCT-08-0549. [DOI] [PubMed] [Google Scholar]
- [150].Fabrias G, Munoz-Olaya J, Cingolani F, Signorelli P, Casas J, Gagliostro V, Ghidoni R. Dihydroceramide desaturase and dihydrosphingolipids: debutant players in the sphingolipid arena. Prog Lipid Res. 2012;51:82–94. doi: 10.1016/j.plipres.2011.12.002. [DOI] [PubMed] [Google Scholar]
- [151].Maurer BJ, Melton L, Billups C, Cabot MC, Reynolds CP. Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl)retinamide and modulators of ceramide metabolism. J Natl Cancer Inst. 2000;92:1897–1909. doi: 10.1093/jnci/92.23.1897. [DOI] [PubMed] [Google Scholar]
- [152].Radin NS. Chemotherapy by slowing glucosphingolipid synthesis. Biochem Pharmacol. 1999;57:589–595. doi: 10.1016/s0006-2952(98)00274-3. [DOI] [PubMed] [Google Scholar]
- [153].Morad SAF, Levin JC, Kester M, Shanmugavelandy SS, Cabot MC. Ceramide-antiestrogen nanoliposomal combinations - novel impact of hormonal therapy in hormone-insensitive breast cancer. Mol Cancer Ther. 2012 doi: 10.1158/1535-7163.MCT-12-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Morad SAF, Bridges LC, Cabot MC. Abstract A31: Revisited use of tamoxifen for improving melanoma therapy. Clinical Cancer Research. 2015;21:A31. [Google Scholar]
- [155].Awad AB, Barta SL, Fink CS, Bradford PG. beta-Sitosterol enhances tamoxifen effectiveness on breast cancer cells by affecting ceramide metabolism. Mol Nutr Food Res. 2008;52:419–426. doi: 10.1002/mnfr.200700222. [DOI] [PubMed] [Google Scholar]
- [156].Roberts CG, Gurisik E, Biden TJ, Sutherland RL, Butt AJ. Synergistic cytotoxicity between tamoxifen and the plant toxin persin in human breast cancer cells is dependent on Bim expression and mediated by modulation of ceramide metabolism. Mol Cancer Ther. 2007;6:2777–2785. doi: 10.1158/1535-7163.MCT-07-0374. [DOI] [PubMed] [Google Scholar]
- [157].Devalapally H, Duan Z, Seiden MV, Amiji MM. Modulation of drug resistance in ovarian adenocarcinoma by enhancing intracellular ceramide using tamoxifen-loaded biodegradable polymeric nanoparticles. Clin Cancer Res. 2008;14:3193–3203. doi: 10.1158/1078-0432.CCR-07-4973. [DOI] [PubMed] [Google Scholar]
- [158].O’Day SJ, Boasberg PD, Kristedja TS, Martin M, Wang HJ, Fournier P, Cabot M, DeGregorio MW, Gammon G. High-dose tamoxifen added to concurrent biochemotherapy with decrescendo interleukin-2 in patients with metastatic melanoma. Cancer. 2001;92:609–619. doi: 10.1002/1097-0142(20010801)92:3<609::aid-cncr1361>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- [159].Bergan RC, Reed E, Myers CE, Headlee D, Brawley O, Cho HK, Figg WD, Tompkins A, Linehan WM, Kohler D, Steinberg SM, Blagosklonny MV. A Phase II study of high-dose tamoxifen in patients with hormone-refractory prostate cancer. Clin Cancer Res. 1999;5:2366–2373. [PubMed] [Google Scholar]
- [160].Johnston SR, Haynes BP, Sacks NP, McKinna JA, Griggs LJ, Jarman M, Baum M, Smith IE, Dowsett M. Effect of oestrogen receptor status and time on the intra-tumoural accumulation of tamoxifen and N-desmethyltamoxifen following short-term therapy in human primary breast cancer. Breast Cancer Res Treat. 1993;28:241–250. doi: 10.1007/BF00666585. [DOI] [PubMed] [Google Scholar]
- [161].Lucci A, Han TY, Liu YY, Giuliano AE, Cabot MC. Multidrug resistance modulators and doxorubicin synergize to elevate ceramide levels and elicit apoptosis in drug-resistant cancer cells. Cancer. 1999;86:300–311. doi: 10.1002/(sici)1097-0142(19990715)86:2<300::aid-cncr14>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- [162].Mimeault M, Venkatraman G, Johansson SL, Moore E, Henichart JP, Depreux P, Lin MF, Batra SK. Novel combination therapy against metastatic and androgen-independent prostate cancer by using gefitinib, tamoxifen and etoposide. Int J Cancer. 2007;120:160–169. doi: 10.1002/ijc.22268. [DOI] [PubMed] [Google Scholar]
- [163].Lucci A, Han TY, Liu YY, Giuliano AE, Cabot MC. Modification of ceramide metabolism increases cancer cell sensitivity to cytotoxics. Int J Oncol. 1999;15:541–546. doi: 10.3892/ijo.15.3.541. [DOI] [PubMed] [Google Scholar]