

Abstract
Reduced expression of the GAD1 gene-encoded 67-kD protein isoform of glutamic acid decarboxylase (GAD67) is a hallmark of the schizophrenia. GAD67 downregulation occurs in multiple interneuronal subpopulations, including the parvalbumin positive (PVALB+) cells. To investigate the role of the PV-positive GABA-ergic interneurons in behavioral and molecular processes, we knocked down the Gad1 transcript using a miRNA engineered to specifically target Gad1 mRNA under the control of Pvalb bacterial artificial chromosome. Verification of construct expression was performed by immunohistochemistry. Follow-up electrophysiological studies revealed a significant reduction in GABA release probability without alterations in postsynaptic membrane properties or changes in glutamatergic release probability in prefrontal cortex pyramidal neurons. Behavioral characterization of our transgenic mice uncovered that the Pvalb/Gad1 Tg mice have pronounced sensorimotor gating deficits, increased novelty seeking and reduced fear extinction. Furthermore, NMDA receptor antagonism by ketamine had an opposing dose-dependent effect, suggesting that the differential dosage of ketamine might have divergent effects on behavioral processes. All behavioral studies were validated using a second cohort of animals. Our results suggest that reduction of GABA-ergic transmission from PVALB+ interneurons primarily impacts behavioral domains related to fear and novelty seeking and that these alterations might be related to the behavioral phenotype observed in schizophrenia.
Keywords: Pvalb, Gad1, GAD67, transgenic, miRNA, ketamine, behavior, interneuron, schizophrenia, GABA release
INTRODUCTION
γ-Aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the mammalian central nervous system. GABA synthesis is primarily performed by the 67-kDa and 65-kDa isoforms of glutamic acid decarboxylase enzymes (GAD67 and GAD65), which are respectively encoded by the Gad1 and Gad2 genes 1. They differentially contribute to GABA production 1, and in mice, deletion of the Gad1 gene (and resulting lack of GAD67) results in ~90% reduction of brain GABA levels and is lethal 2.
GABA-ergic interneurons are diverse 3, 4 with >20 types of interneurons regulating the function of only three types of glutamatergic cells in the hippocampus 5. They can be classified based on their laminar location, molecular content, electrical properties, synaptic targets, and many other criteria 3, 4, 6. Perhaps the most important and distinguishing feature of the various interneuronal cell types is their molecular content: GABA-ergic cells types typically express either calcium binding proteins parvalbumin (PVALB), calretinin, or calbindin or the neuropeptides cholecystokinin (CCK), neuropeptide Y (NPY), somatostatin (SST), or vasointestinal peptide (VIP) in a mostly non-overlapping pattern 3, 4, 6. It appears that these interneurons serve different functions and fine-tune complex neuronal networks.
PVALB-expressing interneurons make up approximately 50% of the neocortical interneuron population and come in two main varieties: fast-spiking basket and chandelier cells that innervate pyramidal cell soma and axon initial segments, respectively 1, 6. PVALB+ interneurons also inhibit other interneuron population that target the proximal dendrites of projection neurons, providing a complex control of neural networks 7. Neocortical PVALB+ cells are essential for driving cortical gamma oscillations in mice, which human studies suggest are essential for normal working memory 1.
GABA-ergic, especially GAD1/GAD67, expression disturbances are an integral part of schizophrenia pathology and pathophysiology 8–10. Levels of GAD1 mRNA 11 and protein 12, 13 have been found consistently decreased in the neocortex and hippocampus of subjects with schizophrenia and this deficit appears to be present in multiple interneuronal cell types 9, 14, 15. In particular, the GAD67 deficit is prominent in PVALB-positive interneurons 1 with approximately 50% of these cell showing non-detectable GAD67 levels 1, 16.
To examine the behavioral consequences of Gad1 gene reduction in vivo, we developed a Pvalb bacterial artificial chromosome-driven, Gad1-silencing miRNA construct expressing transgenic (Tg) mouse model (Pvalb/Gad1 in further text)17–19. After validation by immunohistochemistry (IHC) and electrophysiology, we subjected these mice to a broad battery of behavioral tests. Furthermore, as GABA-ergic interneurons are disproportionately more sensitive to NMDA antagonism than projection neurons 20–22, we assessed the behavioral response of our transgenic animals to sub-anesthetic doses of NMDA receptor antagonist ketamine.
MATERIALS AND METHODS
All animal procedures were performed in accordance with the guidelines of the American Association for Laboratory Animal Science and approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Pvalb/Gad1 mouse generation
RP24-306A6 BAC, containing the mouse parvalbumin (mPvalb) locus (Chr15: 78,126,251 – 78,255,940, NCBI Build 38.1), was purchased from the BACPAC Resource at the Children’s Hospital of Oakland Research Institute (https://bacpac.chori.org/). The presence of the mPvalb locus in RP24-306A6 was verified by restriction enzyme digest mapping. The mPvalb gene itself is located on the negative strand of Chr15: 78,191,117 – 78,206,351. Besides mPvalb, RP24-306A6 BAC carried an additional gene, mRabl4. mRabl4 knock-out BAC was generated by removing 2670 bp (exon 2,3 and 4) via homologous recombination. The mRabl4− BAC was transformed into EL250 E. coli cells (kind gift of Dr. Neil Copeland, NCI). A BAC targeting construct was inserted into pSTBlue-1 plasmid vector (Novagen, Madison) in two steps. First, Pvalb 5′ (170 bp) and 3′ (180 bp) homology arms were PCR generated and cloned into pSTBlue-1. Next, a β-globin minigene containing a Gad1 targeting miRNA in an intronic location was released from a previously engineered construct 17 and inserted at the 3′ end of tdTomato into ptdTomato-N1 vector (Clontech, Mountain View, CA). The adjacent tdTomato and β-globin minigenes were then released from ptdTomato-N1 and inserted between the 5′ and the 3′ Pvalb homology arms into pSTBlue-1. The final mPvalb targeting construct carried Pvalb 5′ and 3′ homology arms surrounding tdTomato, β-globin minigene and an FRT-flanked neomycin resistance cassette. The targeting fragment was then released using AgeI restriction enzyme (New England BioLabs, Ipswich, MA) and electroporated into EL250 cells containing mRabl4− RP24-306A6 BAC for homologous recombination into mPvalb. The resulting BACs were screened by PCR and confirmed with restriction mapping and sequence analysis for correct modifications. Finally, the E. coli strain containing the modified BAC was treated with arabinose to induce the expression of FLP recombinase, which removed the FRT-flanked neomycin resistance cassette. Proper recombination was confirmed with restriction mapping and sequence analysis of the region of interest. The modified RP24-306A6 BAC was isolated with alkaline lysis and purified with Sepharose CL-4B chromatography as described previously 23. Transgenic mice were generated by injection of circular modified BAC into fertilized C57BL/6 mouse oocytes by the University of California Irvine Transgenic Mouse Facility. Transgenic founder mice were identified by PCR by using construct-specific primer pairs. Mouse genotyping of subsequent generations was performed at weaning (P21) from 2 mm tail samples. The samples were digested in 245 μl DirectPCR (Tail) (Viagen Biotech, Los Angeles, CA) and 5 μl Proteinase K (Clontech, Mountain View, CA) overnight at 55°C, then incubated at 85°C for 45 min. PCR genotyping of samples was performed using GTGAACGTGGATGAAGTTGG (forward) and GCAGGCAACGATTCTGTAAA (reverse) primers at 60°C annealing temperature, yielding a 178 bp amplicon. For simplicity, we will refer to the transgenic animals as Pvalb/Gad1 mice in the further text.
Immunohistochemistry (IHC)
For IHC, 4 animals were anesthetized with isoflurane and transcardially perfused with cold Ringer’s solution containing 2% lidocaine HCl (20 mg/mL) and heparin (1000 USP units/mL) followed by 4% paraformaldehyde. Brains were harvested as previously described 19. Immunostaining for tdTomato was performed with a 1:200 diluted mouse anti-DsRed antibody (Clontech, Mountain View, CA). For PVALB immunostaining, we used a 1:5,000 dilution of rabbit anti-parvalbumin primary antibody (Swant Ltd., Marly, Switzerland). CCK-stained sections were incubated with rabbit anti-proCCK (a generous gift from Dr Andrea Varro; 1:1000) for 72 h at 4°C, while NPY staining was performed using rabbit anti-NPY primary antibodies (Sigma, St Louis, MO, USA; 1:1000) . For fluorescence visualization, goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 568 secondary antibodies were used (Life Technologies, Carlsbad, CA, USA) at 1:250 dilution. DAPI staining was performed using 3 min incubation in 300 nM DAPI (Sigma, St Louis, MO, USA). Rinsed brain slices where mounted on slides and cover-slipped in ImmunoMount (Fisher Scientific, Pittsburg, PA, USA). Imaging was performed using the EVOS imaging system and microscope (Life Technologies, Carlsbad, CA, USA).
Behavioral experiments
Animals
Male C57Bl/6J mice (3–4 months of age at start of testing) were employed in all experiments. All animals were housed in groups of two to five. Food and water were available ad libitum. All mice were kept on a 12 hr light-dark cycle.
Behavioral assessment
Behavioral evaluation was performed in the Vanderbilt Murine Neurobehavioral Laboratory. 3–4 month old male mice were handled for 5 days prior to the beginning of behavioral testing. Prior to each testing session, mice were brought from the animal housing room into an anteroom outside each testing room and acclimated for 1 hour under red light. Consecutive tests were at least 24 hours apart. Experimenters were blinded to genotypes. All equipment was cleaned with Vimoba solution (Quip Laboratories, Wilmington, DE) between animals to reduce odor contamination. Two independent cohorts of Pvalb/Gad1 transgenic (Tg) and wild type (Wt) littermates were used for all behavior experiments (first cohort: n= 12 Tg and n= 11 Wt; second cohort; n=12 Tg and n= 10 Wt). Mice were evaluated on the following battery of tests as described previously 18, 19: (1) 10 min open field exploration, (2) Irwin screen (including 90dB acoustic Preyer reflex), and battery of neuromuscular measures (grip strength, rotor rod and swim speed), (3) fear conditioning. Briefly, trace fear conditioning was assessed using a well-validated protocol for mice 24 that was modified to include extinction measures 19. 24 hours after a 12 min habituation session, mice returned to the training chamber and received six tone/footshock pairings (70dB, 20s tone; 0.5mA, 2s shock). 24 hours later, they were placed back into the training environment and tested for context fear conditioning in the absence of tone or shock, then placed in an alternative context where the tone was presented multiple times in the absence of footshock to evaluate cued fear conditioning and extinction. Cued fear extinction was defined as the magnitude of reduction in freezing behavior from the first to the last testing trial. Detailed methods for these tasks can be found in 18, 19. (4) 0-maze and y-maze, (5) sensorimotor gating - prepulse inhibition (PPI), (6) social interaction and social odor investigation, (7) locomotor activity and exploration in response to 3mg/kg amphetamine (AMPH) challenge were recorded as described previously 18, 19. The 3 mg/kg dose was chosen based on previous studies in mice 19, 25. Additional tests included (i) novel object investigation, (ii) light-dark exploration, (iii) water maze, and (iv) locomotor and exploration in response to ketamine (Ket).
Novel object investigation: A novel object was secured in the center of a white plastic box (50 x 50 x 40 cm); lighting in the room was 600 lux. Mice were allowed to explore for 10 min. Locomotor activity was video-recorded and analyzed by ANY-maze software (Stoelting Co., Wood Dale, IL).
Light-dark exploration: Mice were placed into the light side of a two-chambered box. The clear plastic light side (15 x 30 x 20 cm) connected to a dark plastic chamber through a 5 x 7 cm opening. Boxes were housed inside ventilated sound-attenuating chambers and lit with overhead lights. Infrared photocells across each side detected the location of the mouse and time spent in each compartment, locomotor activity, and number of transitions between boxes. These parameters were scored by MED-PC software (MED Associates, Georgia, VT, USA).
Water maze. This test was performed with both cued, place, and probe trials over the course of 7 days as previously established 26. In brief, all trials were no more than 1 min long, with each mouse receiving 4 trials per day - two back-to-back with 1 hour between the two blocks of trials. Days one and two were cue trials where the submerged platform was marked by a stick at its center. Days 3–7 were place trials using room cues to find the platform and a single probe trial was performed on days 5 and 7.
Ketamine challenge experiments. Transgenic mice and wild-type littermate controls received single intraperitoneal injections of either 2.5 mg/kg or 5 mg/kg ketamine dissolved in sterile 0.9% NaCl (Patterson Veterinary Supply Inc. MA, USA) and immediately placed in beam-break chambers for evaluation. Locomotor and exploratory activity, including rearing, was collected over 15 min habituation and 75 min post-drug-injection phases. Each activity chamber consisted of a transparent (30 x 30 x 20 cm) polystyrene enclosure surrounded by a frame containing a 4 × 8 matrix of photocell pairs. MED Activity software (MED Associates, Georgia, VT, USA) tracked total ambulation (whole body movements) and rearing counts.
Statistical analyses of behavioral data
The data are a single cohort of 11 Wt and 12 Tg animals, however all statistically significant findings (p ≤ 0.05) were also replicated in an independent second cohort 10 Wt and 12 Tg mice. All experiments were analyzed in a blinded fashion using Any-maze or Graph Pad software (GraphPad Software Inc., La Jolla, CA, USA). As described in our previous studies 18, 19, ANOVA models containing one between-class variable and at least one within-class variable were used to analyze the behavioral data. We determined appropriateness of ANOVA models by considering the distributions of the variables studied and by the sample’s homogeneity of variance. Greenhouse-Geisser adjustment was used for all within-subjects effects containing more than two levels in order to protect against violations of the sphericity/compound symmetry assumptions when repeated measures ANOVAs are used. Bonferroni correction or Tukey’s test was used to maintain prescribed alpha levels (p=0.05) when multiple comparisons were conducted. Percent extinction was calculated as % chance from the first to the last extinction trial.
Electrophysiology
Brain slice preparation for electrophysiology
Three-week-old male Wt and Tg mice were deeply anesthetized with isoflurane, then transcardially perfused with ice-cold high sucrose, low Na+-containing ACSF and sacrificed by decapitation. Following decapitation, the brain was quickly removed, and a 3 mm coronal block containing the frontal cortex was cut using an ice-chilled, coronal brain matrix. Thereafter, hemisected coronal slices (250 μm) were made using a Leica VT1000S vibratome (Leica Microsystems, Bannockburn, IL) in a 1–4°C oxygenated (95% v/v O2, 5% v/v CO2) high sucrose, low Na+-containing ACSF composed of (in mM): 208 sucrose, 2.5 KCl, 1.6 NaH2PO4, 1 CaCl2·2H2O, 4 MgCl2·6H2O, 4 MgSO4·7H2O, 26 NaHCO3, 1 ascorbate, 3 Na-pyruvate, and 20 glucose. Once cut, slices were transferred to a 32°C oxygenated recovery buffer composed of (in mM): 100 sucrose, 60 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.1 CaCl2·2H2O, 3.2 MgCl2·6H2O, 2 MgSO4·7H2O, 22 NaHCO3, 1 ascorbate, 3 Na-pyruvate, and 20 glucose for 20 minutes followed by a minimum of 30 minutes in 24°C, oxygenated ACSF (in mM): 113 NaCl, 2.5 KCl, 1.2 MgSO4·7H2O, 2.5 CaCl2·2H2O, 1 NaH2PO4, 26 NaHCO3, 1 ascorbate, and 3 Na-pyruvate, and 20 glucose. Thereafter, slices were placed in a submerged recording chamber where they were continuously perfused with oxygenated ACSF (30–32°C) at a flow rate of 2–3 ml/min. For all electrophysiology experiments, except for those examining sEPSCs, the ACSF was supplemented with 2-amino-5-phosphonopentanoic acid sodium salt (AP-5; 50 μM) and 6-cyano-7-nitroquinoxaline-2,3-dione disodium salt (CNQX ; 20 μM) to block NMDA and AMPA receptor mediated transmission, respectively. For these experiments, 3 mice (Bregma 1.90–2.40 mm) were used per experimental condition. To isolate sEPSCs, the ACSF was supplemented with the GABAA receptor antagonist, picrotoxin (50μM) (Bregma 1.98 mm). 2 WT and 1 Tg mice (Bregma 1.98 mm) were used for sEPSC analyses.
Whole-Cell Recordings
Whole-cell recordings were obtained from pyramidal cells in layer V of the prelimbic (PL) region of the prefrontal cortex (PFC). Cells were visualized using Nikon microscopes equipped with differential interference contrast videomicroscopy. sIPSCs and membrane properties were recorded using pipettes pulled from borosilicate glass (3–5 MΩ resistance) filled with K+-based high [Cl]i pipette solution containing (in mM): 70 K+-gluconate, 4 NaCl, 10 HEPES, 60 KCl, 4 Mg-ATP, 0.3 Na-GTP, 10 Na-phosphocreatine, and 0.6 EGTA (285 mOsm, adjusted to pH 7.30–7.35 with KOH). For voltage clamp experiments, access resistance (Ra) was monitored online and cells that demonstrated a >20% change in Ra were excluded from analysis. sEPSCs were recorded using the following pipette solution: 125 K+-gluconate, 4 NaCl, 10 HEPES, 20 KCl, 4 Mg-ATP, 0.3 Na-GTP, and 10 Na-phosphocreatine (pH 7.25–7.35, adjusted with KOH).
For sIPSC and sEPSC recordings, pyramidal cells were held at −70 mV. For current clamp experiments, membrane potential was maintained near −70 mV with current injection. Offline data analysis was performed using Clampfit 10.2(Molecular Devices, Sunnyvale, CA) and Mini Analysis (Synaptosoft) programs.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism 6.01 and Mini Analysis (Synaptosoft, Decatur GA). Statistical significance between the means of two independent groups was assessed using two-tailed unpaired t-test. Cumulative probability plots were analyzed by Kolmogorov-Smirnov (KS) test. Statistical significance is indicated as follows: ****p<0.0001 or N.S. (not significant). Averaged data are presented as means ± S.E.M.
Drugs and Chemicals
AP-5 and CNQX are gifts from the National Institute on Mental Health Drug Supply Program.
RESULTS
Immunohistochemical (IHC) validation of Pvalb/Gad1transgenic animals
The Gad1 miRNA silencing construct has been extensively validated in previous studies 17–19. IHC double-labeling for parvalbumin (PVALB) and the construct reporter (tdTomato Red) showed a >95% co-expression across multiple brain regions (Supplemental Material 1A–C), including the hippocampus, dentate gyrus, and neocortex, validating that the construct was expressed only in PVALB+ interneurons. Furthermore, PVALB+ interneurons show reduced GAD67 staining (Supplemental Materials 2–3). Immunostaining of Pvalb/Gad1 transgenic and Wt littermates for cholecystokinin (CCK) and neuropeptide Y (NPY) revealed comparable densities of these two distinct interneuronal classes, suggesting that inhibition of PVALB+ interneurons did not result in compensatory increase of other GABA-ergic cell types (Supplemental Material 4).
Pvalb/Gad1 transgenic animals show reduced GABAergic synaptic transmission
To test the functional consequences of the molecular inactivation of the PVALB+ interneurons, we performed whole-cell voltage- and current-clamp recordings from pyramidal neurons found in Layer V prelimbic prefrontal cortex (PL-PFC) prepared from Tg and Wt littermates. This region was chosen for recording as PVALB+ interneurons play a critical role in working memory, and PVALB interneuron dysfunction in the frontal cortex has been well-documented in both schizophrenia and corresponding animal models 1, 27–29. Initially, we recorded spontaneous inhibitory postsynaptic currents (sIPSCs) in the presence of CNQX and AP5 to isolate GABAergic transmission. Our examination revealed a significant reduction (p=0.0001; n=13/condition) in sIPSC frequency in Tg as compared to Wt animals (Figure 1A,B). However, there was no significant change in the amplitude of measured sIPSCs (p>0.05; n=13/condition). These results suggest a reduction in the probability of GABA release or the number of release sites/number of GABAergic synapses onto Layer V PL-PFC pyramidal neurons as a result of Gad1 mRNA knockdown. One concern with any genetic manipulation is the possibility of compensation masking the true role of your gene of interest. However, loss of Gad1 transcript does not affect the kinetics of sIPSCs currents (Figure 1C,D n=13/condition) nor does it significantly change measures of membrane excitability as determined by resting membrane potential (Figure 1E; n= 10–11/condition) and input resistance, as well as neuronal excitability, rheobase current, or action potential latency measurements (Figure 1F–I; n=11–12/condition). Notably, Gad1 mRNA knockdown had no effect on the frequency or amplitude of Layer V PL-PFC sEPSCs, further confirming that our genetic manipulations had their primary effect on the GABAergic system (p>0.05; n=8–9/condition; Supplemental Material 5A,B).
Figure 1. Electrophysiological properties of layer V prelimbic prefrontal cortical (PL-PFC) pyramidal neurons from wildtype (Wt) and Pvalb/Gad1 transgenic (Tg) mice.<.

br>(A) Representative sIPSC traces for Wt (black) and Tg (grey) PL-PFC neurons. Calibration bars represent 20 pA and 100 ms. (B) Cumulative probability distributions of sIPSC inter-event interval (left) and amplitude (right) recorded from Wt (black) and Tg (grey) PL-PFC neurons. Insets show that average sIPSC frequency (Hz), but not amplitude (pA) is significantly reduced in Tg as compared to Wt PL-PFC pyramidal neurons (p = 0.0006 and p = 0.3939, respectively; n=13/condition). (C–I) Reduction of GAD1 does not change the kinetic IPSC properties or the intrinsic excitability of layer V PL-PFC pyramidal neurons. No significant changes were observed in the average (C) sIPSC rise (ms; p=0.9297, n=13/condition ) or (D) decay time (ms; p=0.6919, n=13/condition), (E) resting membrane potential (mV; p=0.3141, n=10–11), (F) input resistance (MΩ; p=0.7469, n=11–12), (G) action potential firing frequency (Hz; p>0.05 n=11–12), (H) rheobase current (pA; p=0.8465, n=11–12), and (I) latency to first action potential (ms; p=0.6713, n=11–12). ***p<0.001; Number of cells per experimental conditions is also indicated in bar graphs.
N.S. denotes not statistically significant. Error bars represent standard error of means (SEM).
In summary, the combined IHC and electrophysiology data suggest that the Gad1 transcript reduction in PVALB-expressing interneurons results in a functional reduction of GABAergic transmission at PL-PFC GABAergic synapses.
Pvalb/Gad1 transgenic animals have pronounced sensorimotor gating deficits
Pre-pulse inhibition (PPI) is a behavioral task used to assess sensorimotor gating in both rodents and humans 30. Patients with schizophrenia show a heightened sensitivity to stimuli and a deficit in pre-pulse inhibition 30, 31 and this endophenotype can also be assessed in rodent animal models 32. Pvalb/Gad1 transgenic animals, when compared to their wild-type littermates, showed heightened startle responses at low sound intensities (70 dB; p= 0.01) and did not respond with a normal increase in PPI with progressively louder stimulus intensities (76–88 dB; p= 0.001) (Figure 2A). Importantly, this initial sensitivity to stimuli did not appear to be caused by a general increase in anxiety, as Tg were indistinguishable from Wt littermate in both 0-Maze and light-dark field exploration tests (Figure 2B,C). Basal acoustic startle levels (Supplemental Figure 6C) were preserved and hearing deficits between the Tg or Wt animals were not detected in the Irwin Screen. Finally, Tg and Wt animals had a similar performance of the Y-Maze alternation test (Figure 2D).
Figure 2. Pre-pulse inhibition is altered in Pvalb/Gad1 mice.

(A) Wt show the expected curve to increased pre-pulse tone intensity, while Tg littermates show a constant minimal response to the entire range of tones presented (p< 0.001), with an already high startle percentage at low tone intensities (p<0.001). (B) 0-maze testing indicates normal preference for closed areas over open areas and similar activity levels. (C) Light-dark box activity shows comparable results in Wt and Tg animals, with the expected avoidance of the lighted area. (D) Both Wt and Tg littermates demonstrate ~70% Y-maze alternations, which is the established number for C57 mice with intact spatial working memory. The overall results suggest that Pvalb/Gad1 Tg mice have an unchanging response to different tone intensity, and this deficit is not a result of altered baseline anxiety or memory disruption.
Pvalb/Gad1 transgenic animals demonstrate increased novelty seeking
In standard open field tests Tg and Wt animals showed comparable locomotor activity and preference for the periphery. Likewise, both groups showed normal balance and grip strength (Supplemental Material 6A,B). In contrast, when a novel object was introduced to the center of the open field, Tg animals spent significantly more time investigating the object (p=0.05) (Figure 3A,B). Furthermore, the time the Tg animals spent investigating the object, was also qualitatively different (movie in Supplemental Material 7–8), with the Tg animals physically touching and interacting with the object far more frequently than the Wt littermates. This increased novelty seeking was not limited to novel object investigation, but was also seen during social interaction tests: Tg animals spent significantly more time investigating the novel mouse (p= 0.05) compared to Wt, as well as compared to time spent investigating a familiar mouse (p=0.01) (Figure 3C). This increase in novelty seeking was not the result of olfactory system disturbances, as response to odors was comparable between the Tg and Wt mice (Figure 3D). Similar novelty-seeking behavior has been previously described in patients with schizophrenia 33, 34, and literature findings suggest that this behavior might be controlled via dopamine signaling in PVALB expressing interneurons 35–37.
Figure 3. Pvalb/Gad1 mice show heightened novelty seeking.

In an open field when novel object is placed in the center, Tg animals spend significantly more time in the center (p<0.05) (A) and spend significantly more time investigating the object (p<0.05) (B) than Wt littermate controls. See also Supplemental Material 1 for movie. (C) In social interaction tests, Tg animals showed a heightened preference for the novel mouse over the familiar one compared to Wt, (p<0.05). (D) Baseline investigation of neutral and socially-relevant odors is comparable between the Tg and Wt animals and cannot account for their heightened interest in novel stimuli.
Pvalb/Gad1 animals show reduced fear extinction
During classical fear conditioning, animals quickly learn to associate a discrete cue (tone) with a noxious stimulus (foot shock), and will exhibit freezing behavior in response to the cue, before the shock is applied during training or in the absence of shock during testing. During the learning phase of conditioned fear, Tg animals demonstrated normal fear acquisition (Figure 4A). However, the transgenic animals showed altered fear extinction: over the course of 9 re-exposures to the conditioning stimulus alone (tone) with no foot shock, Wt animals showed a 50% reduction in the freezing behavior whereas Tg animals only reduced their freezing behavior by 20% over the extinction trial (p= 0.001) (Figure 4B). Importantly, contextual and cue based memory were not affected in Tg animals (data not shown). These data suggest that GABA-egic inhibition of PVALB+ interneurons is required for normal fear extinction and it raises the possibility that the loss of PVALB+ interneuron-mediated inhibition can lead to the persistence of fearful behavior.
Figure 4. Transgenic animals show impaired fear extinction.
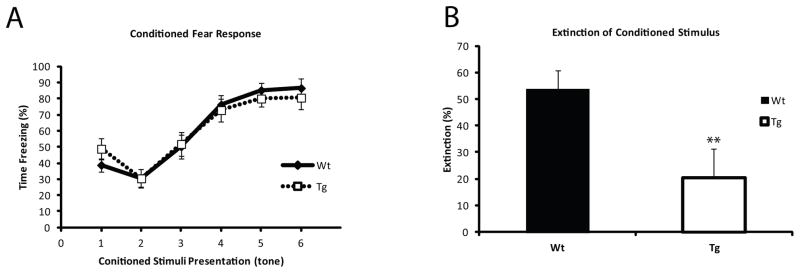
(A) Acquisition of conditioned fear was unaffected in Tg animals, with all animals reaching ~ 80% freezing by the final tone presentation. (B) Extinction percentage over the course of 9 tone presentations with no shock was significantly reduced in transgenic animals compared to Wt littermates (p< 0.001). Extinction was defined as change in freezing behavior from first tone presentation to the last one compared to total freezing at the end of the training phase.
Pvalb/Gad1 animals show an intriguing response to ketamine challenge
Pharmacological challenges are useful (and often necessary) to reveal the deficits in neurotransmitter systems that underlie the various behavioral disturbances 15. To reveal complex deficits in behavior, and assess the glutamatergic and dopaminergic status in the animals, we challenged them with ketamine and amphetamine. Ketamine was chosen based on previous associations between NMDA hypofunction and PVALB interneurons 28, 38 while amphetamine was tested due to previously documented molecular-behavioral interactions between the GABA-ergic and dopaminergic systems 19, 39–41. Tg and Wt mice showed comparable response to 3 mg/kg amphetamine 19, 25, 42, 43 challenge (data not shown), suggesting that the observed behavioral changes were not driven by GABA-ergic modulation of the dopaminergic system.
Ketamine, an NMDA antagonist, has been used to model some of the symptoms of schizophrenia 44–47. As PVALB+ interneurons are known to regulate projection neurons expressing NMDA receptors 20, 48, we exposed the Tg and Wt animals to sub-anesthetic doses of ketamine (2.5 and 5 mg/kg IP injection). Tg and Wt animals showed a similar ambulatory response (Figure 5A,B), indicating normal overall locomotor function. However, the Tg group showed an intriguing rearing response to ketamine. At a dose of 2.5 mg/kg Tg animals revealed a higher rearing behavior (p= 0.0001) (Figure 5C,E), while at a higher dose of 5 mg/kg, this response was reversed: the Tg animals showed significantly lower rearing compared to Wt littermates (p= 0.001) (Figure 5D,F). These findings suggest that the behavioral effects of NMDA receptor antagonism are dose-dependent in our mice and that the differential dosage of ketamine might have divergent effects on behavioral processes.
Figure 5. Transgenic mice show divergent response to ketamine depending on the dose.

Animals were treated with two sub-anesthetic doses of ketamine (2.5 mg/kg and 5 mg/kg). Total ambulation was unaffected by either dose (A, B). However, rearing behavior was strongly reduced in transgenic animals over Wt (p< 0.0001) (C, E) in response to 2.5 mg/kg ketamine, while a higher dose of 5 mg/kg resulted in a significant increase in cumulative rearing events in the Tg animals (p<0.001) (D, F). These findings argue that NMDA receptor antagonism might have dose-dependent effects on behavioral processes in the context of PVALB+ interneuron dysfunction.
DISCUSSION
In the current study we generated and validated an interesting animal model that is relevant for understanding the behavioral control by PVALB+ interneurons and might advance our understanding of mechanisms that underlie behavioral disturbances in schizophrenia. In summary, our data revealed that Pvalb/Gad1 transgenic animals show 1) reduced GABAergic synaptic transmission in layer V PL-PFC, 2) altered sensorimotor gating deficits, 3) increased novelty seeking phenotype, 4) diminished fear extinction; and 5) divergent dose dependent responses to ketamine.
Interneuronal and GABA-ergic disturbances in schizophrenia are complex 9, 29, 49, affect multiple interneuronal cell types 14, and do not appear to be a consequence of disease treatment or progression 1. More specifically, disturbances in PVALB+ interneurons 16 appear to be a hallmark of the schizophrenia disease process 1, and multiple indirect lines of evidence suggest that the reduction of GAD67 expression in this interneuronal subpopulation is (at least partially) responsible for cognitive deficits seen in patients 1, 27, 28, 50–52. However, it is important to realize that our Pvalb/Gad1 transgenic model (just like no other animal model) does not fully recapitulate all the phenotypic features of schizophrenia. Yet, we believe that understanding the individual cellular and molecular building blocks of behavior is a promising approach to gain disease-relevant knowledge 19 and the findings of our current study might be related to behavioral features of schizophrenia in several ways. First, patients with schizophrenia show increased impulsivity and altered novelty seeking 33, 34, 53, and related behaviors were also altered in the Pvalb/Gad1 animals. Second, our Tg mice show reduced fear extinction, which might be linked to the increased fearfulness of patients with schizophrenia, and potentially paranoic ideation 54–57. In a broader context, fear extinction measures adaptive learning and cognitive flexibility, which is also disrupted in schizophrenia 58. Third, altered sensorimotor gating is observed in both the human patient population and in the Pvalb/Gad1 mice 32, 59, 60. These findings underscore the view that deficits of PVALB+ interneurons are directly related to certain phenotypic features of the disease and do not merely represent a consequence of altered glutamatergic drive, which is also altered in the disease 61–63. Finally, these data complement the recent findings that interneuron precursor transplants in adult hippocampus reverse schizophrenia-relevant features in a mouse model of hippocampal disinhibition 64.
Our results are also interesting in a mechanistic context: inhibition is a key feature necessary for refining behavior to provide appropriate response to the environment 65. Recently, in light of human postmortem findings of GAD67 deficits across multiple interneuronal subpopulations 14, and using the same BAC-driven /miRNA silencing technology 17, we generated three other transgenic animals, each silencing Gad1 in a different subpopulations of interneurons (NPY; CCK and cannabinoid receptor 1 – CNR1) 18, 19. The results of these studies, combined with the data presented here, paint a very intriguing and complex picture. Npy/Gad1 animals displayed reduced anxiety-like behavior in using the elevated zero maze and light-dark box paradigms and reduced aversion to the light box compared with littermate controls. Furthermore, they showed increased preference for social novelty and a trend toward investigation of social odors. Importantly, Npy/Gad1 animals show extremely increased responses to amphetamine over that observed in littermate controls 19. Yet, the same animals had preserved sensorimotor gating and hippocampal learning and memory, including contextual fear conditioning. In contrast, Cck/Gad1 Tg animals exhibited decreased locomotor activity, increased interest in several non-social odors, and reduced amphetamine response, with no other domains of behavior affected 19. In addition, Gad1 inhibition in CNR1+ interneurons results in altered amygdalar fear extinction, heightened preference for social novelty and reduced amphetamine response. Although it is clearly an oversimplified view, based on the combined data one might argue that PVALB+ interneurons are primarily coupled to the glutamate system, while NPY+ and CCK+/CNR1+ interneurons are primarily linked to behavior through the dopamine/serotonin systems in an opposing fashion. These discoveries underscore the concept that neurotransmitter systems cannot be studied independently and that disrupting GABA-ergic signaling might results in behavioral readouts related to glutamatergic, dopaminergic, serotonergic and perhaps other neurotransmitter signaling mechanisms. In addition, an important question remains unanswered by our studies – when do the observed changes arise? Is the developmental trajectory disrupted from early developmental stages leading to diverse changes in the brain and behavior or are the results produced by a state of GABA system dysfunction in adulthood? These important questions deserve comprehensive developmental studies and should be assessed across the various BAC-driven Gad1 silenced models that we have generated (PVALB, NPY, CCK, SST, CNR1) 15, 17–19.
These overall findings argue that the hypothesis of Herrick, almost a century ago, was correct: fundamental anatomical building blocks govern complex behavioral responses 66. Yet, while we are starting to understand the contribution of individual cell types to modulation of behavioral domains, it would be wrong to assume that their combined effect will result in a simple summation: the interaction between deficits in the various anatomical “building blocks” will almost certainly result in qualitatively novel, and perhaps unexpected behaviors. Furthermore, understanding the interactions between neurotransmitter systems, various cell types, and the behavioral domains they control and testing them in the context of environmental challenges 67, 68 during susceptible periods of development might be critical for understanding and treating major psychiatric disorders since many of these factors may converge to produce common dysfunction 9 or transitions between disorders along a spectrum 69. Deciphering these interactions will remain a significant biological, clinical, and bioinformatics challenge for a considerable time.
Supplementary Material
Dual-immunohistochemistry was performed using primary antibodies against the construct reporter (td Tomato; red) and PVALB (Alexa Fluor 488; green). Nuclei are DAPI labeled (blue). Note that both in the neocortex (A) and the dentate gyrus of the hippocampus (B,C) show co-localization between the construct and PVALB expression. Circles in (C) denote cells of the same field of view under green (top panel, PVALB staining) and red fluorescence (bottom panel, td-Tomato staining).
Fluorescent micrograph images of the neocortex from Tg (top panels) and Wt (bottom panels) mice. Roman numerals denote cortical laminae. Left column – triple-labeling. Columns on right denote GAD67, dt-Tomato/PVALB and DAPI staining of the same visual field. Note that GAD67 stating is diminished in the Tg animals compared to control Wt littermates.
Micrographs of immunostained sections for dt-Tomato (left column) and PVALB (right column) from a Pvalb/Gad1 Tg mouse. Top panels – hippocampus; middle panels – supragranular cortical layers; bottom panels – nucleus reticularis thalami. Note the interneuronal distribution of labels, and the high, specific expression in the nucleus reticularis thalami.
CCK immunostaining was performed using alexa-568 (red) fluorescence, NPY staining was carried out using alexa-488 (green). Representative micrographs denote hippocampus for CCK (top panels) and frontal cortex for NPY staining (bottom panels). The CCK and NPY subpopulations showed comparable staining pattern across the various brain regions of the Pvalb/Gad1 Tg and Wt mice.
Neither the (A) frequency nor (B) amplitude of Layer V PL-PFC sEPSCs is significantly different between PL-PFC WT or Pvalb/Gad1 Tg experimental conditions. (Panel A: Frequency: WT 4.28±0.51Hz vs. Tg 4.32±0.60Hz, p>0.05); (Panel B: Amplitude: WT 20.4±1.0 pA vs. Tg 20.6±1.34 pA, p>0.05). Number of cells per experimental condition is noted within bar graph. N.S. denotes not statistically significant. Statistical comparison performed using unpaired t-test. Error bars represent standard error of means (SEM).
(A) coordination, (B) grip strength, (C) acoustic startle assessed before PPI testing, and (D) Y-maze alternation test.
Supplemental Material 7. MOVIE: Wt animals are hesitant investigating the novel object.
Supplemental Material 8. MOVIE: Pvalb/Gad1 Tg animals spend more time investigating the novel object and this interaction is qualitatively different.
Acknowledgments
This work was supported by National Institutes of Health R01 MH067234 (KM), R21-MH103515 and K08 MH090412 (SP), and by the NICHD P30 HD15052 grant awarded to the Vanderbilt Kennedy Center for Research on Human Development. JAB is supported by the 2T32MH065215-11 T32 NIH fellowship, while MJS and TSR were supported by the Vanderbilt Neuroscience Scholars Award. RB is supported by the generosity of Rosztoczy Foundation. The authors would like to thank the Vanderbilt Murine Neurobehavioral Laboratory, especially Gregg Stanwood and John Allison, for consultation on behavioral tasks and equipment.
This work was supported by National Institutes of Health R01 MH067234 (KM), R01 R21-MH103515 and K08 MH090412 (SP) and by the NICHD P30 HD15052 grant awarded to the Vanderbilt Kennedy Center for Research on Human Development. JAB is supported by the 2T32MH065215-11 T32 NIH fellowship, while MJS was supported by the Vanderbilt Neuroscience Scholars Award. RB is supported by the generosity of Rosztoczy Foundation. The authors would like to thank the Vanderbilt Murine Neurobehavioral Laboratory, especially Gregg Stanwood and John Allison, for consultation on behavioral tasks and equipment.
Footnotes
CONFLICT OF INTEREST:
The authors declare no competing financial interest.
References
- 1.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35(1):57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, et al. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1997;94(12):6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeFelipe J, Lopez-Cruz PL, Benavides-Piccione R, Bielza C, Larranaga P, Anderson S, et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci. 2013;14(3):202–216. doi: 10.1038/nrn3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, Burkhalter A, et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci. 2008;9(7):557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321(5885):53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5(10):793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- 7.Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, et al. Regulation of neuronal input transformations by tunable dendritic inhibition. Nat Neurosci. 2012;15(3):423–430. S421–423. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]
- 8.Horvath S, Janka Z, Mirnics K. Analyzing schizophrenia by DNA microarrays. Biol Psychiatry. 2010;69(2):157–162. doi: 10.1016/j.biopsych.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt MJ, Mirnics K. Neurodevelopment, GABA System Dysfunction, and Schizophrenia. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faludi G, Mirnics K. Synaptic changes in the brain of subjects with schizophrenia. Int J Dev Neurosci. 2011;29(3):305–309. doi: 10.1016/j.ijdevneu.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52(4):258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 12.Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, et al. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry. 2011;168(9):921–929. doi: 10.1176/appi.ajp.2011.11010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57(11):1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 14.Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2008;13(2):147–161. doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt MJ, Mirnics K. Modeling interneuron dysfunction in schizophrenia. Dev Neurosci. 2012;34(2–3):152–158. doi: 10.1159/000336731. [DOI] [PubMed] [Google Scholar]
- 16.Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23(15):6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garbett KA, Horvath S, Ebert PJ, Schmidt MJ, Lwin K, Mitchell A, et al. Novel animal models for studying complex brain disorders: BAC-driven miRNA-mediated in vivo silencing of gene expression. Mol Psychiatry. 2010;15(10):987–995. doi: 10.1038/mp.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown JA, Horvath S, Garbett KA, Schmidt MJ, Everheart M, Gellert L, et al. The role of cannabinoid 1 receptor expressing interneurons in behavior. Neurobiol Dis. 2013;63:210–221. doi: 10.1016/j.nbd.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt MJ, Horvath S, Ebert P, Norris JL, Seeley EH, Brown J, et al. Modulation of behavioral networks by selective interneuronal inactivation. Mol Psychiatry. 2013;19(5):580–587. doi: 10.1038/mp.2013.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2009;13(1):76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, et al. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16(6):2034–2043. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Clark S, Lewis DV, Wilson WA. NMDA receptor antagonists disinhibit rat posterior cingulate and retrosplenial cortices: a potential mechanism of neurotoxicity. J Neurosci. 2002;22(8):3070–3080. doi: 10.1523/JNEUROSCI.22-08-03070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong S, Yang XW. Modification of bacterial artificial chromosomes (BACs) and preparation of intact BAC DNA for generation of transgenic mice. Curr Protoc Neurosci. 2005;Chapter 5(Unit 5):21. doi: 10.1002/0471142301.ns0521s31. [DOI] [PubMed] [Google Scholar]
- 24.Smith DR, Gallagher M, Stanton ME. Genetic background differences and nonassociative effects in mouse trace fear conditioning. Learn Mem. 2007;14(9):597–605. doi: 10.1101/lm.614807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122(2):261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 26.Wozniak DF, Xiao M, Xu L, Yamada KA, Ornitz DM. Impaired spatial learning and defective theta burst induced LTP in mice lacking fibroblast growth factor 14. Neurobiol Dis. 2007;26(1):14–26. doi: 10.1016/j.nbd.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez-Burgos G, Hashimoto T, Lewis DA. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr Psychiatry Rep. 2010;12(4):335–344. doi: 10.1007/s11920-010-0124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull. 2012;38(5):950–957. doi: 10.1093/schbul/sbs010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6(4):312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 30.Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry. 1990;47(2):181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- 31.Velasques B, Machado S, Paes F, Cunha M, Sanfim A, Budde H, et al. Sensorimotor integration and psychopathology: motor control abnormalities related to psychiatric disorders. World J Biol Psychiatry. 2011;12(8):560–573. doi: 10.3109/15622975.2010.551405. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Du Y, Li N, Wu X, Wu Y. Top-down modulation of prepulse inhibition of the startle reflex in humans and rats. Neurosci Biobehav Rev. 2009;33(8):1157–1167. doi: 10.1016/j.neubiorev.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Miralles C, Alonso Y, Verge B, Seto S, Gaviria AM, Moreno L, et al. Personality dimensions of schizophrenia patients compared to control subjects by gender and the relationship with illness severity. BMC Psychiatry. 2014;14(1):151. doi: 10.1186/1471-244X-14-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy LF, Lee J, Davis MC, Altshuler L, Glahn DC, Miklowitz DJ, et al. Impulsivity and risk taking in bipolar disorder and schizophrenia. Neuropsychopharmacology. 2013;39(2):456–463. doi: 10.1038/npp.2013.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebstein RP, Novick O, Umansky R, Priel B, Osher Y, Blaine D, et al. Dopamine D4 receptor (D4DR) exon III polymorphism associated with the human personality trait of Novelty Seeking. Nat Genet. 1996;12(1):78–80. doi: 10.1038/ng0196-78. [DOI] [PubMed] [Google Scholar]
- 36.Mrzljak L, Bergson C, Pappy M, Huff R, Levenson R, Goldman-Rakic PS. Localization of dopamine D4 receptors in GABAergic neurons of the primate brain. Nature. 1996;381(6579):245–248. doi: 10.1038/381245a0. [DOI] [PubMed] [Google Scholar]
- 37.de Almeida J, Mengod G. D2 and D4 dopamine receptor mRNA distribution in pyramidal neurons and GABAergic subpopulations in monkey prefrontal cortex: implications for schizophrenia treatment. Neuroscience. 2010;170(4):1133–1139. doi: 10.1016/j.neuroscience.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 38.Rotaru DC, Lewis DA, Gonzalez-Burgos G. The role of glutamatergic inputs onto parvalbumin-positive interneurons: relevance for schizophrenia. Rev Neurosci. 23(1):97–109. doi: 10.1515/revneuro-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10(1):40–68. doi: 10.1038/sj.mp.4001558. image 45. [DOI] [PubMed] [Google Scholar]
- 40.Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25(1):1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 41.Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260. doi: 10.1146/annurev.pharmtox.41.1.237. [DOI] [PubMed] [Google Scholar]
- 42.Ren J, Xu H, Choi JK, Jenkins BG, Chen YI. Dopaminergic response to graded dopamine concentration elicited by four amphetamine doses. Synapse. 2009;63(9):764–772. doi: 10.1002/syn.20659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhardwaj SK, Baharnoori M, Sharif-Askari B, Kamath A, Williams S, Srivastava LK. Behavioral characterization of dysbindin-1 deficient sandy mice. Behav Brain Res. 2009;197(2):435–441. doi: 10.1016/j.bbr.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 44.Amann LC, Gandal MJ, Halene TB, Ehrlichman RS, White SL, McCarren HS, et al. Mouse behavioral endophenotypes for schizophrenia. Brain Res Bull. 2010;83(3–4):147–161. doi: 10.1016/j.brainresbull.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 45.Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL, et al. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther. 2010;128(3):419–432. doi: 10.1016/j.pharmthera.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 46.Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13(1):9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- 47.Driesen NR, McCarthy G, Bhagwagar Z, Bloch MH, Calhoun VD, D’Souza DC, et al. The impact of NMDA receptor blockade on human working memory-related prefrontal function and connectivity. Neuropsychopharmacology. 2013;38(13):2613–2622. doi: 10.1038/npp.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coyle JT, Tsai G, Goff D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann N Y Acad Sci. 2003;1003:318–327. doi: 10.1196/annals.1300.020. [DOI] [PubMed] [Google Scholar]
- 49.Horvath S, Mirnics K. Schizophrenia as a Disorder of Molecular Pathways. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yi F, Ball J, Stoll KE, Satpute VC, Mitchell SM, Pauli JL, et al. Direct excitation of parvalbumin-positive interneurons by M1 muscarinic acetylcholine receptors: roles in cellular excitability, inhibitory transmission and cognition. J Physiol. 2014 doi: 10.1113/jphysiol.2014.275453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, et al. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci. 2011;14(3):297–299. doi: 10.1038/nn.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68(3):557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 53.Ouzir M. Impulsivity in schizophrenia: A comprehensive update. Aggress Viol Behav. 2013;18:247–254. [Google Scholar]
- 54.Holt DJ, Weiss AP, Rauch SL, Wright CI, Zalesak M, Goff DC, et al. Sustained activation of the hippocampus in response to fearful faces in schizophrenia. Biol Psychiatry. 2005;57(9):1011–1019. doi: 10.1016/j.biopsych.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 55.Mukherjee P, Whalley HC, McKirdy JW, McIntosh AM, Johnstone EC, Lawrie SM, et al. Lower effective connectivity between amygdala and parietal regions in response to fearful faces in schizophrenia. Schizophr Res. 2011;134(2–3):118–124. doi: 10.1016/j.schres.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 56.Behere RV, Venkatasubramanian G, Arasappa R, Reddy NN, Gangadhar BN. First rank symptoms & facial emotion recognition deficits in antipsychotic naive schizophrenia: Implications for social threat perception model. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(7):1653–1658. doi: 10.1016/j.pnpbp.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 57.Green MJ, Phillips ML. Social threat perception and the evolution of paranoia. Neurosci Biobehav Rev. 2004;28(3):333–342. doi: 10.1016/j.neubiorev.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 58.Addington J, Addington D. Neurocognitive and social functioning in schizophrenia. Schizophr Bull. 1999;25(1):173–182. doi: 10.1093/oxfordjournals.schbul.a033363. [DOI] [PubMed] [Google Scholar]
- 59.Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology (Berl) 2008;199(3):331–388. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giakoumaki SG. Cognitive and prepulse inhibition deficits in psychometrically high schizotypal subjects in the general population: relevance to schizophrenia research. J Int Neuropsychol Soc. 2012;18(4):643–656. doi: 10.1017/S135561771200029X. [DOI] [PubMed] [Google Scholar]
- 61.Woodruff AR, McGarry LM, Vogels TP, Inan M, Anderson SA, Yuste R. State-dependent function of neocortical chandelier cells. J Neurosci. 2011;31(49):17872–17886. doi: 10.1523/JNEUROSCI.3894-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woodruff AR, Anderson SA, Yuste R. The enigmatic function of chandelier cells. Front Neurosci. 2010;4:201. doi: 10.3389/fnins.2010.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Javitt DC. Twenty-five years of glutamate in schizophrenia: are we there yet? Schizophr Bull. 2012;38(5):911–913. doi: 10.1093/schbul/sbs100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gilani AI, Chohan MO, Inan M, Schobel SA, Chaudhury NH, Paskewitz S, et al. Interneuron precursor transplants in adult hippocampus reverse psychosis-relevant features in a mouse model of hippocampal disinhibition. Proc Natl Acad Sci U S A. 2014;111(20):7450–7455. doi: 10.1073/pnas.1316488111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Humby T, Wilkinson LS. Assaying dissociable elements of behavioural inhibition and impulsivity: translational utility of animal models. Curr Opin Pharmacol. 2011;11(5):534–539. doi: 10.1016/j.coph.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 66.Herrick CJ. Anatomical patterns and behavior patterns. Physiol Zool. 1929;II:439–448. [Google Scholar]
- 67.Horvath S, Mirnics K. Immune system disturbances in schizophrenia. Biol Psychiatry. 2013;75(4):316–323. doi: 10.1016/j.biopsych.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michel M, Schmidt MJ, Mirnics K. Immune system gene dysregulation in autism and schizophrenia. Dev Neurobiol. 2012;72(10):1277–1287. doi: 10.1002/dneu.22044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adam D. Mental health: On the spectrum. Nature. 2013;496(7446):416–418. doi: 10.1038/496416a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dual-immunohistochemistry was performed using primary antibodies against the construct reporter (td Tomato; red) and PVALB (Alexa Fluor 488; green). Nuclei are DAPI labeled (blue). Note that both in the neocortex (A) and the dentate gyrus of the hippocampus (B,C) show co-localization between the construct and PVALB expression. Circles in (C) denote cells of the same field of view under green (top panel, PVALB staining) and red fluorescence (bottom panel, td-Tomato staining).
Fluorescent micrograph images of the neocortex from Tg (top panels) and Wt (bottom panels) mice. Roman numerals denote cortical laminae. Left column – triple-labeling. Columns on right denote GAD67, dt-Tomato/PVALB and DAPI staining of the same visual field. Note that GAD67 stating is diminished in the Tg animals compared to control Wt littermates.
Micrographs of immunostained sections for dt-Tomato (left column) and PVALB (right column) from a Pvalb/Gad1 Tg mouse. Top panels – hippocampus; middle panels – supragranular cortical layers; bottom panels – nucleus reticularis thalami. Note the interneuronal distribution of labels, and the high, specific expression in the nucleus reticularis thalami.
CCK immunostaining was performed using alexa-568 (red) fluorescence, NPY staining was carried out using alexa-488 (green). Representative micrographs denote hippocampus for CCK (top panels) and frontal cortex for NPY staining (bottom panels). The CCK and NPY subpopulations showed comparable staining pattern across the various brain regions of the Pvalb/Gad1 Tg and Wt mice.
Neither the (A) frequency nor (B) amplitude of Layer V PL-PFC sEPSCs is significantly different between PL-PFC WT or Pvalb/Gad1 Tg experimental conditions. (Panel A: Frequency: WT 4.28±0.51Hz vs. Tg 4.32±0.60Hz, p>0.05); (Panel B: Amplitude: WT 20.4±1.0 pA vs. Tg 20.6±1.34 pA, p>0.05). Number of cells per experimental condition is noted within bar graph. N.S. denotes not statistically significant. Statistical comparison performed using unpaired t-test. Error bars represent standard error of means (SEM).
(A) coordination, (B) grip strength, (C) acoustic startle assessed before PPI testing, and (D) Y-maze alternation test.
Supplemental Material 7. MOVIE: Wt animals are hesitant investigating the novel object.
Supplemental Material 8. MOVIE: Pvalb/Gad1 Tg animals spend more time investigating the novel object and this interaction is qualitatively different.