

Abstract
CMTX, the X-linked form of Charcot-Marie-Tooth disease, is an inherited peripheral neuropathy arising in patients with mutations in the gene encoding the gap junction protein connexin 32 (Cx32). In this communication, we describe the expression levels and biophysical parameters of seven mutant forms of Cx32 associated with CMTX, when expressed in paired Xenopus oocytes. Paired oocytes expressing the R15Q and H94Q mutants show junctional conductances not statistically different from that determined for Cx32WT, though both show a trend toward reduced levels. The S85C and G12S mutants induce reduced levels of junctional conductance. Three other mutants (R15W, H94Y and V139M) induce no conductance above baseline when expressed in paired oocytes. Analysis of the conductance voltage relations for these mutants shows that the reduced levels of conductance are entirely (H94Y and V139M) or partly (S85C and R15W) explicable by a reduced open probability of the mutant hemichannels. The R15Q and H94Q mutations also show alterations in the conductance voltage relations that would be expected to minimally (H94Q) or moderately (R15Q) reduce the available gap junction communication pathway. The reduction in G12S induced conductance cannot be explained by alterations in hemichannel open probability and are more likely due to reduced junction formation. These results demonstrate that many CMTX mutations lead to loss of function of Cx32. For these mutations, the loss of function model is likely to explain the pathogenesis of CMTX.
Keywords: Connexin 32, Loss of function, Charcot-Marie-Tooth disease, Peripheral neuropathy, Conductance-Voltage relations, Xenopus oocyte expression system
1. Introduction
CMTX, the X-linked form of Charcot-Marie-Tooth disease, is an inherited peripheral neuropathy that arises in patients with mutations in the gene encoding the gap junction protein connexin 32 (Cx32) [4]. Cx32 has been directly demonstrated in myelinating Schwann cells, where it is believed to form intracellular channels between adjacent myelin loops at the Schmidt–Lantermann incisures and in the paranodal regions [39]. These channels may shorten the distance between the Schwann cell nucleus and adaxonal cytoplasm at least 350-fold [32].
Both loss-of-function and gain-of-function models have been advanced to explain the pathogenesis of CMTX. Some experimental evidence supports the (toxic) gain of function model. First, abnormalities in trafficking of several mutants lead to accumulation of protein in the cytoplasm of transfected mammalian cell lines expressing these proteins. Deschenes et al. [14] suggest that this, in turn, may lead to interference with trafficking of other essential proteins. However, three of three mutants examined are efficiently dealt with by the cellular quality control apparatus [45]. Second, transgenic mice expressing both the R142W mutation and wild-type Cx32 (32WT) develop a demyelinating neuropathy [16]. Third, the coinjection of mRNA for Cx32*R142W and wild-type Cx26 (26WT) in paired Xenopus oocytes leads to a reduction in levels of Cx26 mediated intercellular communication when compared to paired oocytes injected with 26WT alone or in combination with 32WT [7].
Several lines of evidence suggest that loss of the Cx32 mediated reflexive pathway is sufficient to cause CMTX. First, several CMTX mutations are predicted to lead to either no expression of protein (e.g., mutations in the promoter region) [25] or very truncated forms of the protein, such as R22STOP [24]. Second, a family with a complete loss of the coding portion of Cx32 has been identified and the disease in this cohort is very similar to that seen in other moderately or severely affected patients [2]. Third, several other mutations lead to either no or very little detectable protein when expressed exogenously [14]. Fourth, a transgenic mouse expressing 32WT and the 175 frameshift mutation under control of the P0 promoter shows no clinical or pathological abnormalities [1].
Cx32 forms gap junction channels that are gated by transjunctional voltage and that allow the passage of molecules smaller than ∼7 Å in radius [32]. Second messengers such as cAMP, Ca2+ and IP3 permeate wild-type Cx32 junctions [5,30]. In this communication, we describe the biophysical parameters of seven mutant forms of Cx32 which participate in the formation of functional cell–cell channels and which are associated with CMTX. For six of these mutants, the functional alterations as described are likely sufficient to lead to a substantial reduction of or change in the Cx32 mediated gap junction communication pathway. These results support the notion that alteration of biophysical properties is a common cause of partial or complete loss of function of Cx32 leading to CMTX.
2. Methods
2.1. Production of mutants
Mutants were produced using a construct consisting of the human Cx32 that was amplified by polymerase chain reaction (PCR) from human chromosomal DNA. The data for S85C and a small portion of the data reported for G12S were generated using mutations generated on a rat Cx32 template. Mutants were produced by oligonucleotide directed PCR, screened by restriction digestion, and sequenced using primers encompassing the entire cloned PCR product and restriction sites.
2.2. In vitro transcription of RNA for expression in oocytes
RNA was transcribed using the Message-Machine™ kit with T7 RNA Polymerase (AMBION, Austin, TX) according to manufacturer's instructions. RNA was column purified and eluted in nuclease free H20 containing anti-sense phosphorothiote oligonucleotide complementary to Cx38 at a concentration of 0.3 pmoles/nl. Cx38 is the only known endogenous gap junction channel expressed in oocytes that forms cell–cell channels and the antisense oligonucleotide blocks all endogenous coupling between paired oocytes within 48–72 h. RNA quality was evaluated by gel electrophoresis and concentration was quantitated by absorbance at 260 nm and evaluation of relative intensity on ethidium stained agarose gels. The RNA was then diluted as needed, aliquoted for single use and stored at −70°C.
2.3. Oocyte preparation and injection
Xenopus oocytes were harvested and prepared as described previously [47]. Cells were allowed to rest 6 h at 18°C in MND96 with Ca2+ (96 mM NaCl, 2 mM KCl, 2 mM MgCl2, 2.5 mM Na-pyruvate, 1.8 mM CaCl2, 5 mM HEPES pH.7.6), then injected with 100 μl of a solution containing 0.22 μg/μl of RNA and 0.3 pmoles/nl of antisense oligonucleotide to Cx38 in nuclease free H20. For wild type Cx26 (26WT), wild type Cx32 (32WT) and mutants expressing at high levels, RNA was further diluted to reduce junctional conductance and minimize series resistance artifacts during determination of conductance voltage relations [52]. After 24 h at 18°C in MND96 with Ca2+, oocytes were placed into a hypertonic solution (200 mM K-aspartate, 20 mM KCl, 1 mM MgCl2, 10 mM HEPES pH 7.6), manually devitilinized, and paired in MND96 with Ca2+. Cells were allowed to rest for another 24–48 h at 18°C and then evaluated for development of junctional conductance. For experiments to measure conductance (Table 2) RNA concentration was 0.22 μg/μl and measurements were made between 22 and 28 h after pairing.
Table 2. Conductances of homotypically paired oocytesa.
| n | Mean | S.E.M. | P value | |
|---|---|---|---|---|
| 32WT | 36 | 23 | 4.8 | P<0.001 |
| G12S | 26 | 0.030 | 0.0089 | P<0.001 |
| R15Q | 31 | 14 | 2.5 | ns |
| R15W | 24 | 0.024 | 0.0053 | P<0.001 |
| S85C | 58 | 2.5 | 0.98 | P<0.001 |
| H94Q | 30 | 15 | 3.4 | ns |
| H94Y | 31 | 0.038 | 0.0094 | P<0.001 |
| V139M | 22 | 0.015 | 0.0029 | P<0.001 |
| Anti Cx38 | 42 | 0.026 | 0.0054 | P<0.001 |
Xenopus oocytes were injected with the respective mRNA's and paired homotypically. Conductances were measured at 22–28 h after pairing as described in the Methods. Comparison of values for 32WT injected cells with mutant and Cx38 antisense injected cells was made using the Kruskal–Wallis test with Dunn's multiple comparisons. Each pairing was examined in at least three independent sets of experiments. In most cases RNA was injected at a concentration of 0.22 μg/μl as determined spectroscopically. For some of the V139M injected pairs, the amounts of RNA were quantitated by comparison of intensity of bands produced by agarose gel electrophoresis. Inclusion of these data does effect the overall conclusions stated in the Results. ns — not significant at P=0.05.
2.4. Oocyte in vivo expression and dual two electrode voltage clamping
Determination of junctional conductances between paired oocytes is accomplished using the dual two electrode voltage clamp as described previously [37,47,22]. Both cells of a pair were voltage clamped at an identical potential near to their resting potentials (usually −30 mV) to yield a transjunctional voltage gradient (Vj) of zero. Conductance measurements (Table 2) were made by pulsing from Vj=0 to±50 mV and±100 mV. For determination of steady state conductance voltage relations, cells were held between each pulse at Vj=0 for an interval long enough for the system to reach steady state, typically 45 to 60 s, but up to three min for some mutants. Cell 1, defined as the cell being pulsed, was pulsed in 10 or occasionally 20 mV increments from −120 to +120 mV. Any resulting change in current in cell 2, the apposed cell, is attributable to junctional current. Currents are displayed with positive up for both cells; thus a positive Vj step in cell 1 causes a downward deflection in cell 2. The term ‘homotypic’ refers to pairs of cells in which both cells are expressing the same connexin, while ‘heterotypic’ is used to denote that each cell in the pair is expressing a different connexin. Heterotypic pairings are described as “connexin expressed in cell 2/connexin expressed in cell 1” with the second cell in the pairing designation being the cell pulsed. ‘32’ or ‘26’ in pairing designations refer to the 32WT or 26WT respectively. Because total number of junctional channels can increase during the long interval required for recording 24 traces each lasting up to 4 min, each voltage pulse was generally preceded by a short (∼500 mS) pulse to −10 or −20 mV for normalization. Normalization to a prepulse is not possible when the open probability at Vj=0 is very low. In these cases, comparison of current responses at the beginning and end of the pulse sequence insured a lack of significant change in the total number of channels available for gating. Data was collected using the pClamp acquisition program (Axon Instruments, Foster City, CA) and Labmaster/TL-1 Software (Scientific Solutions, Mentor, OH), filtering at 100 Hz and digitizing the initial rapid relaxation at 500 Hz and the balance of the acquisition at 125 Hz. Instantaneous and steady state conductances were determined by fitting each current trace to a sum of exponentials of the form A1 e−t/τ(1) + … An e−t/τ(n)+C, where C is the steady state current, A1…n are initial current amplitudes of the component exponentials and τ(1) to τ(n) are the time constants of the component exponentials. Typically, 2 or 3 exponentials were required to fit a trace. Dividing the current at t=0 or t=∞ by the applied voltage gives the instantaneous or steady state junctional conductance. Conductance–voltage (Gj–Vj) relations were plotted and displayed as shown in the Results. Where possible, steady-state plots were fit to a Boltzmann equation of the form: Gss(V)=(Gmax–Gmin)/{1+e[A(V–V0)]} +Gmin where Gss is the steady state junctional conductance normalized to Vj=0, Gmax is the maximal normalized conductance, and Gmin is the normalized residual conductance, which in macroscopic recordings is approached as the absolute value of Vj is increased. V0 is the voltage at which the conductance is 1/2 of the difference between Gmin and Gmax and roughly corresponds to the voltage at which a single connexin hemichannel has an open probability of 50%. A is a parameter which reflects the slope of the Gj–Vj plot and is a measure of voltage sensitivity. A=nq/kT where n is the effective gating charge and q, k and T have their usual meanings. The Boltzmann distribution applies to a two state gating process where the energy difference between the two states is directly proportional to the applied voltage; gating of WT and mutant forms of Cx32 deviates from this model. However, the parameters generated provide a useful basis for comparison among channels produced by various pairing configurations of mutant and WT connexins. All points forming a given limb (left or right) of the steady state Gj–Vj relations were used for fitting to each Boltzmann distribution.
3. Results
3.1. Description of mutants and functional expression in xenopus oocytes
The accepted membrane topology of Cx32 has four transmembrane domains (TM1 to TM4), a cytoplasmic COOH-terminus (CT) and NH2-terminus (NT), a cytoplasmic loop (CL) connecting TM2 and TM3 and extracellular loops connecting TM1 to TM2 (E1) and TM3 to TM4 (E2). The locations in the Cx32 sequence of the mutants selected for evaluation are shown in Fig. 1. Three of the mutants (G12S, R15Q and R15W) are located in NT, a region of the molecule shown to contain charges involved in response to transjunctional voltage, Vj [47]. One mutation (S85C) is located in TM2, a region of the molecule recently shown to be involved in voltage gating [36]. Two mutations (H94Q and H94Y) are located at the junction between TM2 and CL. The CL domain is thought to be an important determinant of pH sensitivity of Cx32 [49,48,50]. One mutation is in TM3 (V139M). Table 1 summarizes the clinical data reported for CMTZ patients harboring these mutations and lists the corresponding references in the literature.
Fig. 1.
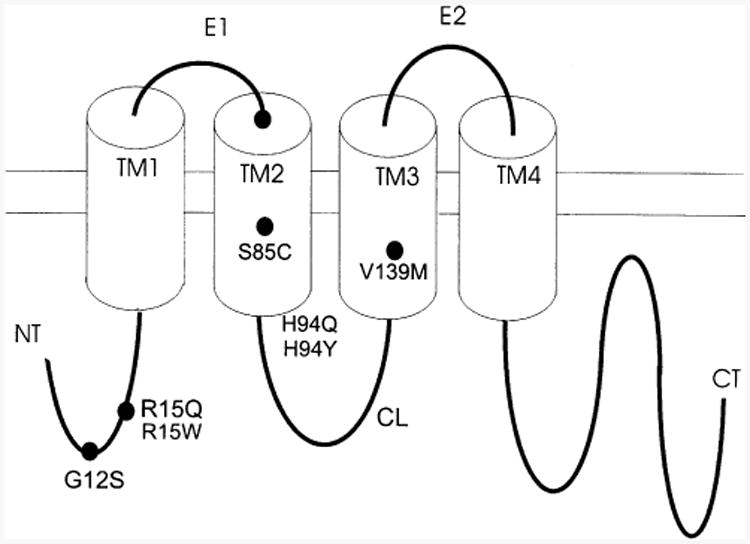
Topography of the connexin 32 subunit and location of mutants described in this communication. See Results for further details.
Table 1. Description of clinical phenotypes associated with selected mutationsa.
| Mutant | Severity in males | Severity in female heterozygotes | Median motor ncv/peroneal ncv | Footnote References |
|---|---|---|---|---|
| G12S | Severe (clinical details not given)b,c | Mild (but may have missed non-manifesting carriers)b | moderate reduction (f)b marked reduction (m)b |
b–f |
| R15Q | Mild to moderate (clinical details not given) c | 0/3 Symptomaticc | 20(m), 30(f) nerve not indicatedc | c,g,h |
| Moderate to severeg | ||||
| R15W | Mild to moderatei | Asymptomatic or | 33–42/22(f)l | i–n |
| Severej | 25–54/31 (m)i | |||
| Mild to? moderatel | 34–36/24–42(f)i 27.7/NA(m) | |||
| Mild to moderatej | Asymptomatic to moderatej | 42–49/37.2–40 (f)j | ||
| S85C | Mildl | 49/51(f)l | l | |
| H94Q | Mildo | o | ||
| H94Y | Moderate? (little clinical data given)p | Mildp | p | |
| V139M | Moderatec | 9/15 Symptomaticc | 28–32/NA(m)C | c,d,g,h,l,m,q–s |
| Mildh | 24/NA(f)h |
Where clinical descriptions in the references are sufficiently detailed, we summarize by using mild to denote disease causing signs without symptoms, moderate to denote difficulty with ambulation possibly requiring use of ankle braces, and severe to denote marked impairment of gait requiring the use of a walking aid or wheelchair. Where data is not adequate for stratification but severity is summarized, that summary is simply reproduced here: ncv — nerve conduction velocity in meters/sec; m — male, f — female.
Deschenes et al. [14].
Bergoffen et al. [4].
Fischbeck et al. [17].
Martin et al. [28].
Hahn et al. [21].
Fairweather et al. [15].
Gutierrez et al. [20].
Senderek et al. [41]. One affected male child with a severe phenotype also has a deletion of exon 3–7 of dystrophin.
Wicklein et al. [51].
Janssen et al. [26].
Nelis et al. [29].
Senderek et al. [40].
H. Midani, P. Kelkar and G.J.G. Parry, personal communication.
Garbern et al. [19].
Bone et al. [6].
Omori et al. [34].
To evaluate the channel forming ability of the various CMT mutants, we injected equal amounts of mRNA into Xenopus oocytes and measured the junctional conductance approximately 24 h after homotypic pairing. The data summarized in Table 2 (and recapitulated in Table 6) show that these mutants varied in their ability to induce electrical coupling between homotypically paired oocytes. Two mutants (R15Q and H94Q) expressed junctional conductances at levels that were not significantly different from those of WT Cx32, though there appeared to be a trend toward lower levels of induced conductance. One mutant (S85C) expressed at levels significantly lower than those seen in paired Cx32 injected cells but much higher than those in the Cx38 anti-sense oligonucleotide injected cells. Four mutants (H94Y, R15W, V139M, and G12S) induced conductances that were statistically indistinguishable from those noted for the Cx38 antisense injected cells. These differences among the 32WT, mutant, and antisense injected cells all persisted when cell pairs were examined at 48 to 72 h. As shown below, most of the differences between these mutants and WT can be accounted for by differences in the macroscopic conductance voltage relations of the channels, without invoking large effects on protein expression, trafficking, assembly, or alterations in single channel conductance.
Table 6. Summary of findingsa.
| Mutant | Average Homotypic conductance | Instantaneous Gj–Vj | Steady-State Gj–Vj | Changes in conductance explained by Gj–Vj |
|---|---|---|---|---|
| G12S | ↓↓↓ | like WT | like WT | No |
| R15W | ↓↓↓ | Not examined | Very altered | Perhaps |
| R15Q | ↓ | like WT | moderately altered | Yes |
| S85C | ↓↓ | Not examined | moderately altered | No |
| H94Q | ↓ | like WT | moderately altered | Yes |
| H94Y | ↓↓↓ | Not examined | Very altered | Yes |
| V139M | ↓↓↓ | Not examined | Very altered | Yes |
The effects of various mutations on the behavior of the Cx32 channel is summarized in this table. ↓ minimally reduced, not statistically significant; ↓↓ moderately reduced. ↓↓↓ statistically indistinguishable from antisense injected. N/A — Not Applicable.
3.2. Instantaneous and steady state junctional conductance–voltage relations for 32WT, R15Q, S85C, and H94Q recorded in the homotypic
Oocytes injected with mRNA for 32WT and three of the mutants (H94Q, R15Q, and S85C) consistently showed junctional conductances above baseline when paired homotypically. To further characterize these mutants, we evaluated the macroscopic instantaneous and steady state junctional conductance–transjunctional voltage (Gj–Vj) characteristics of homotypic channels formed between paired oocytes expressing these proteins. Representative current traces and average instantaneous and steady state Gj–Vj relations are shown in Fig. 2. As shown previously for 32WT [38], junctional currents decayed with time when the absolute value of the transjunctional voltage (Vj) was increased to 40 mV or more, and the rate of decay increased with increasing absolute values of Vj (Fig. 2a). The steady state junctional conductance (Fig. 2b) declined symmetrically about Vj=0 as the magnitude of Vj increased, and even at the largest values of Vj tested, conductance never declined below approximately 20% of its maximal value. This residual conductance likely corresponds to the subconductance states observed in single channel records of 32WT when the absolute value of Vj is increased above 60 to 70 mV [10,32].
Fig. 2.
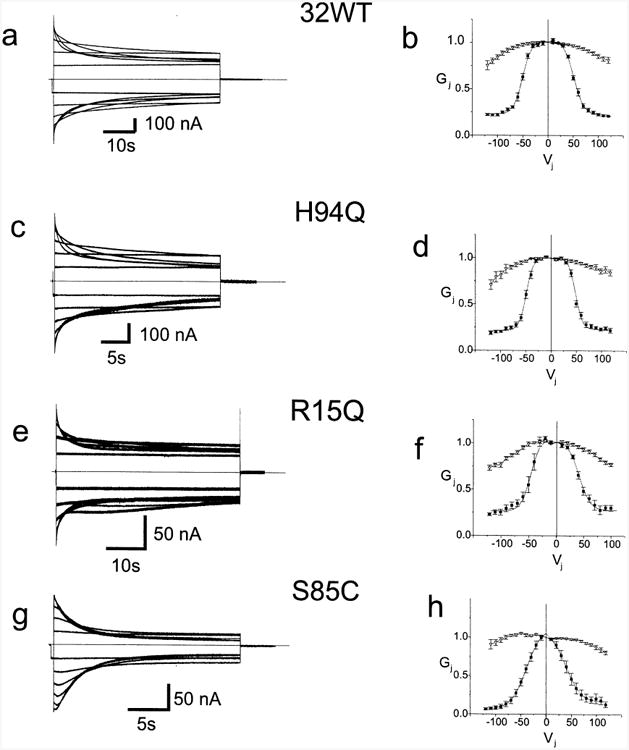
Representative current traces and average Gj–Vj relations for 32WT and three mutants. Fig. 2a, c, e, and g. Xenopus oocytes were injected with the noted mRNA's and paired homotypically as described in the Methods. Both cells were voltage clamped at −30 mV and cell 2 was stepped between −120 and +120 mV in 10 mV increments. Junctional currents for 32WT, H94Q, R15Q, and S85C recorded from cell 1 are shown. Only traces in 20 mV increments from ±20 to ±120 are shown for clarity. Current traces were analyzed as described in the Methods to allow for determination of the instantaneous and steady state Gj–Vj relations for each cell pair. Fig. 2b, d, f, and h. Average instantaneous and steady state Gj–Vj relations for 32WT and three mutants. Data such as shown in Fig. 2a, c, e, g, and i were analyzed as described in the Methods, averaged and plotted as mean±S.E.M. The smooth curves approximating the steady state data correspond to the curves generated by fitting the data to a Boltzmann distribution as described in the Methods. Each point in each Gj–Vj plot is the average value as determined in three to eight independent experiments. Filled squares — steady state conductances; hollow triangles — instantaneous conductances.
The effects of these mutations on current transients and steady state Gj–Vj relations varied. The H94Q mutation had no effect on the homotypic steady state Gj–Vj relation compared to 32WT (Fig. 2d). However, the kinetics of the decay in the current transients (Fig. 2c) were substantially more rapid than for 32WT (Fig. 2a). The current transients for channels formed by the R15Q mutation (Fig. 2e) showed a rapid voltage dependent decay for Vj>±40 mV. At higher Vj's this was followed by a slower, small increase and subsequent decrease in current. The complex pattern can be explained by contingent gating [22], if a fraction of the hemichannels are in the closed state at Vj=0. Contingent gating assumes that if one hemichannel is fully closed, Vj does not affect the open hemichannel; likewise, if the hemichannel is closed to a substate the effect of Vj on the open hemichannel may be reduced. When Vj is stepped to −100 mV, open channels will close by gating of the hemichannel in the cell being pulsed. At the same time, channels closed by the hemichannel in the apposed cell at Vj=0 will open and then close by gating of the hemichannel in the pulsed cell. If the initial closure is faster than the opening, these two competing processes could explain the observed gating, where conductance rapidly decreases then increases and decreases again.
Nonetheless, the R15Q mutation has only a minimal effect on the steady state Gj–Vj relation (Fig. 2f). There is a small asymmetric inward shift of the Gj–Vj relation in which Gj is slightly smaller at positive than at negative Vj's. If voltage dependent gating of connexin channels is a function only of the voltage between the two cells expressing the connexins, termed Vj dependence, a symmetric steady state relation should result. An asymmetric Gj–Vj relation for a symmetric homotypic channel suggests dependence of Gj on the membrane potential of the stepped cell (Vi–0 or Vm). Effects of membrane potential on junctional conductance in the absence of Vj, termed Vm or Vi–0 dependence [46,11] were originally described in insect gap junctions by Obaid et al. [31] and have also been described in vertebrate gap junctions [3].
Introduction of the S85C mutation induced greater alterations in both the current transients (Fig. 2g) and the Gj–Vj relation (Fig. 2h). First, when pulsed from the steady state at Vj =0 the current transients were slightly asymmetric with a prominent increase followed by a decrease with positive pulses (shown as downward currents in cell 2) and a less prominent increase followed by a decrease with negative pulses. As with R15Q, this transient opening followed by closure indicates that a fraction of the channels are not fully open at Vj=0 and that the open probability at Vj=0 is less than 1.0. The asymmetry of the current transients indicates a degree of Vm dependence; asymmetry is also evident in the steady state Gj–Vj relation. Second, the S85C channel is more sensitive to voltage than 32WT, with substantial closure at voltages as low as ±0 mV and an inward shift of both limbs of the steady state Gj–Vj plot. Third, at large Vj, the currents decline to lower values for Vj's induced by hyperpolarizing one cell (shown as upward current deflections in cell 2) than for equal depolarization of the same cell, again indicating Vm dependence. The small value of Gmin seen in S85C junctions may reflect that these channels close fully rather than to a substate. Transitions to full closure are seen rarely in 32WT [32,33] and Cx43 [8] cell–cell channels in response to voltage, prominently with voltage in Cx46 hemichannels [43] and with pharmacological agents in both hemichannels [43] and cell–cell channels [12]. This closure is distinct from the closure to a residual state typically seen in voltage dependent gating of 32WT [32,33]. The slow gate has been provisionally called the loop gate [44] because of the resemblance of these slow gating transitions to the docking currents seen during de novo formation of gap junction cell–cell channels [13] and the presumed requirement for docking of apposed gap junction hemichannels via the extracellular loop domains when forming cell–cell channels. To distinguish the two gates, that which closes channels to a substate in response to transjunctional voltage will be referred to as the Vj gate.
The instantaneous conductance voltage relations for the mutants recorded homotypically (R15Q, S85C, and H94Q) are shown as inverted hollow triangles in Fig. 2. With the exception of that for S85C, these relations are very similar to those determined for WT (Fig. 2b). The instantaneous Gj–Vj relation for channels containing the S85C mutation (Fig. 2d) is slightly asymmetric. This likely reflects the contribution of a very fast component of gating beyond our resolution in paired oocytes. As discussed above, the asymmetry reflects Vm dependence of this channel.
Current traces and steady state Gj–Vj relations for homotypic pairs reflect of the net effect of voltage on both, oppositely oriented hemichannels. The quantitative Boltzmann analysis attempts to isolate the effect of voltage on each individual hemichannel. Values for the Boltzmann fits for the various mutants are shown in Table 3. These confirm the general observations noted above. Specifically, the values for H94Q are nearly identical to those for WT, while the values for R15Q deviate slightly from those of WT in that V0 is reduced asymmetrically, indicating greater sensitivity to Vj induced by depolarizing one cell than by hyperpolarizing the other. The values for S85C show a reduced V0 indicating increased sensitivity to Vj and an asymmetrically reduced Gmin Although the Gj–Vj relation for S85C is asymmetric, the calculated value for V0 is symmetric.
Table 3. Boltzmann parameters for homotypic pairingsa.
| Type | (+) Vj (depolarizing cell 1) | (−)Vj (hyperpolarizing cell1) | ||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Gmax | Gmin | V0 | A | Gmax | Gmin | V0 | A | |
| 32WT | 1.0 | 0.22 | 51 | 9.4 | 1.0 | 0.23 | −51 | 8.2 |
| R15Q | 1.0 | 0.29 | 40 | 9.6 | 1.0 | 0.27 | −47 | 8.5 |
| S85C | 1.1 | 0.16 | 39 | 14 | 1.1 | 0.062 | −39 | 15 |
| H94Q | 1.0 | 0.25 | 46 | 6.6 | 1.0 | 0.21 | −49 | 7.5 |
Boltzmann parameters for homotypic pairings of 32WT and mutant forms of Cx32. Steady state Gj–Vj relations were determined for each type of homotypic junction, as described in the Methods. Steady state Gj–Vj relations were fit to Boltzmann distributions. Data for negative (−) and positive (+) Vj are given separately. Gmin, Gmax, V0 and A have their usual meanings and are defined in the Methods.
The estimation of open probabilities of connexin hemichannels based solely on macroscopic data obtained for homotypic pairings of wild-type and mutant connexins may be confounded in two-ways. First, when both hemichannels have a high probability of being closed at Vj=0, extrapolation to Gmax is less reliable. Second, underestimates of Gmax in this kind of analysis may lead to errors of determination of Gmin which may in turn compound errors in the estimation of hemichannel open probability at Vj=0. Heterotypic pairings (see below) may help to circumvent these limitations.
3.3. Instantaneous and steady state junctional conductance–voltage relations for R15Q, S85C, and H94Q recorded in the heterotypic configuration with CX32WT and CX26WT
To further evaluate the steady state properties of R15Q, S85C, and H94Q we paired them heterotypically with 32WT (Fig. 3a–c). This simplifies the evaluation of the voltage dependence of the mutant hemichannel by providing an apposed WT hemichannel that has an open probability that is near 1.0 at Vj=0 and does not decrease until Vj is more negative than about −40 mV with respect to the cell expressing 32WT. Data are plotted so that positive Vj is relative positivity in the cell expressing the connexin indicated on the right side of the pairing designation, e.g. for H94Q/32WT positive Vj is positivity on the 32WT side. Since Cx32 closes on relative negativity, the decreases in Gj for Vj<0, i.e. on the left side of Vj=0 in Fig. 3a–c are due to gating of 32WT hemichannels. This analysis assumes that alterations seen in a particular heterotypic pairing of mutant with 32WT are due to effects of the mutation of interest on the hemichannel that contains the mutation and are not due to alterations in the behavior of the apposed 32WT hemichannel [47].
Fig. 3.
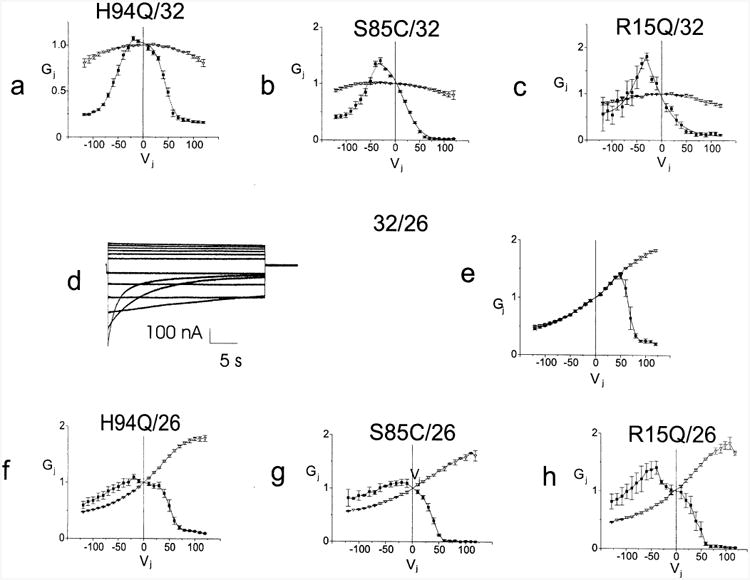
Average Gj–Vj relations for four mutants paired heterotypically with 32WT or 26WT. Figs. 3a to c. Xenopus oocytes were injected with the noted mRNA's and paired heterotypically with Cx32WT. Both cells were voltage clamped at −30 mV and the cell expressing the 32WT was stepped between −120 and +120 mV in 10 mV increments. Current traces were analyzed as described in the Methods, averaged and plotted as mean±S.E.M. The smooth curves approximating the steady state data correspond to the curves generated by fitting the data to a Boltzmann distribution. Figs. 3d and e. Representative current traces and average instantaneous and steady state conductance voltage relations for 32WT paired heterotypically with 26WT were determined as noted above in Fig. 2. Figs. 3f to h. Xenopus oocytes were injected with the noted mRNA's and paired heterotypically with 26WT. Junctional currents were obtained and analyzed as noted above. The resulting conductance voltage relations were averaged and plotted as mean±S.E.M. Individual points are connected for clarity. Each point in each Gj–Vj plot is the average value as determined in three to eight independent experiments. Filled squares — steady state conductances; hollow triangles — instantaneous conductances.
The H94Q/32WT Gj–Vj relation (Fig. 3a) is minimally asymmetric with a slight increase in steady state conductance when pulsing from Vj=0 to Vj=−10 or −20 (with respect to the cell expressing the 32WT) and a slight inward (leftward) shift of the limb corresponding to the H94Q hemichannel. A reasonable explanation for the increase in steady state conductance when pulsing from Vj=0 to negative Vj's is that the open probability (PO) of the H94Q hemichannel at Vj=0 is somewhat less than 1.0, and that its entire Gj–Vj relation is shifted in the positive direction relative to its cytoplasmic side. Similarly, the asymmetries in the S85C/32WT (Fig. 3b) and R15Q/32WT (Fig. 3c) heterotypic Gj–Vj relations suggest that the PO–Vj relations of the R15Q and S85C hemichannels are shifted to the left and that PO is substantially less than 1.0 at Vj=0. An additional feature of the S85C/32WT and, to a lesser extent, the R15Q/32WT heterotypic pairings is the reduced residual conductance compared with 32WT at Vj>50 mV (gating on the mutant side). This suggests that either the majority of the channels are in the fully closed, non-conducting state (i.e. that the ‘loop gate’ has closed) or that they have entered a very low conductance substate.
Parameters for Boltzmann fits for the heterotypic pairings with 32WT quantify the inferences obtained by inspection (Table 4). The parameters for H94Q/32WT show that the H94Q hemichannels differ only minimally from those for 32WT with a V0 ∼10 mV less than WT and a slightly reduced Gmin/Gmax of ∼0.15. V0 is shifted progressively in S85C and R15Q, and A, which reflects the effective gating charge, is increased. The parameters for the apposed WT hemichannel shift progressively with changes in mutant parameters, which may reflect measurement error rather than a change in hemichannel properties. For example, the increases in A compared with parameters obtained for the homotypic WT channel likely reflect that PO of the mutant hemichannel was increasing when PO of the WT was decreasing. Parameters for the S85C/32WT channel reveal marked alterations in the S85C hemichannel with a V0∼35 mV less than WT and a markedly reduced Gmin/Gmax of ∼0. Parameters for the R15Q/32WT channel reveal marked alterations in the R15Q hemichannel with a V0∼50 mV less than WT and a markedly reduced Gmin/Gmax of ∼0.05. For each of these heterotypic pairings of a mutant with a WT hemichannel, the behavior of the WT hemichannel appears relatively preserved, suggesting that the major effects of the mutations are within the mutant hemichannels themselves.
Table 4. Boltzmann parameters for heterotypic pairing with Cx32WTa.
| Pairing |
Vj>0 (depolarizing the cell expressing 32WT, negative on the mutant side) |
(−)Vj (hyperpolarizing the cell expressing 32WT, negative on the WT side) |
||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Gmax | Gmin | V0 | A | Gmax | Gmin | V0 | A | |
| R15Q/32 | 2.0 | 0.11 | −2.9 | 21 | 2.5 | 0.53 | −43 | 20 |
| R15W/32 | NF | 9.9 | 0.26 | −51 | 21 | |||
| S85C/32 | 1.4 | 0.0023 | 18 | 17 | 1.7 | 0.39 | −51 | 17 |
| H94Q/32 | 1.0 | 0.16 | 43 | 10 | 1.2 | 0.22 | −52 | 15 |
| H94Y/32 | NF | 7.3 | 0.34 | −49 | 20 | |||
| V139M/32 | NF | 44 | 0.51 | −100 | 11 | |||
Steady state Gj–Vj relations were determined for each type of heterotypic junction, as described in the Methods. Steady state Gj–Vj relations were fit to Boltzmann distributions. Parameters for negative (−) and positive (+) Vj are given separately. Gmin, Gmax, V0 and A have their usual meanings and are defined in the Methods. NF — not fit.
To confirm the findings seen for heterotypic pairings with 32WT, these three mutants and 32WT were paired heterotypically with Cx26WT (26WT). 26WT hemichannels have the opposite gating polarity from 32WT hemichannels, and close only when Vj>70 mV [37,47]. If the hemichannels retain their gating properties in the heterotypic junctions, pairing each mutant with 26WT allows for observation of the gating of the mutant Cx32 hemichannel in a voltage range where the 26WT hemichannel remains open. When pairing 32WT with 26WT and stepping the cell expressing 26WT, there was instantaneous rectification, which is a single channel property [9,33]. No detectable gating occurred when pulsing through the negative voltages (negative with respect to the 26WT hemichannel and positive with respect to the apposed 32WT hemichannnel). However, when positive voltages of 60 mV or more were applied, closure of both the 26WT hemichannel (closing on positive polarity) and the 32WT hemichannel (closing on negative polarity) were likely to have occurred (See Fig. 3d and e).
The effects of the three mutations on channels formed by heterotypic pairings with 26WT are consistent with those seen for pairings with 32WT (Fig. 3a–c and Table 4). The steady state Gj–Vj relation for H94Q/26WT (Fig. 3f) differs from that for 32WT/26WT in that the plot is shifted to the left, consistent with a shift of V0 toward more positive values relative to the mutant hemichannel. In addition, when pulsing from Vj=0 to negative Vj the steady state conductance exceeded instantaneous conductance. This indicates that PO at Vj=0 is <1.0. The Gj–Vj relations for S85C/26WT (Fig. 3g) and R15Q/26WT (Fig. 3h) show similar but more marked alterations than those seen for H94Q/26. Both Gj–Vj relations show leftward shifts; when pulsing to negative Vj from Vj=0 both show increasing steady state conductances, with the R15Q mutation exhibiting the greater effect. In addition, the residual conductance for R15Q/26WT is markedly reduced and that for S85C/26WT is very close to zero. Overall, the qualitative properties of mutant hemichannels inferred from their behavior in heterotypic pairings with 26WT and 32WT are in good agreement.
Because of the large magnitude of the instantaneous rectification of junctional currents, Boltzmann fits were performed after first normalizing the steady state conductance to the initial conductance at that voltage (Table 5). Only a single Boltzmann, corresponding to gating on positive polarity with respect to the cell expressing 26WT, was fit. Compared to the parameters for the heterotypic 32WT/26WT cell–cell channels, the parameters for H94Q/26WT and S85C/26WT differ markedly, and parameters for R15Q/26WT differ to the greatest degree. The values for V0 (Table 5) indicate that the relative shift in this parameter in the positive direction relative to the cell expressing the mutant hemichannel is greatest for R15Q and successively less S85C and H94Q. This progression is identical to that determined using values for V0 determined by pairing the mutants heterotypically with 32WT (Table 4).
Table 5. Boltzmann parameters for heterotypic pairing with Cx26WTa.
| Pairing | Gmax | Gmin | V0 | A |
|---|---|---|---|---|
| 32/26 | 0.98 | 0.13 | 67 | 5.4 |
| R15Q/26 | 1.9 | −0.029 | 5.5 | 23 |
| S85C/26 | 1.4 | −0.029 | 21 | 16 |
| H94Q/26 | 1.4 | 0.011 | 29 | 21 |
Instantaneous and steady state Gj–Vj relations were determined for each type of heterotypic junction, as described in the Methods. Steady-state values at each voltage were then normalized to their instantaneous counterparts and each normalized steady state plot was fit to a single Boltzmann distribution. Gmin, Gmax, V0 and A have their usual meanings and are defined in the Methods. These parameters were determined only for those mutants for which reliable instantaneous Gj–Vj relations were available.
3.4. One mutant (G12S) induces very low levels of junctional conductance
Very low levels of junctional conductance were seen when oocytes expressing the G12S mutant were paired homotypically or heterotypically with 26WT. On average these values were not statistically different than those for Cx38 antisense injected cells. However, occasional pairs of oocytes did show junctional conductances high enough to allow for determination of Gj–Vj relations. Fig. 4a shows representative current traces and Fig. 4b shows instantaneous and steady state Gj–Vj relations for channels formed between oocytes expressing the G12S mutant. Figs. 4c and 4d show representative current traces and instantaneous and steady state Gj–Vj relations for G12S paired heterotypically with 26WT. It is evident that G12S can form channels with macroscopic properties very similar to those produced by 32WT, either when paired homotypically (Fig. 4a and b) or heterotypically (Fig. 4c and d) with Cx26. This suggests that the reduced levels of conductance seen for this mutant when compared with 32WT (see Table 2) are not a result of altered Vj sensitivity which would lead to a reduced in the open probability at Vj=0. Reduced expression or increased degradation of connexins are possible contributing mechanisms. Reduction in single channel conductance alone seems unlikely to account for the very low currents induced by injection of mRNA for G12S since the predicted single channel conductance for the cell–cell channel (assuming no other differences between G12S and 32WT) would have to be reduced to 0.18 pS and because substantial alterations in single channel conductance would likely be accompanied by significant alterations in instantaneous rectification when paired with 26WT. The reduced conductance levels may be largely attributable to altered trafficking or processing, or a reduced ability of G12S subunits to oligomerize into hexameric hemichannels or to interact normally with apposed hemichannels.
Fig. 4.
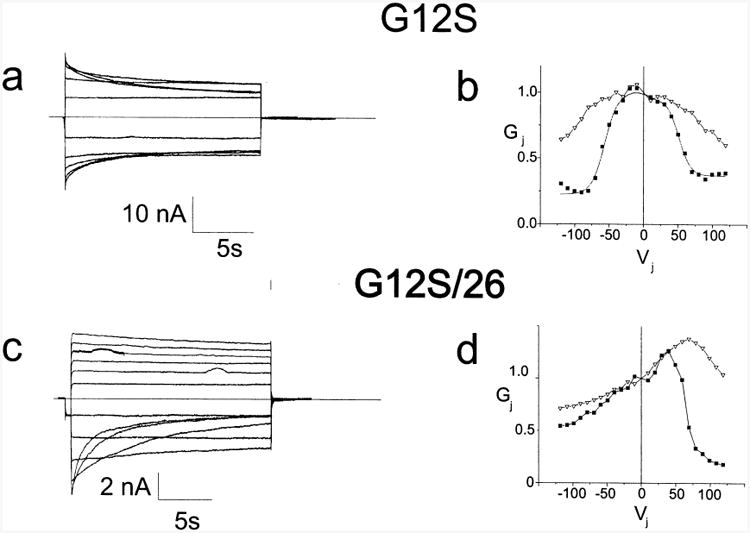
Representative current traces and Gj–Vj relations for G12S paired homotypically and heterotypically with 26WT. Figs. 4a and b. Representative current traces and Gj–Vj relations for G12S paired homotypically. Current traces were analyzed as described in the Methods; the smooth curve approximating the steady state data corresponds to the curve generated by fitting the data to a Boltzmann distribution. Figs. 4c and d. Representative current traces and Gj–Vj relations for G12S paired heterotypically with 26WT. Individual points are connected for clarity. For a and c only traces in 20 mV increments from ±20 to ±120 are shown. Filled squares-steady state conductances; hollow triangles-instantaneous conductances. Current traces and Gj–Vj relations are representative of findings in at least three independent experiments.
3.5. Properties of mutants which fail to induce junctional conductances above baseline when paired homotypically
Three mutants (V139M, R15W and H94Y) did not induce measurable conductances when paired in the homotypic configuration. We have evidence that junctional conductance is negligible because at least one of the two oppositely directed hemichannels has a high probability of being closed regardless of the transjunctional voltage.
Fig. 5a–d shows the representative current traces and the conductance voltage relations for H94Y and R15W paired heterotypically with 32WT. The steady state Gj–Vj relations are markedly shifted to the left with steady state conductances at Vj=0 that are <15% of the total conductance, consistent with substantially reduced open probabilities at Vj=0. Steady state Gj–Vj relations for both R15W/32WT and H94Y/32WT show very small Gmin, suggesting that these heterotypic channels gate to a non-conducting (or very low conductance) state. Since the Cx32 hemichannel is predicted to be fully open at Vj>−40 mV, the small Gmin at large positive Vj's and leftward shift of the Gj–Vj relations can be reasonably ascribed to the mutant hemichannel. These qualitative observations are reflected in the parameters for Boltzmann fits shown in the steady state Gj–Vj plots (Table 3).
Fig. 5.
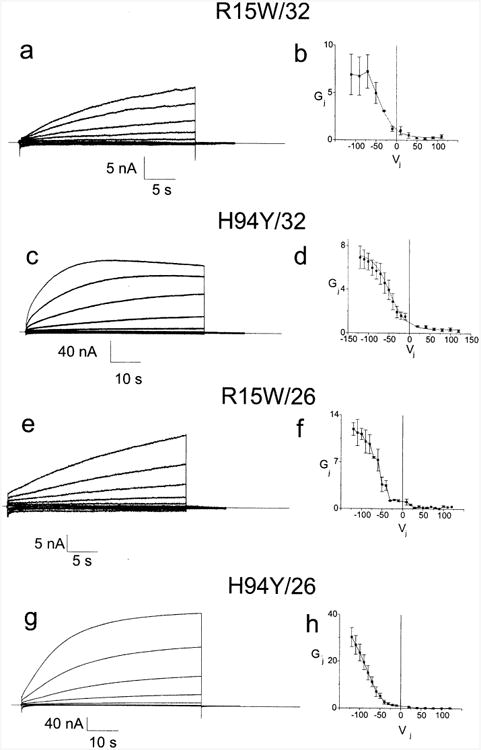
Representative current traces and average Gj–Vj relations for two mutants paired heterotypically with 32WT or 26WT. Figs. 5a and c. Junctional current traces for R15W and H94Y paired heterotypically with Cx32. For clarity, only traces in 20 mV increments from ±20 to ±120 (or ±10 to ±110 for R15W) are shown. Current traces were analyzed as described in the Methods section to allow for determination of the Gj–Vj relations for each cell pair. Figs. 5b and d. Average Gj–Vj relations for R15W and H94Y paired heterotypically with Cx32. Data such as shown in Figs. 5a and c was analyzed as described in the Methods, averaged and plotted as mean±S.E.M. Each point in each Gj–Vj plot is the average value as determined in three to eight independent experiments. The smooth curves approximating the steady state data correspond to the curves generated by the Boltzmann fits. Figs. 5e and g. Junctional current traces for R15W and H94Y paired heterotypically with 26WT. For clarity, only traces in 20 mV increments from ±20 to ±120 are shown. Current traces were analyzed as described in the Methods section to allow for determination of the Gj–Vj relations for each cell pair. Figs. 5f and h. Average Gj–Vj relations for R15W and H94Y paired heterotypically with Cx32. Current traces were analyzed as described in the Methods, averaged and plotted as mean±S.E.M. Individual points are connected for clarity. Each point in each Gj–Vj plot is the average value as determined in three to eight independent experiments.
To confirm the effects of the R15W and H94Y mutations, we paired both of these mutants heterotypically with 26WT. As shown in Fig. 5e to h, two major effects are seen when compared to the 32WT/26WT heterotypic pairing (Fig. 3d and e): (1) the steady state Gj–Vj relations are shifted leftward suggesting a substantially reduced PO of the mutant hemichannel at Vj=0, and (2) the residual conductances are extremely low. Qualitatively, there is good agreement between the results for the heterotypic pairings with 32WT and those with 26WT. However, the normalized conductances at a given large negative voltage are substantially higher for the heterotypic pairings with 26WT than they are for 32WT. This difference could result from closure of the 32WT hemichannel at large negative Vj potentials at which the 26WT hemichannel remains open. The actual increase in PO for heterotypic pairings with 26WT may be somewhat underestimated because of rectification of 32WT/26WT junctions, with current decreasing as Vj becomes more negative with respect to the Cx26WT side (Fig. 3F).
To sum up, data from heterotypic junctions indicate that hemichannels containing the R15W or H94Y mutations have extremely low open probabilities at Vj=0; in homotypic pairs, Vj of either sign will further reduce PO. This in and of itself is sufficient to explain our failure to observe H94Y and R15W in the homotypic configuration. The reasonable nature of this conclusion is demonstrated by the following simple calculation. The open probability of the H94Y hemichannel at Vj=0 can be calculated by assuming that the 30-fold difference in Gj between 0 mV and −120 mV in the H94Y/26WT heterotypic cell–cell channel (Fig. 5d) is due solely to a 30-fold lower open probability of the H94Y hemichannel at Vj=0. Then the predicted open probability of the homotypic H94Y cell–cell channel is 1/302≈0.001. Multiplying 0.001 by the average junctional conductance of homotypically paired oocytes expressing 32WT yields a predicted conductance of 0.001*25.91 μS≈26 nS, a value comparable to the background conductance between paired Cx38 antisense injected cells. Thus, alterations in open probability of the H94Y hemichannel are sufficient to explain our failure to observe this mutant in homotypically paired oocytes. (This analysis neglects the contribution of rectification of the H94Y/26WT cell–cell channel, which should further reduce the predicted PO of the homotypic channel.)
Hemichannels containing the R15W mutation have a reduced open probability at Vj=0, but this alone may not be sufficient to account for the failure to detect them in the homotypic configuration. This failure may be partly attributable to the factors outlined for G12S. A calculation similar to that above shows that the R15W hemichannel open probability at Vj=0 is ∼1/12 of Gmax (see Fig. 5b), and the predicted average cell–cell conductance is 180 nS, a value roughly 10 times that seen for the junctional currents in homotypically paired oocytes expressing R15W. However, this discrepancy may be due to overestimating the open probability at Vj=0 if the open probability at Vj=−120 is substantially less than 1.0; alternatively, neglecting the potential contribution of instantaneous rectification to the calculation of relative open probability at Vj=0 and Vj=−120 could lead to an overestimate of the open probability at Vj=0.
The ability of the R15W and H94Y mutants to participate in the formation of heterotypic junctions demonstrates that these mutant connexins can oligomerize into hexamers, insert into the plasma membrane and dock with 32WT and 26WT hemichannels. Presumably they can dock homotypically as well, since the mutations are on the cytoplasmic side of the molecule. This conclusion is supported by examination of stably transfected Neuro2a cell lines expressing chimeric forms of connexin 32 which shows that the Cx32H94Y*EGFP chimera forms intercellular junctional plaques of size and number similar to those formed by the Cx32WT*EGFP chimera (data not shown).
Although V139M oocyte pairs show no coupling above background, pairing with 32WT or 26WT did lead to significant coupling when the V139M side was made sufficiently positive (Fig. 6). The current responses were similar to those of H94Y and R15W heterotypic junctions with 32WT and 26WT, but differed in that the increase in junctional conductance for positivity on the mutant side reversed very slowly at Vj=0. For Fig. 6a–d, the 32WT or 26WT side was stepped successively from +120 mV to −120 mV in 10 mV or 20 mV increments, with 135 s between pulses. In Fig. 6c the current at the beginning of each of the last three traces is very nearly equal to the current at the end of the previous trace, indicating negligible recovery at Vj=0. In Fig. 6a, the current relaxations at the beginning of the responses to negative Vj's (upward deflections) are ascribable to closure of the 32WT hemichannel with more or less wild-type kinetics. Extrapolation back to the beginning of the pulse shows that the change in conductance from the end of one pulse to the beginning of the next is completely ascribable to gating of the 32WT hemichannel; at Vj=0 little or no recovery of the V139M hemichannel is evident. The slow time course of recovery appears to be a property of the V139M hemichannel, because the slow recovery at Vj=0 was seen with heterotypic pairing with both 32WT and 26WT and because the kinetics of the 32WT hemichannel appear relatively preserved in the V139M/32WT heterotypic pairing. With enough time the conductance does drop to levels similar to those seen in paired antisense injected oocytes, suggesting a very low open probability for the channel at Vj=0. Furthermore, when first voltage clamped at Vj=0 the conductance of V139M/26WT and V139M/32WT oocyte pairs was similar to that for antisense injected cells. (The resting potentials of paired oocytes seldom differ by more than 15 mV, making the initial measurement of junctional conductance when voltage clamping a fairly good indication of the steady state conductance at or near Vj=0.)
Fig. 6.
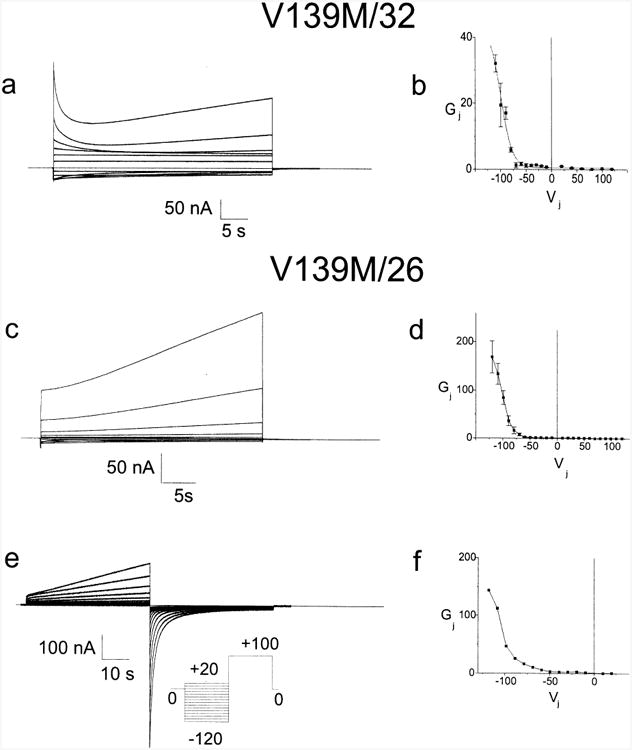
Determination of the steady state Gj–Vj relation for the cell–cell channel formed by heterotypic pairing of oocytes expressing V139M with oocytes expressing 32WT or 26WT. Fig. 6a. Both cells were voltage clamped at −30 mV and the cell expressing the 32WT was stepped between +120 and −120 mV in 10 mV increments as described in the Results. Fig. 6b. Current traces such as those shown in 6a were analyzed as described in the Methods, averaged and plotted as mean±S.E.M. The smooth curves approximating the steady state data correspond to the curves generated by fitting the data to a Boltzmann distribution. Because of very slow closure at Vj=0, current traces could not be normalized to a prepulse prior to being analyzed. Fig. 6c Xenopus oocytes were injected with mRNA for either 26WT or V139M and paired heterotypically. Both cells were voltage clamped at 230 mV and the cell expressing 26WT was successively stepped from +120 to −120 mV in 10 mV increments as described in the Results. Fig. 6d. Current traces such as those shown in Fig. 6c were analyzed as described in the Methods, averaged and plotted as mean±S.E.M. Individual points are connected for clarity. Fig. 6e. Junctional current traces recorded in response to the voltage paradigm pictured in the inset. Both cells were initially clamped to −30 mV. Prior to each test pulse the cell expressing 26WT was subjected to a 45 seconds conditioning pulse such that Vj=+100 mV, closing virtually all cell–cell channels, followed by a 45 s interval at Vj=0 to allow the 26WT hemichannels to reopen. 40 seconds activating test pulses from Vj=−120 mV to Vj=+20 mV were applied. Fig. 6f. Conductance voltage relation determined for the data shown in Fig. 6e. Each activating trace was fit to a sum of exponentials as described in the Methods, to determine the steady state current at that voltage. These currents were then divided by the Vj and plotted to yield Fig. 6g. Individual points are connected for clarity. For a, c and e only traces in 20 mV increments from ±20 to ±120 are shown.
Steady state Gj–Vj relations for each set of current traces were generated as described in the methods to determine steady state currents at each voltage level. The average steady state Gj–Vj relation for V139M/32WT is shown in Fig. 6b. As can be seen, steady state conductance drops to very low levels for Vj>−60 mV. Qualitatively similar steady state current voltage relations were obtained when the V139M hemichannel was paired with 26WT (Fig. 6c and d). As for the H94Y and R15W mutants, there is a significant difference between the results for the heterotypic pairing with 32WT versus 26WT. The explanation for this difference is also likely to be the same as for the H94Y and R15W mutants (see above).
The very slow closure at Vj=0 and slow opening at activating Vj's makes it difficult to determine steady state currents. To accelerate closing of V139M hemichannels and better ascertain the voltage dependence of the steady state conductance voltage relations we used conditioning pulses of +100 mV with respect to Cx26, which would tend to close both WT and mutant hemichannels and reduced Gj to near starting levels. After the conditioning pulse, Vj was returned to 0 mV for 45 s, followed by a 40 second activating pulse of −120 mV, immediately followed by another conditioning pulse. The cycle was then repeated incrementing the activating pulses up to +20 mV in 10 mV steps. The steady state conductance voltage relation generated by this paradigm (Fig. 6f) is similar to that for the steady state conductance voltage relation generated as shown in Fig. 6d, providing further support for the conclusion that V139M has a very low open probability at Vj=0.
Lack of coupling in homotypically paired oocytes expressing V139M can be explained by the reduced open probability of the V139M hemichannel and a negligible residual conductance for the closed hemichannel. A calculation similar to those made for H94Y and R15W predicts that the average conductance of the paired V139M homotypic channels would be <1 nS, a value substantially smaller than that seen in antisense injected oocytes.
4. Discussion
In this communication, we describe the extent of junction formation and the parameters of transjunctional voltage gating of seven mutant forms of Cx32 found in patients with the X-linked form of Charcot-Marie-Tooth disease. All seven missense mutants described here participated in the formation of functional gap junction channels, though three only did so when paired heterotypically with 32WT or 26WT. As described in the Results section, all but one induce alterations in gating of the cell–cell channel. These alterations lead to a reduction in the open probability of the gap junction channels, which in turn would lead to a reduction in gap junction mediated communication. Two mutations (G12S and S85C) lead to a reduction in junctional conductance that is unlikely to be fully explained by alterations in PO; in one case (R15W), factors in addition to alteration in PO may contribute to reduced junctional conductance.
The 32WT mediated gap junction pathway may facilitate communication between the Schwann cell nucleus and the axon; even partial loss of this pathway may have detrimental consequences for either the Schwann cell or the axon it invests. Data presented here suggest that Schwann cells expressing these mutant forms of Cx32 may suffer from either partial or complete loss of 32WT mediated communication. Other connexins may be present in human myelinating cells. However, only 43WT has been identified to date in this cell type, and it does not colocalize with 32WT [27,53]. For this reason, because 43WT does not pair with 32WT (unpublished observations), and because the mutations are not in the extracellular domains, no attempt was made to pair 43WT with the mutants being evaluated.
The mutants described in this communication may be divided into several categories based on (1) their overall efficiency for induction of macroscopic conductances in paired oocytes, (2) their instantaneous conductance voltage relations, and (3) their steady state conductance voltage relations. Table 6 summarizes the alterations of these seven mutants. Cx32 is thought to form ‘reflexive’ junctions between successive cytoplasmic loops in the perinodal regions or at Schmidt–Lanterman incisures. Since there is unlikely to be any significant voltage gradient across these reflexive channels, only the steady state properties at Vj=0 are likely to be physiologically significant. However, significant alterations in the steady state Gj–Vj relations for large Vj and the kinetics of changes in Gj may indicate that significant structural changes are induced by these mutations. Disruption of the normal structure of Cx32 may in turn lead to physiologically significant alterations in either open probability or pore structure. Altered permeability, such as that described for the S26L mutation [32] may also lead to loss of Cx32 function by restricting the passage of critical signaling molecules. In this mutant little change was seen in macroscopic electrical properties or single channel conductance.
The G12S mutation induces only small conductances between paired oocytes. The discrepancy with previous reports that this mutation fails to induce junctional currents [32] may be related to differences in the amount of mRNA injected or to the fact that the conductances induced by expression of this mutation are often near the threshold for detection in oocytes. Interestingly, these small amplitude junctional currents appear to have macroscopic properties very similar to those of 32WT. This suggests that the defect in G12S is not in gating but in expression, trafficking, assembly, insertion or docking of the Cx32 hemichannels. Martin and coworkers [28] recently showed that with G12S there is no defect in oligomerization into hexamers but membrane insertion is defective. Deschenes et al. [14] examined this mutant in transfected PC12 cells and found that the protein was expressed only at very low levels. They did not observe plasma membrane associated G12S, consistent with our finding of very low junctional conductances in paired oocytes.
S85C and H94Q show only minimal alterations in steady state conductance voltage relations. The probability that a homotypic channel will be completely open is the square of the open probability of each apposed hemichannel. S85C is predicted to have an open probability of roughly 0.5 at Vj=0 based on the hemichannel open probability of 0.7 estimated from heterotypic pairings. It seems unlikely that a reduction of this magnitude would lead to significant Schwann cell dysfunction. However, the S85C mutation leads to a marked reduction in the conductance induced in paired oocytes. A decrease in functional expression of this protein might also occur in Schwann cells, though this will clearly require further study. H94Q also shows marked alterations in its kinetic properties, suggesting that structural alterations in the channel may be more extensive than what might be surmised from the steady state Gj–Vj plots alone.
The R15Q mutant shows moderate alterations in its steady state conductance voltage relation. R15Q has previously been shown to have a completely normal pattern of cellular distribution in transfected cell lines [14], making alterations in biophysical properties a particularly attractive mechanism of dysfunction. However, based on calculation of R15Q hemichannel open probabilities from heterotypic pairings, the open probability of a homotypic R15Q channel is about 0.25 at Vj=0. This implies that either (1) the myelinating Schwann cell is sensitive to a 75% reduction in open Cx32 channels, or (2) there are other alterations to the permeability properties of this channel that contribute to its overall dysfunction.
R15W, H94Y, and V139M show profound alterations in steady state conductance voltage relations sufficient, in and of themselves, to lead to a nearly complete absence of open gap junction channels. The V139M mutation has previously been shown to form plaques at appositions of transfected cells and to be incompetent to provide a pathway for diffusion of dye between pairs of cells expressing this mutation [14,34]. These findings are consistent with ours. Interestingly, this mutation is reported to confer additional abnormalities in cellular distribution of this protein in transfected cell lines [14]. Whether or not the altered distribution leads to a reduction in junction formation, the reduced open probability would block communication through whatever channels did form.
Each of these mutations has been described in one or more families afflicted with CMTX. The clinical reports are summarized in Table 1. Correlating the effects of various mutations on the manifestations of the disease with effects on the functional properties of Cx32 is confounded by a great degree by variability both between and within families with the same Cx32 mutations. This variability suggests that there are other yet unidentified factors, either environmental or hereditary, which impact on the severity of the disease. Interestingly, the G12S mutation is the only one among the mutations studied here which has been reported to lead to a severe phenotype. The severity might be predicted from its markedly reduced levels of functional expression in oocytes and its markedly reduced levels of expression in transfected mammalian cell lines as assayed by Western Blot and immunocytochemistry [14]. Thus G12S appears to produce an essentially null phenotype and suggests the hypothesis that null mutations will produce a very severe form of the disease. This hypothesis is made somewhat less tenable by the R15W, H94Y, and V139M mutants. Though they induce severe alterations in the steady state Gj–Vj relation, they are reported to cause mild to moderate disease. The poor correlation between clinical manifestations and in vitro data suggests that we may not have yet identified all potential relevant functional properties of Cx32 in the Schwann cell. These may include interactions between Cx32 and other cellular constituents such as calmodulin [42,35,35] and potential contributions of Cx32 to structural stability of the myelin.
In this communication we have presented evidence that four of seven mutant connexins will form homotypic gap junction channels. However, two of these do so with reduced efficiency in the oocyte expression system. The three mutants unable to induce homotypic junctional conductances lead to dramatic alterations in the Gj–Vj relations of the channels, sufficient to prevent communication. These findings support the hypotheses that CMTX is a disease that arises due to either partial or complete loss of function of Cx32. The exact nature of the crucial message transmitted by the gap junction pathway and the function of that message have not yet been identified, and remain a central question in the pathogenesis of CMTX.
Acknowledgments
This work was supported by NIH Grant 1K08 NS0214901 and a Muscular Dystrophy Association Research Grant to CKA, by NIH grant NS-07512 to MVLB (the Sylvia and Robert Olnick Professor of Neuroscience and by NIH grant GM46889 to TAB. Special thanks to Angela Bukaus-kiene for technical assistance.
References
- 1.Abel A, Bone LJ, Messing A, Scherer SS, Fischbeck KH. Studies in transgenic mice indicate a loss of connexin32 function in X-linked Charcot-Marie-Tooth disease. J Neuropathol Exp Neurol. 1999;58:702–710. doi: 10.1097/00005072-199907000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Ainsworth PJ, Bolton CF, Murphy BC, Stuart JA, Hahn AF. Genotype/phenotype correlation in affected individuals of a family with a deletion of the entire coding sequence of the connexin 32 gene. Hum Genet. 1998;103:242–244. doi: 10.1007/s004390050812. [DOI] [PubMed] [Google Scholar]
- 3.Barrio L, Capel J, Jarillo J, Castro C, Revilla A. Species-specific voltage-gating properties of connexin-45 junctions expressed in Xenopus oocytes. Biophys J. 1997;73:757–769. doi: 10.1016/S0006-3495(97)78108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 5.Bevans CG, Kordel M, Rhee SK, Harris AL. Isoform composition of connexin channels determines selectivity among second messengers and uncharged molecules. J Biol Chem. 1998;273:2808–2816. doi: 10.1074/jbc.273.5.2808. [DOI] [PubMed] [Google Scholar]
- 6.Bone LJ, Dahl N, Lensch MW, Chance PF, Kelly T, Le Guern E, Magi S, Parry G, Shapiro H, Wang S, et al. New connexin32 mutations associated with X-linked Charcot-Marie-Tooth disease. Neurology. 1995;45:1863–1866. doi: 10.1212/wnl.45.10.1863. [DOI] [PubMed] [Google Scholar]
- 7.Bruzzone R, White TW, Scherer SS, Fischbeck KH, Paul DL. Null mutations of connexin 32 in patients with X-linked Charcot-Marie-Tooth disease. Neuron. 1994;13:1253–1260. doi: 10.1016/0896-6273(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 8.Bukauskas F, Jordan K, Bukauskiene A, Bennett M, Lampe P, Laird D, Verselis VK. Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc Natl Acad Sci USA. 2000;97:2556–2561. doi: 10.1073/pnas.050588497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bukauskas FF, Elfgang C, Willecke K, Weingart R. Biophysical properties of gap junction channels formed by mouse connexin 40 in induced pairs of transfected human HeLa cells. Biophys J. 1995;68:2289–2298. doi: 10.1016/S0006-3495(95)80411-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bukauskas FF, Elfgang C, Willecke K, Weingart R. Heterotypic gap junction channels (connexin26–connexin32) violate the paradigm of unitary conductance. Pflugers Arch. 1995;429:870–872. doi: 10.1007/BF00374812. [DOI] [PubMed] [Google Scholar]
- 11.Bukauskas FF, Kempf C, Weingart R. Cytoplasmic bridges and gap junctions in an insect cell line (Aedes albopictus) Exp Physiol. 1992;77:903–911. doi: 10.1113/expphysiol.1992.sp003657. [DOI] [PubMed] [Google Scholar]
- 12.Bukauskas FF, Peracchia C. Two distinct gating mechanisms in gap junction channels: CO2-sensitive and voltage-sensitive. Biophys J. 1997;72:2137–2142. doi: 10.1016/S0006-3495(97)78856-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukauskas FF, Weingart R. Multiple conductance states of newly formed single gap junction channels between insect cells. Pflugers Arch. 1993;423:152–154. doi: 10.1007/BF00374973. [DOI] [PubMed] [Google Scholar]
- 14.Deschenes SM, Walcott JL, Wexler TL, Scherer SS, Fischbeck KH. Altered trafficking of mutant connexin 32. J Neurosci. 1997;17:9077–9084. doi: 10.1523/JNEUROSCI.17-23-09077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fairweather N, Bell C, Cochrane S, Chelly J, Wang S, Mostacciuolo ML, Monaco AP, Haites NE. Mutations in the connexin 32 gene in X-linked dominant Charcot-Marie-Tooth disease (CMTX1) Hum. Mol Genet. 1994;3:29–34. doi: 10.1093/hmg/3.1.29. [DOI] [PubMed] [Google Scholar]
- 16.Fischbeck KH, Abel A, Lin GS, Scherer SS. X-linked Charcot-Marie-Tooth disease and connexin 32. Ann NY Acad Sci. 1999;883:36–41. [PubMed] [Google Scholar]
- 17.Fischbeck KH, Rushdi N, Pericak-Vance M, Rozear M, Roses AD, Fryns JP. X-linked neuropathy: gene localization with DNA probes. Ann Neurol. 1986;20(4):527–532. doi: 10.1002/ana.410200414. [DOI] [PubMed] [Google Scholar]
- 18.Fryns JP, Van den Berghe H. Sex-linked recessive inheritance in Charcot-Marie-Tooth disease with partial clinical manifestations in female carriers. Hum Genet. 1980;55:413–415. doi: 10.1007/BF00290228. [DOI] [PubMed] [Google Scholar]
- 19.Garbern J, Chen L, Kant J. Identification of a novel connexin 32 mutation associated with X-Linked dominant Charcot-Marie-Tooth disease. Neurology. 1996;46:A210. [Google Scholar]
- 20.Gutierrez A, England JD, Sumner AJ, Ferer S, Warner LE, Lupski JR, Garcia CA. Unusual electrophysiological findings in X-linked dominant Charcot-Marie-Tooth disease. Muscle Nerve. 2000;23:182–188. doi: 10.1002/(sici)1097-4598(200002)23:2<182::aid-mus6>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 21.Hahn AF, Bolton CF, White CM, Brown WF, Tuuha SE, Tan CC, Ainsworth PJ. Genotype/phenotype correlations in X-linked dominant Charcot-Marie-Tooth disease. Ann NY Acad Sci. 1999;883:366–382. [PubMed] [Google Scholar]
- 22.Harris AL, Spray DC, Bennett MVL. Kinetic properties of a voltage-dependent junctional conductance. J Gen Physiol. 1981;77:95–117. doi: 10.1085/jgp.77.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heimler A, Friedman E, Rosenthal AD. Naevoid basal cell carcinoma syndrome and Charcot-Marie-Tooth disease: two autosomal dominant disorders segregating in a family. J Med Genet. 1978;15:288–291. doi: 10.1136/jmg.15.4.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ionasescu V, Searby C, Ionasescu R. Point mutations of the connexin32 (GJB1) gene in X-linked dominant Charcot-Marie-Tooth neuropathy. Hum Mol Genet. 1994;3:355–358. doi: 10.1093/hmg/3.2.355. [DOI] [PubMed] [Google Scholar]
- 25.Ionasescu VV, Searby C, Ionasescu R, Neuhaus IM, Werner R. Mutations of the noncoding region of the connexin32 gene in X-linked dominant Charcot-Marie-Tooth neuropathy. Neurology. 1996;47:541–544. doi: 10.1212/wnl.47.2.541. [DOI] [PubMed] [Google Scholar]
- 26.Janssen EA, Kemp S, Hensels GW, Sie OG, de Die-Smulders CE, Hoogendijk JE, de Visser M, Bolhuis PA. Connexin 32 gene mutations in X-linked dominant Charcot-Marie-Tooth disease (CMTX1) Hum Genet. 1997;99:501–505. doi: 10.1007/s004390050396. [DOI] [PubMed] [Google Scholar]
- 27.Mambetisaeva ET, Gire V, Evans WH. Multiple connexin expression in peripheral nerve, Schwann cells and Schwannoma cells. J Neurosci Res. 1999;57:166–175. doi: 10.1002/(SICI)1097-4547(19990715)57:2<166::AID-JNR2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 28.Martin PE, Mambetisaeva ET, Archer DA, George CH, Evans WH. Analysis of gap junction assembly using mutated connexins detected in Charcot-Marie-Tooth X-linked disease. J Neurochem. 2000;74:711–720. doi: 10.1046/j.1471-4159.2000.740711.x. [DOI] [PubMed] [Google Scholar]
- 29.Nelis E, Haites N, Van broeckhoven C. Mutations in the peripheral myelin genes and associates genes in inherited peripheral neuropathies. Human Mutat. 1999;13:11–28. doi: 10.1002/(SICI)1098-1004(1999)13:1<11::AID-HUMU2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 30.Niessen H, Willecke K. Strongly decreased gap junctional permeability to inositol 1,4, 5-trisphosphate in connexin32 deficient hepatocytes. FEBS Lett. 2000;466:112–114. doi: 10.1016/s0014-5793(99)01770-6. [DOI] [PubMed] [Google Scholar]
- 31.Obaid AL, Socolar SJ, Rose B. Cell-to-cell channels with two independently regulated gates in series: analysis of junctional conductance modulation by membrane potential, calcium, and pH. J Membr Biol. 1983;73:69–89. doi: 10.1007/BF01870342. [DOI] [PubMed] [Google Scholar]
- 32.Oh S, Ri Y, Bennett MV, Trexler EB, Verselis VK, Bargiello TA. Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron. 1997;19:927–938. doi: 10.1016/s0896-6273(00)80973-3. [DOI] [PubMed] [Google Scholar]
- 33.Oh S, Rubin JB, Bennett MV, Verselis VK, Bargiello TA. Molecular determinants of electrical rectification of single channel conductance in gap junctions formed by connexins 26 and 32. J Gen Physiol. 1999;114:339–364. doi: 10.1085/jgp.114.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Omori Y, Mesnil M, Yamasaki H. Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: functional defects and dominant negative effects. Mol Biol Cell. 1996;7:907–916. doi: 10.1091/mbc.7.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peracchia C, Wang XG, Peracchia LL. Chemical gating of gap junction channels. Methods. 2000;20:188–195. doi: 10.1006/meth.1999.0936. [DOI] [PubMed] [Google Scholar]
- 36.Ri Y, Ballesteros JA, Abrams CK, Oh S, Verselis VK, Weinstein H, Bargiello TA. The role of a conserved proline residue in mediating conformational changes associated with voltage gating of Cx32 gap junctions. Biophys J. 1999;76:2887–2898. doi: 10.1016/S0006-3495(99)77444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubin JB, Verselis VK, Bennett MV, Bargiello TA. A domain substitution procedure and its use to analyze voltage dependence of homotypic gap junctions formed by connexins 26 and 32. Proc Natl Acad Sci USA. 1992;89:3820–3824. doi: 10.1073/pnas.89.9.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubin JB, Verselis VK, Bennett MV, Bargiello TA. Molecular analysis of voltage dependence of heterotypic gap junctions formed by connexins 26 and 32. Biophys J. 1992;62:183–193. doi: 10.1016/S0006-3495(92)81804-0. Discussion 193-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scherer SS, Deschenes SM, Xu YT, Grinspan JB, Fisch-beck KH, Paul DL. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci. 1995;15:8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senderek J, Bergmann C, Quasthoff S, Ramaekers VT, Schroder JM. X-linked dominant Charcot-Marie-Tooth disease: nerve biopsies allow morphological evaluation and detection of connexin32 mutations (Arg15Trp, Arg22Gln) Acta Neuropathol (Berl) 1998;95:443–449. doi: 10.1007/s004010050823. [DOI] [PubMed] [Google Scholar]
- 41.Senderek J, Hermanns B, Bergmann C, Boroojerdi B, Bajbouj M, Hungs M, Ramaekers VT, Quasthoff S, Karch D, Schroder JM. X-linked dominant Charcot-Marie-Tooth neuropathy: clinical, electrophysiological, and morphological phenotype in four families with different connexin 32 mutations. J Neurol Sci. 1999;167:90–101. doi: 10.1016/s0022-510x(99)00146-x. [DOI] [PubMed] [Google Scholar]
- 42.Torok K, Stauffer K, Evans WH. Connexin 32 of gap junctions contains two cytoplasmic calmodulin-binding domains. Biochem J. 1997;326:479–483. doi: 10.1042/bj3260479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trexler EB, Bennet MV, Bargiello TA, Verselis VK. Rapid and direct effects of pH on connexins revealed by the connexin46 hemichannel preparation. J Gen Physiol. 1999;113(5):721–742. doi: 10.1085/jgp.113.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trexler EB, Bennett MV, Bargiello TA, Verselis VK. Voltage gating and permeation in a gap junction hemichannel. Proc Natl Acad Sci USA. 1996;93:5836–5841. doi: 10.1073/pnas.93.12.5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.VanSlyke JK, Deschenes SM, Musil LS. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol Biol Cell. 2000;11:1933–1946. doi: 10.1091/mbc.11.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verselis VK, Bennett MV, Bargiello TA. A voltage-dependent gap junction in Drosophila melanogaster. Biophys J. 1991;59:114–126. doi: 10.1016/S0006-3495(91)82204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verselis VK, Ginter CS, Bargiello TA. Opposite voltage gating polarities of two closely related connexins. Nature. 1994;368:348–351. doi: 10.1038/368348a0. [DOI] [PubMed] [Google Scholar]
- 48.Wang XG, Peracchia C. Connexin 32/38 chimeras suggest a role for the second half of inner loop in gap junction gating by low pH. Am J Physiol. 1996;271:C1743–C1749. doi: 10.1152/ajpcell.1996.271.5.C1743. [DOI] [PubMed] [Google Scholar]
- 49.Wang XG, Peracchia LL, Peracchia C. Chimeric evidence for a role of the connexin cytoplasmic loop in gap junction channel gating. Pflugers Arch. 1996;431:844–852. doi: 10.1007/s004240050076. [DOI] [PubMed] [Google Scholar]
- 50.Wang XG, Peracchia C. Molecular dissection of a basic COOH-terminal domain of Cx32 that inhibits gap junction gating sensitivity. Am J Physiol. 1998;275:C1384–C1390. doi: 10.1152/ajpcell.1998.275.5.C1384. [DOI] [PubMed] [Google Scholar]
- 51.Wicklein EM, Orth U, Gal A, Kunze K. Missense mutation (R15W) of the connexin32 gene in a family with X chromosomal Charcot-Marie-Tooth neuropathy with only female family members affected. J Neurol Neurosurg Psychiatry. 1997;63:379–381. doi: 10.1136/jnnp.63.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilders R, Jongsma HJ. Limitations of the dual voltage clamp method in assaying conductance and kinetics of gap junction channels. Biophys J. 1992;63:942–953. doi: 10.1016/S0006-3495(92)81664-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshimura T, Satake M, Kobayashi T. Connexin 43 is another gap junction protein in the peripheral nervous system. J Neurochem. 1996;67:1252–1258. doi: 10.1046/j.1471-4159.1996.67031252.x. [DOI] [PubMed] [Google Scholar]