

Abstract
Mitochondrial dysfunction contributes to cell death after cerebral ischemia/reperfusion (I/R) injury. Cannabinoid CB1 receptor is expressed in neuronal mitochondrial membranes (mtCB1R) and involved in regulating mitochondrial functions under physiological conditions. However, whether mtCB1R affords neuroprotection against I/R injury remains unknown. We used mouse models of cerebral I/R, primary cultured hippocampal neurons exposed to oxygen-glucose deprivation/reoxygenation (OGD/R) and Ca2+-induced injury in purified neuronal mitochondria to investigate the role of mtCB1R in neuroprotection. Our results showed selective cell-permeant CB1 receptor agonist, arachidonyl-2-chloroethylamide (ACEA), significantly up-regulated the expression of mtCB1R protein in hippocampal neurons and tissue. In vitro, ACEA restored cell viability, inhibited generation of reactive oxygen species (ROS), decreased lactate dehydrogenase (LDH) release and reduced apoptosis, improved mitochondrial function. In vivo, ACEA ameliorated neurological scores, diminished the number of TUNEL-positive neurons and decreased the expression of cleaved caspase-3. However, ACEA-induced benefits were blocked by the selective cell-permeant CB1 receptor antagonist AM251, but just partially by the selective cell-impermeant CB1 receptor antagonist hemopressin. In purified neuronal mitochondria, mtCB1R activation attenuated Ca2+-induced mitochondrial injury. In conclusion, mtCB1R is involved in ACEA-induced protective effects on neurons and mitochondrial functions, suggesting mtCB1R may be a potential novel target for the treatment of brain ischemic injury.
Cardiac arrest-induced neurologic injury is a vital cause of death among the patients with a successful cardiopulmonary resuscitation1. American Heart Association and related organizations have made long-term efforts to improve resuscitation guidelines; however, the hospital mortality remains proximate 70% in patients resuscitated successfully2. Unfortunately, about 2/3 of the survivors may have moderate to severe cognitive deficits3. Therefore, searching for effective therapeutics which can alleviate neurologic injury induced by cerebral I/R is of great importance.
Mitochondria are cellular energy factories, participating in oxidative stress, calcium homeostasis, generation of reactive oxygen species (ROS) and programmed cell death, which are crucial in regulating brain functions4,5. In cerebral I/R injury, mitochondrial dysfunction leads to neuronal death6. More importantly, several studies showed that mitochondrial damage occurs within 4–6 hours after the onset of cerebral ischemia, which provides a promising strategy for targeting mitochondria to develop cerebral I/R injury therapeutics7,8,9. However, approaches that regulate mitochondria effectively after cerebral I/R injury are still poorly understood.
Cannabinoid CB1 receptor is a G protein-coupled receptor expressed mainly in neuronal plasma membrane, modulating neuronal metabolism, activity and functions strictly10,11,12. Early studies indicated that the marijuana-derivative cannabis Δ9-tetrahydrocannabinol (THC) could induce neuroprotection by up-regulating CB1 receptor and maintain mitochondrial function10,13,14. In addition, recent evidence also showed that CB1 receptor can adjust mitochondrial biogenesis in non-neural tissues15,16. However, THC may be of limited use due to its side effects, including addiction, psychoactivity, tolerance and cytotoxicity17,18. In 2012, Marsicano et al. found that CB1 receptor is also expressed in neuronal mitochondrial membranes (mtCB1R), and up-regulation of which regulates mitochondrial respiration and energy metabolism under physiological conditions19. However, whether up-regulation of mtCB1R modulates mitochondrial functions and exerts neuroprotection against I/R injury is still unknown.
In the present study, we used mouse models of global cerebral I/R, primary cultured hippocampal neurons OGD/R and Ca2+-induced injury in purified intact mitochondria from hippocampal neurons to investigate the role of mtCB1R in neuroprotection and improvement of mitochondrial function.
Results
ACEA-induced up-regulation of mtCB1R was blocked by AM251, but not by hemopressin
First, we attempted to detect the expression of mtCB1R at different time points after cerebral I/R. Mitochondria protein were isolated at 2 h, 6 h, 24 h, 48 h and 72 h after reperfusion respectively (Fig. 1). We observed that mtCB1R expression was significantly increased at 2 h after reperfusion compared with Sham group (P = 0.046) and ACEA increased the mtCB1R expression more obviously (P < 0.001). ACEA also up-regulated the mtCB1R expression at 6 h after reperfusion compared with Sham group (P < 0.001). However, there was no significant difference in mtCB1R protein expression at the other time points.
Figure 1. Expression of mtCB1R protein in mouse hippocampus at different time points after reperfusion.
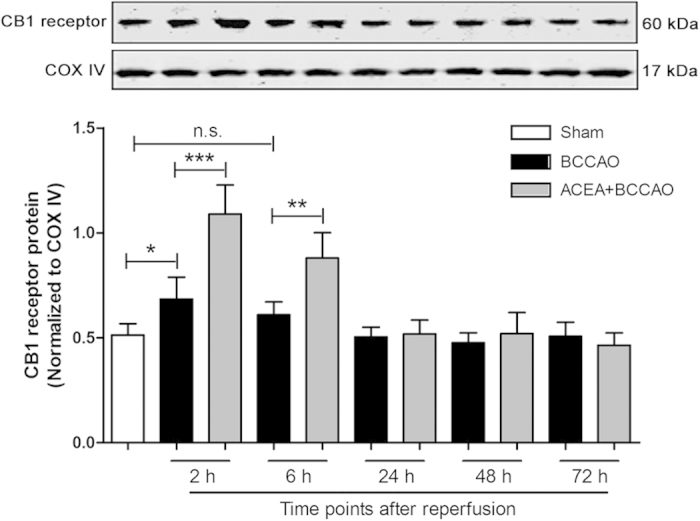
Western blot showing mtCB1R protein expression at 2 h, 6 h, 24 h, 48 h and 72 h after reperfusion respectively. Data represent mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; n.s.: no significance. COX IV: cytochrome c oxidase; BCCAO: bilateral common carotid artery occlusion.
Subsequently, we investigated the effect of ACEA on mtCB1R expression. Previous studies have reported that cannabinoid could increase the expression of CB1 receptor protein and the increased CB1 receptor was functional20,21. In the present study, we isolated mitochondria protein at 2 h after ACEA administration to detect the expression of mtCB1R. As shown in Fig. 2A,C, mtCB1R expression was significantly increased at 2 h after ACEA treatment compared with Control group in hippocampal neurons and mouse hippocampus (P < 0.001). The effects were reversed by the cell-permeant CB1 receptor antagonist, AM251 (P < 0.001), but not by the cell-impermeant CB1 receptor antagonist, hemopressin (P = 0.24 and P = 0.105). There were no differences in mtCB1R expression between Control group and Vehicle group (P = 0.333 and P = 0.18). In the remaining samples without mitochondria protein, CB1 receptor expression increased just in ACEA group (Fig. 2B,D, P < 0.001), but not in AM251 + ACEA, Hemo + ACEA or Vehicle groups, compared with Control group (P = 0.449 and 0.125, P = 0.735 and 0.979, P = 0.237 and 0.054, respectively).
Figure 2. Expression of CB1 receptor protein in primary cultured hippocampal neurons and mouse hippocampus.

(A, C) Western blot showing mtCB1R protein expression at 2 h after administration in vitro and in vivo (n = 5). (B, D) Western blot showing CB1 protein expression in the remaining samples without mitochondria protein at 2 h after administration in vitro and in vivo (n = 5). (E) Immunogold electron microscopy showing mtCB1R protein expression. (F) Density of mtCB1R immunoparticles were calculated per area of mitochondria (n = 3). Scale bars = 200 nm. Data represent mean ± SD. **P < 0.01; ***P < 0.001; n.s.: no significance. COX IV: cytochrome c oxidase; Hemo: hemopressin.
CB1 receptor immunoreactivity was detected by immunogold electron microscopy in the mouse CA1 hippocampal neurons (Fig. 2E). We observed intracellular CB1 receptor was expressed on the mitochondrial membrane, and the density of mtCB1R was significantly increased in ACEA and Hemo + ACEA groups compared with Control group (Fig. 2F, P < 0.001), but not in AM251 + ACEA and Vehicle groups (P = 0.424 and P = 0.603).
To detect the CB1 receptor expression after OGD/R or I/R, mitochondria were isolated at 2 h after reoxygenation or reperfusion to detect the expression of mtCB1R. As shown in Fig. 3A,C, mtCB1R expression was significantly increased in OGD group compared with Control group (P = 0.01). ACEA increased the mtCB1R expression further compared with OGD and BCCAO groups respectively (P = 0.002 and P < 0.001). The effects were abolished by AM251 (P < 0.001), but not by hemopressin (P = 0.575 and P = 0.47). In the remaining samples without mitochondria protein, CB1 receptor expression was also increased at 2 h after reoxygenation or reperfusion (Fig. 3B,D, P = 0.001 and P = 0.003), and ACEA increased the expression further (P = 0.014 and P = 0.001). While treatment with AM251 or hemopressin abolished the effects of ACEA (P < 0.001).
Figure 3. Expression of CB1 receptor protein in primary cultured hippocampal neurons at 2 h after reoxygenation and mouse hippocampus at 2 h after reperfusion.

(A, C) Western blot showing mtCB1R protein expression in vitro and in vivo (n = 5). (B, D) Western blot showing CB1 protein expression in the remaining samples without mitochondria protein in vitro and in vivo (n = 5). Data represent mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; n.s.: no significance. COX IV: cytochrome c oxidase; OGD: oxygen-glucose deprivation; Hemo: hemopressin; BCCAO: bilateral common carotid artery occlusion.
ACEA-induced cytoprotection against OGD/R injury was reversed by AM251, but just partially by hemopressin
Next, we asked whether up-regulation of mtCB1R could induce neuroprotection in vitro. To find a suitable ACEA concentration, hippocampal neurons were exposed to ACEA at 10 nM, 100 nM, 0.5 μM, 1 μM and 2 μM at the onset of reoxygenation. Cell viability was significantly increased in the presence of ACEA at 0.5 μM, 1 μM and 2 μM (Fig. 4A, P = 0.003, 0.001 and 0.002) and the dose of 1 μM was used in the subsequent experiments.
Figure 4. ACEA-induced cytoprotection against OGD/R injury in primary cultured hippocampal neurons.

(A) The effective dose of ACEA against OGD/R evaluated by WST-8 (n = 8). Cell injury was evaluated in terms of WST-8 cell viability (B), (n = 8), LDH release (C), (n = 6), apoptosis rate (D, E), (n = 5) and intracellular ROS level (F, G), (n = 6) at 24 h after reoxygenation. Scale bars = 20 μm. Data represent mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; n.s.: no significance. OGD/R: oxygen-glucose deprivation/reoxygenation; WST-8: 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt; Hemo: hemopressin; LDH: lactate dehydrogenase; ROS: reactive oxygen species.
The injury of neurons was assessed at 24 h after reoxygenation. The apoptotic rate of neurons was analyzed by flow cytometry and the lower right quadrant represented the percentage of early apoptotic cells. ACEA increased cell viability, attenuated LDH release, decreased apoptotic rate and inhibited generation of intracellular ROS in the cells exposed to OGD/R (Fig. 4B–G, P < 0.001). The protective effects of ACEA were abolished by the cell-permeant CB1 receptor antagonist, AM251 (P < 0.001, P < 0.001, P = 0.011 and P < 0.001), while not or just partially reversed by the cell-impermeant CB1 receptor antagonist, hemopressin. There were no statistical differences in the cell viability, LDH release, apoptotic rate or intracellular ROS between OGD and Vehicle + OGD groups (P = 0.502, P = 0.782, P = 0.063 and P = 0.892, respectively).
ACEA-induced neuroprotection against cerebral I/R injury was reversed by AM251, but just partially by hemopressin
Subsequently, we investigated the effect of mtCB1R up-regulation on neuroprotection in mice. All animals survived until the final neurological assessment at 72 h after reperfusion. The neurological scores in ACEA + BCCAO group were significantly higher than those of BCCAO group at 24, 48, and 72 h after reperfusion respectively (Fig. 5A, P < 0.001, P = 0.002 and P < 0.001). The cell-permeant CB1 receptor antagonist AM251 reversed the ACEA-induced benefits at the three time-points (P = 0.004, P = 0.001 and P < 0.001), while the cell-impermeant CB1 receptor antagonist hemopressin just partially reversed the benefits. No significant differences in the neurological scores were observed between BCCAO and Vehicle + BCCAO groups at the three time-points (P = 0.912, P = 0.796, P = 0.766, respectively).
Figure 5. ACEA-induced neuroprotection against cerebral ischemia/reperfusion injury in mice.

(A) Neurological assessment at 24 h, 48 h and 72 h after reperfusion (n = 13). Data represent the median. (B) Representative photomicrographs of TUNEL staining in hippocampal CA1 region. Scale bars = 50 μm. (C) Quantitative analysis of the number of TUNEL-positive cells in hippocampal CA1 region at 72 h after reperfusion (n = 5). (D) Western blot showing representative the expression of cleaved caspase-3 (Cl. Cas-3) protein in the hippocampus at 72 h after reperfusion (n = 5). Data represent mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001. BCCAO: bilateral common carotid artery occlusion; Hemo: hemopressin.
The number of TUNEL-positive neurons in the hippocampal CA1 region (Fig. 5B,C) and the expression of cleaved caspase-3 in the hippocampus (Fig. 5D) were significantly increased at 72 h after reperfusion in BCCAO group compared with that of Sham group (P < 0.001). ACEA diminished the number of TUNEL-positive neurons and decreased the expression of cleaved caspase-3 induced by BCCAO (P < 0.001). The benefits of ACEA were blocked by the cell-permeant CB1 receptor antagonist, AM251 (P < 0.001), but just partially by the cell-impermeant CB1 receptor antagonist, hemopressin. No significant differences were found in the number of TUNEL-positive neurons or the cleaved caspase-3 expression between BCCAO and Vehicle + BCCAO groups (P = 0.818, P = 0.461, respectively).
ACEA-induced improvement of mitochondrial function was blocked by AM251, but just partially by hemopressin
In order to determine whether ACEA-induced neuroprotection was accompanied by the improvement of mitochondrial function, we observed the mitochondrial ultrastructure in mouse CA1 hippocampal neurons, measured the activities of complexes I, II, and IV of mitochondrial electron transport chain and the mitochondrial membrane potential (MMP) in purified mitochondria from primary cultured hippocampal neurons.
The mitochondria in Sham group were elongated or round and had numerous cristae with parallel alignment. The outer and inner membranes of mitochondria were clearly distinguishable. Mitochondria from BCCAO, AM251 + ACEA + BCCAO and Vehicle + BCCAO groups showed swelling, vacuolization, disruption and loss of cristae. In ACEA + BCCAO and Hemo + ACEA + BCCAO groups, the mitochondrial structure was almost normal, but the cristae were slightly disrupted (Fig. 6).
Figure 6. Transmission electron micrographs showing mitochondrial ultrastructure.

Representative electron microphotographs show mitochondrial ultrastructure in mouse CA1 hippocampal neurons at 72 h after the reperfusion (n = 3). Mitochondrial ultrastructures are indicated by arrows. Scale bars = 200 nm. BCCAO: bilateral common carotid artery occlusion; Hemo: hemopressin.
At 2 and 24 h after reoxygenation (Fig. 7), the activities of complexes I and IV and the MMP showed significant reduction in OGD group, compared with Control group (2 h: P = 0.001, P < 0.001 and P < 0.001; 24 h: P < 0.001). And ACEA improved the mitochondrial functions significantly (2 h: P = 0.006, P = 0.017 and P = 0.001; 24 h: P < 0.001). The benefits of ACEA were completely reversed by the cell-permeant CB1 receptor antagonist, AM251 (2 h: P = 0.019, P = 0.012 and P = 0.006; 24 h: P < 0.001, P < 0.001 and P = 0.001), while not or just partially by the cell-impermeant CB1 receptor antagonist, hemopressin. No significant differences were observed between OGD and Vehicle + OGD groups (2 h: P = 0.788, P = 0.787 and P = 0.499; 24 h: P = 0.225, P = 0.652 and P = 0.565). The OGD-induced decrease in complex II activity was observed at 24 h after reoxygenation (Fig. 7F, P < 0.001), but not at 2 h (Fig. 7B, P = 0.102); however, ACEA did not alleviate the decrease of complex II activity (P = 0.288).
Figure 7. Effects of ACEA on mitochondrial function at 2 and 24 h after reoxygenation.

Neuronal mitochondria were purified at 2 and 24 h after reoxygenation respectively, and the activities of complexes I (A, E), II (B, F) and IV (C, G) were measured spectrophotometrically; the mitochondrial membrane potential (MMP) (D, H) was evaluated by fluorescence spectrophotometry. Data represent mean ± SD (n = 6). *P < 0.05; **P < 0.01; ***P < 0.001; n.s.: no significance. OGD: oxygen-glucose deprivation; Hemo: hemopressin.
ACEA attenuates Ca2+-induced mitochondrial dysfunction in purified intact mitochondria from hippocampal neurons
To further clarify the protection of mtCB1R activation against mitochondrial injury, intact mitochondria were obtained from normal hippocampal neurons to investigate the direct protective effects of ACEA-induced mtCB1R activation on mitochondrial functions19. Purified mitochondria were treated with a high concentration of Ca2+ to mimic the disorder of Ca2+ homeostasis, which is thought to be an important inducer in cerebral I/R injury22,23. Mitochondrial suspensions were divided into five groups (n = 6, Fig. 8A) and the purified mitochondria were round or oval with the morphological integrity of membrane and cristae structures (Fig. 8B). Except for the Control and the Ca2+ only groups, the other three groups of mitochondrial suspensions received 0.1 μM, 1 μM and 10 μM ACEA, respectively, at 30 min before Ca2 + administration. Ca2+ induced a noticeable mitochondrial swelling, as evidenced by the reduction in absorbance at 520 nm (Fig. 8C, P < 0.001), which was followed by a decline of MMP (Fig. 8D, P < 0.001). ACEA markedly prevented Ca2+-induced mitochondrial swelling (P < 0.001) and increased the MMP at the doses of 1 and 10 μM (P = 0.002 and 0.004).
Figure 8. Effects of ACEA on Ca2 + -induced mitochondrial injury in vitro.

(A) Protocol. (B) Representative electron micrograph of purified intact mitochondria from normal hippocampal neurons. Scale bars = 200 nm. Mitochondrial swelling was measured spectrophotometrically (C) and mitochondrial membrane potential (MMP) was evaluated by a fluorescence spectrophotometer (D). Data represent mean ± SD (n = 6). **P < 0.01; ***P < 0.001.
Discussion
In the present study, we found that mtCB1R expression was significantly up-regulated after the administration of ACEA, leading to neuroprotection against OGD/R and cerebral I/R injury. These benefits of ACEA were abolished by the selective cell-permeant CB1 receptor antagonist, AM251, but not or just partially by the selective cell-impermeant CB1 receptor antagonist, hemopressin. Furthermore, in purified intact mitochondria from normal hippocampal neurons, ACEA-induced mtCB1R activation markedly prevented Ca2+-induced mitochondrial swelling and increased the membrane potential. These findings suggest that up-regulation of mtCB1R is involved in ACEA-induced protective effects on neurons and mitochondrial functions, indicating mtCB1R could be a potential novel target for the treatment of brain ischemic injury.
Early studies presumed that CB1 receptor is mainly expressed in neuronal plasma membrane and CB1 receptor activation can induce neuroprotection against ischemic brain injury24,25. A recent research showed that CB1 receptor is also present in neuronal mitochondrial membrane, where the receptor directly regulates mitochondrial respiration and energy metabolism under physiological conditions19. However, whether the neuroprotection is mediated by mtCB1R is still not clear. After the administration of cell-permeant CB1 receptor agonist ACEA, a high-selective CB1 receptor agonist26,27, the expression of mtCB1R protein was significantly increased in hippocampal neurons and mouse hippocampus. By using immunogold electron microscopy, we found that ACEA obviously up-regulated mtCB1R expression on the mitochondrial membrane of mouse CA1 hippocampal neurons. Previous studies have showed that ACEA increased the expression of CB1 receptor protein and the increased CB1 receptor was functional20,21. Thus, it is generally considered that activation of CB1 receptor is positive correlation with the up-regulation of CB1 receptor expression. Moreover, we found that ACEA-induced mtCB1R protein up-regulation was reversed by the selective cell-permeant CB1 receptor antagonist AM25127,28, but not by the selective cell-impermeant CB1 receptor antagonist hemopressin29, suggesting mtCB1R may be up-regulated selectively.
We then investigated whether increased expression of mtCB1R can induce neuroprotection. C57BL/6 mouse has been reported to be highly prone to neuronal damage after cerebral ischemia because of the poorly developed anastomosis between the carotid artery (posterior communicating artery)30,31 and the bilateral common carotid artery occlusion (BCCAO) in C57BL/6 mouse was used widely as a model of transient global cerebral ischemia32,33. The BCCAO mimics the systemic decrease in cerebral blood flow, affects the entire brain especially in hippocampus34. During the operation procedure, regional cerebral blood flow (rCBF) measurement was taken and only the mice that had <10% residual cortical microperfusion after BCCAO were included for further study. In the present study, we found that ACEA protected hippocampal neurons exposed to OGD/R in vitro and improved the neurological scores and eliminated apoptosis induced by BCCAO in vivo. Moreover, the cell-permeant CB1 receptor antagonist AM251 reversed the ACEA-induced benefits, while the cell-impermeant CB1 receptor antagonist hemopressin just partially did. These findings suggest that ACEA-induced reduction of neuronal damage may be partially mediated by up-regulating mtCB1R.
Mitochondrial dysfunction is thought to be an inhibition of mitochondrial ATP production, leading to neuronal cell death after cerebral I/R injury6,7,35. Besides the early damage such as mitochondrial transition pore opening, calcium accumulation and decreased glucose utilization36, delayed mitochondrial damage may occur within 4–6 h after I/R and affect the activities of mitochondrial complexes, which results in permanent mitochondrial failure7,37. The mitochondrial electron transport chain (ETC) includes two sites for electron entry. The major one is complex I (NADH dehydrogenase) and the minor one is complex II (succinate dehydrogenase). Electrons offered to either of the two complexes are transferred to complex III (ubiquinol cytochrome c reductase) and subsequently transferred to complex IV (cytochrome c oxidase, CcO) which is the rate-limiting enzyme in electron transfer38. In the present study, we chose to detect the activities of three representative complexes, complexes I, II and IV, after OGD/R. The activities of complexes I and IV were significantly decreased at 2 and 24 h after reoxygenation, while the activity of complex II was reduced just at 24 h after reoxygenation; these findings are basically in accordance with a previous study39. ACEA ameliorated the reduction of complexes I and IV activities after OGD/R, but had no significant effect on improving complex II activity. MMP establishes a proton gradient across the mitochondrial membrane. During cerebral ischemia, mitochondrial membrane destabilization disturbs the proton gradient and results in the opening of mitochondrial permeablity transition pore (mPTP), releasing mitochondrial components to initiate the apoptotic cascade40,41. In this study, ACEA improved the mitochondrial membrane potential depolarization after OGD/R. The cell-permeant CB1 receptor antagonist AM251 significantly reversed the ACEA-induced benefits on mitochondrial complexes activities and membrane potential, while the cell-impermeant CB1 receptor antagonist hemopressin did not or just partially reversed the benefits, suggesting that ACEA-induced amelioration of mitochondrial functions may be mediated by up-regulating mtCB1R.
To further clarify the protection of mtCB1R against mitochondrial injury, we employed a Ca2+-induced injury model in purified intact mitochondria isolated from normal hippocampal neurons. Ca2+-induced mPTP opening has become a widely used model for assessing the effects of drugs on the mPTP42. Opening of mPTP can increase mitochondrial inner membrane permeability to solutes with a molecular mass less than 1.5 kDa, causing mitochondrial swelling, which can be evidenced by the reduction in absorbance at 520 nm43. In this study, Ca2+ induced a noticeable mitochondrial swelling which was followed by a decline of MMP. ACEA-induced mtCB1R activation markedly prevented Ca2+-induced mitochondrial swelling and increased the MMP.
However, some limitations of our research should be noted. First, because there are no selected mtCB1R knock-out mouse at present, we just used the regular methods to prove our hypothesis in this study. Second, the usage of cannabis to induce neuroprotection may be limited due to its side effects, including addiction, psychoactivity, tolerance and cytotoxicity17,18. In the present study, we up-regulated mtCB1R selectively and observed neuroprotection. However, whether the selective up-regulation of mtCB1R brings about fewer side effects still needs further investigation.
In conclusion, this study demonstrated that mtCB1R is involved in ACEA-induced protective effects on neurons and mitochondrial functions, suggesting the mtCB1R may be a potential novel target for the treatment of brain ischemic injury.
Methods
Animals and drugs
All animal procedures used in this study were approved by the Ethics Committee for Animal Experimentation of the Fourth Military Medical University and was conducted according to the Guidelines for Animal Experimentation of the Fourth Military Medical University (Xi’an, China). Male C57BL/6 mice (age: 12 weeks; weight: 20–24 g) were provided by the Experimental Animal Center of the Fourth Military Medical University. Mice were kept in cages under the following controlled conditions: a 12-hour light/dark cycle, 21 ± 2 °C, 60–70% humidity, and free access to rodent diet and water.
The selective CB1 receptor agonist, arachidonyl-2-chloroethylamide (ACEA), was dissolved in dimethylsulfoxide (DMSO). The drug was administered intraperitoneally at a dose of 1.5 mg/kg according to a previous study24. Hemopressin trifluoroacetate salt, a selective CB1 receptor antagonist, is unable to penetrate plasma membranes19,44. Therefore, hemopressin (dissolved in H2O) can be used as a cell-impermeant CB1 receptor antagonist, which is administered intraperitoneally at a dose of 1 mg/kg for mouse and 10 μM for cells19,45. All of these agents were obtained from Sigma-Aldrich (St. Louis, USA).
Another selective CB1 receptor antagonist, AM251, which is lipophilic and can penetrate plasma membranes, was dissolved in DMSO before use19. AM251 was used at doses of 1 mg/kg for mouse and 10 μM for cells based on previous studies46. The agent was obtained from Tocris Bioscience (Bristol, UK). 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8) was purchased from Sigma-Aldrich (St. Louis, USA). The lactate dehydrogenase (LDH) kit was provided by the Jiancheng Bioengineering Institute (Nanjing, China). The ROS detection reagents were obtained from Invitrogen (Carlsbad, USA).
Experimental protocols
For in vitro experiments, to determine the effect of drugs on CB1 receptor expression, five groups of hippocampal neurons (n = 5) were defined as follows: Control, ACEA, AM251 + ACEA, Hemo + ACEA, and Vehicle groups (Fig. 9A). The control cells were cultured in drug-free medium, while cells in other four groups received 1 μM ACEA, 10 μM AM251 + 1 μM ACEA, 10 μM hemopressin + 1 μM ACEA, and vehicle (5% DMSO, diluted by medium), respectively. Mitochondrial proteins were extracted from all groups of cells for Western blot analysis at 2 h after drugs administration. To evaluate the effect of drugs on cell injury and CB1 receptor expression, there were six groups of hippocampal neurons (Fig. 9B). Apart from Control group, other five groups of cells were exposed to OGD for 3 h. The cells in the ACEA + OGD, AM251 + ACEA + OGD, Hemo + ACEA + OGD, and Vehicle + OGD groups received 1 μM ACEA, 10 μM AM251 + 1 μM ACEA, 10 μM hemopressin + 1 μM ACEA, and vehicle (5% DMSO diluted by medium), respectively, at the onset of reoxygenation. Mitochondrial protein was extracted from neurons for Western blot analysis at 2 h after reoxygenation (n = 5). Cell injury was then evaluated in terms of WST-8 cell viability (n = 8), and LDH release (n = 6), apoptotic rate (n = 5) and intracellular ROS level (n = 6) at 24 h after reoxygenation. To detect the mitochondrial function after OGD/R, neuronal mitochondria were purified at 2 and 24 h after reoxygenation respectively (Fig. 9B, n = 6).
Figure 9. Experimental protocol and regional cerebral blood flow (rCBF).

(A) Protocol 1: Detection of CB1 receptor expression in vitro and in vivo. (B) Protocol 2: ACEA treatment in vitro. ①: at 2 h after reoxygenation, the neuronal mitochondria were purified for the detection of CB1 receptor expression and mitochondrial function assay; ②: at 24 h after reoxygenation, cell viability was measured and the neuronal mitochondria were purified for mitochondrial function assay. (C) Protocol 3: ACEA treatment in vivo. ①: at 2 h after reperfusion, mitochondria were isolated for the measurement of CB1 receptor expression; At 24 h (②) and 48 h (③) after reperfusion respectively, neurological tests were conducted; at 72 h (④) after reperfusion, neurological tests were performed, and neuronal apoptosis and apoptosis markers were assessed. (D) Changes of rCBF in C57BL/6 mice. The rCBF was reduced to <10% of the pre-ischemic baseline value immediately after ischemia and was maintained for 20 min. The rCBF returned to the pre-ischemic value at 5 min after reperfusion. OGD: oxygen-glucose deprivation; BCCAO: bilateral common carotid artery occlusion; Hemo: hemopressin.
For in vivo experiments, to detect the expression of mtCB1R at different time points after I/R, fifty-five C57BL/6 mice were randomly divided into three groups: Sham (n = 5), BCCAO (n = 25) and ACEA + BCCAO (n = 25) groups. Mitochondria protein were isolated at 2 h, 6 h, 24 h, 48 h and 72 h after reperfusion respectively and the expression of mtCB1R was assessed by Western blot analysis. To determine the effect of drugs on CB1 receptor expression, forty C57BL/6 mice were randomly divided into five groups (n = 8): Control, ACEA, AM251 + ACEA, Hemo + ACEA and Vehicle groups (Fig. 9A). Mice in the Control group were untreated, the other four groups received 1.5 mg/kg ACEA, 1 mg/kg AM251 + 1.5 mg/kg ACEA, 1 mg/kg hemopressin + 1.5 mg/kg ACEA, and 0.1 ml vehicle (5% DMSO diluted by saline), respectively. The expression of mtCB1R was assessed by Western blot analysis (n = 5) and immunocytochemistry for electron microscopy (n = 3) at 2 h after the administration. To evaluate the effect of drugs on cerebral I/R injury and CB1 receptor expression, one hundred and eight C57BL/6 mice were randomly divided into six groups (n = 18) (Fig. 9C). Except for Sham group, mice from the other five groups were subjected to bilateral common carotid artery occlusion (BCCAO) for 20 min and only the mice whose regional cerebral blood flow (rCBF) remained <10% of pre-ischemic baseline were included (Fig. 9D). The same surgical procedure was performed in Sham group; however, the occlusion was omitted. Mice in the ACEA + BCCAO, AM251 + ACEA + BCCAO, Hemo + ACEA + BCCAO, and Vehicle + BCCAO groups received 1.5 mg/kg ACEA, 1 mg/kg AM251 + 1.5 mg/kg ACEA, 1 mg/kg hemopressin + 1.5 mg/kg ACEA, and 0.1 ml vehicle (5% DMSO diluted by saline), respectively, at the onset of reperfusion. The expression of CB1 receptor was assessed by Western blot analysis (n = 5). Neurological tests were conducted at 24, 48 and 72 h after reperfusion (n = 13). Neuronal apoptosis was assessed by TUNEL staining (n = 5) and apoptosis markers (n = 5) at 72 h after reperfusion. To observe the mitochondrial structure, the mouse CA1 hippocampal neurons were observed by transmission electron microscopy at 72 h after reperfusion (n = 3).
A supplementary file shows the detailed materials and methods concerning cell culture, OGD/R model, cell viability analysis, global cerebral ischemia and reperfusion, neurological scores, TUNEL staining, isolation of mitochondria, Western blot, immunocytochemistry for electron microscopy, semi-quantification of mtCB1R, transmission electron microscopy, mitochondrial enzyme assays and MMP, mPTP detection (see Supplementary Information).
Statistical analysis
SPSS 13.0 for Windows (SPSS Inc., Chicago, USA) was used to conduct statistical analyses. All values, except for neurological scores, were presented as the mean ± SD, and were analyzed by one-way ANOVA. Between-group differences were detected with post-hoc Student-Newman-Keuls tests. The neurological scores were expressed as the median and were analyzed with Kruskal-Wallis tests followed by the Mann-Whitney U tests with Bonferroni correction. P < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Ma, L. et al. Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions. Sci. Rep. 5, 12440; doi: 10.1038/srep12440 (2015).
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant 81473488), the Overseas, Hong Kong & Macao Scholars Collaborated Researching Fund (Grant 81228022), and the National Key Basic Research and Development Program (973, Grant 2014 CB543202).
Footnotes
Author Contributions L.M. conducted the experimental work and drafted the manuscript. J.J. participated in the study coordination and helped to conduct the laboratory work. W.N. participated in the design of the study and performed the statistical analysis, T.J., Q.Z., L.Y. and F.H.B. helped to perform the study and the data analysis. Q.W. and L.Z.X. conceived of the study, coordinated and supervised the experiments, participated in the study design and revision of the manuscript. All authors read and approved the final manuscript.
References
- Laver S., Farrow C., Turner D. & Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med 30, 2126–2128 (2004). [DOI] [PubMed] [Google Scholar]
- Nichol G. et al. Regional systems of care for out-of-hospital cardiac arrest: A policy statement from the American Heart Association. Circulation 121, 709–729 (2010). [DOI] [PubMed] [Google Scholar]
- Roine R. O., Kajaste S. & Kaste M. Neuropsychological sequelae of cardiac arrest. JAMA 269, 237–242 (1993). [PubMed] [Google Scholar]
- MacAskill A. F. & Kittler J. T. Control of mitochondrial transport and localization in neurons. Trends Cell Biol 20, 102–112 (2010). [DOI] [PubMed] [Google Scholar]
- Mattson M. P., Gleichmann M. & Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo M. F., Ikuta I., Zhu X., Casadesus G. & Jordán J. Mitochondrial biology in Alzheimer’s disease pathogenesis. J Neurochem 114, 933–945 (2010). [DOI] [PubMed] [Google Scholar]
- Vosler P. S., Graham S. H., Wechsler L. R. & Chen J. Mitochondrial targets for stroke: focusing basic science research toward development of clinically translatable therapeutics. Stroke 40, 3149–3155 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anne Stetler. R., Leak R. K., Gao Y. & Chen J. The dynamics of the mitochondrial organelle as a potential therapeutic target. J Cereb Blood Flow Metab 33, 22–32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinzon M. A., Stetler R. A. & Fiskum G. Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab 32, 1362–1376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G. et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88 (2003). [DOI] [PubMed] [Google Scholar]
- Iversen L. Cannabis and the brain. Brain 126, 1252–1270 (2003). [DOI] [PubMed] [Google Scholar]
- Kano M., Ohno-Shosaku T., Hashimotodani Y., Uchigashima M. & Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89, 309–380 (2009). [DOI] [PubMed] [Google Scholar]
- Gilbert G. L., Kim H. J., Waataja J. J. & Thayer S. A. Delta9-tetrahydrocannabinol protects hippocampal neurons from excitotoxicity. Brain Res 1128, 61–69 (2007). [DOI] [PubMed] [Google Scholar]
- Bartova A. & Birmingham M. K. Effect of delta9-tetrahydrocannabinol on mitochondrial NADH-oxidase activity. J Biol Chem 251, 5002–5006 (1976). [PubMed] [Google Scholar]
- Tedesco L. et al. Cannabinoid receptor stimulation impairs mitochondrial biogenesis in mouse white adipose tissue, muscle, and liver: the role of eNOS, p38 MAPK, and AMPK pathways. Diabetes 59, 2826–2836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquila S. et al. Human sperm anatomy: ultrastructural localization of the cannabinoid 1 receptor and a potential role of anandamide in sperm survival and acrosome reaction. Anat Rec (Hoboken) 293, 298–309 (2010). [DOI] [PubMed] [Google Scholar]
- Sim-Selley L. J. & Martin B. R. Effect of chronic administration of R-(+)-[2,3-Dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl)methanone mesylate (WIN55,212-2) or delta(9)-tetrahydrocannabinol on cannabinoid receptor adaptation in mice. J Pharmacol Exp Ther 303, 36–44 (2002). [DOI] [PubMed] [Google Scholar]
- Tomiyama K. & Funada M. Cytotoxicity of synthetic cannabinoids on primary neuronal cells of the forebrain: the involvement of cannabinoid CB1 receptors and apoptotic cell death. Toxicol Appl Pharmacol 274, 17–23 (2014). [DOI] [PubMed] [Google Scholar]
- Bénard G. et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat Neurosci 15, 558–564 (2012). [DOI] [PubMed] [Google Scholar]
- Proto M. C. et al. Interaction of endocannabinoid system and steroid hormones in the control of colon cancer cell growth. J Cell Physiol 227, 250–258 (2012). [DOI] [PubMed] [Google Scholar]
- Laprairie R. B., Kelly M. E. & Denovan-Wright E. M. Cannabinoids increase type 1 cannabinoid receptor expression in a cell culture model of striatal neurons: implications for Huntington’s disease. Neuropharmacology 72, 47–57 (2013). [DOI] [PubMed] [Google Scholar]
- Ye R. D. et al. Sevoflurane preconditioning improves mitochondrial function and long-term neurologic sequelae after transient cerebral ischemia: role of mitochondrial permeability transition. Crit Care Med 40, 2685–2693 (2012). [DOI] [PubMed] [Google Scholar]
- Sun J. et al. Inhibition of mitochondrial permeability transition pore opening contributes to the neuroprotective effects of ischemic postconditioning in rats. Brain Res 1436, 101–110 (2012). [DOI] [PubMed] [Google Scholar]
- Nagayama T. et al. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 19, 2987–2995 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leker R. R., Gai N., Mechoulam R. & Ovadia H. Drug-induced hypothermia reduces ischemic damage: effects of the cannabinoid HU-210. Stroke 34, 2000–2006 (2003). [DOI] [PubMed] [Google Scholar]
- Hillard C. J. et al. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J Pharmacol Exp Ther 289, 1427–1433 (1999). [PubMed] [Google Scholar]
- Pacher P. & Haskó G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br J Pharmacol 153, 252–262 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N. et al. Contribution of hypothermia and CB1 receptor activation to protective effects of TAK-937, a cannabinoid receptor agonist, in rat transient MCAO model. PLoS One 7, e40889 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimann A. S. et al. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc Natl Acad Sci USA 104, 20588–20593 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M. et al. Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke 28, 1805–1810 (1997). [DOI] [PubMed] [Google Scholar]
- Yang G. et al. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res 752, 209–218 (1997). [DOI] [PubMed] [Google Scholar]
- Murakami Y. et al. Increases in tumor necrosis factor-alpha following transient global cerebral ischemia do not contribute to neuron death in mouse hippocampus. J Neurochem 93, 1616–1622 (2005). [DOI] [PubMed] [Google Scholar]
- Cho J. Y., Kim I. S., Jang Y. H., Kim A. R. & Lee S. R. Protective effect of quercetin, a natural flavonoid against neuronal damage after transient global cerebral ischemia. Neurosci Lett 404, 330–335 (2006). [DOI] [PubMed] [Google Scholar]
- Huang Y. & McNamara J. O. Ischemic stroke: “acidotoxicity” is a perpetrator. Cell 118, 665–666 (2004). [DOI] [PubMed] [Google Scholar]
- Schon E. A. & Przedborski S. Mitochondria: the next (neurode) generation. Neuron 70, 1033–1053 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev 79, 1431–1568 (1999). [DOI] [PubMed] [Google Scholar]
- Sims N. R. & Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta 1802, 80–91 (2010). [DOI] [PubMed] [Google Scholar]
- Sanderson T. H., Reynolds C. A., Kumar R., Przyklenk K. & Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol 47, 9–23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racay P. et al. Mitochondrial calcium transport and mitochondrial dysfunction after global brain ischemia in rat hippocampus. Neurochem Res 34, 1469–1478 (2009). [DOI] [PubMed] [Google Scholar]
- Iijima T. Mitochondrial membrane potential and ischemic neuronal death. Neurosci Res 55, 234–243 (2006). [DOI] [PubMed] [Google Scholar]
- Li J. et al. Reperfusion promotes mitochondrial dysfunction following focal cerebral ischemia in rats. PLoS One 7, e46498 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elimadi A., Jullien V., Tillement J. P. & Morin D. S-15176 inhibits mitochondrial permeability transition via a mechanism independent of its antioxidant properties. Eur J Pharmacol 468, 93–101 (2003). [DOI] [PubMed] [Google Scholar]
- Wu L. et al. The neuroprotection conferred by activating the mitochondrial ATP-sensitive K+ channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci Lett 402, 184–189 (2006). [DOI] [PubMed] [Google Scholar]
- Rozenfeld R. & Devi L. A. Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. FASEB J 22, 2311–2322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd G. T., Mancini G., Lutz B. & Luckman S. M. The peptide hemopressin acts through CB1 cannabinoid receptors to reduce food intake in rats and mice. J Neurosci 30, 7369–7376 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. et al. Activation of epsilon protein kinase C-mediated anti-apoptosis is involved in rapid tolerance induced by electroacupuncture pretreatment through cannabinoid receptor type 1. Stroke 42, 389–396 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.