

Abstract
Individuals presenting with primary hyperparathyroidism (PHPT) at a young age commonly have an underlying germline gene mutation in one of the following genes: MEN1, CASR, or CDC73. A small number of families with primary hyperparathyroidism have been identified with germline mutations in CDKN1B and those patients with primary hyperparathyroidism have almost exclusively been women who present in middle age suggesting that the age of onset of PHPT in MEN4 may be later than that of MEN1. We present a case of apparently sporadic PHPT presenting in adolescence with single gland disease associated with a novel CDKN1B germline mutation (heterozygote for a missense mutation in exon 1 of the CDKN1B gene (c.378G>C) (p.E126D)). The implication from this case is that CDKN1B germline mutations may be associated with PHPT at an earlier age than previously thought.
1. Introduction
Primary hyperparathyroidism (PHPT) is a relatively common endocrine disorder with a prevalence of approximately 6.7/1000 in the adult population [1]. PHPT is more common in women and is typically diagnosed between the ages of 40 and 75 years [1, 2]. Most cases of PHPT are sporadic, that is, not familial or part of a syndrome. Primary hyperparathyroidism presenting at a young age is uncommon and needs to be differentiated from familial hypocalciuric hypercalcaemia (FHH) in order to avoid unnecessary surgery for the latter disorder. Young patients with PHPT are more likely to have an underlying germline mutation in genes such as MEN1, CASR, RET, or CDC73 [3]. Germline mutations in cyclin-dependent kinase inhibitor 1B (CDKN1B) have also recently been identified to be associated with a Multiple Endocrine Neoplasia (MEN) syndrome which may include PHPT, termed MEN4 (OMIM number 610755) [4]. In MEN1 syndrome, tumours occur in the parathyroid glands, pancreas, and pituitary and less commonly elsewhere, whereas in the very rare MEN4 syndrome a broader spectrum of organ pathology including PHPT, pituitary, and pancreatic tumours occurs, with neuroendocrine tumours also described at a variety of other sites (e.g., cervix, bronchus, and stomach) [5–9]. CDKN1B encodes p27(kip1), a cyclin-dependent kinase 2 inhibitor involved with the control of the cell cycle at G1 [10]. To date, only a small number of families have been identified with germline mutations in CDKN1B and of those cohorts, patients with primary hyperparathyroidism have almost exclusively been women who present in middle age [4–9, 11, 12]. This suggests that the age of onset of PHPT in MEN4 may be later than that of MEN1 [5]. The questions are raised as to at what age screening for PHPT in CDKN1B carriers should start and whether assessment of germline mutations in CDKN1B should be considered in patients with familial or young onset PHPT in the absence of other gene mutations predisposing to PHPT.
We report a case of primary hyperparathyroidism presenting with renal stones at age 15 associated with a novel germline heterozygous missense mutation in CDKN1B.
2. Case Presentation
A previously fit and well 15-year-old girl presented with recurrent renal calculi. Her serum calcium was elevated at 2.9 mmol/L (reference range (RR) 2.1–2.55 mmol/L), with a reduced phosphate of 0.63 mmol/L (RR 0.7–1.5 mmol/L) and increased PTH at 11.6 pmol/L (RR 1.3–6.8 pmol/L). Twenty-four-hour urine calcium was elevated at 16.3 mmol/d (RR 2.5–7.5 mmol/d) and urine calcium/creatinine clearance ratio was 0.025, consistent with primary hyperparathyroidism. There was no clinical or biochemical evidence of any other endocrinopathy. There was no family history of calcium or other endocrine disorders and both of her parents had a normal serum calcium level.
Initial sestamibi imaging suggested an enlarged left lower parathyroid gland but at surgery only brown fat was present in this location and four-gland exploration failed to identify a parathyroid adenoma or the left inferior parathyroid. The other parathyroid glands appeared normal. Additional imaging studies, including 4D CT scanning, repeat sestamibi, and MRI, were inconclusive. Selective venous sampling demonstrated markedly elevated levels of PTH in the thymic vein consistent with an adenoma in the anterior mediastinum. This was unable to be removed by a cervical approach and was resected via an upper hemisternotomy resulting in normalization of her calcium and parathyroid hormone status. Histology demonstrated a parathyroid adenoma surrounded by normal thymic parenchyma. Parafibromin staining of the adenoma was positive and PGP9.5 negative making a CDC73 germline mutation unlikely.
Despite the lack of a family history of calcium disorders, given her young age genetic testing was offered to determine whether there was a predisposing germline mutation. PCR and Sanger sequencing of leukocyte DNA for the MEN1, CASR (all coding regions and flanking intronic regions), and RET (exons 10–16 including splice junctions) genes did not identify a mutation. However she was found to be heterozygote for a novel missense mutation in exon 1 of the CDKN1B gene (c.378G>C) resulting in an amino acid substitution (p.E126D). Her mother (aged 46) and maternal grandfather (aged 74 years) also carried the same missense mutation but both were normocalcemic with normal PTH levels. The immunohistochemical staining pattern of the adenoma for p27 (mouse monoclonal antibody clone SX53G8, Cell Marque) demonstrated normal nuclear staining (Figure 1).
Figure 1.
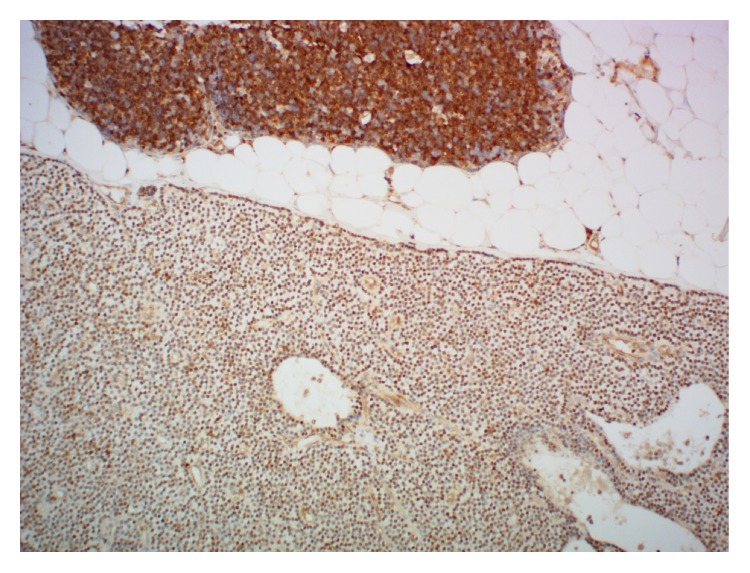
Immunohistochemical staining for p27 (original magnification ×100). Positive nuclear staining for parathyroid adenoma in lower half and positive nuclear staining for thymic lymphocytes in upper half.
3. Discussion
We present a case of apparently sporadic PHPT presenting in adolescence with single gland disease and a novel CDKN1B germline mutation.
Primary hyperparathyroidism is rare in children and adolescents. In a Scottish epidemiological study in which adults aged 20+ years with probable or definite PHPT were identified from community biochemistry data, based on an elevated serum calcium level with inappropriately normal or elevated PTH levels, only 1.4% of PHPT occurred in patients under the age of 30 years [1]. In that study, patients aged <20 years were excluded because of the increased likelihood of FHH; however, FHH was not excluded in those patients aged between 20 and 30 years [1]. As not all patients in that study had confirmed PHPT based on detailed clinical assessment and surgical outcomes, the true rate of definite PHPT is likely to be less than the 1.4% reported. In a large American study of surgically treated patients with PHPT only 0.86% (88/10190) of patients were aged <20 years [2]. A higher rate of 2.1% of patients <20 years with PHPT (21/1000) was found in another US study of surgically treated PHPT but the authors reported that this proportion was probably falsely increased due to referral bias [13]. As such, it is likely that if patients with FHH are excluded, <2% of cases of PHPT occur in patients <30 years. Interestingly, young patients who do develop PHPT are reported to be more liable to be symptomatic than their adult counterparts, that is, more likely to have renal stones, fatigue, depression, and weakness [13].
PHPT may be sporadic (accounting for the vast majority of cases) or occur as part of a familial syndrome associated with a germline mutation in one of the predisposing genes such as MEN1, RET, CASR, CDC73, or CDKN1B. Young onset PHPT has been reported to be more frequently associated with an underlying germline mutation [3]. In a recent study patients who had undergone parathyroid surgery and were aged <45 years at the time of surgery were offered germline mutation testing of MEN1, RET, CDC73, and CASR [3]. Of the 102 patients, 16 patients had familial PHPT identified either preoperatively or as part of the work-up (11 MEN1, 4 MEN2a, and 1 HPT-JT). Of the remaining 86 patients who had nonsyndromic, apparently sporadic disease 8 further patients were identified as having relevant germline mutations (4 MEN1, 3 CASR, and 1 CDC73). Overall, in that cohort of patients aged <45 years at the time of surgery, 24% of those tested had an underlying germline mutation identified [3]. Conversely, in a study from a different unit of surgically treated young PHPT patients aged <20 years, 18/21 had single gland disease and of the 3 patients with multigland disease only 1 was identified as having an underlying germline mutation (MEN1) [13]. However, it is unclear from this paper as to whether all patients underwent germline testing and which genes were assessed [13]. In a Northern Finnish cohort where testing for MEN1, CDC73, CASR, CDKN1B, and AIP genes was offered to all patients presenting with PHPT aged <40 years or with multigland or recurrent disease or family history of PHPT or MEN1 only 1/29 had a mutation detected (which was in MEN1) [14].
Guidelines from the Proceedings of the Fourth International Workshop on Asymptomatic PHPT recommend testing those who present at a young age (although the age is not specified), the presence of multigland disease, parathyroid carcinoma, or atypical adenoma, and those with a family history or evidence of syndromic disease [15]. In the absence of syndromic features the recommended guidelines for sequence of gene testing based on order of likely frequency are MEN1, CASR, AP2S1, GNA11, CDC73, and CDKN1A/1B/2C genes, RET, and PTH [15]. Screening is useful in identifying carriers who can continue to be monitored for both PHPT and other tumours associated with the syndrome for which they carry a gene mutation. Investigating family members for specific gene mutations also allows those who do not carry the germline mutation to be reassured and avoids the expense of ongoing assessment and testing of these individuals.
In our unit, due to financial constraint, in the absence of familial or syndromic features we offer genetic testing only to those with an onset of PHPT aged <30 years (MEN1, RET, CDKN1B, CASR, and CDC73 (with germline mutation testing for the latter being guided by parafibromin staining of the parathyroid lesion [16])). Until more clinical data is available to suggest a different approach, we also limit testing for AP2S1 and GNA11 to those with an FHH phenotype. This practice is likely to miss some cases in which the penetrance is low or if a detailed family history is not available.
Germline CDKN1B mutations are associated with MEN4 [4]. To date, only a few families have been reported [4–9, 11, 12]. Based on the limited published data, the penetrance of primary hyperparathyroidism in MEN4 is assumed to be fairly high, although there remains a lack of large well-documented families to confirm this concept. Interestingly, the diagnosis of primary hyperparathyroidism in MEN4 appears to occur later than MEN1 and predominantly in women with the average age reported being 56 years compared to 20–25 years for MEN1 [5]. In our case, we have identified early onset primary hyperparathyroidism, with its associated complications (renal stones) present at age 15 associated with a germline CDKN1B variant but no other features of MEN4. If the E126D CDKN1B variant is pathogenic, it would suggest that CDKN1B mutations may need to be considered in young patients presenting with PHPT when other mutations are not identified. Detailed assessment of the family we describe has not yet revealed any other endocrinopathies in affected members or their untested relatives suggesting that the disease penetrance for this particular missense mutation may be low.
The missense mutation identified in the proband (c.378G>C) has not previously been reported. The base change results in an amino acid substitution of glutamic acid to aspartic acid at position 126 (E126D) in exon 1. The region in which this change occurs is a highly conserved area in a c-Jun activation domain-binding protein-1 (JAB1) binding domain. JAB1 is important in shuttling p27 from the nucleus to the cytoplasm and in p27 degradation [17]. PolyPhen-2, a predictive software tool which predicts the possible impact of an amino acid substitution on the structure and function of a human gene [18], predicts the E126D variant as probably damaging (score 0.996, sensitivity 0.55, and specificity 0.98). In this case normal p27 immunohistochemical staining was present in the parathyroid adenoma. CDKN1B is somewhat atypical for a tumour suppressor gene as, rather than two “hits” being necessary for the development of disease [19], haploinsufficiency is thought to potentially be sufficient [20]. A limitation of this paper is that functional studies have not been performed to confirm that this variant is pathological; however despite extensive testing no other cause for the patient's PHPT was identified making it possible that this germline variant was a predisposing factor.
4. Conclusions
Young onset PHPT (aged <30 years) is uncommon and even in the absence of syndromic features and multigland disease an underlying germline mutation in one of the PHPT predisposing genes should be considered. This case suggests that CDKN1B mutations may be associated with early onset PHPT.
Acknowledgments
The authors would like to thank Associate Professor Anthony Gill for performing parafibromin staining of the parathyroid adenoma and Drs. Carolyn Tysoe and Martina Owens for performing genetic testing. Immunohistochemistry for p27 staining was funded by an Auckland University PBRF grant (awarded to Marianne S. Elston).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Yu N., Donnan P. T., Murphy M. J., Leese G. P. Epidemiology of primary hyperparathyroidism in Tayside, Scotland, UK. Clinical Endocrinology. 2009;71(4):485–493. doi: 10.1111/j.1365-2265.2008.03520.x. [DOI] [PubMed] [Google Scholar]
- 2.Miller B. S., Dimick J., Wainess R., Burney R. E. Age- and sex-related incidence of surgically treated primary hyperparathyroidism. World Journal of Surgery. 2008;32(5):795–799. doi: 10.1007/s00268-007-9427-2. [DOI] [PubMed] [Google Scholar]
- 3.Starker L. F., Åkerström T., Long W. D., et al. Frequent germ-line mutations of the MEN1, CASR, and HRPT2/CDC73 genes in young patients with clinically non-familial primary hyperparathyroidism. Hormones & Cancer. 2012;3(1-2):44–51. doi: 10.1007/s12672-011-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pellegata N. S., Quintanilla-Martinez L., Siggelkow H., et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(42):15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tonelli F., Giudici F., Giusti F., et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. European Journal of Endocrinology. 2014;171(2):K7–K17. doi: 10.1530/eje-14-0080. [DOI] [PubMed] [Google Scholar]
- 6.Georgitsi M., Raitila A., Karhu A., et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. Journal of Clinical Endocrinology and Metabolism. 2007;92(8):3321–3325. doi: 10.1210/jc.2006-2843. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal S. K., Mateo C. M., Marx S. J. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. Journal of Clinical Endocrinology and Metabolism. 2009;94(5):1826–1834. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molatore S., Marinoni I., Lee M., et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Human Mutation. 2010;31(11):E1825–E1835. doi: 10.1002/humu.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Occhi G., Regazzo D., Trivellin G., et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotyp. PLoS Genetics. 2013;9(3) doi: 10.1371/journal.pgen.1003350.e1003350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polyak K., Kato J.-Y., Solomon M. J., et al. P27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes and Development. 1994;8(1):9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 11.Costa-Guda J., Marinoni I., Molatore S., Pellegata N. S., Arnold A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. Journal of Clinical Endocrinology and Metabolism. 2011;96(4):E701–E706. doi: 10.1210/jc.2010-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malanga D., De Gisi S., Riccardi M., et al. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. European Journal of Endocrinology. 2012;166(3):551–560. doi: 10.1530/eje-11-0929. [DOI] [PubMed] [Google Scholar]
- 13.Pashtan I., Grogan R. H., Kaplan S. P., et al. Primary hyperparathyroidism in adolescents: the same but different. Pediatric Surgery International. 2013;29(3):275–279. doi: 10.1007/s00383-012-3222-3. [DOI] [PubMed] [Google Scholar]
- 14.Vierimaa O., Villablanca A., Alimov A., et al. Mutation analysis of MEN1, HRPT2, CASR, CDKN1B, and AIP genes in primary hyperparathyroidism patients with features of genetic predisposition. Journal of Endocrinological Investigation. 2009;32(6):512–518. doi: 10.1007/bf03346498. [DOI] [PubMed] [Google Scholar]
- 15.Eastell R., Brandi M. L., Costa A. G., D'Amour P., Shoback D. M., Thakker R. V. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the fourth international workshop. Journal of Clinical Endocrinology and Metabolism. 2014;99(10):3570–3579. doi: 10.1210/jc.2014-1414. [DOI] [PubMed] [Google Scholar]
- 16.Gill A. J., Clarkson A., Gimm O., et al. Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidism-jaw tumor (HPT-JT) syndrome-related adenomas from sporadic parathyroid adenomas and hyperplasias. The American Journal of Surgical Pathology. 2006;30(9):1140–1149. doi: 10.1097/01.pas.0000209827.39477.4f. [DOI] [PubMed] [Google Scholar]
- 17.Tomoda K., Kubota Y., Kato J.-Y. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398(6723):160–165. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- 18.Adzhubei I. A., Schmidt S., Peshkin L., et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knudson A. G., Jr. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fero M. L., Randel E., Gurley K. E., Roberts J. M., Kemp C. J. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396(6707):177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]