

Zinc and ifenprodil induce similar conformational changes in the NMDA receptor, suggesting a conserved mechanism of inhibition independent of receptor subtype and the site at which the inhibitor binds.
Abstract
N-methyl-d-aspartate (NMDA) receptors, one of the three main types of ionotropic glutamate receptors (iGluRs), are involved in excitatory synaptic transmission, and their dysfunction is implicated in various neurological disorders. NMDA receptors, heterotetramers typically composed of GluN1 and GluN2 subunits, are the only members of the iGluR family that bind allosteric modulators at their amino-terminal domains (ATDs). We used luminescence resonance energy transfer to characterize the conformational changes the receptor undergoes upon binding ifenprodil, a synthetic compound that specifically inhibits activation of NMDA receptors containing GluN2B. We found that ifenprodil induced an overall closure of the GluN2B ATD without affecting conformation of the GluN1 ATD or the upper lobes of the ATDs, the same mechanism whereby zinc inhibits GluN2A. These data demonstrate that the conformational changes induced by zinc and ifenprodil represent a conserved mechanism of NMDA receptor inhibition. Additionally, we compared the structural mechanism of zinc inhibition of GluN1–GluN2A receptors to that of ifenprodil inhibition of GluN1–GluN2B. The similarities in the conformational changes induced by inhibitor binding suggest a conserved structural mechanism of inhibition independent of the binding site of the modulator.
INTRODUCTION
Ionotropic glutamate receptors mediate the majority of excitatory synaptic transmission in the central nervous system. There are three subtypes of ionotropic glutamate receptors: AMPA, kainate, and NMDA receptors. NMDA receptors mediate the slower component of the excitatory transmission and are also calcium permeable (Traynelis et al., 2010). Their activation triggers several intracellular downstream processes that underlie synaptic remodeling and long-term potentiation (Traynelis et al., 2010). Aberrant NMDA receptor activation is implicated in a variety of neurological disorders including ischemic stroke, schizophrenia, Parkinson’s disease, and Alzheimer’s disease (Paoletti et al., 2013). Given this role, there is intense interest in understanding the mechanisms of NMDA receptor modulation with a view to guiding drug development (Paoletti et al., 2013; Yuan et al., 2015).
NMDA receptors are obligate heterotetramers, typically composed of GluN1 and GluN2 subunits. The GluN2 subunits can be one of four subtypes (A–D). The GluN2 subtype dictates the biophysical properties of the channel, apparent agonist affinity, and allosteric modulation of the receptor (Gielen et al., 2009; Yuan et al., 2009). Each subunit of the receptor is organized into distinct domains: an intracellular carboxyl-terminal domain, the pore-forming transmembrane domain, extracellular ligand-binding domain (LBD), and amino-terminal domains (ATDs). The ATDs of NMDA receptors serve as the binding sites for allosteric modulators. Zinc and ifenprodil are two such inhibitory modulators that bind to the ATDs with GluN2 subtype–dependent affinity (Gallagher et al., 1996; Rachline et al., 2005).
Ifenprodil, which is specific for GluN2B-containing receptors, increases the energy barrier for activation and results in increased mean closed times and decreased mean open times of the receptor (Amico-Ruvio et al., 2012). Ifenprodil binds at the interface between the GluN1 and GluN2B ATDs (Fig. 1) (Karakas et al., 2011; Tomitori et al., 2012). Cross-links introduced between the lower lobe of the GluN2B ATD and the upper lobe of the GluN1 ATD resulted in a loss of ifenprodil inhibition, suggesting that the mobility of the lower lobe of GluN2B is critical to the ability of ifenprodil to inhibit the NMDA receptor (Karakas et al., 2011). However, given the lack of an apo-state structure, the conformational changes associated with its binding and hence its mechanism of modulation are still unknown.
Figure 1.

Ifenprodil-binding site and LRET label sites. The structure of the NMDA receptor is shown with the GluN1 subunits in cyan and the GluN2B subunits in red. The expanded view is of a GluN1/GluN2B ATD dimer with residues mutated and labeled in LRET analysis highlighted in pink and the ifenprodil compound shown in gold in its binding site between the two ATDs.
Zinc binds between the upper and lower lobes of the GluN2 ATD with higher affinity for GluN2A- than GluN2B-containing receptors (Choi and Lipton, 1999; Paoletti et al., 2000; Rachline et al., 2005). Previous studies demonstrated that zinc inhibits the NMDA receptor by stabilizing a closed conformation of the GluN2 ATD (Gielen et al., 2009; Sirrieh et al., 2013). Conversely, spermine potentiation occurs by stabilizing open conformations of both the GluN1 and GluN2B ATDs (Sirrieh et al., 2015). Here, we have investigated the conformational changes associated with ifenprodil binding to the NMDA receptor using luminescence resonance energy transfer (LRET). Additionally, we compare the changes observed caused by ifenprodil binding to GluN1/GluN2B receptors to those induced by zinc binding to GluN1/GluN2A receptors and highlight their conserved mechanism of action.
MATERIALS AND METHODS
Molecular biology
All full-length receptor constructs were in pcDNA3.1 vectors. GluN1, GluN2A, and GluN2B were provided by S. Nakanishi (Osaka Bioscience Institute, Osaka, Japan). To specifically label receptors with maleimide-derived fluorophores, all non-disulfide–bonded cysteines, as identified from existing crystal structures and previous work in our laboratory (Karakas et al., 2009; Rambhadran et al., 2010; Sirrieh et al., 2013), were mutated to serines: C22S and C459S for GluN1 (GluN1*); C231S, C399S, and C461S for GluN2A (GluN2A*); and C232S, C399S, and C495S for GluN2B (GluN2B*). The numbering of residues includes the signal peptide. Mutations were added to these base constructs unless an inherent cysteine was retained for labeling. Table 1 lists the mutations in each construct, construct coexpression, and the acceptor fluorophore used for LRET measurements. The locations of the introduced cysteines in the receptor are shown in Fig. 1. Mutations were made using standard PCR methods, and the integrity of the plasmid and insert was verified by sequencing. To measure distance changes in full-length receptors in intact membranes without protein purification, the thrombin (LVPRGS) recognition sequence was introduced between donor and acceptor fluorophores, as described previously (Gonzalez et al., 2008; Rambhadran et al., 2010; Sirrieh et al., 2013). Subtraction of the fluorescence signal after cleavage from that before cleavage allows for the isolation of LRET arising from the relevant labeled sites in NMDA receptors.
Table 1.
Mutations made and acceptor fluorophores
| Construct name | Mutations | Construct coexpressed with | Acceptor fluorophore |
| GluN1* | C22S, C459S | GluN2Bcleft | Ni(NTA)2Cy3 |
| GluN1*C22 | C459S | GluN2B*H30 | Ni(NTA)2Cy3 |
| GluN2B*232 | Alexa Fluor 555 | ||
| GluN2A*231 | Alexa Fluor 555 | ||
| GluN1cleft | S224C, C459S | GluN2B* | Alexa Fluor 555 |
| GluN2A*231 | C395S, C461S | GluN1*C22 | Alexa Fluor 555 |
| GluN2B* | C232S, C399S, C495S | GluN1cleft | Alexa Fluor 555 |
| GluN2B*H30 | C232S, C399S, C495S, insertion of hexa-his tag followed by amino acids LVPRGS after residue 30 | GluN1*C22 | Ni(NTA)2Cy3 |
| GluN2Bcleft | C399S, C495S, insertion of hexa-his tag followed by amino acids LVPRGS after residue 30 | GluN1* | Ni(NTA)2Cy3 |
| GluN2B*231 | C399S, C495S | GluN1*C22 | Alexa Fluor 555 |
This table describes all the mutations present in each construct, the construct it was coexpressed with, and the acceptor fluorophore used in the LRET experiments.
Sites chosen to be labeled for LRET measurements are surface exposed so that the fluorophores can be freely rotating. The rotation of the fluorophores is on a timescale faster than the time resolution of the LRET, so any changes in the LRET lifetime reflect a movement or conformational change in the protein to which the fluorophores are attached. The measurements made actually reflect the distance between the cone of rotation of the acceptor fluorophore and the isotropic terbium atom. As such, the LRET measurements are not precise indicators of absolute distances. However, the distance resolution for conformational changes is quite high, especially because the measurements for the apo receptor and the ligand-bound receptor are done on the same receptors in the same cells.
Fluorophores and reagents
In all experiments, terbium chelate was the donor fluorophore with the acceptor fluorophores being Alexa Fluor 555 (Sigma-Aldrich) or Ni(NTA)2Cy3. Ni(NTA)2Cy3 was prepared as described previously (Kapanidis et al., 2001) using bis-reactive Cy3 purchased from GE Healthcare. DL-2-amino-5-phosphonopentanoic acid (DL-APV) and 5,7-dichlorokynurenic acid (DCKA), competitive antagonists of the NMDA receptor, were purchased from Abcam. Zinc and ifenprodil tartrate were purchased from Sigma-Aldrich.
Chinese hamster ovary (CHO) cell maintenance and protein expression
CHO-K1 cells (ATCC) were maintained in Ham’s F12 Nutrient Mix (Invitrogen) supplemented with 10% FBS (Sigma-Aldrich) and penicillin/streptomycin (Invitrogen). Cells were passaged once they reached a confluence of 80–90%, approximately every 2 d. CHO cells were transfected at 50–80% confluency using Lipofectamine 2000 (Invitrogen) with 5–12 µg DNA per 10-cm dish, at a GluN1/GluN2 microgram ratio of 1:3 (Brimecombe et al., 1999) and a DNA/Lipofectamine ratio of 1:2. To induce expression of the NMDA receptor, cells were maintained in glutamine-free DMEM (Invitrogen) (Chazot et al., 1999) for 1–2 h before transfection through their harvest for use in spectroscopic studies. To further facilitate expression, cells were maintained for at least 40 h after transfection in 300 µM DL-APV before LRET experiments. Cells were collected in extracellular buffer containing 1 mM CaCl2, 150 mM NaCl, 2.8 mM KCl, and 10 mM HEPES, pH 7.3–7.4 (HCl). CHO cells were labeled for 1 h at room temperature on a rotator in the dark using 200 nM each of the donor and acceptor fluorophores in a 2–3-ml volume. Cells were then washed twice with extracellular buffer before spectroscopic analysis. Ifenprodil measurements were obtained in the presence of saturating ifenprodil (10 µM) (Perin-Dureau et al., 2002). Zinc measurements were performed in the presence of 1 µM zinc chloride, which is saturating for the allosteric binding site on GluN2A-containing receptors (Rachline et al., 2005).
LRET
Labeled cells were probed in a cuvette-based LRET analysis using a QuantaMaster (model QM3-SS) with Fluorescan software (Photon Technology International) (Dolino et al., 2014). Data were analyzed with Origin 8.6 (OriginLab Corporation). The lifetime of the sensitized acceptor emission was measured upon directly exciting the donor. Donor-only samples were excited at 337 nm, and emission was collected at 545 nm. Donor acceptor–labeled samples were excited at 337 nm, and the emission was collected at 565 nm for Alexa Fluor 555–labeled receptors and at 572 nm for NI(NTA)2Cy3-labeled receptors. A Peltier TE temperature controller (Photon Technology International) maintained the temperature at 15–20°C for all recordings. To acquire measurements of the true apo state in GluN2A-containing receptors, 10 mM tricine was included in the buffer for apo measurements to chelate contaminating zinc (Paoletti et al., 1997; Sirrieh et al., 2013).
Distances were calculated using the Förster equation:
| (1) |
The n values represent the number of biological repeats. Each sample (n) was scanned a minimum 297 times, the resulting scans were averaged, and the subtracted NMDA receptor–specific signal was fit with a single exponential. The LRET lifetimes reported here are the averages of the resulting time constants with the error representing the SEM. The error in the distance estimates was calculated by propagating the errors in the donor and acceptor lifetimes using the Error Propagation Calculator developed by Thomas Huber in the Physics Department of Gustavus Adolphus College.
Electrophysiology
CHO cells were transfected using Lipofectamine 2000 (Invitrogen) with wild-type or mutant GluN1, GluN2A, and enhanced GFP at a microgram ratio of 1.25:3.75:1, respectively, with 6 µg of total DNA per 10 ml of media, or with wild-type or mutant GluN1, GluN2B, and enhanced GFP at a microgram ratio of 1.5:4.5:1, respectively, with 7 µg of total DNA per 10 ml of medium. After a 10–12-h incubation with transfection reagents, cells were replated at low density onto Petri dishes coated with poly-d-lysine. 300–400 µM DL-APV and 30 µM DCKA were present in the media during and after transfection. Whole-cell patch-clamp recordings were performed 24–48 h after transfection using borosilicate glass pipettes with 3–5-MΩ resistance, coated with dental wax, fire polished, and filled with the following solution (mM): 135 CsF, 33 CsOH, 2 MgCl2, 1 CaCl2, 11 EGTA, and 10 HEPES, pH 7.4. The external solution was (mM): 140 NaCl, 2.8 KCl, 1 CaCl2, and 10 HEPES, pH 7.4. For zinc experiments, 10 mM tricine was added and free Zn2+ solutions up to 1 µM were made as described by Paoletti et al. (1997), with 10 µM zinc added to an external solution lacking tricine. Control, agonist, and agonist plus Zn2+ solutions were locally applied to isolated cells using a solenoid valve system (VC-6; Warner Instruments) and modified triple-barrel tubing as described previously (Tang, 2001). 100 µM glycine was present continuously and 100 µM glutamate was applied for 12 s every 20 s. To determine extent of inhibition, 100 µM each of glutamate and glycine were applied for 5 s, before switching into a solution that contained the agonists with 10 µM ifenprodil or the agonists with 1 µM zinc. All recordings were performed using an amplifier (Axopatch 200B; Molecular Devices), acquired at 10 kHz using pCLAMP10 software (Molecular Devices), and filtered online at 3 kHz (eight-pole Bessel; Frequency Devices). All experiments were performed at room temperature.
Statistics
For electrophysiology experiments, a standard two-tailed Student’s t test was used with a p-value of 0.05 taken to be significant. For LRET experiments, a one-tailed t test on the donor–acceptor lifetimes was used with a p-value of 0.05 taken to be significant.
Online supplemental material
Supplemental figures show two additional control experiments performed for the LRET measurements. First, cells were labeled with only the acceptor fluorophore, and the intensity of the acceptor fluorophore upon directly exciting it was measured (Fig. S1). The intensity was then measured in the presence of ifenprodil to ensure that the ifenprodil did not quench or otherwise alter the fluorescent properties of the acceptor fluorophore (Fig. S1). Second, untransfected cells were labeled with donor and acceptor fluorophore, as with the LRET experiments on transfected cells, and the sensitized emission of the acceptor was measured in extracellular buffer and in the presence of ifenprodil to ensure that the CHO cells themselves were not affected by ifenprodil (Fig. S2). These controls showed that ifenprodil does not affect acceptor fluorophore fluorescence and that the CHO cells themselves do not contribute to the changes in lifetime detected in the LRET. The online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201511422/DC1.
RESULTS
To investigate the distances between the upper and lower lobes of the GluN2B ATD in the apo and ifenprodil-bound states, we measured the LRET lifetime between a hexa-histidine tag inserted after residue 30 and C232, an inherent cysteine, in the GluN2B ATD lower lobe. The his-tag was labeled with Ni(NTA)2Cy3, and the cysteine was labeled with terbium chelate. The specific introduction of the donor and acceptor fluorophores in this case allowed for the isolation of a specific signal from within an ATD, without cross-talk across the subunits. Consistent with this, the LRET lifetime, as measured by the sensitized emission of the acceptor, was well represented by a single-exponential decay with a time constant of 252 ± 12 µs for the apo state (Fig. 2 A, black trace). This corresponds to a distance of 48.8 ± 0.4 Å (Table 2) between the two fluorophores, using Eq. 1 and the lifetime of the donor alone. The apo measurements here and throughout the rest of this paper are in the absence of ifenprodil and any agonists, corresponding to the resting state of the receptor. One of the advantages of LRET is that it allows us to probe the resting state of the receptor, which is difficult to investigate with electrophysiological measurements as it is an electrically “silent” state. Upon ifenprodil binding, the acceptor lifetime decreases to 203 ± 12 µs (Fig. 2), P = 0.03, which reflects a distance of 46.0 ± 0.5 Å. The difference in the distances between the apo and ifenprodil-bound states is ∼2 Å, indicating a movement of the upper and lower lobes of the GluN2B ATD toward each other upon ifenprodil binding (Table 2). Such a movement would be consistent with a cleft closure like conformational change. Importantly, the receptors used for the LRET measurements were inhibited by saturating (10 µM) ifenprodil to the same degree as the wild-type receptor (extent of inhibition: for mutant, 0.82 ± 0.03, n = 4; for wild type, 0.82 ± 0.03, n = 5; P = 1.0; Fig. 3 B).
Figure 2.

Ifenprodil effects on ATD cleft conformations. (A) LRET measurements in the GluN2B cleft reveal that ifenprodil induces a cleft closure. The acceptor fluorophore used was Ni(NTA)2Cy3. (B) LRET measurements of the GluN1 cleft are shown; the acceptor fluorophore used was Alexa Fluor 555. (C and D) The donor-only lifetimes for the GluN2B and GluN1 clefts, respectively. In all panels, the black curve is from the apo receptor, and the teal curve is from the ifenprodil-bound receptor.
Table 2.
LRET lifetimes and measurements
| Ligated State (n) | Donor-only lifetime | Donor-acceptor lifetime | Distance |
| µs | µs | Å | |
| GluN2B Cleft – GluN1* + GluN2Bcleft (Ni(NTA)2Cy3) (51.0 Å) | |||
| Apo (3) | 1,847 ± 1 | 252 ± 12 | 48.8 ± 0.4 |
| Ifenprodil (4) | 1,826 ± 1 | 203 ± 12 | 46.0 ± 0.5 |
| GluN1 Cleft – GluN1cleft + GluN2B* (Alexa Fluor 555) (47.2 Å) | |||
| Apo (2) | 1,740 ± 1 | 275 ± 4 | 49.2 ± 0.1 |
| Ifenprodil (2) | 1,747 ± 1 | 263 ± 2 | 48.7 ± 0.1 |
| GluN1–GluN2B Upper Lobes – GluN1*C22 + GluN2B*H30 (Ni(NTA)2Cy3) (64.0 Å) | |||
| Apo (5) | 1,738 ± 1 | 362 ± 26 | 52.1 ± 0.6 |
| Ifenprodil (2) | 1,807 ± 1 | 374 ± 26 | 52.0 ± 0.6 |
| GluN1 Upper Lobe–GluN2B Lower Lobe – GluN1*C22 + GluN2B*232 (Alexa Fluor 555) (35.2 Å) | |||
| Apo (3) | 1,674 ± 16 | 228 ± 9 | 47.8 ± 0.3 |
| Ifenprodil (3) | 1,694 ± 4 | 194 ± 8 | 46.2 ± 0.3 |
| GluN1 Upper Lobe–GluN2A Lower Lobe – GluN1*C22 + GluN2A*C231 (Alexa Fluor 555) | |||
| Apo (3) | 1,692 ± 1 | 187 ± 6 | 45.9 ± 0.2 |
| Zinc (2) | 1,683 ± 1 | 156 ± 9 | 44.4 ± 0.4 |
Shown are the lifetimes and distances from measurements in GluN1/GluN2B receptors in the apo, ifenprodil-bound, and zinc-bound states. The distance next to the construct name is measured between the alpha carbons of the residues labeled from the ifenprodil-bound structure of the NMDA receptor (Protein Data Bank accession no. 4PE5).
Figure 3.

Functional characterization of LRET constructs. (A) A sample whole-cell recording showing ifenprodil inhibition of NMDA receptors. (B) The extent of ifenprodil inhibition is plotted in comparison to the wild-type receptor. (C) The extent of inhibition caused by the application of 1 µM zinc to CHO cells expressing the indicated NMDA receptor construct. The constructs used for LRET measurements are normally inhibited by zinc, as compared with the wild-type receptor. Error bars represent the SEM.
Recently, we have shown that the GluN1 ATD adopts distinct conformations when coassembled with different GluN2 subunits (Sirrieh et al., 2015). Additionally, although zinc does not affect the conformation of the GluN1 ATD, the GluN2B-specific potentiator spermine induces an opening of the GluN1 ATD cleft (Sirrieh et al., 2015). These results raise the question of whether the conformation of the GluN1 ATD is only affected by modulators that bind at the interface between the GluN1 and GluN2 ATDs, such as spermine and ifenprodil, but not by modulators binding within the GluN2 ATD cleft, such as zinc. To investigate the conformational changes in the GluN1 ATD, we measured LRET lifetimes with receptors tagged at sites C22 and S224C (Table 1). The LRET lifetime for the apo receptor was 275 ± 4 µs, and this lifetime corresponds to a distance of 49.2 ± 0.1 Å (Fig. 2 B and Table 2). The lifetime for the ifenprodil-bound receptor was 263 ± 2 µs (Fig. 2 B), P = 0.05, which corresponds to a distance of 48.7 ± 0.1 Å (Table 2). The LRET data show that ifenprodil has no effect on the overall conformation of the GluN1 ATD. The construct used for this LRET experiment also showed undisrupted ifenprodil inhibition (extent of inhibition: 0.80 ± 0.03, n = 8; P = 0.67 vs. wild type; Fig. 3 B).
In addition to the conformational changes within the individual subunits, we also measured distances between the subunits in the presence and absence of ifenprodil. Previous studies with zinc and spermine revealed that the upper lobes of the ATDs were stable and did not undergo significant conformational changes upon modulator binding (Sirrieh et al., 2013, 2015). To investigate the movements between subunits, LRET lifetimes were obtained between the cysteine introduced at site 22 on GluN1 and a histidine tag at site 30 on GluN2B. The receptors were labeled with the thiol-reactive terbium chelate and Ni(NTA)2Cy3 (Table 1). The LRET lifetime for the apo receptor was 362 ± 26 µs (Fig. 4 A), which corresponded to a distance of 52.1 ± 0.6 Å. The lifetime when the receptor was bound by ifenprodil was 347 ± 26 µs (Fig. 4 A), P = 0.17, corresponding to a distance of 52.0 ± 0.6 Å. The LRET lifetimes indicate no significant changes between the upper lobes of the GluN1 and GluN2B subunits upon the binding of ifenprodil. The receptor used for this measurement was also normally inhibited by ifenprodil (extent of inhibition: 0.82 ± 0.02, n = 5; P = 1.0 vs. wild type; Fig. 3 B).
Figure 4.

Intersubunit measurements with ifenprodil. (A) The LRET measurement between the upper lobes of the GluN1 and GluN2B ATDs, using Ni(NTA)2Cy3 as the acceptor fluorophore. In black is the apo receptor, and in teal is the ifenprodil-bound receptor. Measurements were made between C22 on GluN1 and a his-tag introduced after K30 on GluN2B. (B) Donor-only lifetimes for the GluN1–GluN2B upper lobe measurements are shown.
Previous cross-linking studies between sites L341C on GluN1 and D210C on GluN2B suggested that mobility of the ATDs is required for ifenprodil inhibition (Karakas et al., 2011). If there are conformational changes taking place within an ATD (Fig. 2) yet the upper lobes of the ATDs are not moving with respect to each other (Fig. 4 A), this indicates that the lower lobes must be the source of motion. To investigate such a motion, we measured LRET lifetimes between the cysteine at site 22 on the upper lobe of GluN1 and the cysteine at site 232 on GluN2B’s lower lobe, tagged with terbium chelate and Alexa Fluor 555. The sensitized acceptor lifetime of the apo receptor was 229 ± 9 µs (Fig. 5 A), which corresponds to a distance of 47.8 ± 0.4 Å (Table 2). The lifetime decreased to 194 ± 8 µs when the receptor was bound by ifenprodil, P = 0.03, corresponding to a distance of 46.2 ± 0.3 Å. These measurements indicate that a slight decrease in the distance by 1.6 Å between the fluorophores tagged at site 22 on GluN1 and site 232 on GluN2B is induced by ifenprodil binding. As with prior constructs, the extent of ifenprodil inhibition was not distinguishable from wild type (extent of inhibition: 0.75 ± 0.03, n = 7; P = 0.14 vs. wild type; Fig. 3 B).
Figure 5.

LRET measurements showing movement of lower lobe of GluN2 ATD. (A) Changes in the LRET lifetime induced by ifenprodil are shown as measured between C22 on GluN1 and C232 on GluN2B. In black is the LRET lifetime from the apo receptor, and in teal is the lifetime with ifenprodil. (B) Zinc similarly induces a reduction in the distance between the GluN1 ATD upper lobe and the GluN2A ATD lower lobe, as measured between C22 on GluN1 and C231 on GluN2A. The lifetimes from apo receptor are in green, and the zinc-bound receptor is in cyan. The acceptor fluorophore used for the LRET in A and B was Alexa Fluor 555. (C) The donor-only lifetimes from the apo (black) and ifenprodil-bound (teal) receptor. The donor-only lifetimes from the GluN1*C22-GluN2A*231 (D) receptor in the presence (cyan) and absence (green) of zinc are shown.
Collectively, these data illustrate that upon ifenprodil binding, the GluN2B ATD cleft closes with the lower lobe rotating slightly toward the upper lobe of the GluN1 ATD, as demonstrated by the decrease in the distance measured between the lower lobe of the GluN2B ATD and the upper lobe of the GluN1 ATD (Fig. 5 A).
The structural changes accompanying ifenprodil and zinc binding appear similar. Both do not affect the GluN1 ATD but induce a cleft closure of the GluN2 ATD, which, at least in the case of ifenprodil, arises from the lower lobe of the GluN2 ATD moving upwards and inwards. We wanted to determine if zinc binding to the GluN2A ATD induces a similar such rotation as ifenprodil in the GluN2B ATD. To test this, measurements were made at identical sites in GluN2A as those used in GluN2B in a construct with intact zinc inhibition (Fig. 3 C). The LRET between C231 in GluN2A and C22 in GluN1 reveals a decrease in the lifetime (Fig. 5 B) corresponding to a decrease in the distance from 45.9 ± 0.2 Å to 44.4 ± 0.4 Å when zinc binds (Table 2). On average, the distance between these two sites decreased by 1.5 Å (P = 0.04). These measurements can be directly compared with the distance between C232 in GluN2B and C22 in GluN1, as the cysteine is conserved in both GluN2A and GluN2B (C231 and C232, respectively), and the same GluN1 construct was either coexpressed with GluN2A or GluN2B. The distances were measured using the same pair of fluorophores for both GluN2A- and GluN2B-containing receptors. The change in this distance, when zinc or ifenprodil binds, is small; spermine binding induced a 3-Å reduction between these same two sites (Sirrieh et al., 2015). This, and our previous findings (Sirrieh et al., 2013), demonstrates that subtype-specific allosteric modulators similarly affect the conformation of their respective ATDs and therefore suggests that the ATDs of the functional distinct GluN2A and GluN2B share the same operational mechanism.
DISCUSSION
The LRET measurements of the NMDA receptor described here are in good agreement with the crystal structure of the ifenprodil-bound NMDA receptor (Protein Data Bank accession no. 4PE5) (Table 2). Ifenprodil binding to the NMDA receptor induces a decrease in distance between the upper and lower lobes of the GluN2B ATD, without affecting the overall conformation of the GluN1 ATD (Fig. 2). Further, the distances between the GluN1 and GluN2B upper lobes of the ATDs do not undergo any significant conformational changes (Fig. 4), suggesting that they are stable during the process of ifenprodil inhibition. Measurements from the site 232 in the lower lobe of the GluN2B ATD to the upper lobe of the GluN1 ATD (C22) also show a decrease in the distance upon ifenprodil binding (Fig. 5 A). The most parsimonious explanation is that the lower lobe of the GluN2B ATD rotates toward the upper lobe of the GluN1 ATD. As the distances between the subunits do not change in the presence of modulators, confirming that the upper lobes of the ATDs are stable, the data suggest that the upper lobe interactions serve as an anchor to allow the conformational changes within the ATDs to propagate downward toward the LBDs and ultimately the pore of the receptor to affect function. During inhibition, the GluN2 ATD is the moiety undergoing a conformational change, but the presence of the GluN1 ATD is required, as its deletion results in altered inhibition. If the GluN1 ATD is not undergoing any conformational changes during allosteric inhibition, it suggests that the GluN1 ATD may function as a wedge that supports the conformation of the GluN2 ATD. If interactions did not exist between the GluN1 and GluN2 ATDs, then conformational changes induced within the GluN2 ATD could result in a movement of the domain as a whole, rather than affecting agonist binding or open probability of the receptor. The GluN1 ATD serves to hold the GluN2 ATD in a particular configuration. It is interesting to note that similar movements were observed in the GluN1/GluN2A ATDs upon zinc binding (Fig. 5 and Madry et al., 2007). Considering the results of these studies, we suggest that the ATD cleft–closure mechanism is conserved across NMDA receptor subtypes (Fig. 6).
Figure 6.
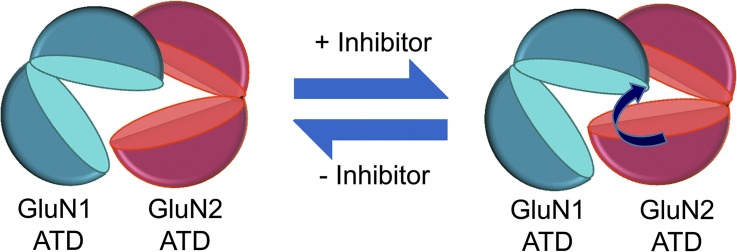
Schematic of inhibition. The inhibitors of the NMDA receptor ifenprodil and zinc both induce cleft-closure conformational changes in the GluN2 ATD. Presumably, proton inhibition, which is affected by the binding of ifenprodil or zinc, influences the conformation of the GluN2 ATD as well.
Consistent with a common structural pathway between the ATD and LBD induced by both zinc and ifenprodil, there are several common effects of zinc and ifenprodil on the receptor, in addition to inhibition. First, both zinc and ifenprodil have been shown to increase glutamate affinity (Kew et al., 1996; Zheng et al., 2001; Erreger and Traynelis, 2005), indicating a cooperativity between the ATD and LBD in the GluN2 subunit. Although the mechanism for this cooperativity remains poorly understood, it is not surprising that the two modulators share a common effect on agonist potency at the LBD. Zinc and ifenprodil both seem to increase desensitization of the NMDA receptor (Zheng et al., 2001), although they push to the receptor to occupy kinetically different desensitized states (Amico-Ruvio et al., 2011, 2012). The difference in the desensitized states the receptor occupies when bound by zinc or ifenprodil could arise from the different GluN2 subtype or from differences in the structural state of the LBD of the receptor. Importantly, zinc and ifenprodil’s effects on the respective diheteromeric receptors are similar when recordings are made from neurons (Tovar and Westbrook, 2012). Finally, the inhibition by both ifenprodil and zinc increases at more acidic pHs (Pahk and Williams, 1997; Mott et al., 1998; Low et al., 2000). Recently, a new pair of carboxylate residues in the ATD of the GluN2B subunit was suggested to be these proton sensors (Glu106 and Glu235) (Yuan et al., 2015). These residues are located in the upper and lower lobes of the GluN2B ATD, respectively, with pKa’s corresponding to the range of the NMDA receptor’s proton sensitivity (Yuan et al., 2015). The conformational changes we observe accompanying ifenprodil, zinc, and spermine modulation (Sirrieh et al., 2013, 2015); the pH sensitivity of their action (Traynelis et al., 1995; Pahk and Williams, 1997, Mott et al., 1998; Low et al., 2000); and the locations of these new putative proton sensors can all be incorporated into a single molecular framework. At lower pHs, the protonation of one of these residues may result in a noncovalent interaction with its partner’s side chain, which stabilizes the closed conformation of the ATD. Because ifenprodil and zinc also induce a closure of the GluN2 ATD (Sirrieh et al., 2013), the interaction between these two carboxylates at lower pHs likely augments the modulators’ stabilization, giving rise to a synergistic interaction between inhibitor and protons. Conversely, spermine, which potentiates the receptor by inducing an opening of the GluN2 ATD (Sirrieh et al., 2015), disrupts the possible interaction of these two carboxylate residues by increasing the distance between them.
However, one objection to this conservation of ATD closure hypothesis is the apparently discrepant magnitudes of modulator effects. GluN2B experiences a slightly smaller conformational change when ifenprodil binds, at least as measured at the above sites, than GluN2A does upon zinc binding (2.8-Å change with ifenprodil vs. a 3.9-Å change with zinc) (Sirrieh et al., 2013). However, the extent of ifenprodil inhibition of the GluN2B-containing receptors is actually greater than that of zinc inhibition of GluN2A-containing receptors. Part of the difference can be accounted for by the inherent difference in the ATD cleft between GluN2A and GluN2B subtypes, where the GluN2A ATD adopts a more open conformation than the GluN2B ATD (Sirrieh et al., 2015). As a result, the ifenprodil-induced change in the GluN2B ATD is enough to inhibit the receptor more than the zinc-induced change is in the GluN2A ATD. Although the difference between these two changes appears small, this difference is comparable to that measured in the AMPA receptor LBD between full agonist glutamate and weak partial agonist kainate-bound states as well as between glutamate-bound AMPA receptor LBDs alone or in the presence of stargazin (MacLean et al., 2014). In the future, our experimental efforts would do well to move downward, toward the ATD–LBD linkers, and examine how, precisely, the well-mapped conformational changes of the ATD exert their influence on the LBD and the pore, as there are likely other differences between GluN2 subtypes.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01GM094245-01A1 and 1R01GM113212-01 to V. Jayaraman and by NIH-2T32GM008280 to R.E. Sirrieh.
The authors declare no competing financial interests.
Author contributions: R.E. Sirrieh designed and performed LRET experiments, analyzed data, and wrote the paper. D.M. MacLean performed the electrophysiology experiments, analyzed the data, and helped write the paper. V. Jayaraman designed experiments, analyzed data, and wrote the paper.
Richard W. Aldrich served as editor.
Footnotes
Abbreviations used in this paper:
- ATD
- amino-terminal domain
- CHO
- Chinese hamster ovary
- LBD
- ligand-binding domain
- LRET
- luminescence resonance energy transfer
References
- Amico-Ruvio S.A., Murthy S.E., Smith T.P., and Popescu G.K.. 2011. Zinc effects on NMDA receptor gating kinetics. Biophys. J. 100:1910–1918. 10.1016/j.bpj.2011.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amico-Ruvio S.A., Paganelli M.A., Myers J.M., and Popescu G.K.. 2012. Ifenprodil effects on GluN2B-containing glutamate receptors. Mol. Pharmacol. 82:1074–1081. 10.1124/mol.112.078998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimecombe J.C., Potthoff W.K., and Aizenman E.. 1999. A critical role of the N-methyl-d-aspartate (NMDA) receptor subunit (NR) 2A in the expression of redox sensitivity of NR1/NR2A recombinant NMDA receptors. J. Pharmacol. Exp. Ther. 291:785–792. [PubMed] [Google Scholar]
- Chazot P.L., Cik M., and Stephenson F.A.. 1999. Transient expression of functional NMDA receptors in mammalian cells. Methods Mol. Biol. 128:33–42. [DOI] [PubMed] [Google Scholar]
- Choi Y.-B., and Lipton S.A.. 1999. Identification and mechanism of action of two histidine residues underlying high-affinity Zn2+ inhibition of the NMDA receptor. Neuron. 23:171–180. 10.1016/S0896-6273(00)80763-1 [DOI] [PubMed] [Google Scholar]
- Dolino D.M., Ramaswamy S.S., and Jayaraman V.. 2014. Luminescence resonance energy transfer to study conformational changes in membrane proteins expressed in mammalian cells. J. Vis. Exp. 91:51895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K., and Traynelis S.F.. 2005. Allosteric interaction between zinc and glutamate binding domains on NR2A causes desensitization of NMDA receptors. J. Physiol. 569:381–393. 10.1113/jphysiol.2005.095497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher M.J., Huang H., Pritchett D.B., and Lynch D.R.. 1996. Interactions between ifenprodil and the NR2B subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem. 271:9603–9611. 10.1074/jbc.271.16.9603 [DOI] [PubMed] [Google Scholar]
- Gielen M., Siegler Retchless B., Mony L., Johnson J.W., and Paoletti P.. 2009. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 459:703–707. 10.1038/nature07993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J., Rambhadran A., Du M., and Jayaraman V.. 2008. LRET investigations of conformational changes in the ligand binding domain of a functional AMPA receptor. Biochemistry. 47:10027–10032. 10.1021/bi800690b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis A.N., Ebright Y.W., and Ebright R.H.. 2001. Site-specific incorporation of fluorescent probes into protein: Hexahistidine-tag-mediated fluorescent labeling with (Ni2+:nitrilotriacetic acid)n-fluorochrome conjugates. J. Am. Chem. Soc. 123:12123–12125. 10.1021/ja017074a [DOI] [PubMed] [Google Scholar]
- Karakas E., Simorowski N., and Furukawa H.. 2009. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 28:3910–3920. 10.1038/emboj.2009.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E., Simorowski N., and Furukawa H.. 2011. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 475:249–253. 10.1038/nature10180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew J.N., Trube G., and Kemp J.A.. 1996. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J. Physiol. 497:761–772. 10.1113/jphysiol.1996.sp021807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low C.-M., Zheng F., Lyuboslavsky P., and Traynelis S.F.. 2000. Molecular determinants of coordinated proton and zinc inhibition of N-methyl-d-aspartate NR1/NR2A receptors. Proc. Natl. Acad. Sci. USA. 97:11062–11067. 10.1073/pnas.180307497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean D.M., Ramaswamy S.S., Du M., Howe J.R., and Jayaraman V.. 2014. Stargazin promotes closure of the AMPA receptor ligand-binding domain. J. Gen. Physiol. 144:503–512. 10.1085/jgp.201411287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madry C., Mesic I., Betz H., and Laube B.. 2007. The N-terminal domains of both NR1 and NR2 subunits determine allosteric Zn2+ inhibition and glycine affinity of N-methyl-d-aspartate receptors. Mol. Pharmacol. 72:1535–1544. 10.1124/mol.107.040071 [DOI] [PubMed] [Google Scholar]
- Mott D.D., Doherty J.J., Zhang S., Washburn M.S., Fendley M.J., Lyuboslavsky P., Traynelis S.F., and Dingledine R.. 1998. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat. Neurosci. 1:659–667. 10.1038/3661 [DOI] [PubMed] [Google Scholar]
- Pahk A.J., and Williams K.. 1997. Influence of extracellular pH on inhibition by ifenprodil at N-methyl-d-aspartate receptors in Xenopus oocytes. Neurosci. Lett. 225:29–32. 10.1016/S0304-3940(97)00176-6 [DOI] [PubMed] [Google Scholar]
- Paoletti P., Ascher P., and Neyton J.. 1997. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 17:5711–5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P., Perin-Dureau F., Fayyazuddin A., Le Goff A., Callebaut I., and Neyton J.. 2000. Molecular organization of a zinc binding n-terminal modulatory domain in a NMDA receptor subunit. Neuron. 28:911–925. 10.1016/S0896-6273(00)00163-X [DOI] [PubMed] [Google Scholar]
- Paoletti P., Bellone C., and Zhou Q.. 2013. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14:383–400. 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- Perin-Dureau F., Rachline J., Neyton J., and Paoletti P.. 2002. Mapping the binding site of the neuroprotectant ifenprodil on NMDA receptors. J. Neurosci. 22:5955–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachline J., Perin-Dureau F., Le Goff A., Neyton J., and Paoletti P.. 2005. The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J. Neurosci. 25:308–317. 10.1523/JNEUROSCI.3967-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambhadran A., Gonzalez J., and Jayaraman V.. 2010. Subunit arrangement in N-methyl-d-aspartate (NMDA) receptors. J. Biol. Chem. 285:15296–15301. 10.1074/jbc.M109.085035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirrieh R.E., MacLean D.M., and Jayaraman V.. 2013. Amino-terminal domain tetramer organization and structural effects of zinc binding in the N-methyl-d-aspartate (NMDA) receptor. J. Biol. Chem. 288:22555–22564. 10.1074/jbc.M113.482356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirrieh R.E., MacLean D.M., and Jayaraman V.. 2015. Subtype-dependent N-methyl-d-aspartate receptor amino-terminal domain conformations and modulation by spermine. J. Biol. Chem. 290:12812–12820. 10.1074/jbc.M115.649723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C.M. 2001. Rapid solution application methods. Curr. Protoc. Neurosci. 6:6.9. [DOI] [PubMed] [Google Scholar]
- Tomitori H., Suganami A., Saiki R., Mizuno S., Yoshizawa Y., Masuko T., Tamura Y., Nishimura K., Toida T., Williams K., et al. 2012. Structural changes of regulatory domain heterodimer of N-methyl-d-aspartate receptor subunits GluN1 and GluN2B through the binding of spermine and ifenprodil. J. Pharmacol. Exp. Ther. 343:82–90. 10.1124/jpet.112.192286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar K.R., and Westbrook G.L.. 2012. Amino-terminal ligands prolong NMDA receptor-mediated EPSCs. J. Neurosci. 32:8065–8073. 10.1523/JNEUROSCI.0538-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis S.F., Hartley M., and Heinemann S.F.. 1995. Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science. 268:873–876. 10.1126/science.7754371 [DOI] [PubMed] [Google Scholar]
- Traynelis S.F., Wollmuth L.P., McBain C.J., Menniti F.S., Vance K.M., Ogden K.K., Hansen K.B., Yuan H., Myers S.J., and Dingledine R.. 2010. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 62:405–496. 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H., Hansen K.B., Vance K.M., Ogden K.K., and Traynelis S.F.. 2009. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 29:12045–12058. 10.1523/JNEUROSCI.1365-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H., Myers S.J., Wells G., Nicholson K.L., Swanger S.A., Lyuboslavsky P., Tahirovic Y.A., Menaldino D.S., Ganesh T., Wilson L.J., et al. 2015. Context-dependent GluN2B-selective inhibitors of NMDA receptor function are neuroprotective with minimal side effects. Neuron. 85:1305–1318. 10.1016/j.neuron.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F., Erreger K., Low C.-M., Banke T., Lee C.J., Conn P.J., and Traynelis S.F.. 2001. Allosteric interaction between the amino terminal domain and the ligand binding domain of NR2A. Nat. Neurosci. 4:894–901. 10.1038/nn0901-894 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.