

H+ not only activates the TRPV1 cation channel but inhibits ion permeation, yielding a prominent OFF current response.
Abstract
The TRPV1 cation channel is a polymodal nociceptor that is activated by heat and ligands such as capsaicin and is highly sensitive to changes in extracellular pH. In the body core, where temperature is usually stable and capsaicin is normally absent, H+ released in response to ischemia, tissue injury, or inflammation is the best-known endogenous TRPV1 agonist, activating the channel to mediate pain and vasodilation. Paradoxically, removal of H+ elicits a transient increase in TRPV1 current that is much larger than the initial H+-activated current. We found that this prominent OFF response is caused by rapid recovery from H+ inhibition of the excitatory current carried by H+-activated TRPV1 channels. H+ inhibited current by interfering with ion permeation. The degree of inhibition is voltage and permeant ion dependent, and it can be affected but not eliminated by mutations to acidic residues within or near the ion selectivity filter. The opposing H+-mediated gating and permeation effects produce complex current responses under different cellular conditions that are expected to greatly affect the response of nociceptive neurons and other TRPV1-expressing cells.
INTRODUCTION
TRPV1 ion channel is a polymodal nociceptor (Caterina et al., 1997; Tominaga et al., 1998; Jordt and Julius, 2002; Yang et al., 2015). Its activation by capsaicin and heat has attracted intensive attention (Clapham, 2003; Zheng, 2013). Nonetheless, in the body core where TRPV1 is expressed in many organs and tissues, temperature is stable and capsaicin is normally absent. Extracellular H+ serves as the best-known endogenous agonist for TRPV1 (Tominaga et al., 1998; Julius and Basbaum, 2001). Proton release upon ischemia, tissue injury, and inflammation, which has been indicated to activate TRPV1 (Hoover, 1987; Oroszi et al., 1999; Hu et al., 2002), is suggested to play important roles in inducing vasodilation (to mitigate ischemia) and producing pain (to signal tissue damage) (Bevan and Geppetti, 1994). It is thus both physiologically significant and biophysically intriguing to understand how TRPV1 sensitively responds to <10-fold changes in extracellular H+ concentration that generally occur during ischemia and inflammation (Stevens et al., 1991; Pan et al., 1999).
TRPV1 is a member of the tetrameric cation channel superfamily. Its molecular structure has been revealed at near-atomic resolutions by cryo-electron microscopy (Cao et al., 2013; Liao et al., 2013). The channel complex closely resembles a voltage-gated potassium channel (Long et al., 2007), with the four subunits arranged symmetrically around a central ion permeation pore formed mainly by S6 and the pore-loop region (Zheng and Ma, 2014). Interestingly, the ion selectivity filter sequence of TRPV1 (TIGMGD) is very similar to that of highly potassium-selective channels (typically TVGYGD), even though TRPV1 is a nonselective cation-permeable channel. There are numerous acidic residues in the outer pore region as well as the peripheral extracellular loops that may serve as potential protonation sites for channel gating. Among them, mutations to E601 and E649 (amino acid numbers according to mouse TRPV1 used in the present study) were found to have dramatic effects on H+-induced channel activation (Jordt et al., 2000), although mutations to many other residues including nontitratable residues could also alter or even eliminate H+-induced channel activation (Welch et al., 2000; Wang et al., 2010; Aneiros et al., 2011; Boukalova et al., 2013). It thus remains unclear how extracellular H+ promotes TRPV1 activation gating.
As expected from allosteric coupling, the response of TRPV1 to H+ can be strongly influenced by the presence of other gating stimuli such as voltage, heat, capsaicin, and intracellular Ca2+ (Tominaga et al., 1998; Ryu et al., 2003; Aneiros et al., 2011). In this study, we demonstrated that even in the absence of these confounding factors, H+ induced complex responses from TRPV1. We observed that H+ exhibited two opposite effects: it potently activated the channel but at the same time inhibited ion permeation in a concentration- and voltage-dependent manner. Our results suggest that inhibition of permeation is achieved by H+ binding inside or near the ion-conducting pore. The combination of the potentiating gating effect and the inhibitory permeation effect yielded complex current responses from TRPV1 under different recording conditions that are expected to greatly influence physiological and pathological behaviors of the host cells.
MATERIALS AND METHODS
cDNA constructs and cell transfection
Mouse TRPV1 cDNA fused with enhanced YFP cDNA at the C terminus (Cheng et al., 2007) was used in the present study. Point mutants were generated using the QuickChange II Site-Directed Mutagenesis kit (Agilent Technologies). HEK293T cells were cultured in a DMEM medium supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% (vol/vol) nonessential amino acids at 37°C with 5% CO2. Cells were passaged 18–24 h before transfection by plating onto glass coverslip. Transient transfection was conducted 18–24 h before patch recording using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Electrophysiology
Macroscopic and single-channel currents from TRPV1-expressing cells were recorded in whole-cell or outside-out configuration using an amplifier (EPC10; HEKA) controlled with PatchMaster software (HEKA). Patch pipettes were pulled from thin-wall borosilicate glass (A-M Systems) and fire-polished to a resistance of ∼2 MΩ. For whole-cell recordings, the capacity current was minimized by amplifier circuitry, and the series resistance was compensated by 30–65%. Membrane potential was held at 0 mV from which steps to various voltages were used. For the ramp protocol, the membrane potential was first stepped from 0 to −250 mV and then ramped up to 250 mV in 100 ms. Data were filtered at 2.25 kHz and sampled at 12.5 kHz. All recordings were performed under room temperature (∼24°C). Temperature variation was <1°C as monitored by a thermometer.
Solutions
During the course of the present study, we observed a prominent endogenous current when cells were challenged by a low pH solution (Fig. S1). This endogenous current was strongly outward rectifying and exhibited chloride dependence. Replacing chloride with glutamate almost completely eliminated this current (Fig. S1). Therefore, for all experiments used to obtain quantitative measurements, chloride in the solution was replaced with glutamate. Symmetrical bath and pipette solutions were used, except that the pH level of extracellular solution was adjusted according to experimental needs. An agar bridge was used when needed. To prevent rapid inactivation, all experiments were done in Ca2+-free solutions containing 0.2 mM EGTA. For solutions at pH 7.2 to 6.0, HEPES was used as the H+ buffer, whereas for solutions at pH 5.5 to 4.0, MES was used. The compositions of four different solutions are shown in Table 1.
Table 1.
Composition of solutions used in this study
| No. | Sodium chloride | Magnesium chloride | Potassium chloride | Sodium glutamate monohydrate | HEPES or MES | EGTA |
| I | 130 | 0 | 0 | 0 | 3 | 0.2 |
| II | 30 | 100 | 0 | 0 | 3 | 0.2 |
| III | 0 | 0 | 130 | 0 | 3 | 0.2 |
| IV | 0 | 0 | 0 | 130 | 3 | 0.2 |
All values indicate the final concentration (mM) after adjustment of the pH level. For Solutions I, II, and III, pH was adjusted by hydrochloric acid or sodium hydroxide, whereas for Solution IV, pH was adjusted by l-glutamic acid or sodium hydroxide.
Rapid solution switching
A rapid solution changer (RSC-200; Biological Science Instruments) was used for normal solution switching. This method was preferable in that switching among many solutions could be accommodated, and there was no mixing of these solutions throughout the perfusion system. For experiments requiring a higher solution switching speed, a solution-switching system based on a pulled Θ-glass capillary driven by a step motor (Perfusion Fast-Step; model SF-77B; Warner Instruments) was used. This approach allowed switching between two solutions in <1 ms (Zheng and Sigworth, 1997). As was done previously, the rate of solution switching was measured by monitoring the time course of change in liquid junction potential between the pipette solution and the perfused solutions. The transition time from 10 to 90% change in junction potential was 0.8 ms in a typical experiment.
Data analysis
The H+ concentration–response relationship was fitted to the Hill equation:
where Ix is the total or OFF response current in the presence of extracellular H+ at concentration [x]; Imin and Imax are the current amplitude at neutral pH (7.2) and at that of the maximal current amplitude, respectively; EC50 is the H+ concentration at which activation is half-maximal; and n is the Hill coefficient. Although at pH 7.2 there was a finite concentration of free H+, the error in Imin associated with this is negligible (for example, causing an estimated 0.7% shift to the pKa value).
The G-V relationship was constructed from steady-state currents elicited by voltage steps. G-V and inhibition–voltage relationships were fitted to a single-Boltzmann function:
where G/Gmax is the normalized conductance, Inhibition is the percentage of inhibited conductance, q is the equivalent gating charge, Vhalf is the half-activation or half-inhibition voltage, T is temperature, R is the gas constant, and F is the Faraday’s constant.
Single-channel conductance was estimated from all-point histograms constructed from current events recorded using the outside-out configuration at 80 mV. A double-Gaussian function was used for fitting of the histograms. From the positions of the Gaussian peaks, the closed- and open-current levels were identified. Single-channel current amplitude and the corresponding conductance were calculated.
All statistical values are given as mean ± SEM for the number of measurements indicated (n). Statistical significance was determined using the Student’s t test, and indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Online supplemental material
Fig. S1 shows H+-induced endogenous currents that could be eliminated by replacing chloride with glutamate in the recording solutions. The online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201511386/DC1.
RESULTS
Steady-state activation of TRPV1 by extracellular H+
We overexpressed mouse TRPV1 in HEK293T cells and compared channel currents at low and high extracellular H+ concentrations by patch-clamp recording in the whole-cell configuration (Fig. 1). At the normal physiological pH level, depolarization beyond 0 mV weakly activated the channel, with the half-activation voltage, V1/2, estimated at 129.9 ± 20.2 mV (n = 4; Fig. 1, A, left, and C). Raising H+ concentration substantially increased channel open probability and the current amplitude (Fig. 1 A, right), and shifted V1/2 to lower voltages (Fig. 1 C), reflecting a strong potentiation effect of H+ on the gating of TRPV1. At pH 5.0, the V1/2 value was reduced to −93.2 ± 36.5 mV (n = 4), a level below the resting membrane potential of most cells. Both an increase in amplitude and a shift in V1/2 could be clearly observed using a voltage-ramp protocol (Fig. 1 B). Therefore, extracellular H+ strongly potentiates TRPV1 and is able to cause channel activation in a resting neuron in the absence of other activation stimuli.
Figure 1.

Extracellular H+ strongly activates TRPV1. (A) Representative whole-cell patch-clamp current traces recorded in Solution I in response to voltage steps at pH 7.2 (left) and pH 5.0 (right). Voltage steps were applied from a holding potential of 0 mV to various membrane potentials from −200 to 300 mV in 20-mV intervals. Dotted lines indicate zero current level. (B) Representative outside-out patch-clamp current traces recorded in Solution I in response to voltage ramps at different pH levels. Voltage ramps from −250 to 250 mV in 100 ms were applied from a holding potential of 0 mV. (C) H+-dependent shift of the G-V relationship. Different symbols represent separate recordings, whereas their colors match those of the pH levels. Superimposed are fits of a Boltzmann function with the following Vhalf and q values: pH 7.2, 129.9 mV and 0.5 e0 (n = 4); pH 6.5, 99.1 mV and 0.5 e0 (n = 3); pH 6.0, −6.1 mV and 0.4 e0 (n = 3); pH 5.5, −33.3 mV and 0.4 e0 (n = 5); pH 5.0, −93.2 mV and 0.3 e0 (n = 4).
Response of TRPV1 to changes in extracellular H+ concentration
The temporal response of TRPV1 to changes in H+ concentration was recorded at a constant voltage of 40 mV. Switching from a neutral pH solution to an acidic solution caused the channel to activate (Fig. 2 A). The current amplitude increased in a H+ concentration-dependent manner at the pH range from 6.5 to 5.5; at even lower pH levels, the current amplitude started to decrease. As expected for a ligand-driven process, the current rising rate was H+ concentration dependent (Fig. 2 B), whereas upon switching back to the neutral pH solution, the current decline rate was concentration independent, with an estimated time constant at 100 ± 3 ms (Fig. 2 C).
Figure 2.

H+ activates TRPV1 in a concentration-dependent manner. (A) Representative outside-out patch-clamp current traces at 40 mV in Solution IV in response to various pH levels from pH 6.5 to pH 4.5 (indicated by a bar on the top). Dotted lines indicate zero current level and the peak current level. Time constants of the current rising phase (B) and the current decline phase (C) are plotted against the extracellular pH level. n = 6 each. Error bars indicate mean ± SEM.
A striking feature of H+-induced TRPV1 activation is that upon removal of the acidic solution, there was a large transient increase in current amplitude (Fig. 2 A). This prominent OFF response also increased in a H+ concentration-dependent manner, reaching its peak at pH 5.0. Note that the OFF response peaked at a lower pH level than the steady-state current did. Furthermore, unlike the steady-state current, the OFF response only slightly dipped from its peak level when the pH level was further decreased (see also Fig. 4). The existence of a large OFF response and its distinct pH dependence from the steady-state current illustrated the complexity of the TRPV1 response to H+.
Figure 4.

The OFF response reveals two distinct H+-induced processes in TRPV1. The total OFF response current (I1, red squares) and the rising phase of the OFF response (I2, blue circles) were quantified in Solution IV. Curves represent fits of a Hill equation with the following EC50 and slope factor values: total current (I1), 0.3 µM, pH 6.5, and 1.6; rising phase current (I2), 1.9, pH 5.7, and 0.9. n = 6 each. Error bars indicate mean ± SEM.
Rapid solution switching revealed two H+-induced processes
The transient OFF response rose rapidly at the solution-changing rate and declined at a rate of ∼10 s−1 (Fig. 2 C). Therefore, to accurately record the amplitude of the OFF response, the solution-switching speed had to be fast. We set up a rapid solution-switching system using a step motor–controlled Θ-glass capillary (Fig. 3 A). It permitted switching between a pair of solutions in <1 ms (Fig. 3, B and C). Using this approach, we were able to accurately follow both H+-induced activation and the OFF response (Fig. 3, D and E). The speed of the OFF response rising phase was found to be still determined by the solution-switching rate and could not be resolved under our experimental condition; however, the decline phase was perfusion and concentration independent and could be reliably measured (Fig. 3 E).
Figure 3.

A rapid solution-switching method allows reliable recording of the OFF response. (A) Schematic diagram of rapid solution switching. (B) Representative current-clamp recording using solutions that had different junction potentials. (C) Example of solution-switching speed measured from the region indicated by the box in B. (D) Representative outside-out patch-clamp current induced by Solution IV at pH 4.5. (E) Expanded presentation of the OFF response marked by the box in D. The red dotted trace represents a single-exponential fit with a time constant of 0.10 ± 0.01 s−1 (n = 6).
We analyzed the complex H+-induced response in two ways (Fig. 3 D). The peak OFF response was defined as I1, and the difference between the peak OFF response and the steady-state current, e.g., the rising phase of the OFF response, was defined as I2. As shown in Fig. 4, the two currents exhibited distinct pH dependence. Fitting of the concentration dependence to a Hill equation revealed an apparent pKa value of 6.5 for the peak OFF response, I1. The peak response was also highly sensitive to changes in H+ concentration, with a Hill slope of 1.6 ± 0.5 (n = 6) that indicates the binding of more than one H+ ion. In comparison, the rising phase of the OFF response, I2, exhibited a much lower pKa value of 5.7, suggesting that a much weaker binding of H+ gave rise to the OFF response. It also had a much shallower H+ concentration-dependent curve, with a Hill slope of 0.9 ± 0.1 (n = 6) that was consistent with binding of a single H+ ion. Therefore, the OFF response revealed that H+ induced two distinct processes in TRPV1.
Single-channel recordings revealed both gating and permeation effects of H+
To better understand how TRPV1 responded to H+, we conducted single-channel recording from outside-out patches in different pH solutions (Fig. 5 A). We observed that as the pH level dropped, the channel spent much longer times in the open state, consistent with the potentiation effect on gating. Similar to previous findings (Liu et al., 2009), the single-channel current amplitude gradually decreased upon an increase in H+ concentration (Fig. 5, A and B), reducing conductance by about one third when the pH level reached 5.0 (Fig. 5 C). From the pH dependence of single-channel conductance, the apparent pKa value associated with H+ inhibition of conductance was estimated to be 5.7 (Fig. 5 D). It is noticed that this pKa value coincides with the pKa value of the OFF response (I2). Single-channel recordings thus revealed that H+ has two opposite effects on TRPV1: it strongly potentiates gating of the channel; however, when the channel opens, it inhibits ion permeation.
Figure 5.

Single-channel recording reveals two opposing effects of H+ on TRPV1. (A) Representative single-channel current traces recorded in outside-out configuration at 80 mV in Solution I at the indicated extracellular pH levels. The presented current traces were further processed by a digital filter to a final cutoff frequency of 0.41 kHz. (B) All-point histogram of single-channel events at the indicated pH levels. The superimposed curve represents a fit of a double-Gaussian function. (C) Box-and-whisker plot of single-channel conductance versus the corresponding pH level. The whisker top, box top, line inside the box, box bottom, and whisker bottom represent the maximum, 75th percentile, median, 25th percentile, and minimum value of each pool of conductance measurements, respectively. n = 5–11. *, P < 0.05; ***, P < 0.001. (D) Inhibition of conductance is plotted against the extracellular H+ concentration. Superimposed is a fit of a Hill function, with the following parameters: IC50, 2.2 µM; slope factor, 0.2. n = 5–11. Error bars indicate mean ± SEM.
The opposite gating and permeation effects of H+ provide a natural explanation for the transient OFF response observed in macroscopic currents. When H+ activated TRPV1, the open-channel current was instantly reduced by H+ inhibition of permeation. Upon removal of H+, the channel first recovered from permeation inhibition, yielding an increase in current amplitude that was the OFF response. The H+-gating effect was reversed more slowly, leading to the decline of the OFF response. The whole process can be represented by the following scheme:
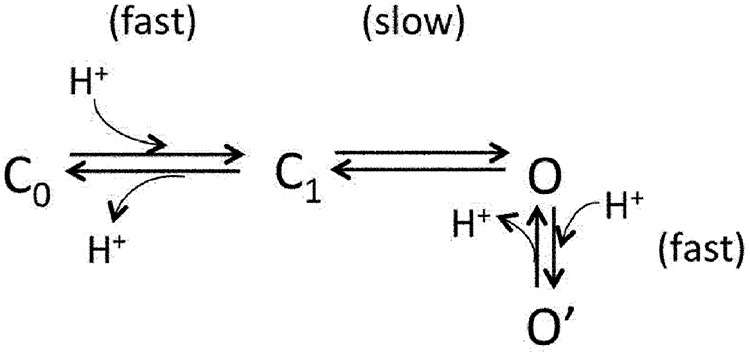 |
(Scheme 1) |
According to Scheme 1, there are two types of H+-binding steps (and hence two pKa’s). The first H+-binding step produces the gating effect (horizontal transitions), whereas the second H+-binding step produces inhibition of permeation (vertical transition). An important prediction of this scheme is that to produce the asymmetrical OFF response seen in macroscopic currents, the O′→O transition and the O→C1 transition must occur at different rates, one being faster than the solution-switching rate and the other being comparable to the decline rate of the OFF response. As described in the next section, we observed that recovery from H+-induced permeation inhibition is the rapid transition. Therefore, the reversal of H+-induced activation must be a slow process, reflecting a substantial energy barrier for the transition.
H+ inhibition of macroscopic current
Our study so far has revealed strong permeation inhibition by H+ that, according to Scheme 1, is produced by a H+-binding step distinct from the H+-binding step that leads to channel activation. If this is the case, it is expected that when the channel is brought into the O state by an activation stimulus other than H+, the current should also be inhibited by H+. We were able to confirm this prediction by the experiment shown in Fig. 6, in which channels were first activated by capsaicin after which solutions at different pH levels were applied to the cell. The open-channel currents were concentration-dependently inhibited by H+. We noticed that in this experiment, both the onset and recovery of H+ inhibition were rapid. Interestingly, H+ inhibition was much more prominent in the inward current measured at −80 mV than in the outward current measured at 80 mV, suggesting that H+ inhibition is a voltage-dependent process.
Figure 6.

H+ inhibition of macroscopic current. (A) Representative current traces from whole-cell recording at 80 mV (black) and −80 mV (red) using Solution IV. Extracellular H+ inhibits current induced by a saturating concentration of capsaicin (10 µM) in a pH-dependent manner. Dotted line indicates zero current level. (B) Percentage of current inhibition at different pH levels at 80 mV (black) and −80 mV (red). n = 5 each. Error bars indicate mean ± SEM.
Voltage dependence of H+ inhibition
We followed up the observation shown in Fig. 6 and conducted detailed tests of the voltage dependence of H+ inhibition. A pH-5.0 solution was applied at different voltages in both whole-cell and outside-out recordings (Fig. 7, A and B, respectively). In both cases, we observed obvious OFF responses. Furthermore, at more negative voltages, the OFF response was much larger compared with the steady-state current. The ratio I2/I1 demonstrated clearly voltage dependence (Fig. 7 C). From a Boltzmann fit of the voltage dependence, we obtained the apparent charge associated with H+ inhibition. The estimated value, at 0.48 e0, is equivalent to moving a H+ ion halfway into the transmembrane electric field from the extracellular side. The voltage-dependent inhibition and the apparent charge are in close agreement with those observed previously from the rat TRPV1 single-channel currents (Liu et al., 2009).
Figure 7.

Voltage-dependent H+ inhibition. Representative current traces from whole-cell recording in Solution I (A) and outside-out patch recording in Solution IV (B) at various voltages and pH 5.0. (C) The ratio I2/I1 measured from outside-out patch recordings in Solution IV exhibits voltage dependence. The superimposed curve represents a fit of a Boltzmann function with 26.6 mV for Vhalf and 0.48 e0 for q. n = 5–9. Error bars indicate mean ± SEM.
As an alternative way to confirm voltage dependence of H+ inhibition, we recorded H+-induced reduction of capsaicin-activated current at different voltages (Fig. 8). Again, H+ inhibition was found to be voltage dependent, being more prominent at negative voltages. Based on a fit of a Boltzmann function to the voltage dependence, the apparent charge was estimated to be 0.75 e0.
Figure 8.

Voltage-dependent inhibition of capsaicin-activated current. (A) Representative currents from outside-out patch recording at various voltages in Solution IV. Arrows indicate the time when capsaicin (3 µM) was applied. Dotted line indicates zero current level. (B) The percentage of current inhibition at pH 5.0 is plotted against voltage. The superimposed curve represents a fit of a Boltzmann function with 48.7 mV for Vhalf and 0.75 e0 for q. The dotted curve represents the Boltzmann fit as shown in Fig. 7 C. n = 2–7. Error bars indicate mean ± SEM.
Inhibition of TRPV1 current by intracellular H+
The voltage dependence of current inhibition by H+, together with its independence of H+-induced channel activation, suggests the possibility that H+ might interfere with permeant ion in the pore to slow down ion conductance. If this is the case, one would expect that intracellularly applied H+ should also affect ion permeation. We found that indeed intracellular H+ was able to substantially inhibit single-channel conductance, reducing its amplitude by 68.4 ± 2.3% (n = 5) at 80 mV when the H+ level was changed from pH 7.2 to pH 5.0 (Fig. 9 A). The H+ concentration dependence indicated an apparent pKa of 1.4 ± 0.5 (n = 5), pH 5.9, very similar to the apparent pKa value estimated with extracellular H+. Macroscopic current recording confirmed the strong intracellular H+ inhibition, which was found again to be independent of H+-induced activation that occurs only from the extracellular side (Fig. 9, B and C). Interestingly, from the voltage dependence it was estimated that the apparent charge associated with intracellular H+ inhibition is 0.49 ± 0.02 e0 (n = 4) (Fig. 9 D), a value that complements well the apparent charge associated with extracellular H+. It is also noteworthy that H+ inhibition from the intracellular side was facilitated by depolarization, whereas H+ inhibition from the extracellular side was facilitated by hyperpolarization. These observations indicate that the same binding site might be involved no matter which side H+ was applied.
Figure 9.

Current inhibition by intracellular H+. (A) Representative single-channel current traces recorded from an inside-out patch at 80 mV with Solution IV at pH 7.2 (left) or pH 5.0 (right) in the bath. (B and C) Representative macroscopic current traces recorded from inside-out patches at stable voltages (B) or in response to voltage ramps (C). (D) Voltage dependence of H+ inhibition (black trace) was fitted to a Boltzmann function (red trace).
The OFF response of Mg2+-induced TRPV1 current
Previously, we observed that extracellular Mg2+, a TRPV1 agonist (Ahern et al., 2005), can also inhibit open-channel current, which we attributed to Mg2+ block of ion permeation by moving into the channel pore as a permeant ion (Cao et al., 2014; Yang et al., 2014). Hence, like H+, Mg2+ also exhibits dual gating and permeation effects. In the present study, we showed that, as anticipated, Mg2+ could also produce a large OFF response when applied to TRPV1 (Fig. 10). The Mg2+-induced OFF response appeared very similar to that induced by H+, except that much higher Mg2+ concentrations (in the millimolar range, comparing to the micromolar range for H+) were needed to both activate the channel and to inhibit ion permeation. The Mg2+-induced OFF response also exhibited strong voltage dependence. Importantly, the apparent charge associated with the Mg2+ OFF response was estimated to be 0.91 e0, roughly double the apparent charge estimated in the same way for the monovalent cation H+.
Figure 10.

The OFF response of Mg2+-induced TRPV1 current. (A) Representative currents from outside-out patch recording in Solution II (containing 100 mM Mg2+) at various voltages. (B) The ratio I2/I1 exhibits voltage dependence in Mg2+-induced inhibition. The superimposed curve represents a fit of a Boltzmann function with 56.3 mV for Vhalf and 0.91 e0 for q. n = 3–6. Error bars indicate mean ± SEM.
Interaction between H+ and permeant ions
Both the rapid onset (and reversal) and the voltage dependence suggested that H+ inhibition of permeation is likely caused by H+ binding inside the pore to interfere with the passage of permeant ions. If this is the case, it is anticipated that permeant ions would in turn exert an influence on H+ binding. Therefore, we compared H+ inhibition in the presence of different permeant ions. We found that indeed the percentage of current inhibition varied slightly when the permeant ion was changed from Na+ to K+ (Fig. 11 A). Although the inhibition itself remained voltage dependent, the difference in percentage of inhibition between Na+ and K+ could be detected at several voltages (Fig. 11 B).
Figure 11.

Interaction between H+ and permeant ions. (A) Representative currents from outside-out patch recording at pH 5.0 using different permeant ions, Na+ (left, Solution I) or K+ (right, Solution III), at various voltages. (B) The ratio I2/I1 exhibits permeant ion dependence. n = 5 each. *, P < 0.05; **, P < 0.01. Error bars indicate mean ± SEM.
Based on all the observations presented so far, we conclude that, when H+ is present in the extracellular solution, it binds and unbinds rapidly at a site ∼50% into the electric field along the ion permeation pore with an apparent affinity of 1.9 ± 0.5 µM (pKa of 5.7). This H+-binding event, which causes inhibition of TRPV1 current, is distinct from the H+-binding event(s) that leads to channel activation.
Mutations of acidic residues E637 and D647 did not prevent H+ inhibition
It was previously reported that a double mutation of titratable residues E636 and D646 completely prevented voltage-dependent inhibition of the conductance of rat TRPV1 by H+ (Liu et al., 2009). Cryo-EM structures of TRPV1 (Cao et al., 2013; Liao et al., 2013) revealed that E636 is located on the pore helix, whereas D646 is located at the entrance of the ion selectivity filter (Fig. 12 A). The distance between E636 residues on subunits across the pore is 21.3 Å in the closed state and 22.5 Å in the toxin-bound state (measured from the hydroxyl group), and the corresponding distance between D646 residues is 14.7 Å in the closed state and 8.6 Å in the toxin-bound state. To determine whether one of these two titratable residues may form the binding site for H+ inhibition of mouse TRPV1 channel used in this study, we examined E637Q and D647N singular mutations as well as E637Q/D647N double mutation.
Figure 12.

Concentration- and voltage-dependent inhibition of D647N current by H+. (A) The cryo-EM pore structures of TRPV1 in the closed (left; Protein Data Bank accession no. 3J5P) and double-knot toxin DkTx–bound (right; Protein Data Bank accession no. 3J5Q) states. Only two of the four subunits are shown. Dotted lines indicate the distances between the hydroxyl groups of E637 and D647. (B) Representative current traces induced by 10 µM capsaicin from whole-cell recording at ±80 mV in Solution IV containing different concentrations of H+ (left) and the percentage of current inhibition (right). Dotted line indicates zero current level. n = 4. Error bars indicate mean ± SEM.
The H+ effect on permeation was measured from capsaicin-activated current in the presence of varying concentrations of H+ at both negative and positive voltages. We observed that capsaicin-induced current from D647N was inhibited effectively by H+ at 80 mV and even more effectively at −80 mV, like the wild-type channel (Fig. 12 B). For example, at pH 5.0, H+ inhibited the wild-type channel current by 26.1 ± 3.0% (n = 5) at 80 mV and 66.6 ± 3.3% (n = 5) at −80 mV (Fig. 6); under the same condition, inhibition of D647N current was 16.8 ± 0.7% (n = 4) at 80 mV and 44.1 ± 5.6% (n = 4) at −80 mV (Fig. 12 B). The inhibition was H+ concentration dependent, ranging from 1.7 ± 0.2% (n = 4) at pH 6.5 to 25.1 ± 1.3% (n = 4) at pH 4.5 (measured at 80 mV), which was also like the wild-type channel. Therefore, both the concentration-dependent property and the voltage-dependent property of H+ inhibition were preserved in D647N mutant channel.
Similar observations were made from E637Q. This mutant channel exhibited an interesting change in capsaicin-induced activation, in that the activation process was extremely slow (Fig. 13 A). Nonetheless, current from E637Q was effectively inhibited by H+ in a concentration- and voltage-dependent manner. The percentage of inhibition was again very close to that of the wild-type channel at all H+ concentrations and voltages. Furthermore, we found that when both E637 and D647 were mutated to a nontitratable residue, the mutant channel was still sensitive to H+ inhibition. The E637Q/D647N double mutant inherited the slow capsaicin activation behavior of E637Q (Fig. 13 B). We found that, like in the wild-type channel, E637Q, and D647N, H+ effectively inhibited the current from E637Q/D647N double mutant in a concentration- and voltage-dependent manner.
Figure 13.

Mutations E637Q and E637Q/D647N do not prevent H+ inhibition. Representative current traces of E637Q (A) or E637Q/D647N (B) from whole-cell recording in Solution IV at ±80 mV (left) and the percentage of current inhibition (right). Dotted lines indicate zero current level. n = 6 each. Error bars indicate mean ± SEM.
To further examine the property of H+ inhibition in the mutant channels, we estimated the apparent pKa values for H+ inhibition from the concentration dependence in reducing the capsaicin-induced currents, which were compared with that of the wild-type channel. As summarized in Fig. 14, all four channel types exhibited comparable voltage dependence in that inhibition was much stronger at −80 mV than at 80 mV. All channel types exhibited H+ concentration dependence in inhibition, with the estimated pKa values at 80 mV varied from 5.6 (wild type) to 4.9 (D647N and E637Q/D647N) and 4.8 (E637Q). Similar variability was observed at −80 mV. These results indicate that mutations at these two positions affected the binding affinity of H+. Nonetheless, titration of the two acidic residues is not required for inhibition of permeation.
Figure 14.

Summary of H+-dependent inhibition of currents from wild type, E637Q, D647N, and E637Q/D647N in whole-cell configuration using Solution IV at 80 mV (A) or −80 mV (B). Curves represent fits of a Hill equation with the following EC50 and slope factor: (A) Wild type, 2.3 µM, pH 5.6, and 0.9; E637Q, 16.8 µM, pH 4.8, and 0.6; D647N, 34.6 µM, pH 4.9, and 0.5; E637Q/D647N, 13.2 µM, pH 4.9, and 0.8. (B) Wild type, 1.7 µM, pH 5.8, and 1.0; E637Q, 0.5 µM, pH 6.3, and 0.2; D647N, 1.4 µM, pH 5.9, and 0.4; E637Q/D647N, 37.7 µM, pH 4.4, and 0.4. n = 4–6. Error bars indicate mean ± SEM.
To further confirm that H+ could indeed inhibit current from the E637Q/D647N double mutant, we performed single-channel recordings using Na+ as the major permeant ion (Fig. 15). We found that the double mutation reduced single-channel conductance to 53.5 ± 0.7 pS (P < 0.001 compared with wild type; n = 5). A similar reduction in single-channel conductance was also observed in the previous study using K+ as the major permeant ion (Liu et al., 2009). Nonetheless, when the recording pH level was dropped to 5.0, we found that the single-channel conductance of the E637Q/D647N double mutant was significantly reduced, to 41.5 ± 1.6 pS (P < 0.005; n = 4).
Figure 15.

Single-channel recordings confirm H+ inhibition of E637Q/D647N mutation channels. (A) Representative single-channel current traces recorded in outside-out configuration at 80 mV in Solution IV at the indicated extracellular pH levels in the presence or absence of capsaicin. The presented current traces were processed by a digital filter to a final cutoff frequency of 0.41 kHz. (B) All-point histograms of single-channel events at the indicated pH levels. The superimposed curve represents the fit of a double-Gaussian function. n = 3–5.
Highly variable TRPV1 responses to H+ under different conditions
The opposing effects of H+ on gating and permeation greatly complicate the response of TRPV1 to changes in extracellular H+ concentration. To better appreciate how this complexity may affect the behavior of sensory neurons, we recorded responses of the exogenously expressed TRPV1 to varying pH levels at two conditions (Fig. 16). In the first condition, a sub-activating concentration of capsaicin (10 nM) was present to roughly mimic a resting neuron at physiological temperature (Fig. 16 A). We observed that increasing H+ concentration first elicited a larger and larger steady-state current response. However, as the H+ concentration kept increasing, the steady-state current started to decline; the OFF response, on the other hand, increased further and then remained at the maximal level. Varying the recording voltage would further change the shape of these current responses. Nonetheless, the overall effect of H+ was to elicit an excitatory current.
Figure 16.

Variable TRPV1 responses to H+ under different conditions. Representative outside-out patch current traces at 40 mV in Solution I containing 10 nM (A) or 3 µM (B) capsaicin in response to H+ at different concentrations. The trend lines indicate gating (red) and permeation (blue) effects of H+ on TRPV1.
When we switched to another experimental condition by using a saturating concentration (3 µM) of capsaicin to mimic an already excited neuron, we observed very different current responses (Fig. 16 B). Increasing H+ concentration was seen to gradually reduce the steady-state current. Upon removal of the H+-containing solution, the OFF response brought the current back to the maximal level. In other words, under this condition, the presence of H+ exhibits an inhibitory effect on the excitatory current, whereas the removal of H+ produces a stronger transient excitatory current. These observations highlight that under physiological or pathological conditions, a cell may respond to changes in extracellular H+ differently depending on the state of the cell membrane and the state of the TRPV1 channel. Full appreciation of the nature of these complex responses requires detailed molecular understanding of interactions between H+ ions and the channel.
DISCUSSION
Results from the present study demonstrate that extracellular H+ is a dual regulator for TRPV1: it potentiates activation gating but strongly inhibits permeation of the open channel. A combination of these two dynamic effects produces a prominent OFF response that phenotypically resembles the transient current of hERG channels (Bers, 2001). For hERG channels, equilibria among the closed, open, and inactivated states are strongly voltage dependent. The transient current is produced by a unique combination of these voltage-dependent transitions between the conducting (open) state and two nonconducting (closed and inactivated) states (Smith et al., 1996). Depolarization moves the channel quickly from the closed state through the open state to the inactivated state; repolarization brings the channel out of the inactivated state and generates a transient current that carries out the channel’s critical roles, such as controlling the duration of cardiac action potential. For TRPV1, the transient OFF response is produced by a combination of a H+-potentiating effect on gating and an inhibitory effect on permeation. The relative contribution of H+-induced steady-state current and the OFF response current to membrane excitability of TRPV1-expressing neurons is expected to depend on both the state of the cell and the state of the channel, as demonstrated in Fig. 16. For example, given the strong voltage dependence of H+ inhibition and weak voltage dependence of TRPV1 gating, a neuron in the resting state at body temperature may respond to a brief H+ challenge mostly by the OFF response. The anticipated complex current response needs to be carefully considered in the situation of stroke and subsequent reperfusion; indeed, reports show that reperfusion injury of ischemic tissues constitutes a major medical issue (Carden and Granger, 2000; Wu and Grotta, 2013). Consistent with observations of the present study, both potentiating and inhibitory effects of H+ were observed on currents recorded from capsaicin-sensitive trigeminal neurons (Baumann and Martenson, 2000).
In terms of molecular mechanism, results of the present study suggest that the inhibitory effect of H+ on TRPV1 ion permeation is produced by H+ entering the ion permeation pore, moving about halfway into the transmembrane electric field where it interferes with permeant ions. These observations argue that binding of H+ is likely supported by ion coordination inside or very near the ion permeation pathway. In agreement with this view, we observed that different permeant ions could also affect H+ inhibition. For BK channels, it was found that H+ inhibits single-channel conductance by directly competing with K+ for the same permeant ion–binding site(s) (Brelidze and Magleby, 2004). A previous study investigating this issue in TRPV1 showed that increasing K+ concentration could not prevent H+ inhibition, arguing against direct competition between H+ and permeant ion for the same binding site (Liu et al., 2009). Given all these observations, it seems that H+ may bind to sites close to permeant ions to exert an electrostatic influence. One such example is found in cyclic nucleotide–gated channels, for which binding of H+ to glutamate residues at the pore entrance (with a pKa of 7.6) causes reductions in single-channel conductance (Root and MacKinnon, 1994). Because both binding and unbinding are at a millisecond time scale with 2H2O in the solution, switching between multiple conductance levels as a result of titration of the glutamates could be directly observed with single-channel recording. If a similar process occurs in TRPV1, H+ unbinding (and binding) could be extremely rapid—beyond the temporal resolution of the patch-clamp recording—to yield reduced current amplitudes that reflect the weighted average of currents in the H+-bound and unbound states. Alternatively, H+ unbinding could be extremely slow to keep the conductance at a stable reduced level. The latter case can happen if the H+-bound state is stabilized by electrostatic interactions or hydrogen bond formation. Indeed, in the cryo-EM structures (Cao et al., 2013; Liao et al., 2013), multiple titratable residues could be seen in the vicinity of the permeation pathway. Although we were originally attracted to this hypothesis, results from the present study showed that mutations of two negatively charged residues closest to the permeation pathway, E637 and D647N, did not eliminate the inhibition effect of H+. Therefore, it remains to be determined where H+ binds to produce current inhibition.
In the present study, we found that the E637Q/D647N double-mutant channel could no longer be directly activated by H+ (not depicted), but the channel conductance could still be inhibited by H+. Lack of H+-induced activation was also observed in rat TRPV1 channel when the two residues were mutated (Liu et al., 2009). In both cases, capsaicin was used to open the channel so that the H+ effect on permeation could be examined. It was further observed that mutation to a nontitratable residue (V538L in rat TRPV1) also prevented H+ gating without eliminating H+ inhibition (Liu et al., 2009). These observations, together with our observation that intracellular H+ could also inhibit permeation, argue that H+ inhibition of conductance and H+ potentiation of activation gating are separate events. Distinct mechanistic bases for H+ effects on gating and permeation are the reason that the channel exhibits the OFF response. This conclusion will provide important guidance for future study of H+ activation of TRPV1.
Because TRPV1 is a polymodal nociceptor, the gating effect of H+ is expected to be substantially affected by the presence of other activation stimuli. Results from the present study demonstrate that the permeation effect of H+ further greatly complicates the situation. Not only is the steady-state response reduced by the H+ inhibitory effect in a concentration- and voltage-dependent manner, but removal of H+ could yield a large transient current that is again sensitive to H+ concentration and voltage. The strong voltage dependence of H+ inhibition is expected to substantially affect how a sensory neuron or other TRPV1-expressing cells respond to H+, which will depend on whether the cell is in the resting state or excited state. The present study focused on the permeation effect of H+ on TRPV1 current. Because the OFF response is produced by a combination of allosteric gating and inhibition of permeation, future study is needed to better understand the characteristics and molecular mechanism of the H+-gating effect.
Supplementary Material
Acknowledgments
We thank our laboratory members for assistance and discussions, in particular Fan Yang and Xu Cao who conducted initial tests of proton on TRPV1. We also thank Jon Sack for comments on the manuscript.
This study was supported by funding from National Institutes of Health (R01NS072377 to J. Zheng) and University of California Davis Superfund Research Program (to B.H. Lee).
The authors declare no competing financial interests.
Richard W. Aldrich served as editor.
References
- Ahern G.P., Brooks I.M., Miyares R.L., and Wang X.B.. 2005. Extracellular cations sensitize and gate capsaicin receptor TRPV1 modulating pain signaling. J. Neurosci. 25:5109–5116. 10.1523/JNEUROSCI.0237-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aneiros E., Cao L., Papakosta M., Stevens E.B., Phillips S., and Grimm C.. 2011. The biophysical and molecular basis of TRPV1 proton gating. EMBO J. 30:994–1002. 10.1038/emboj.2011.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann T.K., and Martenson M.E.. 2000. Extracellular protons both increase the activity and reduce the conductance of capsaicin-gated channels. J. Neurosci. 20:RC80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers D.M. 2001. Excitation-contraction coupling and cardiac contractile force. Kluwer Academic Publishers, Norwell, MA: 427 pp 10.1007/978-94-010-0658-3 [DOI] [Google Scholar]
- Bevan S., and Geppetti P.. 1994. Protons: small stimulants of capsaicin-sensitive sensory nerves. Trends Neurosci. 17:509–512. 10.1016/0166-2236(94)90149-X [DOI] [PubMed] [Google Scholar]
- Boukalova S., Teisinger J., and Vlachova V.. 2013. Protons stabilize the closed conformation of gain-of-function mutants of the TRPV1 channel. Biochim. Biophys. Acta. 1833:520–528. 10.1016/j.bbamcr.2012.11.017 [DOI] [PubMed] [Google Scholar]
- Brelidze T.I., and Magleby K.L.. 2004. Protons block BK channels by competitive inhibition with K+ and contribute to the limits of unitary currents at high voltages. J. Gen. Physiol. 123:305–319. 10.1085/jgp.200308951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao E., Liao M., Cheng Y., and Julius D.. 2013. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature. 504:113–118. 10.1038/nature12823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X., Ma L., Yang F., Wang K., and Zheng J.. 2014. Divalent cations potentiate TRPV1 channel by lowering the heat activation threshold. J. Gen. Physiol. 143:75–90. 10.1085/jgp.201311025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carden D.L., and Granger D.N.. 2000. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 190:255–266. 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- Caterina M.J., Schumacher M.A., Tominaga M., Rosen T.A., Levine J.D., and Julius D.. 1997. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 389:816–824. 10.1038/39807 [DOI] [PubMed] [Google Scholar]
- Cheng W., Yang F., Takanishi C.L., and Zheng J.. 2007. Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J. Gen. Physiol. 129:191–207. 10.1085/jgp.200709731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham D.E. 2003. TRP channels as cellular sensors. Nature. 426:517–524. 10.1038/nature02196 [DOI] [PubMed] [Google Scholar]
- Hoover D.B. 1987. Effects of capsaicin on release of substance P-like immunoreactivity and physiological parameters in isolated perfused guinea-pig heart. Eur. J. Pharmacol. 141:489–492. 10.1016/0014-2999(87)90571-1 [DOI] [PubMed] [Google Scholar]
- Hu C.P., Xiao L., Deng H.W., and Li Y.J.. 2002. The cardioprotection of rutaecarpine is mediated by endogenous calcitonin related-gene peptide through activation of vanilloid receptors in guinea-pig hearts. Planta Med. 68:705–709. 10.1055/s-2002-33794 [DOI] [PubMed] [Google Scholar]
- Jordt S.E., and Julius D.. 2002. Molecular basis for species-specific sensitivity to “hot” chili peppers. Cell. 108:421–430. 10.1016/S0092-8674(02)00637-2 [DOI] [PubMed] [Google Scholar]
- Jordt S.E., Tominaga M., and Julius D.. 2000. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. USA. 97:8134–8139. 10.1073/pnas.100129497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D., and Basbaum A.I.. 2001. Molecular mechanisms of nociception. Nature. 413:203–210. 10.1038/35093019 [DOI] [PubMed] [Google Scholar]
- Liao M., Cao E., Julius D., and Cheng Y.. 2013. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504:107–112. 10.1038/nature12822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Yao J., Wang Y., Li H., and Qin F.. 2009. Proton inhibition of unitary currents of vanilloid receptors. J. Gen. Physiol. 134:243–258. 10.1085/jgp.200910255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S.B., Tao X., Campbell E.B., and MacKinnon R.. 2007. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 450:376–382. 10.1038/nature06265 [DOI] [PubMed] [Google Scholar]
- Oroszi G., Szilvassy Z., Nemeth J., Ferdinandy P., Szolcsanyi J., and Tosaki A.. 1999. Interaction between capsaicin and nitrate tolerance in isolated guinea-pig heart. Eur. J. Pharmacol. 368:R1–R3. 10.1016/S0014-2999(99)00056-4 [DOI] [PubMed] [Google Scholar]
- Pan H.L., Longhurst J.C., Eisenach J.C., and Chen S.R.. 1999. Role of protons in activation of cardiac sympathetic C-fibre afferents during ischaemia in cats. J. Physiol. 518:857–866. 10.1111/j.1469-7793.1999.0857p.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root M.J., and MacKinnon R.. 1994. Two identical noninteracting sites in an ion channel revealed by proton transfer. Science. 265:1852–1856. 10.1126/science.7522344 [DOI] [PubMed] [Google Scholar]
- Ryu S., Liu B., and Qin F.. 2003. Low pH potentiates both capsaicin binding and channel gating of VR1 receptors. J. Gen. Physiol. 122:45–61. 10.1085/jgp.200308847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.L., Baukrowitz T., and Yellen G.. 1996. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 379:833–836. 10.1038/379833a0 [DOI] [PubMed] [Google Scholar]
- Stevens C.R., Williams R.B., Farrell A.J., and Blake D.R.. 1991. Hypoxia and inflammatory synovitis: observations and speculation. Ann. Rheum. Dis. 50:124–132. 10.1136/ard.50.2.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M., Caterina M.J., Malmberg A.B., Rosen T.A., Gilbert H., Skinner K., Raumann B.E., Basbaum A.I., and Julius D.. 1998. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 21:531–543. 10.1016/S0896-6273(00)80564-4 [DOI] [PubMed] [Google Scholar]
- Wang S., Poon K., Oswald R.E., and Chuang H.H.. 2010. Distinct modulations of human capsaicin receptor by protons and magnesium through different domains. J. Biol. Chem. 285:11547–11556. 10.1074/jbc.M109.058727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch J.M., Simon S.A., and Reinhart P.H.. 2000. The activation mechanism of rat vanilloid receptor 1 by capsaicin involves the pore domain and differs from the activation by either acid or heat. Proc. Natl. Acad. Sci. USA. 97:13889–13894. 10.1073/pnas.230146497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.C., and Grotta J.C.. 2013. Hypothermia for acute ischaemic stroke. Lancet Neurol. 12:275–284. 10.1016/S1474-4422(13)70013-9 [DOI] [PubMed] [Google Scholar]
- Yang F., Ma L., Cao X., Wang K., and Zheng J.. 2014. Divalent cations activate TRPV1 through promoting conformational change of the extracellular region. J. Gen. Physiol. 143:91–103. 10.1085/jgp.201311024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F., Xiao X., Cheng W., Yang W., Yu P., Song Z., Yarov-Yarovoy V., and Zheng J.. 2015. Structural mechanism underlying capsaicin binding and activation of the TRPV1 ion channel. Nat. Chem. Biol. 11:518–524. 10.1038/nchembio.1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. 2013. Molecular mechanism of TRP channels. Compr Physiol. 3:221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., and Ma L.. 2014. Structure and function of the thermoTRP channel pore. Curr. Top. Membr. 74:233–257. [DOI] [PubMed] [Google Scholar]
- Zheng J., and Sigworth F.J.. 1997. Selectivity changes during activation of mutant Shaker potassium channels. J. Gen. Physiol. 110:101–117. 10.1085/jgp.110.2.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.