

Abstract
Mitochondrial metabolism is highly responsive to nutrient availability and ongoing activity in neuronal circuits. The molecular mechanisms by which brain cells respond to an increase in cellular energy expenditure are largely unknown. Mild mitochondrial uncoupling enhances cellular energy expenditure in mitochondria and can be induced with 2, 4-dinitrophenol (DNP), a proton ionophore previously used for weight loss. We found that DNP treatment reduces mitochondrial membrane potential, increases intracellular Ca2+ levels and reduces oxidative stress in cerebral cortical neurons. Gene expression profiling of the cerebral cortex of DNP-treated mice revealed reprogramming of signaling cascades that included suppression of the mTOR and insulin – PI3K – MAPK pathways, and up-regulation of tuberous sclerosis complex 2, a negative regulator of mTOR. Genes encoding proteins involved in autophagy processes were up-regulated in response to DNP. CREB (cAMP-response element-binding protein) signaling, Arc and BDNF, which play important roles in synaptic plasticity and adaptive cellular stress responses, were up-regulated in response to DNP, and DNP-treated mice exhibited improved performance in a test of learning and memory. Immunoblot analysis verified that key DNP-induced changes in gene expression resulted in corresponding changes at the protein level. Our findings suggest that mild mitochondrial uncoupling triggers an integrated signaling response in brain cells characterized by reprogramming of mTOR and insulin signaling, and up-regulation of pathways involved in adaptive stress responses, molecular waste disposal and synaptic plasticity.
Keywords: mild mitochondrial uncoupling; TSC2; mTOR; mTORC1; mTORC2; insulin signaling; PI3K; Akt; CREB; BDNF; neurons; autophagy; 2, 4-dinitrophenol
INTRODUCTION
The brain consumes relatively large amounts of energy because of the ongoing electrochemical activity of its neuronal networks. Similar to other cell types, mitochondria generate the majority of energy substrates (ATP and NAD+) in neurons, and energy availability influences cellular metabolism via core regulatory networks including those involving mammalian target of rapamycin (mTOR), AMP-activated protein kinase (AMPK) and insulin signaling pathways (Cheng et al. 2010; Inoki et al. 2012; Salminen and Kaarniranta 2012). The energy demand of neurons is acutely responsive to synaptic activity, and the resulting Ca2+ influx and ion-motive ATPase activities (Gellerich et al. 2003). In addition, long-lasting changes in neuronal energy metabolism are influenced by neurotrophic factor signaling pathways involving phosphatidylinositol 3 (PI3) and mitogen activated protein (MAP) kinases, and transcription factors such as cyclic AMP response element-binding protein (CREB) (Burkhalter et al. 2003; Cheng et al. 2012). Mild intermittent metabolic challenges such as fasting and exercise can be beneficial for neurons and brain health (Mattson 2012), and are neuroprotective in animal models of stroke, Parkinson’s disease and Alzheimer’s disease (Halagappa et al. 2007; Arumugam et al. 2010; Zhang et al. 2011; Griffioen et al. 2013; Meller and Simon 2013). In addition, pharmacological agents that inhibit mTOR or activate AMPK can protect neurons against dysfunction and degeneration in animal models of acute brain injury and neurodegenerative disorders (Culmsee et al. 2001; Tain et al. 2009; Visrosci et al. 2012). A better understanding of such adaptive responses of neurons to bioenergetic challenges may lead to the development of novel approaches for promoting optimal brain function and for preventing and treating neurodegenerative disorders.
Mitochondrial uncoupling imposes an energetic stress on cells by causing a proton leak across the inner membrane which reduces the membrane potential and dissipates substrate oxidation from ADP phosphorylation, thereby increasing energy expenditure (Starkov 2006; Echtay 2007). Mitochondrial uncoupling is a physiologically regulated process that plays important roles in adaptive responses of organisms to changing environmental conditions. Mitochondrial uncoupling proteins regulate multiple physiological processes including thermogenesis, mitochondrial redox balance and free radical production, cellular calcium homeostasis and autophagy/mitophagy (Enerbäck et al. 1997; Starkov and Fiskum 2003; Maragos and Korde 2004; Andrews et al. 2005; Liu et al. 2006; Caldeira da Silva et al. 2008; Mattson, 2010; Mookerjee et al. 2010; Youle and Narendra 2011; Ramsden et al. 2012). Abnormalities in mitochondrial uncoupling are implicated in pathological conditions including obesity, insulin resistance/diabetes, cardiovascular disease and neurodegenerative disorders (Vidal-Puig 2000; Chan and Harper 2006; Colman 2007; Tseng et al. 2010).
Mild mitochondrial uncoupling can be induced by treating cultured cells or animals with low doses of chemical uncouplers such as 2, 4-dinitrophenol (DNP), a proton ionophore previously used in the clinic to treat obesity (Colman, 2007). Low doses of DNP can protect neurons against dysfunction and degeneration in experimental models of ischemic stroke (Korde et al. 2005), traumatic brain injury (Pandya et al. 2007) and peripheral nerve injury (da Costa et al 2010). Several changes occur in neurons exposed to DNP that may contribute to its neuroprective effects including reduced mitochondrial free radical production, a bioenergetic shift (Liu et al. 2006) and stabilization of cellular Ca2+ homeostasis (Chan et al. 2006). However, the molecular mechanisms by which mild mitochondrial uncoupling enhances neuronal resilience remain unknown. To elucidate the ways in which neural cells respond to mild mitochondrial uncoupling, we performed gene array analyses of bioenergetics- and neuroplasticity-related signaling networks on samples of cerebral cortex from adult mice that had been treated with DNP or vehicle for time periods of 6 to 72 hours. The findings suggest that mild mitochondrial uncoupling triggers a complex integrated cellular response in the brain that includes suppression of mTOR and insulin signaling, enhanced autophagy, and up-regulation of cyclic AMP response element binding protein (CREB), a transcription factor of great importance for neuronal plasticity. Analyses of key proteins in these pathways corroborated the mRNA analysis. Moreover, we show that DNP treatment improves performance of mice in a learning and memory task, consistent with the molecular evidence of enhanced synaptic plasticity.
METHODS
Mice and drug administration
All animals were male C57BL/6 mice purchased from either Jackson Laboratories or Daehan Biolink Co. Ltd. and were 6 months old when DNP was administered. Mice were maintained on a 12 h light/12 h dark cycle and were provided water and food ad libitum. 2,4-dinitrophenol (DNP, Sigma-Aldrich, St. Louis, MO) was dissolved in DMSO, and administered to mice by intraperitioneal (i.p.) injection at a dose of 5 mg/kg/day; control mice were injected with vehicle alone. There were 7 mice/group/each time point and brain tissues were collected at 6, 24 and 72 hours after DNP or vehicle administration; brain tissues from control mice were collected at 24 hours after vehicle injection. There was no mortality in any of the groups of mice. The DNP dose was chosen based on neuronal cell survival experiments (Figure S1a) and previous reports showing that it is non-toxic or neuroprotective in rodents (Kaiser 1964; Korde et al. 2005; Pandya et al. 2007; da Costa et al. 2010). All procedures using live mice were approved by the National Institute on Aging Animal Care and Use Committee, and complied with NIH guidelines.
Microarray data analysis
RNA was extracted from cortical brain tissue using a Qiagen RNeasy Mini Kit. As described previously (Stranahan et al. 2010), RNA was converted into single-stranded DNA, then copied to produce double-stranded DNA, which was then transcribed into cRNA during which16-UTP was incorporated. cRNA was then hybridized to Illumina Sentrix MouseRef-8 Expression Bead-Chips containing 24,000 transcripts (Illumina, San Diego, CA) for 16 hours at 58 °C. An Illumina BeadStation 500× Genetic Analysis Systems scanner was used to acquire the images, and the image data was extracted using Illumina BeadStudio software, Version 3.0. Microarray data was analyzed using the DIANE 6.0 program as described previously (Stranahan et al. 2010). Raw microarray data were subjected to z-transformation and filtered by the detection p-value and Z normalization. Sample quality was assessed by scatter plots, principal component analysis, and gene sample z-ratio score-based hierarchy clustering to exclude possible outliers (Figure S4, and see Cheadle et al. 2003). ANOVA analysis was used to eliminate genes with large variances. Significance was set by a z-ratio >1.5 in both directions and analysis of variance p-value <0.05. Gene regulatory network and canonic pathway analysis was performed using Ingenuity Pathway Analysis software. Additional information on the analysis methods is provided in Supplemental Methods.
Reverse transcription and real-time qPCR
Real-time qPCR was performed to validate the expression of selected mRNAs in cerebral cortex samples of DNP-treated and control mice (n=4 individual mice/group). Total RNA was isolated with Trizol reagent and reverse transcribed into cDNA (RT) with random hexamers and SSIII reverse transcriptase (mixture) using SuperScript® III First-strand synthesis SuperMix (Invitrogen). Real-time qPCR was performed using a SYBR Green-based assay (QIAGEN) according to manufacturer’s protocol and performed on an AB 7300 Applied Biosystem instrument. The forward and reverse primers used for mouse BDNF were 5″-ACT CTT TCC CTC CCT CCT CC-3″ and 5″-TCT GCA AAC ACT GTT AGG CCA-3″; and primers for mouse Arc/Arg3.1 were 5′-GAC ATCCTA GGG CAG ATG CT-3′ and 5′-ACT GGT ATG AAT CAC TGC TGG-3′. The GAPDH and β-actin genes were included in each experiment and the data used for normalization. Cycle threshold values were used for data quantification and normalization, as ΔCt per Gene = Ct Gene X- Ct HKGs (Avg).
Immunoblots
Methods used were similar to those described previously (Liu et al. 2010). Brain tissues were homogenized and solubilized in SDS–PAGE sample buffer, and protein concentrations were determined using a protein assay kit (BCA assay, Piece). Equal amount of protein extracts (50 μg/lane) were separated by electrophoresis on pre-cast NuPAGE Bis-Tris Mini gels (Invitrogen), with acrylamide concentrations appropriate for the molecular weight of the protein of interest. After electrophoretic transfer to a nitrocellulose membrane, the membrane was incubated in a blocking buffer containing 5% non-fat milk, and then incubated overnight at 4°C in the presence of one of the following primary antibodies: TSC1 and TSC2 (Cell Signaling); mTOR and phospho-mTOR (Cell Signaling); LC3B (co-purified with microtubule-associated protein 1B, Sigma); Akt (Cell Signaling) and p-Akt (Upstate); Erk1/2 and p-Erk1/2/ MAPK (Cell Signaling); CREB and p-CREB (Cell Signaling); β-actin (Sigma). FoxO1 and FoxO3 (Cell Signaling); HRP-conjugated secondary antibodies were used for all immunoblots (Vector Laboratories) and were detected by enhanced chemiluminescence (Amersham). The densities of protein bands were quantified use Image J software and normalized to the β-actin band intensity for the same sample.
Passive avoidance test
The mice were given i. p. injections of vehicle or DNP at 5 mg/kg daily for 14 days. Mice in the vehicle group were administered same volume of PBS. Passive avoidance test was measured by using a Gemini Avoidance System (San Diego Instrument, San Diego, CA, USA). Passive avoidance learning memory was examined using the step through test, which takes advantage of the natural preference mice show for a dark environment. The apparatus consists of illuminated (bright) and dark compartments. During the training day, mice were placed in the bright compartment and received an electric foot shock (0.25 mA, 2 sec) on entering the dark compartment. On the test day (1 day after training), mice were placed in the bright compartment for a maximum of 300 sec and times taken to enter the dark compartment (step-through latencies) were measured for memory retention. No shock was administered during the test day. Memory performance was measured as the latency for the mouse to move into the dark compartment.
Cortical neuronal cell culture and cell viability assay
Dissociated embryonic rat cerebral cortical cell cultures were established and maintained as described previously (Mattson et al. 1995). The cells were grown in polyethylenimine-coated plastic culture dishes for biochemical assays or glass coverslips for imaging. The cultures were maintained at 37°C (in a 6% CO2/94% air atmosphere) in Neurobasal medium containing B-27 supplements (Invitrogen) plus 2 mM L-glutamine, 2 mg/ml gentamycin, 0.001% gentamycin sulfate and 1 mM HEPES (pH 7.2). Experiments were performed with 7 to 9 day-old cultures.
Cell viability was quantified using the dye Alamar blue (resazurin) as described previously (Liu et al. 2009). Neurons were cultured in 24-well plates for 7 days, and then exposed to experimental treatments. At designated time points cells were incubated in medium with Alamar blue at 37°C and the intensity of fluorescence was measured using a fluorescence plate reader with excitation at 540 nm and emission at 590 nm. Values were normalized to the mean value for vehicle-treated control cultures.
Time-lapse confocal imaging
Changes in mitochondrial membrane potential of cultured neurons were evaluated using the fluorescent probe tetramethylrhodamine ethyl ester (TMRE, Molecular Probes) with methods described previously (Liu et al. 2006). Cells were loaded with TMRE (25 nM) for 20 min at 37°C. Images of TMRE fluorescence were acquired every 10 seconds at excitation and emission wavelengths of 549 nm and 574 nm, respectively, using a Zeiss LSM510 confocal microscope. Levels of cyoplasmic free calcium [Ca2+]i and mitochondrial calcium [Ca2+]m were evaluates using the fluorescent probes Fluo-4 and Rhod-2, respectively as described previously (Liu et al. 2010). The data are presented as the average pixel intensity in the neuronal cell body relative to baseline intensity (ΔF/F0) from neurons (6–10 neurons per culture) in 3–5 separate cultures.
Measurement of lipid peroxidation
Levels of 4-hydroxynonenal (HNE) lysine and histidine adducts (lipid peroxidation products) were measured using tandem mass spectrometry as described previously (Cutler et al. 2002). Total lipids from cultured rat cortical neurons were prepared by homogenizing the cells on ice in 10 volumes of cold deionized water, then adding 3 volumes of cold 100% methanol containing 53 mM ammonium formate. Samples were vortexed, four volumes of ice cold chloroform was added, and the mixture was again vortexed and centrifuged at 1,000 × g for 10 min. The chloroform layer was removed and analyzed by direct injection into a Sciex (Thornhill, ON, Canada) 3000 electrospray tandem mass spectrometer. HNE-lysine (355.4 m/z) and HNE-histidine (371.3m/z) were measured and normalized to the total phosphoethanolamine level.
Statistical analysis
One or two way ANOVA was used for comparisons among the different treatment groups at different time points, and Scheffe or Fisher’s PLSD post-hoc tests were performed for pairwise comparisons among treatment groups. For comparisons involving only two groups, Student’s t test was performed.
RESULTS
DNP treatment down-regulates the TSC2-linked mTOR pathway in the cerebral cortex of adult mice
In preliminary experiments we determined whether direct application of DNP to cortical neurons elicited changes consistent with mitochondrial uncoupling. Neuronal cell survival was not affected at DNP concentrations lower than 80 μM (Figure S1). To evaluate the mitochondrial uncoupling activity of DNP, mitochondrial membrane potential was monitored by time-lapse confocal imaging (Figure S1). DNP concentrations of 10 – 40 μM reduced TMRE fluorescence levels suggesting a reduction of mitochondrial membrane potential (Figure S1). Levels of the lipid peroxidation products HNE-lysine and HNE-histidine were reduced in neurons treated with DNP (20 μM) compared to vehicle-treated neurons (Figure S1), consistent with reduced mitochondrial production of reactive oxygen species. In addition, DNP induced an increase of [Ca2+]i and an associated decrease of [Ca2+]m (Figure S1).
To determine how brain cells respond to mild mitochondrial uncoupling in vivo, DNP was administered to adult mice at a dose of 5 mg/kg/day (i.p.), previously used in human subjects (Kaiser 1964). Total RNA was extracted from cerebral cortical samples of vehicle- and DNP-treated mice at 6, 24 and 72 hours following DNP administration (mice in the 72 hour time point were give 3 doses of DNP separated by 24 hours). There were 7–10 mice/group/time point and cortical tissue samples were processed individually. RNA samples were analyzed using a 24,000 gene microarray for expression profiling and network linkage studies. The entire microarray dataset can be found at http://www.ncbi.nlm.nih.gov/gds. Computational analysis (Supplemental Methods) of the gene expression data revealed that DNP modulated multiple signaling pathways in cortical cells, with mTOR (mammalian target of rapamycin) signaling and its associated networks being prominently affected (Figure 1). mTOR is an evolutionarily conserved serine-threonine kinase that responds to nutrient availability, growth factors and cellular bioenergetic status. mTOR interacts with several proteins and forms two complexes, mTORC1 and mTORC2, initially characterized in yeast (Loewith et al. 2002; Wullachleger et al. 2006). mTORC1 is rapamycin-sensitive and the primary effector for stimulation of protein synthesis in response to increased amino acid and energy substrate availability and growth factors. mTORC1 also regulates autophagy (Laplante and Sabatini 2012; Dibble and Manning 2013). mTORC2 responds to activation of Akt, SGK and PKC, and controls cell survival, size and glycolytic metabolism through FoxO transcription factors (Inoki and Guan 2006; Jacinto et al. 2004; Masui et al., 2013).
Figure 1.
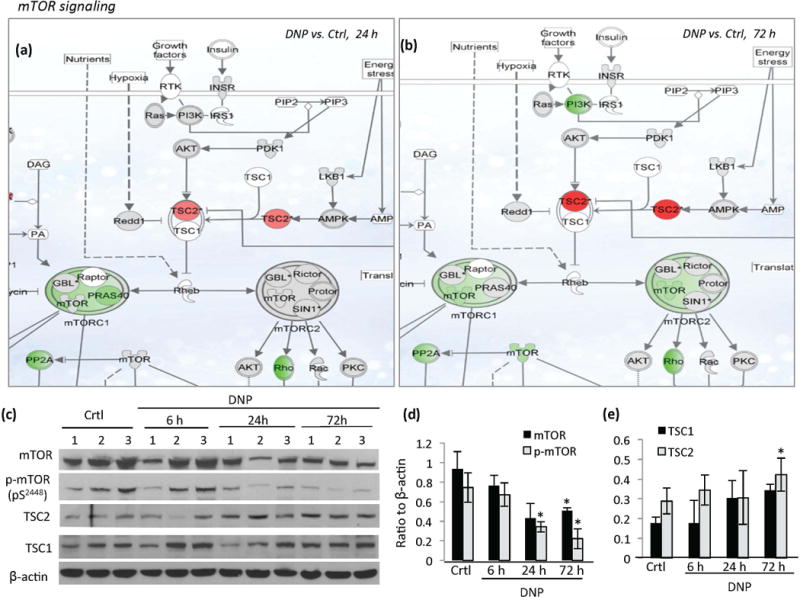
Evidence that mild mitochondrial uncoupling modulates mTOR signaling in the cerebral cortex. (a and b) Schematic diagrams depicting the effects of DNP on the expression of genes in the mTOR signaling pathway showing genes upregulated (red) and downregulated (green) in the cerebral cortex at 24 hours (a) or 72 hours (b) following DNP treatment compared to vehicle-treated control mice. Grey denotes no significant change and white denotes data not available. Genes with a z-score above 1.5 compared to vehicle-treated control were considered significantly changed. (c) Immunoblot analysis of the indicated proteins in cerebral cortex samples from DNP-treated and control mice (n= 7–10 mice/group; samples from 3 animals/group were presented). (d and e). Results of densitometric analysis of the indicated proteins (normalized to β-actin level). Values are mean ± SD. *p<0.05 two tailed compared to the values of DNP to vehicle-treated control mice.
The impact of DNP on the expression of genes in the mTOR signaling is shown in Figure 1a and b. The gene encoding the tumor suppressor protein TSC2 (tuberous sclerosis complex 2), an important upstream regulator of mTOR (Yoshida et al. 2011; Russell et al. 2011), was up-regulated at 24 hours (red, z-ratio =1.59, p<0.05) and became more prominent at 72 hours (z-ratio = 3.47, p<0.01) in DNP-treated mice. TSC2 contains a GTPase-activating protein (GAP) in its carboxyl terminus which inhibits Rheb (Ras homolog enriched in brain), a small GTPase and activator of the mTOR complex (mainly mTORC1). TSC2 negatively regulates mTORC1 by blocking conversion of inactive GDP-bound Rheb into its active GTP-bound state (Inoki et al. 2003). Downstream of TSC2, the expression of the gene encoding the core mTOR kinase was reduced in DNP-treated mice at 72 hours (Figure 1b) (green, z-ratio = −1.5, p<0.01). Suppression of mTORC1 was apparent at 24 and 72 h, and mTORC2 at 72 h (Figure 1a and b). Among mTORC1 complex genes, Raptor (mammalian target of rapamycin) and Gβl/mLst8 (mammalian lethal with sec-13 protein 8) were not changed significantly, whereas Pras40 (proline-rich Akt substrate 40 kDa) was reduced (z-ratio = −1.35, p<0.01). Genes encoding components of the mTORC2 complex, including Rictor (rapamycin-insensitive companion of mTOR), GβL, mSin1 (mammalian stress-activated map kinase-interacting protein 1) and Protor (protein observed with rictor 1 and 2), were not changed significantly. Downstream of mTORC1 signaling, p70S6K expression (p70 ribosomal protein S6 kinase), was not changed significantly (Figure 1a and b). Downstream of mTORC2, expression of the genes encoding Rho GTPase (z-ratio = −4.78, p<0.01) and PP2A (protein phosphatase 2A, z-ratio = −2.3, p<0.01) were down-regulated (Figure 2b).
Figure 2.
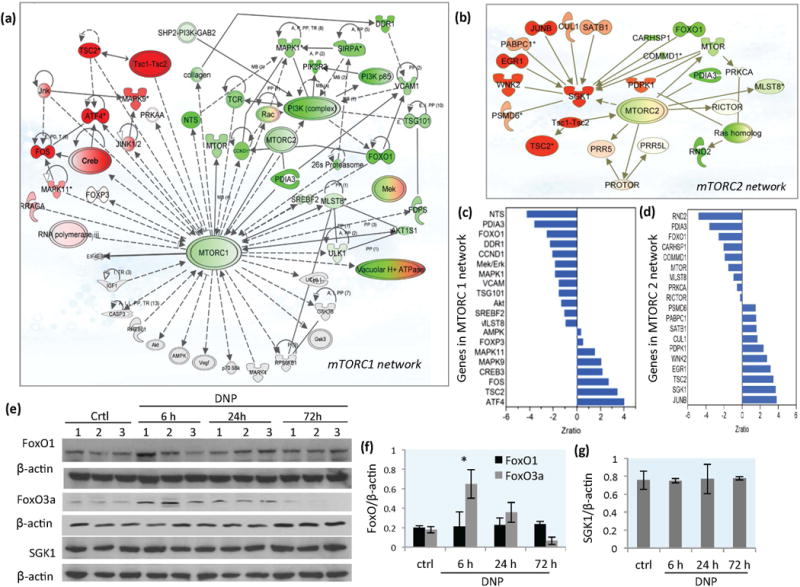
DNP treatment triggers mTORC1 and mTORC2 signaling network re-programming in the cerebral cortex. (a) Gene expression network profile showing genes in the mTORC1 signaling network that were upregulated (red) or downregulated (green) in the cerebral cortex of mice in response to DNP treatment (72 hours). (b) Gene expression profile showing upregulated (red) and downregulated (green) genes in the mTORC2 signaling network from cerebral cortex of mice in response to DNP treatment (72 hours). The attributes that appear both red and green are genes that encode proteins that form complexes; some genes that encode proteins in the complex are up-regulated (red) and some are down-regulated (green). (c) List of genes with z-ratio change in response to DNP treatment in mTORC1 signaling network at 72 hours. (d) List of genes with z-ratio change in response to DNP treatment in mTORC2 signaling network at 72 hours. (e) Immunoblot showing levels of FoxO1, FoxO3a, SGK1 and β-actin in cerebral cortex tissue samples from vehicle-treated control mice and mice that had been treated with DNP for the indicated time periods. (f and g) Densitometric quantification of protein band intensities normalized to β-actin. Values are mean ± SD.
The levels of major proteins involved in mTOR signaling cascade were measured by immunoblot analysis on cortical tissue from control and DNP-treated mouse brains (Figure 1c–e). Levels of TSC2 and TSC1 proteins were increased in cortex samples of DNP-treated mouse brains at 72 hours (Figure 1c and e), while mTOR and p-mTOR (serine2448) were reduced at 24 hours and 72 hours following DNP administration (Figure 1c and d). Similar effects of DNP on TSC2 and mTOR levels were also observed in striatal tissues from DNP treated mice (Figure S2). Protein levels of S6K, phospho-p70S6 kinase and raptor (a scaffold protein regulating the assembly, localization and substrate binding of mTORC1) were not changed significantly by DNP treatment (data not shown).
DNP treatment reprograms mTOR-related signaling networks
mTOR signaling plays key roles in regulating energy metabolism, cell growth, adaptive stress responses and autophagy, and abnormalities in mTOR signaling are implicated in cancers, obesity and age related diseases (Laplante and Sabatini 2012; Johnson et al., 2013). To further determine how mild mitochondrial uncoupling may influence mTOR-associated pathways, we performed mTORC network linkage analysis on microarray data from cortical tissue samples taken at 72 h after DNP treatment. The genes with significant expression changes in the mTORC1network are shown in Figure 2a and z-ratios for each gene are listed (Figure 2c and Table S1). The up-regulated genes included those encoding proteins in the Tsc1/Tsc2 complex, most notably TSC2, and other genes that modulate the mTOR pathway including Creb3 (cAMP-response element binding protein 3), ATF4 (activating transcription factor 4), and Fos (AP-1). ATF4 is a transcriptional regulator that increases in cells exposed to various stressful conditions (Baird and Wek 2012). JNK1/2 (c-Jun N-terminal kinase), a stress-responsive kinase belonging to the mitogen-activated protein kinase family, was linked to upregulation of Mapk9 in response to DNP treatment (Figure 2a). The up-regulation of stress-related genes suggests that mild mitochondrial uncoupling initiates an adaptive bioenergetics-related stress response. Genes down-regulated in the mTORC1 network linkage included Akt, Ddr1 (discoidin domain receptor tyrosine kinase 1), Mapk1, PI3K p85/PIK3R3, Mek/Erk, NTS (neurotensin, PDIA3 (ER60), Srebf2 (sterol regulatory element binding transcription factor 2) and Tsg101 (tumor susceptibility gene 101) (Figure 2a and c). Genes in themTORC1 network that were not significantly affected by mild mitochondrial uncoupling included those encoding AMPK, p70S6K and EIF4E-BP.
Computational prediction on the impact of mild mitochondrial uncoupling on the mTORC2 signaling network is shown in Figure 2b. The major mTORC2 pathway-associated genes with significant expression changes in response to DNP treatment are listed in Figure 2d and Table S2. The Tsc1-Tsc2 complex (mainly TSC2), which plays roles in the integration of cellular responses to energetic stress, was up-regulated upstream of mTORC2 (z-ratio = 3.73, p<0.01). Up-regulated genes included JUNB, EGE1, Sgk1and Egr1. mTORC2 has been reported to activate SGK1(serum and glucocorticoid regulated serine/threonine protein kinase 1) and control cellular ion homeostasis (Garcia-Martinez and Alessi 2008). However, we found that expression of the mTor gene and levels of phosphorylated (active) mTOR protein (p-mTOR) in mTORC2 complex were somewhat reduced in response to DNP (Figure 1a–d), while several other genes encoding proteins upstream of SGK1were upregulated including JunB, EGR1 and Satb1 (Figure 2b) One of the downstream targets of SGK1 is the Forkhead transcription factor FOXO3a (or FKHRL1; Sahin et al. 2013; Brunet et al. 2001). mTORC2 can activate AKT to repress FOXO1 and FOXO3 in mammalian cells (Guertin et al., 2006). We observed an early increase of FoxO1 (z-ratio = 2.29) gene expression (6 hours), but reduced expression of FoxO1 at 72 hours (z-ratio = −1,54). Immunoblot analysis showed that levels of FOXO3a was increased at 6 hours, and FOXO1 and SGK1 were not changed significantly in response to DNP (Figure 2e–g). The increased level of FOXO3a could be related to the reduced inhibition from AKT in the insulin and mTORC2 signaling pathways. Genes down-regulated in the mTORC2 signaling network included Rnd2 (a Rho family GTPase,), and Pdia3 (protein disulfide isomerase family A member 3, also known as ERp57), an activator of the mTORC1 complex (Ramírez et al. 2011), and Ras homolog enriched in brain (Rheb), a GTPase and key activator of mTORC1 that is inhibited by the Tsc1-Tsc2 complex (Inoki et al. 2003). Rictor (rapamycin-insensitive companion of mTOR), a protein involved in the assembly of the mTORC2 complex, was not changed significantly.
DNP treatment down-regulates insulin signaling in the cerebral cortex
Insulin receptor signaling pathways, including those involving PI3K, AKT1 and MEK – ERK, positively regulate mTOR. We found that several key genes in the insulin signaling pathway were down-regulated in the cerebral cortex within 6 hours of DNP administration (Fig. 3a). These genes included Mapk1 (z-ratio = −1.85, p<0.05), PI3k p85-PIk3r3 (z-ratio = −3.04, p< 0.001), Akt/Pkb (z-ratio = −1.24, p<0.05), GSK3β (z-ratio = −4.25, p<0.01), Pdk1 (z-ratio = −3.18, p<0.01) and Mapk/Erk1/2 (z-ratio = −5.27, p<0.01) (Figure 3a). The expression of Pten (phosphatase and tensin homolog deleted on chromosome 10), a negative regulator of insulin signalling, was up-regulated (z-ratio = 2.34. p<0.01) (Figure 3a). PTEN suppresses AKT by catalyzing the conversion of phosphatidylinositol (3, 4, 5)-trisphosphate (PIP3) into phosphatidylinositol (4, 5)-bisphosphate (PIP2) (Huang and Manning 2009). The expression of the genes encoding the insulin receptor and insulin receptor substrate (IRS) were not significantly affected by DNP treatment at the 24 and 72 hour time points (Figure 3b). The protein levels of AKT, p-AKT (Thr308), ERK and p-ERK were examined by immunoblotting which showed that the activate (phosphorylated) forms of these kinases (p-AKT and p-ERK 42/44) were reduced in the cerebral cortex at 24 and 72 hours after DNP treatment (Figure 3c–e). Collectively, these results suggest that insulin receptor signaling is supressed in cerebral cortical cells in response to mild mitochondrial uncoupling.
Figure 3.
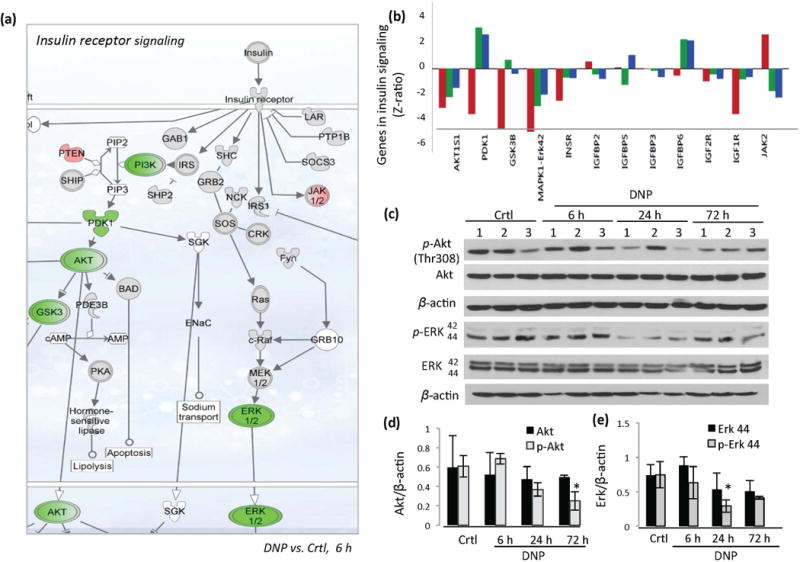
DNP treatment reduces expression of genes in the insulin signaling pathway in the cerebral cortex. (a) Schematic diagram on gene expression in the insulin signaling pathway showing genes upregulated (red) and downregulated (green) in the cerebral cortex within 6 hours of DNP treatment. (b) Genes changed in insulin signaling pathways in the cerebral cortex at 6, 24 and 72 hours following DNP treatment. (c) Immunoblot showing levels of p-Akt, total Akt, p-ERK, total ERK and actin in cerebral cortex tissue samples from vehicle-treated control mice, and mice that had been treated with DNP for the indicated time periods. (d and e) Densitometric quantification of Akt and p-Akt (D), and Erk1/2 and p-Erk1/2 (E) protein band intensities normalized to β-actin. Values are mean ± SD *p<0.05 compared to the control value.
DNP treatment up-regulates calcium – calmodulin – CREB signaling in the cerebral cortex
Our cerebral cortex gene expression profile also revealed that the Ca2+-calmodulin – CREB signaling pathway was upregulated in response to DNP treatment (Figure 4a). Ca2+ and CREB signaling play critical roles in synaptic plasticity and learning and memory (Sakamoto et al. 2011; Chen et al. 2012). The gene expression of Creb3 was significantly up-regulated at 72 h of DNP treatment (Figure 4a, and b). Immunoblot analysis showed that the level of pCREB protein was elevated in the cerebral cortex at 24 and 72 h of DNP treatment (Figure 4b and c). The calcium/calmodulin-dependent protein kinase II (CaMKII) gene complex, mainly CamkIIα (z-ratio = 2.76, p<0.01), was up-regulated in response to DNP treatment (Figure 4a and e), suggesting a possible role for this kinase in CREB phosphorylation in response to mild mitochondrial uncoupling. Other genes with significant changes in this pathway that were responsive to DNP included Atf4 (activating transcription factor 4, z-ratio = 4.03, p<0.01), and calmodulin 1 (Calm1, z-ratio = 1.23, p<0.05) (Figure 4a and e). Because mild mitochondrial uncoupling is known to affect [Ca2+]I and [Ca2+]m in cortical neurons (Figure S1c), it is possible that such a Ca2+ signal mediates CREB activation in response to DNP.
Figure 4.

DNP treatment engages Ca2+ and CREB signaling, up-regulates synaptic plasticity-related genes and enhances memory consolidation. (a) Schematic diagram of Ca2+ and CREB signaling pathways showing genes upregulated (red) and downregulated (green) in the cerebral cortex of mice treated with DNP (72 hours). (b) Immunoblot showing levels of p-CREB, total CREB and Arc/Arg3.1 in cerebral cortex tissue samples from vehicle-treated control mice, and mice that had been treated with DNP for the indicated time periods. (c and d) Densitometric quantification of CREB, p-CREB and Arc/Arg3.1 protein band intensities normalized to β-actin. Values are mean ± SD (n = 7 mice per group). *p<0.05; **p<0.01. (e and f) Parametric analysis of gene set enrichment (PAGE) revealed genes in the CREB signaling pathway, Bdnf and Arc/Arg3.1 upregulated in the cerebral cortex in response to DNP treatment (6 hour time point). Data represent comparisons of 3 control and 3 DNP-treated mice in (c). (g and h) Real-time PCR results show levels of Arc/Arg3.1 and Bdnf mRNAs in cerebral cortex samples from control and DNP-treated mice. (i) Passive avoidance testing was performed to assess memory retention. There was no difference in latency time between vehicle and DNP-treated mice during training. DNP-treated mice exhibited significantly greater retention of the memory of the shock when tested 24 hours after training. Values are mean ± SD (4 mice in the vehicle group and 5 mice in the DNP group). *p<0.05 vs. vehicle (ANOVA with Fisher’s PLSD procedure).
The expression of genes encoding BDNF (z-score= 3.47, p<0.01) and Arc/Arg 3.1 (z-score=3.93, p<0.01), two important activity- responsive proteins that play critical roles in synaptic plasticity and learning and memory, were significantly elevated in the cerebral cortex within 6 h of DNP treatment, and results were validated by real-time PCR analysis (Figure 4f – h). Elevated Bdnf and Arc/Arg 3.1 gene expression in response to DNP suggests that mild mitochondrial uncoupling initiates a plasticity-related adaptive stress response in neurons.
It was reported that rapamycin, an inhibitor of mTORC1, improves cognitive function in a mouse model of Alzheimer’s disease (Spilman et al. 2010; Majumder et al. 2012). To know whether suppression of mTORC1 and activation of the CREB pathway induced by mild mitochondrial uncoupling might influence cognitive function, passive avoidance testing was performed to assess memory retention in mice that had been treated with DNP or vehicle. There was no difference in latency time between vehicle and DNP-treated mice during training, indicating that DNP did not affect the natural preference of the mice for a dark environment (Figure 4i). However, the retention of the memory of the shock after training was significantly greater in the DNP-treated mice compared to the control mice (Figure 4i).
Evidence that autophagy-related processes are responsive to mild mitochondrial uncoupling
mTOR is a negative regulator of autophagy, a process that removes damaged proteins and organelles and recycles intracellular nutrient resources (Ravikumar et al. 2004; Yu et al. 2010). The mechanism whereby mTOR inhibits autophagy involves dephosphorylation of ULKs (autophagy-initiating kinase ULK1, mammalian Atg1 homologs UNC-51-like kinase 1), Atg13 (autophagy-related gene 13) and FIP200 (focal adhesion kinase family–interacting protein of 200 kDa) (Jung et al. 2010).
We investigated genes involved in macroautophagy, chaperone-mediated autophagy, mitophagy and proteasomal degradation to identify those responsive to mild mitochondrial uncoupling (Figure 5). Several genes known to be involved in autophagy and proteosome degradation processes were up-regulated in response to DNP treatment including beclin, Atg8a/Gabarap (GABA-A receptor associated protein, LC3 family), p62 (an E3 ubiquitin ligase), Ulk1 (a protein critical for the formation of the isolation membrane in autophagy), and lysosomal heat shock protein of 70 kDa (hsp70) which plays a key role in chaperone-mediated autophagy (Chiang et al. 2007) (Figure 5a). The genes encoding the proteosome subunit proteins PSMA4 (proteasome alpha 4 subunit) and PSMA7 (proteasome alpha 7 subunit,) were also up-reglulated in the cerebral cortex of DNP-treated mouse brains (Figure 5a). Genes encoding mitochondria-associated proteins involved in apoptosis (Bcl2 and BclxL), mitochondrial fission and fusion (Drp1 and Mfn2), and mitophagy (PINK1, PARK2 and BHIP3L) were not significantly affected by DNP treatment (Figure 5a).
Figure 5.
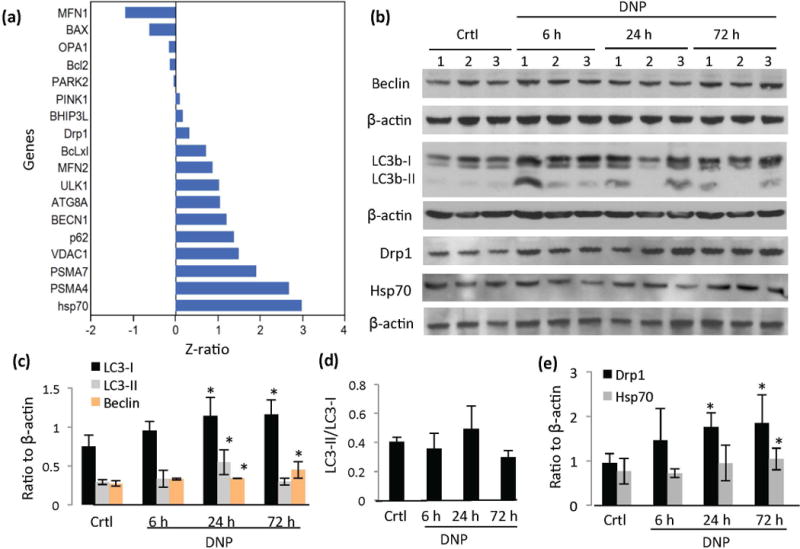
Evidence that mitochondrial uncoupling upregulates autophagy in the cerebral cortex. (a) Relative changes in the expression of genes involved in autophagy, mitophagy and mitochondrial dynamics from the cerebral cortex of mice in response to DNP treatment for 6 hours. (b) Representative immunoblots showing relative levels of the indicated proteins in mouse cerebral cortex samples from the indicated groups. (c) Densitometric analysis of LC3-I, LC3-II and beclin proteins normalized to β-actin. (d) LC3-II/LC3-I ratio in cerebral cortex of mice treated with DNP for the indicated time periods. (e) Densitometric analysis of Drp1 and Hsp70 protein levels in and the cerebral cortex of mice in the indicated treatment groups (n = 7 mice/group). *p<0.05.
We evaluated levels of several autophagy protein markers by immunoblot analysis of cortical tissue samples from control and DNP-treated mice (Figure 5b). LC3b (microtubule-associated protein 1, light chain 3), an ubiquitin-like protein and mammalian ortholog of yeast Atg8 (Nakatogawa et al. 2007), exists in a cytosolic form (LC3b-I) and an autophagosome membrane-associated form (LC3b-II). LC3b-II is covalently attached to phosphatidylethanolamine during autophagosome formation, and is thus an autophagosome marker (Kabeya et al. 2000). Both LC3b-I and LC3b-II levels were significantly increased in DNP-treated mouse cortex at 24 h (Figure 5b and c). The ratio of LC3-II/LC3-I was not significantly affected by DNP treatment (Figure 5d), indicating autophagosome-lysosome system was functioning normally. Beclin/Atg6, a Bcl-2- interacting protein involved in the early stages of the autophagic complex formation (Liang et al. 1999; Kihara et al. 2001), was increased at both the mRNA and protein levels in cortical tissue samples from DNP-treated mice (Figure 5a and c). The levels of Hsp-70 and Drp1 (Dynamin-related protein 1) proteins were also significantly greater in the cortex of DNP-treated mice (Figure 5b and e), suggesting the activation of chaperone-mediated autophagy and mitochondrial dynamics (Lee et al. 2011; Liesa and Shirihai 2013) in response to mild mitochondrial uncoupling.
Finally, we investigated the expression of genes that encode proteins involved in mitochondrial function and biogenesis. Some genes encoding proteins of the mitochondrial respiratory chains were upregulated while others were downregulated (Figure S3). The genes coding for the mitochondrial transcription factor TFAM and PGC-1α were not changed significantly (data not shown). Selected mitochondrial proteins were examined by immunoblotting, including mtDNA coded respiratory chain proteins complex 1 subunit NDUFB8, Fe-S protein 3 (NDUFS3), and nuclear DNA coded protein COX IV and cytochrome c. There were no significant changes in levels of most of the selected proteins, although NDUF8 and NDUFS3 were slightly up-regulated at 24 hours (Figure S3c and d).
DISCUSSION
While mitochondrial uncoupling can generate heat in brown fat cells (Brand 2000), the roles of mild mitochondrial uncoupling in neurons are unclear and likely complex. We found that at concentrations that cause mild mitochondrial uncoupling as indicated by mitochondrial membrane depolarization, DNP mobilizes Ca2+, enhances energy expenditure and reduces lipid peroxidation in cultured cortical neurons. The results of our gene microarray and immunblot analyses of brain tissue samples from control and DNP-treated mice suggest that mild mitochondrial uncoupling suppresses the mTOR pathway, induces reprogramming of the mTORC signaling network and upregulates autophagy (Figure 6). Moreover, pathways involving adaptive stress response and CREB which are known to play important roles in synaptic plasticity were upregulated in response to DNP. These effects of mild mitochondrial uncoupling are similar to those of physiological energetic challenges including exercise and intermittent fasting, which were previously reported to enhance autophagy and stimulate CREB signaling (Mattson 2012; Yang et al. 2014; Altarejos and Montmin 2011; Jamart et al. 2013), even though DNP has the specific effect to enhance energy expenditure through mild mitochondrial uncoupling while the effects of exercise and fasting on neuronal bioenergetics are more complex.
Figure 6.
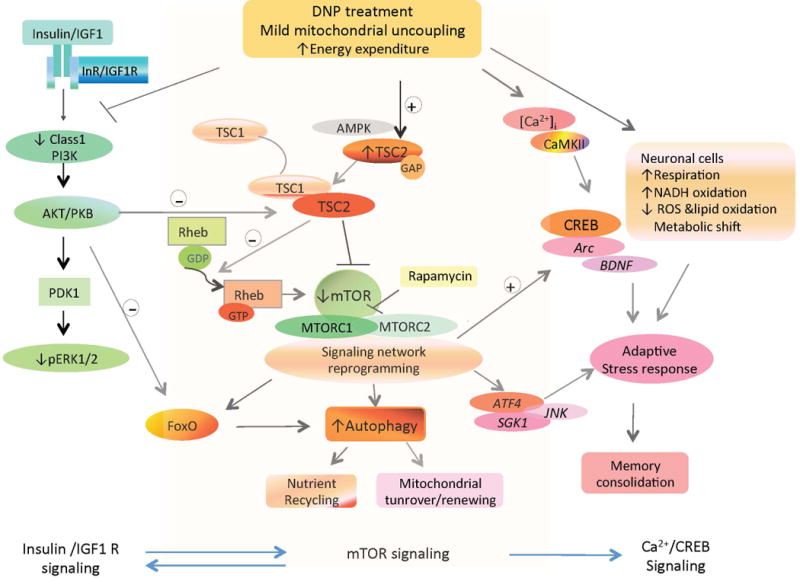
Model for brain signaling network reprogramming in response to mild mitochondrial uncoupling. Mild mitochondrial uncoupling induces a proton leak and enhances energy expenditure. Three major interactive signaling pathways affected by mitochondrial uncoupling are the mTOR and insulin signaling pathways, which are downregulated, and the Ca2+ – CamKII – CREB and autophagy pathways which is upregulated. These changes bolster cellular adaptive stress resistance, and enhance synaptic plasticity by upregulation of Arc/Arg3.1 and BDNF gene transcription. Abbreviations: TSC1/TSC2, tuberous sclerosis complexes 1 and 2; mTOR, mammalian target of rapamycin; CREB, cAMP-response element binding protein; Rheb, Ras homolog enriched in brain; BDNF, brain-derived neurotrophic factor; S6K, ribosomal S6 kinase.
DNP can cross the blood-brain barrier (Perry et al., 2013) and can act directly on neurons to cause mitochondrial uncoupling and increase cellular stress resistance (Figure S1; Korde et al., 2005; da Costa et al., 2010). DNP has been reported to improve brain bioenergetics when administered systemically (Caldeira da Silva et al., 2008). Previous studies of the pharmacokinetics and tissue distribution of DNP in mice and rats demonstrate that DNP readily enters the brain, but the kinetics of its presence in the brain are not fully understood. A study in mice suggested that the half-life of DNP in mice is about 1 hour (Shatzow et al., 1980). In a recent study (Perry et al., 2013), when DNP was administered to rats at the same dose (5 mg/kg) and by the same route (i.p.) as in the present study the peak plasma concentration reached 4–5 μM at 4 h and then dropped to approximately 2 μM at 8 h. In the brain, the DNP concentration reached about 1 μM at 4 h. The latter concentrations of DNP are within the range that we found to be effective in reducing mitochondrial membrane potential and eliciting a Ca2+ response in cultured cortical neurons. While we did not evaluate mitochondrial membrane potential in vivo, it is likely that once daily dosing with DNP results in a transient mitochondrial uncoupling during a period of several hours and then recovers. In this view, the effects of DNP on brain cell gene expression documented in the present study may represent prolonged adaptive responses to a metabolic challenge similar to the adaptive responses of brain cells to vigorous exercise (Tong et al., 2001; Stranahan et al., 2010) and preconditioning ischemia (Stenzel-Poore et al., 2003) reported previously. However, DNP can also reverse diet-induced obesity, hypertriglyceridemia and insulin resistance in diet-induced obesity (Perry et al. 2013; Goldgof et al. 2014). Because such improvements in peripheral energy metabolism can enhance neuronal plasticity and can protect neurons against dysfunction and degeneration in animal models of Alzheimer’s disease and stroke (Mattson, 2012; Searcy et al., 2012), it is possible that some of the molecular responses of brain cells to DNP documented in the present study are secondary to effects of DNP on peripheral organs.
Autophagy is sensitive to cellular energy balance, and mTOR is a major negative regulator of autophagy (Singh and Cuervo, 2011). When nutrients (glucose and amino acids) are prevalent the mTOR pathway is active and cells actively synthesize proteins to enable their growth while autophagy is minimally active. Conversely, under conditions of reduced nutrient availability and/or increased energy expenditure mTOR activity is reduced and autophagy is upregulated. Indeed, many of the major proteins involved in autophagy were discovered because they are up-regulated in response to nutrient deprivation in yeast (He and Klionsky 2009). We found that multiple genes in pathways involved in mTOR activation (PI3K complex, ERKs and Akt) are downregulated by DNP. The mTOR inhibition and upregulation of autophagy in response to mild mitochondrial uncoupling would be expected to protect neurons against the accumulation of damaged/aggregated macromolecules and dysfunctional mitochondria. Previous studies have provided evidence that impaired autophagy occurs in neurons in Alzheimer’s and Parkinson’s diseases (Nixon and Yang 2011; Manzoni and Lewis 2013), and that the mTOR inhibitor rapamycin can upregulate autophagy and protect neurons against dysfunction and degeneration in experimental models of these neurodegenerative disorders (Spilman et al. 2010; Malagelada et al. 2010). It is therefore likely that upregulation of autophagy contributes to the neuroprotective effects of DNP in animal models of stroke (Korde et al. 2005) and traumatic nerve injury (da Costa et al. 2010).
Previous studies have demonstrated a complex interplay between PI3K/Akt, TSC and mTOR complexes (Shah et al. 2004; Huang and Manning 2009). The Tsc1/Tsc2 complex interacts with PI3K/Akt signaling such that inhibition of TSC1/TSC2 activates mTOR and disrupts PI3K-Akt signaling (Zhang et al. 2003; Zhang et al. 2011). On the other hand, insulin receptor signaling inhibits TSC1/TSC2 via Akt, thereby activating the mTOR pathway (Zhang et al. 2003). Ablation of mTORC components in mice reveals that mTORC2 is required for signaling of Akt-FOXO and PKCα (Guertin et al. 2006). However, more recent findings suggest that the effects mTORC2 in regulating glycolytic metabolism may be Akt-independent (Masui et al. 2013). We found that multiple genes in the insulin/ PI3K/Akt signaling pathway were suppressed, and Foxo3a was upregulated transiently in the cerebral cortex in response to DNP. Down-regulation of insulin receptor signaling may contribute to the mechanism whereby caloric restriction extends longevity in animal models of aging (Blüher et al. 2003; Mercken et al. 2013). FoxO3a also plays a role in stimulating autophagy (Mammucari et al., 2007). Interestingly, it was reported that mild mitochondrial uncoupling can also enhance longevity in mice (Caldeira da Silva et al., 2008). It is therefore important to determine the relative contributions of mTOR cascade signaling reprogramming to the neuroprotective actions of DNP in models of neurodegenerative disorders (Korde et al., 2005; Pandya et al., 2007; da Costa et al., 2010).
CREB is activated in response to excitatory synapse activation, BDNF and exercise, and plays important roles in synaptic plasticity and learning and memory (for review see Sakamoto et al., 2011; Benito and Barco, 2010). CREB also mediates adaptive responses to metabolic and oxidative stress, including stimulation of DNA repair (Lee et al., 2009; Sebollela et al., 2010; Yang et al., 2010). We found that CREB-related signaling is upregulated in the cerebral cortex in response to DNP treatment, suggesting that mild mitochondrial uncoupling engages a prominent pathway involved in synaptic plasticity and memory. Consistent with a plasticity-promoting effect of mild mitochondrial uncoupling we found that DNP-treated mice exhibited improved memory retention in the passive avoidance test. However, it was previously reported that DNP can impair discrimination memory in chickens when administered after a training session (O’Dowd et al., 1994). In the latter study, DNP 0.2 mmole was administered intracranially at various times before and after training sessions. They suggested a 3-stage model of memory formation and the second or intermediate stage involves two phases: phase A which lasts up to 30 min following learning, is energy dependent and susceptible to inhibition by DNP; phase B which occurs after phase A, lasts up to 50 min following learning, is energy independent and is not susceptible to inhibition by DNP. Thus, DNP is not simply causing memory deficits, and its effects are energy- and time-related, and dose-related as well. In our study of mice, a low dose of DNP was administrated i.p. once daily for 14 days prior to passive avoidance testing. We found that the DNP-treated mice performed significantly better in the passive avoidance test, a result consistent with up-regulation of signaling pathways involved in synaptic plasticity in response to our once-daily DNP dosing schedule.
We also found that BDNF expression was increased by more than three-fold in the cerebral cortex of DNP-treated mice compared to vehicle-treated mice. Excitatory synaptic activity stimulates CREB by a mechanism involving Ca2+ influx and activation of Ca2+-calmodulin-dependent kinase II (CaMKII) pathway. Previous studies in which the expression of mitochondrial uncoupling proteins (UCP2 and UCP4) were genetically manipulated in neurons provided evidence that mitochondrial uncoupling affects Ca2+ signaling (Chan et al., 2006; Mehta and Li, 2009) suggesting a mechanism whereby DNP treatment could activate the Ca2+ – Arc – CaMKII – CREB pathway. We found that exposure of cultured cortical neurons to DNP resulted in a rapid transient elevation of intracellular Ca2+ levels that peaked within 1–2 minutes and then remained elevated by a modest amount for at least 10 minutes. Similar Ca2+ responses are known to trigger long-lasting (hours to days) changes in CREB-mediated gene expression and associated synaptic plasticity (Benito and Barco, 2010; Suzuki et al., 2011). The activation of the latter pathway is also known to induce BDNF expression which itself likely contributes to the cognition-enhancing effects of DNP/mild mitochondrial uncoupling. It was previously reported that low concentrations of DNP increase cyclic AMP levels in cultured neurons (Wasilewska-Sampaio et al., 2005) which, in addition to Ca2+, would also be expected to activate CREB and upregulate BDNF expression. BDNF regulates neuronal energy metabolism and stress responses (Nakagawa et al., 2002; Arancibia et al., 2003; Larsen et al., 2010; Marosi and Mattson, 2014). BDNF can enhance glucose uptake and mitochondrial biogenesis in neurons, which may contribute to its important roles in synaptic plasticity and neuroprotection (Burkhalter et al., 2003; Cheng et al., 2012). Although not established in the present study, it is possible that BDNF contributes to bioenergetic responses of neurons to DNP as we found that cortical neurons treated with DNP exhibit enhanced glucose uptake (Liu et al., 2008). The Bdnf gene is a target of CREB and, conversely, BDNF signaling can activate CREB (Suzuki et al. 2011). BDNF and CREB signaling pathways may therefore cross-amplify each other to enhance synaptic plasticity in response to mild mitochondrial uncoupling. Moreover, crosstalk between cAMP-mediated CREB signaling and the mTOR pathway have been reported previously, although the underlying molecular mechanisms are unclear (Carlon et al. 2010). In any case, our findings are consistent with a role for Ca2+ signaling in CREB activation and BDNF expression in response to mild mitochondrial uncoupling.
In summary, our findings demonstrate that treatment of mice with a low dose of the mitochondrial uncoupling agent DNP results in major changes in cell signaling networks and metabolism in cerebral cortical cells characterized by downregulation of the mTOR and insulin signaling pathways, and upregulation of autophagy, FoxO3a, and Ca2+, CREB and BDNF signaling. These responses to mild mitochondrial uncoupling suggest a complex adaptive remodeling of the molecular pathways that regulate neuronal stress responses and synaptic plasticity. Further studies may clarify the potential role of mild mitochondrial uncoupling as a therapeutic approach for age-related diseases including neurological disorders.
Supplementary Material
Acknowledgments
The authors thank W. Wood III for help with microarray processing, P. Ghosh, J. Lim and J. Licata for providing antibodies, and Mark Wilson for technical support. This research was supported in part by the Intramural Research Program of the National Institute on Aging, and the Financial Supporting Project of Long-term Overseas Dispatch of PNU’s Tenure-track Faculty.
Footnotes
Conflicts of interests
The authors declare no competing interests.
Author contributions: D. L. planned and performed experiments, and wrote the manuscript. Y. Z. and K. G. B. performed the gene array analysis. R. G., H. R. P., S. S., R. T., M. N., R. C. and H. J. performed assays and helped analyze data. J. L. planned experiments and wrote the manuscript. M. P. M. planned the experiments and wrote the manuscript.
References
- Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Diano S, Horvath TL. Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nat Rev Neurosci. 2005;6:829–40. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- Arancibia S, Rage F, Givaloi L, Tapia-Arancibia L. Neurotrophins in neuroendocrine control: brain derived-neurotrophic factor (BDNF) and somatostatin involvement in the stress response and reproductive physiology. Ann Rev Biomed Sciences. 2003;5:5–28. [Google Scholar]
- Arumugam TV, Phillips TM, Cheng A, Morrell CH, Mattson MP, Wan R. Age and energy intake interact to modify cell stress pathways and stroke outcome. Ann Neurol. 2010;67:41–52. doi: 10.1002/ana.21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird TD, Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism American Society for Nutrition. Adv Nutr. 2012;3:307–321. doi: 10.3945/an.112.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Barco A. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Blüher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Experiment Gerontol. 2000;35:811–820. doi: 10.1016/s0531-5565(00)00135-2. [DOI] [PubMed] [Google Scholar]
- Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Molecu Cellul Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhalter J, Fiumelli H, Allaman I, Chatton JY, Martin JL. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J Neurosci. 2003;23:8212–8220. doi: 10.1523/JNEUROSCI.23-23-08212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell. 2008;7:552–60. doi: 10.1111/j.1474-9726.2008.00407.x. [DOI] [PubMed] [Google Scholar]
- Carlon S, Girelli S, Scopa C, Buonocore G, Longgin M, Balduini W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy. 2010;6:366–377. doi: 10.4161/auto.6.3.11261. [DOI] [PubMed] [Google Scholar]
- Chan CB, Harper ME. Uncoupling proteins: role in insulin resistance and insulin insufficiency. Curr Diabetes Rev. 2006;2:271–283. doi: 10.2174/157339906777950660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Li D, Kyriazis G, Bagsiyao P, Ouyang X, Mattson MP. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store-depletion-induced apoptosis in neural cells. J Biol Chem. 2006;281:37391–37403. doi: 10.1074/jbc.M605552200. [DOI] [PubMed] [Google Scholar]
- Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Bambah-Mukku D, Pollonini G, Alberini CM. Glucocorticoid receptors recruit the CaMKIIα-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci. 2012;15:1707–1714. doi: 10.1038/nn.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HU, Terlecky SR, Plant CP, Fred Dice J. A Role for a 70-kilodaton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;346:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- Colman E. Dinitrophenol and obesity: an early twentieth-century regulatory dilemma. Regul Toxicol Pharmacol. 2007;48:115–117. doi: 10.1016/j.yrtph.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, Mattson MP. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat Commun. 2012;3:1250. doi: 10.1038/ncomms2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. 2010;21:589–598. doi: 10.1016/j.tem.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol. 2002;52:448–457. doi: 10.1002/ana.10312. [DOI] [PubMed] [Google Scholar]
- da Costa RF, Martinez AM, Ferreira ST. 2,4-Dinitrophenol blocks neurodegeneration and preserves sciatic nerve function after trauma. J Neurotrauma. 2010;27:829–841. doi: 10.1089/neu.2009.1189. [DOI] [PubMed] [Google Scholar]
- Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtay KS. Mitochondrial uncoupling proteins–what is their physiological role? Free Radic Biol Med. 2007;43:1351–1371. doi: 10.1016/j.freeradbiomed.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Enerbäck S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, Kozak LP. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 1997;387:90–94. doi: 10.1038/387090a0. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Gainutdinov T, Muth K, Seppet E, Orynbayeva Z, Vielhaber S. The control of brain mitochondrial energization by cytosolic calcium: the mitochondrial gas pedal. IUBMB Life. 2013;65:180–190. doi: 10.1002/iub.1131. [DOI] [PubMed] [Google Scholar]
- Goldgof M, Cuiying Xiao C, Chanturiya T, Jou W, Gavrilova O, Marc L, Reitman ML. The Chemical uncoupler 2,4-dinitrophenol (DNP) protects against diet-induced obesity and improves energy. J Biol Chem. 2014;289:19341–19350. doi: 10.1074/jbc.M114.568204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffioen KJ, Rothman SM, Ladenheim B, Wan R, Vranis N, Hutchison E, Okun E, Cadet JL, Mattson MP. Dietary energy intake modifies brainstem autonomic dysfunction caused by mutant α-synuclein. Neurobiol Aging. 2013;34:928–935. doi: 10.1016/j.neurobiolaging.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Ann Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochemical Society Transactions. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Develop. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Guan KL. Complexity of the TOR signaling network. Trends Cell Biol. 2006;16:206–212. doi: 10.1016/j.tcb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Ann Rev Pharmacol Toxicol. 2012;52:381–400. doi: 10.1146/annurev-pharmtox-010611-134537. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Jamart C, Naslain D, Gilson H, Francaux M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab. 2013;305:E964–974. doi: 10.1152/ajpendo.00270.2013. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Rabinovitch PS, Kaeberlein M. MTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung AH, Ro S, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Let. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser JA. Studies on the toxicity of disophenol (26-diiodo-4-nitrophenol (2,6-diiodo-4-nitrophenol) to dogs and rodents plus some comparisons with 2,4-dinitrophenol. Ther Ggw. 1964;103:232–44. doi: 10.1016/0041-008x(64)90108-5. [DOI] [PubMed] [Google Scholar]
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–335. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korde AS, Pettigrew LC, Craddock SD, Maragos WF. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J Neurochem. 2005;94:1676–1684. doi: 10.1111/j.1471-4159.2005.03328.x. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen MH, Mikkelsen JD, Hay-schmid A, Sandi C. Regulation of brain-derived neurotrophic factor (BDNF) in the chronic unpredictable stress rat model and the effects of chronic antidepressant treatment. J Psychiat Res. 2010;44:808–816. doi: 10.1016/j.jpsychires.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Lee B, Cao R, Choi YS, Cho HY, Rhee AD, Hah CK, Hoyt KR, Obrietan K. The CREB/CRE transcriptional pathway: protection against oxidative stress-mediated neuronal cell death. J Neurochem. 2009;108:1251–1265. doi: 10.1111/j.1471-4159.2008.05864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lee HY, Hanna RA, Gustafsson ÅB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;301:H1924–H1931. doi: 10.1152/ajpheart.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Chan SL, de Souza-Pinto NC, Slevin JR, Wersto RP, Zhan M, Mustafa K, de Cabo R, Mattson MP. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromol Med. 2006;8:389–414. doi: 10.1385/NMM:8:3:389. [DOI] [PubMed] [Google Scholar]
- Liu D, Pitta M, Mattson MP. Preventing NAD+ depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann N Y Acad Sci. 2008;1147:275–282. doi: 10.1196/annals.1427.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Pitta M, Lee JH, Ray B, Lahiri DK, Furukawa K, Mughal M, Jiang H, Villarreal J, Cutler RG, Greig NH, Mattson MP. The KATP channel activator diazoxide ameliorates amyloid-b and tau pathologies and improves memory in the 3xTgAD mouse model of Alzheimer’s disease. J Alzheimers Dis. 2010;22:443–457. doi: 10.3233/JAD-2010-101017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromol Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Caccamo A, Medina DX, Benavide AD, Javors MA, Kraig E, Strong R, Richardson A, Oddo S. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1β and enhancing NMDA signaling. Aging Cell. 2012;11:326–335. doi: 10.1111/j.1474-9726.2011.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Piccolo PD, Burden SJ, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metabol. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Manzoni C, Lewis PA. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 2013;27:3424–3429. doi: 10.1096/fj.12-223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragos WF, Korde AS. Mitochondrial uncoupling as a potential therapeutic target in acute central nervous system injury. J Neurochem. 2004;91:257–262. doi: 10.1111/j.1471-4159.2004.02736.x. [DOI] [PubMed] [Google Scholar]
- Marosi K, Mattson MP. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol Metab. 2014;25:89–98. doi: 10.1016/j.tem.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, Campos C, Zhu S, Yang H, Yong WH, Cloughesy TF, Mellinghoff IK, Cavenee WK, Shaw RJ, Mischel PS. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18:726–39. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Perspective: Does brown fat protect against diseases of aging? Ageing Res Rev. 2010;9:69–76. doi: 10.1016/j.arr.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab. 2012;16:706–722. doi: 10.1016/j.cmet.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SL, Li PA. Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. J Cereb Blood Flow Metab. 2009;29:1069–1078. doi: 10.1038/jcbfm.2009.4. [DOI] [PubMed] [Google Scholar]
- Meller R, Simon RP. Tolerance to Ischemia – an increasingly complex biology. Transl Stroke Res. 2013;4:40–50. doi: 10.1007/s12975-012-0246-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercken EM, Crosby SD, Lamming DW, Jebailey L, Krzysik-Walker S, Villareal DT, Capri M, Franceschi C, Zhang Y, Becker K, Sabatini DM, de Cabo R, Fontana L. Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell. 2013;12:645–51. doi: 10.1111/acel.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee SA, Divakaruni AS, Jastroch M, Brand MD. Mitochondrial uncoupling and lifespan. Mech Ageing Dev. 2010;13:463–472. doi: 10.1016/j.mad.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Ono-Kishino M, Sugaru E, Yamanaka M, Taiji M, Noguchi H. Brain-derived neurotrophic factor (BDNF) regulates glucose and energy metabolism in diabetic mice. Diabetes Metab Res Rev. 2002;18:185–191. doi: 10.1002/dmrr.290. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, Maragos WF, Hall ED, Sullivan PG. Post-injury administration of mitochondrial uncouplers increases tissue sparing and improves behavioral outcome following traumatic brain injury in rodents. J Neurotrauma. 2007;24:798–811. doi: 10.1089/neu.2006.3673. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, Spiegel DA, Shulman GL. Reversal of hypertriglyceridemia, fatty liver disease and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 2013;18:740–748. doi: 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden DB, Ho PW, Ho JW, Liu HF, So DH, Tse HM, Chan KH, Ho SL. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2012;2:468–478. doi: 10.1002/brb3.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of the Huntington’s disease mutation in drosophila and mouse models. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Russell RC, Fang C, Guan KL. An emerging role for TOR signaling in mammalian tissue and stem cell physiology. Develop. 2011;138:3343–56. doi: 10.1242/dev.058230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah OJ, Wan Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Sahin P, McCaig C, Jeevahan J, Murra JT, Hainsworth AH. The cell survival kinase SGK1 and its targets FOXO3a and NDRG1 in aged human brain. Neuropathol Appl Neurobiol. 2013;39:623–633. doi: 10.1111/nan.12023. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Karelina K, Obrietan K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem. 2011;116:1–9. doi: 10.1111/j.1471-4159.2010.07080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Searcy JL, Phelps JT, Pancani T, Kadish I, Popovic J, Anderson KL, Beckett TL, Murphy MP, Chen KC, Blalock EM, Landfield PW, Porter NM, Thibault O. Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2012;30:943–961. doi: 10.3233/JAD-2012-111661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebollela A, Freitas-Corrêa L, Oliveira FF, Mendes CT, Wasilewska-Sampai AP, Camacho-Pereir J, Galina A, Brentani H, Passetti F, De Felice FG, Dias-Neto E, Ferreira ST. Expression profile of rat hippocampal neurons treated with the neuroprotective compound 2,4-dinitrophenol: up-regulation of cAMP signaling genes. Neurotox Res. 2010;18:112–23. doi: 10.1007/s12640-009-9133-y. [DOI] [PubMed] [Google Scholar]
- Shatzow S, U. S. Environmental Protection Agency EPA 440/5-80-063. Ambient water quality criteria for nitrophenols. 1980:C37.
- Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnat J, Gorostiza O, Bredese D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003;86:1101–7. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- Starkov AA. Protein-mediated energy-dissipating pathways in mitochondria. Chemico-Biol Interact. 2006;161:57–68. doi: 10.1016/j.cbi.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003;362:1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Becker KG, Zhang Y, Maudsley S, Martin B, Cutler RG, Mattson MP. Hippocampal gene expression patterns underlying the enhancement of memory by running in aged mice. Neurobiol Aging. 2010;31:1937–1949. doi: 10.1016/j.neurobiolaging.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Fukushima H, Mukawa T, Toyoda H, Wu LJ, Zhao MG, Xu H, Shang Y, Endoh K, Iwamoto T, Mamiya N, Okano E, Hasegawa S, Mercaldo V, Zhang Y, Maeda R, Ohta M, Josselyn SA, Zhuo M, Kida S. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J Neurosci. 2011;31:8786–8802. doi: 10.1523/JNEUROSCI.3257-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Shen H, Perreau VM, Balazs R, Cotman CW. Effects of exercise on gene-expression profile in the rat hippocampus. Neurobiol Dis. 2001;8:1046–1056. doi: 10.1006/nbdi.2001.0427. [DOI] [PubMed] [Google Scholar]
- Tseng YH, Cypess AM, Ronal KC. Cellular bioenergetics as a target for obesity therapy. Nat Rev Drug Discov. 2010;9:465–482. doi: 10.1038/nrd3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Puig AJ. Uncoupling expectations. Nat Genet. 2000;26:387. doi: 10.1038/82489. [DOI] [PubMed] [Google Scholar]
- Wasilewska-Sampaio AP, Silveira MS, Holub O, Goecking R, Gomes FC, Neto VM, Linden R, Ferreira ST, De Felice FG. Neuritogenesis and neuronal differentiation promoted by 2,4-dinitrophenol, a novel anti-amyloidogenic compound. FASEB J. 2005;19:1627–36. doi: 10.1096/fj.05-3812com. [DOI] [PubMed] [Google Scholar]
- Wullachleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yang JL, Tadokoro T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1. J Biol Chem. 2010;285:28191–28199. doi: 10.1074/jbc.M109.082883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JL, Lin YT, Chuang PC, Bohr VA, Mattson MP. BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromolecular Med. 2014;16:161–74. doi: 10.1007/s12017-013-8270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle R, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, Okada M, Guan KL, Inoki K. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J Biol Chem. 2011;286:32651–60. doi: 10.1074/jbc.M111.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wu Y, Jia J. Exercise preconditioning and brain ischemic tolerance. Neuroscience. 2011;177:170–176. doi: 10.1016/j.neuroscience.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.