

Abstract
The rapid spread of dengue is a worldwide public health problem. In two clinical studies of dengue in Managua, Nicaragua, we observed an abrupt increase in disease severity across several epidemic seasons of dengue virus serotype 2 (DENV-2) transmission. Waning DENV-1 immunity appeared to increase the risk of severe disease in subsequent DENV-2 infections after a period of cross-protection. The increase in severity coincided with replacement of the Asian/American DENV-2 NI-1 clade with a new virus clade, NI-2B. In vitro analyses of viral isolates from the two clades and analysis of viremia in patient blood samples support the emergence of a fitter virus in later, relative to earlier, epidemic seasons. In addition, the NI-1 clade of viruses was more virulent specifically in children who were immune to DENV-1, while DENV-3 immunity was associated with more severe disease among NI-2B infections. Our data demonstrate that the complex interaction between viral genetics and population dynamics of serotype-specific immunity contribute to the risk of severe dengue disease. Furthermore, this work provides insights into viral evolution and the interaction between viral and immunological determinants of viral fitness and virulence.
INTRODUCTION
Dengue virus (DENV) is a mosquito-borne, enveloped flavivirus, with an RNA genome of 10.7 kb that is translated into 3 structural (C, prM/M, and E) and 7 nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4A, NS5) proteins. DENV infection results in a range of disease severity, from asymptomatic infection to dengue fever (DF), a debilitating but self-limited febrile illness, to the more severe, potentially fatal dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). DHF and DSS are characterized by increased vascular permeability, low numbers of circulating platelets (thrombocytopenia) and hemorrhagic manifestations; DSS ensues when plasma leakage results in low blood pressure–induced shock that can lead to death (1). DENV infections frequently occur in the context of pre-existing immunity in the host to another one of the four DENV serotypes (2). Immune responses to prior DENV infection play an important role in determining the outcome of subsequent heterologous infection with a serotype distinct from the previous infection (3–9). Studies in human volunteers (10) and in non-human primates (11) have shown that, after an initial infection with one DENV serotype, the individual is protected for a period of time from dengue disease and/or high viral load when infected with a heterologous DENV serotype. However, this immunity is short-lived, and infection with a heterologous DENV serotype after longer time intervals leads to increased risk of a more severe clinical phenotype, a phenomenon termed enhancement (3–6, 9, 12, 13). The precise time intervals of cross-protection and enhancement during heterologous DENV infection are not well defined. This enhanced disease can result from antibody-dependent enhancement (ADE) whereby subneutralizing concentrations of antibodies directed to a DENV serotype from a previous infection result in increased viral replication in Fcγ receptor (FcγR)–bearing myeloid cells (14–17) and/or from potentially harmful T cell responses that are directed primarily toward the prior rather than the current infecting serotype (18–20). Mathematical modeling and analysis of dengue incidence data in endemic populations have suggested both cross-protection from and enhancement of infection by heterologous DENV strains as the main drivers of periodic fluctuations in the incidence of different DENV serotypes within a given geographical location across time (21–24). However, the risk of severe disease upon DENV infection cannot be explained completely by a misdirected host immune response to a prior infecting serotype (3, 5–7, 10, 25–29); rather, disease severity appears to be determined by a combination of multiple host (30–37) and viral factors, including the genetics of the infecting viral strain (38–48).
Each of the four DENV serotypes is composed of several genotypes, which in turn consist of various clades. DENV evolution is characterized by lineage turnover in which an entire clade of circulating viruses is replaced by a new clade (49–52). Certain genotypes within DENV serotypes and certain clades within genotypes have been shown to be more frequently associated with severe disease outcomes (53–55). Fitter viruses (fitness is defined here as the ability to replicate better in a given environment (56)) that replicate more efficiently are often hypothesized to be more pathogenic in the host (53). In support of this concept, high levels of virus in patient sera (viremia) have been associated with severity of dengue disease (57, 58). Consistent with a model in which fitter viruses are more virulent, DENV-2 viruses of Asian origin are capable of more robust replication in vitro relative to the less virulent American DENV-2 genotype viruses (45, 59–64). Further, Asian DENV-2 viruses that are more frequently associated with severe disease are less sensitive than American genotype DENV-2 viruses to antibody-mediated neutralization by sera from individuals previously infected with DENV-1 (65), suggesting that mutations that may alter the neutralization profile of certain DENV genotypes may lead to increased fitness relative to other strains of the same serotype.
The spread of dengue over the past several decades has made this disease a major public health concern worldwide. Previous work has clearly shown that the risk of severe dengue disease is higher in a secondary DENV infection, especially for DENV-2 (3, 4). Further, it has been shown that particular genotypes of DENV increase risk of severe disease (53). However, it is not well understood how the specific interplay between serotype-specific immunity and viral genetics affects risk of severe dengue disease. Such an understanding is not only essential for advancing our understanding of DENV epidemiology and pathogenesis but is also important for the development of safe and effective dengue vaccines. Here we took advantage of a rare observation in two population-based studies in Nicaragua in which a clade replacement in the context of DENV-2 infection occurred concurrently with an increase in dengue disease severity. We combine epidemiological, phylogenetic, virologic, serological, and clinical analyses to provide a more precise understanding of the contributions of DENV genetics and pre-existing immunity to dengue disease severity.
RESULTS
Association of Prior DENV-1 Exposure with Increased Dengue Severity in Later Seasons
Here we report our findings from two independent studies in Managua, Nicaragua (one hospital-based study, termed here the Hospital study and one prospective cohort study, termed here the Cohort study) from the 2004/5 to the 2008/9 epidemic seasons. Children in the Hospital study were enrolled upon presentation to the hospital with relatively more severe disease, allowing us to examine factors related to severe dengue (defined as DHF/DSS according to the WHO 1997 guidelines (1)); in contrast, children in the community-based prospective Cohort study presented earlier and demonstrated a lower rate of severe disease. In addition, annual healthy blood samples collected throughout the Cohort study provided a rich source of material with which to investigate questions related to prior DENV immunity (see Materials and Methods for detailed study descriptions; see Table 1 for demographic information).
Table 1.
Characteristics of DENV-2 cases, Cohort study, 2004/5–2008/9, and Hospital study, 2005/6–2008/9.
| Demographic Characteristics | Cohort study | Hospital study | ||
|---|---|---|---|---|
| 2004/5 – 2005/6 | 2006/7 – 2008/9 | 2005/6 | 2006/7 – 2008/9 | |
| N (%)a | N (%)a | N (%)a | N (%)a | |
| DENV-2 Cases | 30 | 65 | 34 | 102 |
| Sex | ||||
| Female | 12 (40) | 28 (43) | 16 (47) | 48 (47) |
| Male | 18 (60) | 37 (57) | 18 (53) | 54 (53) |
| Age, years (mean) | 5.2 | 7.8 | 8.3 | 8.2 |
| Day of presentation after onset of symptoms (mean) | 2.0 | 2.1 | 4.1 | 4.7 |
| Immune Status | ||||
| Primary | 10 (33) | 23 (35) | 5 (15) | 15 (15) |
| Secondary | 20 (67) | 42 (65) | 29 (85) | 87 (85) |
| Birth Group | ||||
| <1994 | 0 (0) | 0 (0) | 9 (26) | 13 (13) |
| 1994–1995 | 6 (20) | 3 (4) | 7 (21) | 21 (21) |
| 1996–1998 | 11 (37) | 31 (48) | 10 (29) | 23 (23) |
| 1999–2001 | 10 (33) | 17 (26) | 0 (0) | 16 (16) |
| 2002–2004 | 3 (10) | 14 (22) | 5 (15) | 22 (22) |
| 2005+ | 0 (0) | 0 (0) | 3 (9) | 7 (7) |
| WHO Classification | ||||
| Dengue Fever (DF) | 30 (100) | 56 (86) | 24 (71) | 38 (37) |
| Dengue Hemorrhagic Fever (DHF) | 0 (0) | 6 (9) | 7 (21) | 37 (36) |
| Dengue Shock Syndrome (DSS) | 0 (0) | 3 (5) | 3 (9) | 27 (26) |
| Lowest Platelet Count (cells/mL) (mean) | 249,000 | 174,000 | 129,000 | 66,000 |
n = number of patients; % = % of total
In our Hospital study, 29% of DENV-2 hospitalized cases were classified as DHF/DSS in the 2005/6 season, and this proportion increased to 63% across the 2006/7–2008/9 seasons (P = 0.001; Fig. 1A). We observed a similar trend over the same time-frame among symptomatic DENV-2 infections in our Cohort study (P = 0.05; Fig. 1B). In both studies, DENV-2 was the dominant circulating serotype (Fig. 1C, Hospital study; Fig. 1D, Cohort study), which mirrors the serotype circulation dynamics reported for Managua by the Ministry of Health as a part of the Nicaraguan National Surveillance System (fig. S1). Furthermore, most symptomatic DENV-2 infections occurred in children already immune to another DENV serotype (85% of Hospital cases; 65% of Cohort cases; Fig. 1E, Table 1), including the majority of severe cases in both studies (91% of Hospital DHF/DSS cases; 89% of Cohort DHF/DSS cases).
Fig. 1. Increase in severity of DENV-2 infections in two independent studies of pediatric dengue in Nicaragua.
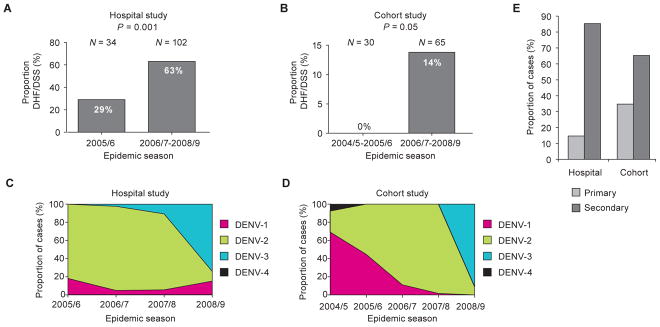
(A, B) The proportion of DENV-2 cases, including both primary and secondary infections, classified as DHF/DSS in the early as compared to later seasons in the Hospital (P = 0.001, Fisher’s exact test) (A) and Cohort (P = 0.05, Fisher’s exact test) (B) studies. The total number (N) of DENV-2 infections included in the analysis is shown above each graph. (C, D) The proportion of each serotype across four epidemic seasons (2005/6–2008/9) in the Nicaraguan Hospital study (C) and five epidemic seasons (2004/5–2008/9) in the Nicaraguan Pediatric Dengue Cohort study (D). Pink = DENV-1; green = DENV-2; blue = DENV-3; black = DENV-4. (E) The proportion of all DENV-2 cases classified as primary (light gray bars) or secondary (dark gray bars) DENV infections in the Hospital and Cohort studies, as defined in Materials and Methods.
Prior to the circulation of DENV-2 in the epidemic seasons described here, DENV-3 (1994–8), DENV-2 (1999–2002) and DENV-1 (2002–5) circulated in Nicaragua, as did very low levels of DENV-4 (48, 66–71) (Fig. 2A). We used this serotype timeline to estimate risk of exposure of children in both studies to specific heterologous serotypes by birth year, as children from the same birth year share a common risk of DENV exposure over time. In the Cohort study, we were able to estimate DENV seroprevalence for the entire cohort for each birth year from the annual samples collected prior to each season of dengue transmission. These data showed that children born before 1999 had higher rates of exposure to DENV than children born in 1999 or later (Fig. 2B). Referring to the serotype timeline, we inferred that children born before 1999 in our studies would likely have been exposed to DENV-3 (“DENV-3 era”), whereas children born in 1999 or later (“post-DENV-3 era”) would have been exposed to either DENV-2, which circulated 1999–2002 (“DENV-2 era”), or DENV-1, which circulated in 2002–2005 (“DENV-1 era”). We tested a panel of DENV-immune sera from our Cohort study to determine the serotype of primary infection prior to subsequent DENV-2 infection and, of the 81% of samples for which we could make a determination, we found that indeed most DENV-immune children born before 1999 had been exposed to DENV-3, while all children born in 1999 or after had been exposed only to DENV-1 (Fig. 2C).
Fig. 2. DENV serotype exposure across seasons in Nicaragua.

(A) The relative circulation of DENV serotypes (DENV-1 in pink; DENV-2 in green; DENV-3 in blue) by year in Nicaragua from 1994 to 2009 as derived from various DENV epidemiological studies across these years (48, 66–69). (B) The proportion of children from the Cohort study who were immune to DENV plotted as a function of birth year. Immunity was measured annually by Inhibition ELISA in samples collected prior to each epidemic season. The dotted lines separate “DENV-3-era” (born before 1999; blue), “DENV-2-era” (born between 1999 and 2002; green) and “DENV-1-era” (born between 2003 and 2006; pink) children as analyzed in our study. Birth year color codes correspond to dominant sertoype as defined in panel A and are shown to the right of the graph. (C) Proportion of patients in the Cohort study in each birth year immune to DENV-1 (pink) or DENV-3 (blue) as determined by assaying neutralizing antibody titers (NT50 determined using RVP assay) to DENV-1 and DENV-3; N is the total number of samples analyzed from each year; the number of samples assayed in each year that did not generate significant titers for either DENV-1 or DENV-3 were 1/5 (1995), 1/8 (1996), 0/10 (1997), 3/5 (1998), 0/1 (1999), 1/3 (2000), 2/8 (2001), 0/2 (2003); nd, not determined.
We also found that the proportion of secondary DENV-2 Hospital cases in “post-DENV-3-era” children, as compared to “DENV-3-era” children, increased significantly after the 2005/6 season (from 14% to 40%; P = 0.02; Fig. 3A). Thus, risk for symptomatic infection changed over time, such that DENV-1-immune children contributed increasingly to Hospital cases in later seasons (2006/7–2008/9), the same epidemic seasons in which we observed increased severity (Fig. 1, A and B). These data suggest that after DENV-1 exposure, cross-reactive immunity decreased the risk of symptomatic infection in the 2005/6 season, but increased the risk of severe disease in later seasons as immunity waned. In the same study, we did not observe a significant increase in severity across seasons in the predominantly DENV-1 immune group (“post-DENV-3-era”; P = 1.00; Fig 3B), although the extremely small number of cases (n = 4) in the 2005/6 season may be masking an increase in severity in later seasons in this group. However, in this study we also noted an increase in severity across seasons in the predominantly DENV-3 immune group (“DENV-3-era”; i.e., children born before 1999) from 28% to 71% (P = 0.0005; Fig. 3C), suggesting that waning DENV-1 immunity did not entirely explain the increase in severity observed across seasons. Therefore, an additional factor, such as viral genetics, must have also played a role.
Fig. 3. Higher frequency of DENV-2 infection in DENV-1–immune children in later (2006/7–2008/9), versus earlier (2005/6) seasons.
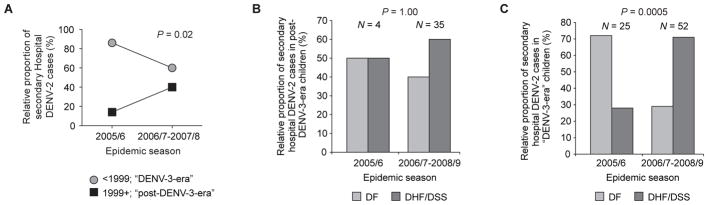
(A) Relative proportion of secondary DENV-2 cases in the Hospital study that occurred across seasons, stratified by birth group (circles, <1999, “DENV-3-era”; squares, 1999+, “post-DENV-3-era”; P = 0.02, Rao Scott chi square test, comparing the proportion of secondary DENV-2 Hospital cases from the “post-DENV-3-era” birth group (as opposed to the “DENV-3-era” birth group) in early versus later seasons, adjusting for the decreased representation of children from the older birth group in later seasons). (B) Relative proportion of secondary DENV-2 cases in “post-DENV-3-era” children (1999+) in the Hospital study classified as DF (light gray bars) or DHF/DSS (dark gray bars) (P = 1.00; Fisher’s exact test, comparing the proportion of secondary DENV-2 Hospital cases that are DHF/DSS in early versus later seasons among “post-DENV-3-era” children). (C) Relative proportion of secondary DENV-2 cases in “DENV-3-era” children (<1999) in the Hospital study classified as DF (light bars) or DHF/DSS (dark gray bars) (P = 0.0005; Fisher’s exact test, comparing the proportion of secondary DENV-2 Hospital cases that are DHF/DSS in early versus later seasons among “DENV-3-era” children).
Association of Increased Viral Fitness with Clade Replacement in Nicaraguan DENV-2 Infection
We derived complete coding-region and partial untranslated-region (UTR) consensus sequences from patients infected with DENV-2 in both the Hospital and Cohort studies from the 2004/5 season through the 2008/9 season, as well as from viruses previously isolated during DENV-2 epidemics in Nicaragua (1999–2002). Through phylogenetic analysis of these complete coding-region sequences, we found that in the context of the American, Asian 1, and Asian/American genotypes, Nicaraguan sequences formed a unique clade (NI) within the Asian/American genotype (Fig. 4A). Of the viruses circulating in the 2004/5 and 2005/6 seasons, the majority formed a single clade defined as NI-1 that was replaced after the 2005/6 season by a related, yet independently derived clade of viruses, NI-2 (Fig. 4, A–C; fig. S2A). NI-2 was composed of two subclades, NI-2A and NI-2B, with NI-2A contributing relatively few sequences and NI-2B rapidly coming to dominance after the 2005/6 epidemic season (Fig. 4, A–C), supported by an increase in relative genetic diversity at the time of clade replacement (fig. S2, B and C). On the basis of nucleotide ancestral node reconstructions, which represent the hypothetical ancestral sequence of a clade as determined by extant sequences, we observed a total of 36 amino acid residues at which Nicaraguan virus sequences showed nonsynonymous variation that altered the encoded amino acid as compared to the American, Asian 1, and Asian-American DENV-2 ancestral sequences (table S1). The NI-1 to NI-2B clade replacement event was associated with nine nonsynonymous mutations, as well as 4 mutations in the viral UTRs that together distinguished NI-1 from NI-2B viruses (Table 2). Three mutations from the ancestral NI sequence occurred in the NI-1 lineage (R97K in capsid (C), K94R in nonstructural protein 1 (NS1) and P245T in nonstructural protein 3 (NS3)), one mutation occurred in the evolution of all NI-2 viruses (N245S in NS4B), and five mutations occurred during the evolution of the NI-2B clade (M492V in envelope (E), L279F in NS1, and K200Q, T290I and R401K in nonstructural protein 5 (NS5); bold in Table 2). Notably, assessment of all publicly available full-length DENV genomes (n=2,660) and available E gene sequences (n=1,650) revealed that several of the nine nonsynonymous mutations that occurred during the evolution of NI-2B viruses have previously been observed only rarely in sampled DENV-2 sequences; specifically, the NI-2B mutations N245S in NS4B and K200Q/T290I in NS5 are novel, with only a single previous observation of T290I in any DENV serotype (table S2). In addition, several different methods were implemented to test for positive selection (see Supplemental Methods), but in all site and branch-site models, log likelihoods of the alternate and null models indicated no significant evidence of selection.
Fig. 4. Association of Nicaraguan DENV-2 clade replacement with increased viral fitness.
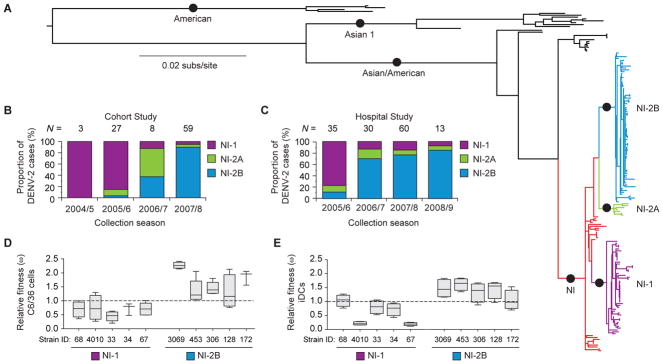
(A) Maximum likelihood tree of 159 complete coding-region nucleotide sequences sampled from the Cohort and Hospital studies, as well as other samples collected in Nicaragua (see Materials and Methods for details) between 1999 and 2008. Twenty-five sequences representing the major genotypes of DENV-2 (American, Asian 1 and Asian/American) were used as outgroup sequences, as they were genetically distinct from the Nicaraguan sequences. Only nodes (circles) that are supported at least 60% of the time by bootstrap re-sampling (1000 replications) of the data are shown. Major Nicaraguan clades are highlighted (NI = red; NI-1 = purple; NI-2A = green; NI-2B = blue). (B, C) Changes over time in the proportion of DENV-2 cases caused by infection with major Nicaraguan clades that occurred during the specified epidemic seasons for virus isolates that were either sequenced or genotyped (B) in the Cohort study (2004/5–2007/8; N = 97) or in (C) the Hospital study (2005/6–2008/9; N = 138). (D, E) Relative fitness (ω) as determined by dual-infection assays of NI-1 isolates in competition with each NI-2B isolate as assayed in (D) mosquito cells (C6/36) and (E) human monocyte-derived immature dendritic cells (iDCs). The proportion of each isolate present in the cell supernatant at a given time-point after infection (fo) was divided by its initial proportion in the inoculum (io) to derive a relative fitness (ω) value (ω = fo/io) (72); fo = 4 days for C6/36 cells; fo = 2 days for iDCs. A relative fitness value of 1, the neutral expectation given equal fitness of viral isolates, is shown as a dotted line. Box plots show the 25th and 75th percentiles with a line indicating the median relative fitness value and error bars showing the minimum and maximum fitness values. Five NI-1 viruses were competed against 5 NI-2B viruses. Viral strain numbers used in each assay are indicated at the bottom of the graph.
Table 2.
Nonsynonymous amino acid mutations and UTR mutations that distinguish Nicaraguan DENV-2 cladesa.
| Gene | Positionb | Annotationc | NId (N=159) | NI-1 (N=50) | NI-2A (N=7) | NI-2B (N=80) |
|---|---|---|---|---|---|---|
| 5′UTRe | -21 | - | TTTTT- | TTTTT- | TTTTTTf | TTTTTTf |
| C | 97 | - | R (AGG) | K (AAG)f | R (AGG) | R (AGG) |
| E | 492 | - | M (ATG) | M (ATG) | M (ATG) | V (GTG)f |
| NS1 | 94 | - | K (AAA) | R (AGA)f | K (AAA) | K (AAA) |
| NS1 | 279 | - | L (CTC) | L (CTC) | L (CTC) | F (TTC)f |
| NS3 | 245 | P | P (CCA) | T (ACA)f | P (CCA) | P (CCA) |
| NS4B | 245 | - | N (AAC) | N (AAC) | S (AGC)f | S (AGC)f |
| NS5 | 200 | C | K (AAA) | K (AAA) | K (AAA) | Q (CAA)f |
| NS5 | 290 | P | T (ACA) | T (ACA) | T (ACA) | I (ATA)f |
| NS5 | 401 | - | R (AGA) | R (AGA) | R (AGA) | K (AAA)f |
| NS5 | 523 | P | G (GGC) | G (GGC) | S (AGC)f | G (GGC) |
| NS5 | 642 | - | A (GCT) | A (GCT) | V (GTT)f | A (GCT) |
| 3′UTRg | 19 | - | T | T | T | Cf |
| 3′UTRg | 132 | - | A | A | Gf | Gf |
| 3′UTRg | 145 | - | T | T | Cf | Cf |
Consensus sequences for each clade, NI-1, NI-2A and NI-2B.
Amino acid numbering based on start of each protein.
C = charge change; P = polarity change.
NI is an ancestral sequence inferred by maximum parsimony.
5′UTR: the base preceding the start AUG is -1.
Amino acids or UTR mutations that distinguish NI-1, NI-2A or NI-2B viruses from the Nicaraguan ancestor (“NI”) are shown in bold.
3′UTR: the first base of the stop codon is 1.
To determine whether a fitness increase in clade NI-2B viruses in either the mosquito vector or human host could explain its dominance in later epidemic seasons, we assessed the relative replication of low-passage NI-1 and NI-2B viral isolates in vitro. We adapted a direct viral competition assay (72), in which the cells were infected with an equal proportion of NI-1 and NI-2B viruses, and the relative proportion of each virus in culture supernatant was measured by direct nucleic acid sequencing and analysis of relative peak height with polySNP software (73), which was used to derive a relative fitness value (ω) for each competition (72). In C6/36 mosquito cells, NI-2B viruses had higher median relative fitness values than did NI-1 viruses in multiple pairwise comparisons (Fig. 4D and fig. S3A). Replication of NI-1 and NI-2B viruses was next assayed in primary human monocyte-derived immature dendritic cells (iDCs), in which the NI-2B viruses again demonstrated higher mean relative fitness (Fig. 4E and fig. S3B). Likewise, in the human monocytic cell line U937/DC-SIGN, which expresses the DENV attachment factor DC-SIGN, viral competition assays revealed higher median relative viral fitness of NI-2B viruses as compared to NI-1 (fig. S3 C and D), and similar results were also obtained in a viral replication assay in the K562 hematopoietic cell line, which requires antibody-mediated entry of DENV (fig. S3E).
In addition to this in vitro evidence of relative NI-2B fitness, we found in vivo evidence from Hospital study serum samples collected during acute DENV infection. Analysis of virus levels in these serum samples (viremia) showed a significant relation among clade identity and disease severity and hospital day (P = 0.0003), such that, among DF cases early in the course of infection, NI-2B viruses reached higher viremia compared to NI-1 and NI-2A viruses (fig. S4). Among DHF/DSS cases, all clades exhibited high viremia (fig. S4). Thus, our in vitro and in vivo data indicate that enhanced replicative fitness of NI-2B viruses might have contributed to the displacement of NI-1 viruses by NI-2B viruses in later dengue epidemic seasons.
Contribution of Viral Genetics to Increased Dengue Severity in the Context of Serotype-Specific Immunity
We next analyzed data from our Hospital study to determine whether the arrival of NI-2B viruses into the population could explain the increased severity in later seasons. However, the observed increase in disease severity across seasons was not simply a direct effect of clade replacement, as we saw the same trend of increased severity among those infected with either NI-1 or NI-2B viruses (Fig. 5A). Specifically, 9/26 (35%) NI-1 infections were severe (DHF/DSS) in the 2005/6 season, increasing to 10/14 (71%) in the later seasons (P = 0.05), a pattern similar to that of NI-2B infections, for which no cases (0/4) of severe disease were documented in the 2005/6 season, but 47/77 (61%) NI-2B infections in later seasons were severe (P = 0.03) (Fig. 5A). NI-2A also had low rates of severe disease (25%) in the early season followed by high rates (64%) in the later seasons (Fig. 5A), but the difference was not statistically significant (P = 0.28), likely due to low numbers of NI-2A-infected patients.
Fig. 5. Association of NI-1 and NI-2A infections with more severe disease outcomes in DENV-1-immune children.
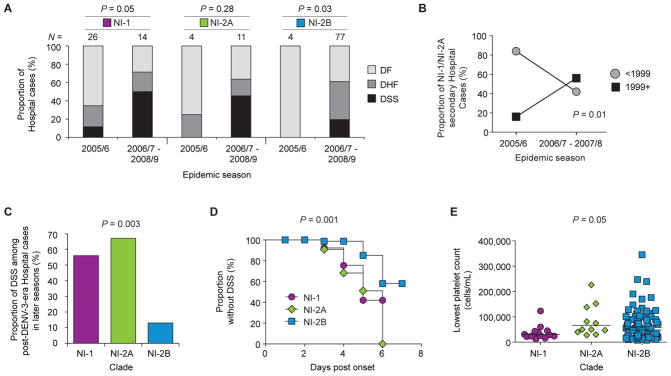
(A) The proportion of dengue cases across seasons by clade—NI-1, NI-2A or NI-2B—classified as DF (light gray), DHF (dark gray) or DSS (black) in the Hospital study. An increase in cases of severe dengue disease (DHF/DSS) was observed in all clades (NI-1, P = 0.05; NI-2A, P = 0.28; NI-2B, P = 0.03; Fisher’s exact test, comparing the proportion of DENV-2 Hospital cases that are DHF/DSS in early versus later seasons for each clade). (B) The proportion of cases of secondary NI-1 and NI-2A infections in the Hospital study across seasons for each birth group (circles, <1999; squares, 1999+; P = 0.01, Rao Scott chi square test, comparing the proportion of NI-1/NI-2A secondary Hospital cases from the “post-DENV-3-era” birth group in early versus later seasons, adjusting for the expected decrease in representation of children from the older birth group in later seasons). (C) The proportion of cases of secondary DENV infections from the 1999+ birth group (DENV-1-immune) in the Hospital study in seasons 2006/7–2008/9 classified as DSS for each clade (NI-1, NI-2A and NI-2B). The proportion of each clade classified as DSS is compared against the null hypothesis that all three clades are at equal proportion (P = 0.003; Fisher’s exact test, comparing the proportion of DENV-2 Hospital cases in the later seasons that are DSS, caused by NI-1 versus NI-2A versus NI-2B, among children from the “post-DENV-3-era” birth group). (D) Kaplan-Meier plot showing the time to DSS critical phase by clade among all cases of DENV-2 infection in the later seasons in the Hospital study plotted as a function of days post-onset of illness. NI-1, circles; NI-2A, diamonds; NI-2B, squares (P = 0.001; Logrank test, comparing the time to develop DSS in the later seasons, NI-1 versus NI-2A versus NI-2B, among children in the Hospital study). (E) Lowest platelet count (cells/mL) measured during illness episode from children infected with each of the DENV-2 clade viruses in the Hospital study in later seasons (2006/7–2008/9). NI-1, circles; NI-2A, diamonds; NI-2B, squares. P = 0.05; Kruskal-Wallis, comparing the lowest platelet count, NI-1 versus NI-2A versus NI-2B, among DENV-2 Hospital cases in the later seasons. Line indicates the geometric mean. For all panels, clade designations are colored as in Figure 4.
Although a direct correlation of clade and disease severity was not initially observed, we found that clade in the context of serotype-specific immunity impacted disease outcome. In the later seasons (2006/7–2007/8), the proportion of all secondary NI-1/NI-2A infections that occurred in “post-DENV-3-era” children (“1999+”; Fig. 5B) increased from 16% to 58% as compared to “DENV-3-era” children (P = 0.01), suggesting an interplay between NI-1/NI-2A viruses and DENV-1 immunity. The effect in “post-DENV-3-era” children appears to be specific to the most severe form of dengue, DSS, as 5/9 (56%) NI-1 and 4/6 (67%) NI-2A cases, as compared to 4/30 (13%) NI-2B cases, were classified as DSS in later seasons (2006/7–2008/9) (P = 0.003) (Fig. 5C). No significant difference in the rate of severe disease was observed when DHF and DSS were analyzed together (P = 0.54; Fig. S5). In further support of the increased severity of NI-1/NI-2A infections as compared to NI-2B infections, we observed that NI-1 cases developed DSS significantly earlier in the course of illness than did NI-2B cases (P = 0.001) (Fig. 5D) and that NI-1 infections in the later seasons resulted in lower platelet counts (a sign of more severe disease (69, 74)) when compared with NI-2A and NI-2B infections (Fig. 5E; P = 0.05). Finally, multivariate analysis showed an association between NI-1 infections with DSS and platelet counts ≤50,000 cells/mL in later seasons (table S3) (74). Therefore, NI-1/NI-2A infections are at higher risk of more severe disease, particularly DSS, in “post-DENV-3-era” children in later seasons. These observations suggest that there is a specific interaction between NI-1/NI-2A viruses and DENV-1 immunity such that NI-1/NI-2A viruses efficiently infected and caused severe disease in DENV-1-immune children after a period of protection had passed.
Next, we observed that patients with NI-2B infections were distributed differently across birth groups compared to NI-1 or NI-2A infections. Specifically, NI-2B Hospital cases came disproportionately from children born before 1999 (“DENV-3-era”), with the “1994–1995”, “1996–1998”, and “<1994” birth groups showing significantly more cases than the “post-DENV-3 era” (“2002–2004” and “1999–2001”) birth groups (P = 0.005; Fig. 6A), and with a similar pattern apparent in the distribution of DHF/DSS cases (P = 0.04; Fig. 6B). The distribution of NI-2B cases in the Cohort study also followed the same pattern (P = 0.004; Fig. 6C). Thus, the relative over-representation of NI-2B infections as compared to NI-1/NI-2A infections in children born before 1999 implies that NI-2B infections were more likely to present as symptomatic cases and as severe cases than were NI-1/NI-2A infections specifically in predominantly DENV3-exposed children.
Fig. 6. Association of prior DENV-3 immunity with subsequent DENV-2 NI-2B infection.

(A–C) Contribution of each clade (NI-1, purple; NI-2A, green; NI-2B, blue) to (A) all dengue cases in the Hospital study, (B) cases of DHF/DSS in the Hospital study, (C) all dengue cases in the Cohort study. The observed distribution of each clade was compared to the expected distribution under the assumption that all children have equal risk of infection. Birth years were grouped into birth groups based on the history of the circulation of particular serotypes in Nicaragua (“pre”, “early” and “late” “DENV-3-era” and “post-DENV-3-era”, including “DENV-2-era”, “DENV-1-era” and “recent DENV-2 era”). Clade NI-2B is not evenly distributed among birth groups (chi square goodness of fit test with Rao Scott correction: (A) P = 0.005; (B) P = 0.04; (C) P = 0.004). (D) Titers of neutralizing antibody (NT50) against DENV-1 (circles) and DENV-3 (triangles) assayed using the RVP neutralization assay in serum samples collected prior to 2006/7 and 2007/8 DENV-2 seasons from DF (n = 21) or DHF/DSS cases (n = 8). The limit of detection of the assay (NT50 = 10) is shown with a dotted line. The solid line indicates the geometric mean. DENV-1, P = 0.72; DENV-3, P = 0.003; Wilcoxon Rank-Sum test, comparing RVP NT50 titers in DF versus DHF/DSS cases in a nested case control study within the Hospital study. For all panels, clade designations are colored as in Figure 4.
To empirically test the association of NI-2B and DENV-3 immunity with more severe disease, we measured the virus neutralization capacity of annual serum samples collected from Cohort DENV-2 cases prior to infection with NI-2B viruses (most Cohort cases in later seasons resulted from infection with NI-2B viruses). Using a nested case control analysis, we matched 8 DHF/DSS Cohort cases (all from the 2007/8 season except for one from January of the 2006/7 season) with 21 randomly selected DF cases from 2007/8, and we measured neutralizing antibody titers to DENV-1 and DENV-3 using Reporter Viral Particle (RVP) and Focus Reduction Neutralization Test (FRNT) neutralization assays. Among the NI-2B cases, the severe cases displayed higher neutralizing antibody titers to DENV-3 (P = 0.003) but not to DENV-1 (P = 0.72), compared to the DF control group (Fig. 6D). Similar results were obtained using the FRNT assay with Nicaraguan viruses, with higher titers among NI-2B severe cases to DENV-3 (P = 0.0005) but not to DENV-1 (P = 0.60), compared to the DF control group (fig. S6). Therefore, the arrival of NI-2B viruses into the population in later seasons and their higher propensity to cause severe disease in DENV-3-exposed children, compared to NI-1/NI-2A viruses, also contributed to the increase in disease severity observed across seasons. Moreover, the greater contribution of NI-2B viruses, compared to NI-1/NI-2A viruses, to cases in DENV-3 immune children could have resulted in a competitive advantage for NI-2B viruses in the population, as they may have been able to replicate more efficiently in the DENV-3-immune environment.
DISCUSSION
Through an interdisciplinary approach, we have demonstrated that two evolving factors, serotype-specific immunity and viral genetics, both contribute to the dynamics of dengue disease severity across time; specifically, the complex interplay between both factors is a major driver in determining dengue disease outcome. Such a dramatic increase in risk of severe dengue disease across several years of epidemics caused by the same serotype, as we observed in Managua, presented a unique opportunity to assess the role of viral genetics in dengue disease severity. Our study was strengthened by the fact that a single DENV serotype generally dominates each epidemic season in Nicaragua to a greater extent than in other locations where dengue is hyperendemic (75), thereby constraining the immunological background of previous DENV infections in the population. Further, the studies were restricted to a small geographic area, ensuring a constant host genetic background, and to a brief temporal period, restricting viral genetic variation.
The 1999–2002 DENV-2 epidemics in Nicaragua demonstrated a rate (~20%) of severe disease (DHF/DSS) in children who were part of our previous study in the same hospital (69) that was similar to the rate we observed in the relatively mild 2005/6 season analyzed here. This previous DENV-2 period was preceded by DENV-3 in 1994–1998 (66) (see Fig. 2A), suggesting that the sequence of DENV-3/DENV-2 infection does not necessarily produce the high levels of severe disease (~60% DHF/DSS) observed in the later 2006/7–2008/9 seasons of the Nicaraguan DENV-2 epidemics. Therefore, factors that are variable across epidemic seasons, such as an evolving adaptive immune response to prior DENV infections and changes in viral genetics, are key candidates for driving dengue disease outcomes across seasons.
Our data indicate that pre-existing immunity to DENV-1 initially provided protection from dengue disease during a subsequent DENV-2 infection in the 2005/6 season, but then waning cross-protection contributed to an increased risk of DENV-2 disease in the 2006/7–2008/9 seasons. A model consisting of a window of protection from disease after heterologous infection is well-supported by previous observations, particularly when the sequence of infection is DENV-1 followed by DENV-2 (10). Studies in nonhuman primates of secondary DENV-2 infection immediately following a primary DENV-1 infection also resulted in reduced viremia and protection from disease in these animals (11). However, epidemiological studies in human populations have demonstrated that when DENV-2 infection follows DENV-1 infection after an interval of several years, individuals are at higher risk of severe dengue disease, as observed in both Cuban and Thai populations (3, 76). Our results suggest a window of DENV-1-induced cross-protection against symptomatic dengue disease due to subsequent DENV-2 infection on the order of one to three years and thus help to more specifically define the timing between infections that contributes to disease-protective versus pathogenic immunity.
We demonstrated that a novel, phylogenetically distinct DENV-2 clade (NI-2B) with increased fitness began to circulate in the study populations concurrently with the observed increase in disease severity. Both in vitro analyses of viral isolates and viremia levels in patient sera suggest that the NI-2B viruses have increased fitness relative to the previously circulating DENV-2 clade, likely explaining their success in overtaking the NI-1 viruses that were circulating in the Nicaraguan population. Therefore, our data are consistent with a model in which DENV clade-replacement events may be associated with increased viral fitness, rather than stochastic events (49). Fitness of NI-1 and NI-2B viruses in vivo in the Aedes aegypti mosquito vector are important to confirm our in vitro findings and are underway.
Nine amino acid changes distinguish the more fit NI-2B viruses from the replaced clade (NI-1). One or more of these mutations may contribute to the observed increased replicative ability of NI-2B isolates. In particular, we are intrigued by the two mutations in the RNA-dependent polymerase (RdRp) domain of the NS5 viral protein. One RdRp domain mutation (R401K) is adjacent to the RNA-binding groove and may affect the efficiency of RNA replication. The other RdRp mutation (T290I) maps to a region implicated in binding to the viral helicase NS3 and is a candidate for modulating the interaction between the DENV NS3 and NS5 proteins that is required for viral replication (77). The single mutation identified in E (M492V) lies adjacent to the membrane and would not be expected to directly alter antigenicity or function of E, although there is a possibility that this mutation could alter the conformation of E at other sites that could affect function.
Mathematical modeling has suggested that both the patterns of circulation of DENV serotypes across seasons and clade-replacement events are linked to cross-reactive immunity in the population (78). In fact, we found evidence of interplay between clade genetics and specific immune backgrounds. The Nicaraguan clades — for which relative circulation in the population changed across seasons (more NI-1/NI-2A in early seasons; more NI-2B in later seasons) — differentially affected risk of disease in children who were previously exposed to specific DENV serotypes. NI-1 and NI-2A infections were associated with more severe disease (specifically, DSS) than were NI-2B infections in children primarily exposed to DENV-1. Therefore, the few NI-1 and NI-2A viruses circulating in later seasons appear to have contributed to the increase in severity across seasons specifically after protective DENV-1 immunity was lost. In addition, we found that, compared to NI-1 and NI-2A viruses, NI-2B viruses disproportionately caused disease in DENV-3-immune children. Our analysis of the neutralization capacity of sera collected prior to NI-2B infection in the Cohort study demonstrated that cases of severe disease were significantly more likely to be immune to DENV-3 than a related control group of DF cases from the same season. The observed increase in severity across years after NI-2B infection may result specifically from interactions between NI-2B genetics and the immune response to DENV-3, which is different in infection with NI-1 or NI-2A viruses. Therefore, specific mutations (such as those outlined in Table 2) may contribute not only to the increased fitness of NI-2B viruses relative to NI-1 viruses, but also to the enhanced virulence of NI-2B viruses in DENV-3 immune children. Supporting the potential importance of these mutations in the observed immunological backgrounds is the observation that several of these mutations are unique to the Nicaraguan strains (table S2).
Our study has several possible limitations. First, we were not able to obtain complete DENV genome sequences from all of the patient samples. However, we developed a genotyping assay to distinguish among the Nicaraguan clades and were then able to obtain clade information for ~90% of the viruses studied. Also, our analyses were limited to a subset of cases, from both studies, that had complete clade data and other clinical and demographic information; however, only ~11% of cases were missing clade or other data, and thus we do not believe these omissions significantly affected our results. In addition, the shift in clade occurred completely in tandem with changing epidemic seasons in the Cohort study, preventing association analyses of either variable with disease severity. However, clade shift was not as tightly linked with dengue season in the Hospital study; thus, we were able to perform robust association analyses that demonstrated associations of both clade and dengue season with disease severity. Further, whereas the Cohort study design permitted analysis of samples collected prior to DENV-2 infection due to the existence of annual healthy blood samples, this material was not available in the Hospital study. Therefore, we relied on birth year of children in the Hospital study as a proxy measure of infection history; this is a valid approach because of the serotype timeline in Nicaragua wherein one serotype dominates in each dengue season, and we provide experimental support using data from the Cohort study (Fig. 2C). Finally, we cannot rule out unmeasured factors that may have contributed to our observation of increased severity across seasons.
Our study provides insight into the complex dynamics that determine dengue disease outcomes across epidemics. The population-level dynamics of immunity to specific DENV serotypes can be a primary driver of observed fluctuations in disease manifestations and may explain a significant portion of the variation in disease outcomes observed in endemic populations over time (75). We find that changes in the immunological profile of the population and changes in viral genetics, such as those detailed here for both NI-1/2A and NI-2B infections, contributed to the abrupt increase in DENV-2 disease severity that occurred after the 2005/6 season. Our data and samples should enable the deciphering of biological processes and mechanisms that underlie the switch between a protective versus a pathogenic immune response.
Notably, the fact that we did not observe an apparent link between viral genetics and disease severity until the host immunological background was accounted for highlights the importance of phylodynamic studies that combine high-resolution genomic information with associated epidemiological information (79). This interplay between viral genetics and serotype-specific immunity that determines risk of dengue disease has important implications for our understanding of DENV epidemiology and pathogenesis as well as for dengue vaccine design. In addition, our results strongly support the notion that genetic diversity in RNA viruses, given their rapid evolution and relatively recent common ancestry, is predominantly driven by the immune structure of the host population they infect. Finally, our work addresses fundamental questions in viral evolution and the interaction between viral and immunological determinants of viral fitness and virulence.
MATERIALS & METHODS
Nicaraguan DENV cohort and hospital studies
The protocols for both studies were reviewed and approved by the Institutional Review Boards (IRBs) of the University of California, Berkeley (CPHS 2004-6-178 and CPHS 2005-5-35, respectively) and of the Nicaraguan Ministry of Health (CIRE-02/05/06-008 and CIRE-02/05/06-012, respectively); in addition, the Cohort study protocol was reviewed and approved by the IRB of the International Vaccine Institute in Seoul, South Korea (IRB 2004-010). Parents or legal guardians of all subjects in both studies provided written informed consent, and subjects 6 years of age and older provided assent.
The Nicaraguan Pediatric Dengue Cohort Study (PDCS) (70, 80, 81)
The Cohort study is a community-based prospective study that was initiated in August of 2004 in low- to middle-income District II of Managua, Nicaragua. The cohort population consists of children from 2 to 14 years of age with yearly enrollment of new 2-year olds and withdrawal of 15-year olds to maintain the age structure. The catchment area as well as study design and methods have been described previously (81). Our study has a high case capture rate (82), and we have not observed significant population movement (81). Briefly, participants are encouraged to present at the first sign of illness to the Health Center Sócrates Flores Vivas (HCSFV), in which study physicians use standardized forms to collect data on the signs and symptoms of dengue. Subjects were followed daily during the acute phase of illness. Acute and convalescent (~14 days after onset of symptoms) blood samples were drawn for dengue diagnostic testing (see below), and complete blood count (CBC) and blood chemistry tests were performed as indicated by the physicians. Participants requiring hospitalization were treated at the study hospital (Hospital Infantil Manuel de Jesús Rivera, HIMJR), in which medical data is collected on systematic forms. Each year in July-August, a healthy blood sample was drawn from all subjects to identify silent DENV transmission during the year and to determine dengue immune status.
The Nicaraguan Dengue Hospital Study (69, 83)
This study was based in the Infectious Disease Ward of the Hospital Infantil Manuel de Jesús Rivera (HIMJR), the national pediatric reference hospital in Nicaragua, and has been ongoing since 1998. During the peak dengue season (August–January), children between six months and 14 years of age with suspected dengue disease (acute febrile illness with at least one of the following accompanying symptoms: headache, arthralgia (bone/joint pain), myalgia (muscle pain), retro-orbital (eye) pain, positive tourniquet test, petequiae (small red spots, caused by broken capillaries), signs of bleeding (1)) who presented to the hospital within 7 days of symptom onset were eligible to participate in the study. Blood samples were collected at enrollment, during the acute phase, and 2 weeks after symptom onset (convalescent phase) for clinical laboratory tests and dengue diagnostic tests. Clinical data, including vital signs, symptoms, fluid balance and treatment, platelet count, hematocrit and ultrasound and/or X-ray results were recorded on standardized data collection forms by a team of study physicians and nurses. Participants who required more intensive therapies were transferred to the intensive care unit (ICU). In both studies, a study physician completed case report forms (CRFs), which were then reviewed by a second physician, after which the CRFs were systematically monitored, and the information was entered into an Access 2003 database by double-data entry, with quality control checks performed daily and weekly.
A suspected dengue case was considered dengue-positive if it met one or more of the following criteria (68): DENV was isolated (66, 84); DENV RNA was detected by reverse transcriptase-polymerase chain reaction (RT-PCR) (68, 85); seroconversion of DENV-specific IgM antibodies was detected in paired acute- and convalescent-phase samples (86, 87); or antibody titer by Inhibition ELISA (67, 88) demonstrated a ≥4-fold increase in titer between acute and convalescent sera. Primary DENV infections were those in which acute antibody titer was <10 or convalescent antibody titer was <2,560 and secondary infections were those in which antibody titers were ≥10 (acute) or ≥2,560 (convalescent) as determined by Inhibition ELISA (68). In paired annual blood samples, seroconversion from a titer of <1:10 to ≥1:10 by Inhibition ELISA indicated a primary DENV infection, while a ≥4-fold increase in anti-DENV antibody titer indicated a secondary DENV immune response (81).
Genome sequencing and genotyping
The clade of Nicaraguan DENV-2 sequences derived from patient samples was assigned based on either full-length genome sequencing (N=159) or a clade genotyping assay (N=107) (see http://www.broadinstitute.org/annotation/viral/Dengue/GlobalPopulationStructure.html for a description of Nicaraguan sample sets). For genome sequencing, viral RNA was isolated (QIAmp viral RNA mini kit, Qiagen) from serum or first-passage cell culture supernatant (C6/36) and reverse-transcribed to cDNA using Superscript III RT (Invitrogen), random hexamers (Roche), and a specific oligonucleotide that targeted the 3′ end of the target genome (5′-AGAACCTGTTGATTCAACAGCAC-3′; nucleotides 10701-10723 for DENV-2). cDNA was then amplified using a high-fidelity DNA polymerase (pfu Ultra II, Stratagene) and a pool of specific primers to produce 14 overlapping amplicons of 1.5 to 2 kb in size, for a physical coverage of 2X. Amplicons were then generated using primer panels consisting of 96 specific primer pairs tailed with M13 forward and reverse primer sequences, which produced 500- to 700-bp amplicons from the target viral genomes. These amplicons were sequenced in the forward and reverse directions using M13 primers, with a total post-sequencing coverage of 8-fold. Resulting sequence reads were assembled de novo using the Broad Institute’s AV454 assembly algorithm, followed by genome annotation using the Broad Institute’s in-house viral annotation algorithm and quality-checking using the annotation and supporting reads to resolve any base call ambiguities and frame-shifts. Resulting genome sequences were deposited in GenBank (see accession numbers in References and Notes). To establish a more complete data set beyond that for which we had complete genome sequence, we used an RT-PCR genotyping assay that enabled classification of DENV-2 strains as NI-1, NI-2A or NI-2B. Addition of the RT-PCR genotyping information to the complete genome data permitted identification of clade in ~90% of DENV-2 infections across both studies. Viral RNA was extracted (QIAmp Viral RNA Mini) and nested RT-PCR targeting either of 2 regions (one in NS2A and another in NS3; see table S4 for primer sequences) was performed. Reverse transcription and PCR were performed with 0.2μL AMV-RT (Invitrogen), 0.25μL GoTaq (Promega), 5X GoTaq Buffer, 0.5μL 10mM dNTPs, 2.5μL 5M betaine, 0.75μL 1M tetramethylammonium chloride (TMAC), 1.25μL each 10μM primer, 1μL RNaseOUT (Invitrogen), 1.25μL 1mM DTT and 5μL viral RNA. Cycling conditions were 50°C for 60 minutes (min) followed by 30 cycles of 94°C for 30 seconds (sec), 54°C for 30 sec and 72°C for 1 min, followed by a final extension at 72°C for 3 min. First-round product was diluted 1:10 and 5μL amplified by a second, nested PCR including 0.125μL GoTaq, 5μL 5X GoTaq Buffer, 2.5μl 5M betaine, 0.75μL TMAC, 2.5μL each 10μM primer and 0.5μL 10mM dNTPs by cycling at 95°C for 2 min, 30 cycles of 94°C for 30 sec, 54°C for 30 sec, and 72°C for 1 min, followed by a final extension at 72°C for 3 min. Amplification of PCR products was verified by gel electrophoresis, and amplicons were sequenced directly using an internal primer (table S4) for each amplicon. Between 3 and 6 positions (3659, 3672, 3704, 5667, 5685 and 5757; numbering starting at AUG initiation codon) distinguishing NI-1, NI-2A and NI-2B were evaluated for each viral RNA, allowing for the DENV-2 clade to be determined.
Phylogenetic analysis
Maximum likelihood (ML) and Bayesian Maximum Clade Credibility (MCC) phylogenetic trees were inferred from the coding sequences of 184 complete DENV-2 genome sequences. Twenty-five additional sequences representing the American, Asian 1, and Asian/American genotypes of DENV-2 were also included in the analysis (table S1). UTRs were not used for phylogenetic reconstructions in order to eliminate alignment artifacts resulting from unconfirmed indels in these regions. Sequences were aligned using ClustalW 1.83 (89) and manually curated. ML trees were generated using RAxML 7.0.4 (90) and PhyML 2.2 (91) incorporating the GTR+Γ4 model of nucleotide substitution, as determined by ModelTest v3.7 (92), and bootstrap resampling of 1000 and 100 replications respectively. For PhyML, we invoked the invariant sites. All resulting trees were compared across the two algorithms using in-house scripts to confirm that all major nodes were consistent across the algorithms. For final display, branches with <60% bootstrap support in the final tree were collapsed.
We used the pamp module of PAML 4 (93) to infer ancestral states by maximum parsimony for best trees and all bootstrap trees from RAxML and PhyML. We computed final ancestral sequences by comparing the calls from the RAxML and PhyML best trees and the amount of ancestral variation represented in the bootstrap trees for each position and making a decision about which sequence was most probable. Ambiguity codes were assigned where no clear most probable ancestral base existed.
Cells and viruses
C6/36 cells (gift from P. Young, University of Queensland, Australia) were grown in M199 (Invitrogen) with 10% Fetal Bovine Serum (FBS) (Denville Scientific), 100 units/mL penicillin/100 μg/mL streptomycin (P/S), and 1X HEPES at 28°C with 5% CO2. Immature monocyte-derived dendritic cells (iDCs) were grown in RPMI/10% FBS plus P/S at 37°C in 5% CO2. Vero-76 cells (gift from Aravinda de Silva, University of North Carolina, Chapel Hill) were grown in DMEM/10% FBS plus P/S at 37°C in 5% CO2. iDCs were derived from a Leukopak donor (All Cells, Inc.) as previously described (14). For infection, cells were thawed and plated in 10-cm dishes in 10mL of RPMI/10% plus P/S at 2.5 × 106 cells/mL. Cells were allowed to adhere for 1 hour (h) at 37°C, and non-adherent cells were removed by washing 5–10 times in warm RPMI. Ten mL of RPMI/10% FBS plus P/S was added to each plate, supplemented with 20,000U each of IL-4 (R&D Systems) and GM-CSF (Peprotech). Cytokines were supplemented 4–5 days post-plating with an additional 10,000U of each cytokine. iDCs were harvested at day 6 or 7 and phenotyped by flow cytometry to confirm DC phenotype (FSC/SSC; CD209-PerCP).
Low-passage (P1-P4) stocks of virus isolates from Nicaraguan patient sera were generated by inoculating acute patient sera onto C6/36 cells as previously described (66), serial-passaging in C6/36 cells and storing at −80°C. Re-sequencing of five isolates after 2 passages in C6/36 cells did not reveal the introduction of any spurious mutations (sequences available upon request). The quantity of virus present in each stock (genome equivalents; GE) was determined by quantitative RT-PCR targeting the NS5 gene as described below. The ratio of GE to PFU was not significantly different among clades (fig. S3F).
Viral competition assays
C6/36 cells or iDCs were infected with low-passage (<P4) viral isolates from each clade (5 NI-1 and 5 NI-2B pairwise; total N=25) mixed at equal GE ratios (1:1). C6/36 cells were plated at 3 × 105 cells/mL in 125μL in a flat-bottom 96-well plate one day prior to infection with 1.9 × 107 GE/well of each virus isolate (3.8 × 107 total GE/well) in triplicate. Supernatants were removed 3 h post-infection, wells washed 1X in PBS, and 250μL media added back to each well. To establish Day 0 values, 20μL supernatant was removed and pooled from triplicate wells before washing. All supernatant was removed at each time-point, frozen at −80°C, and replaced with 250μL fresh media. For iDC competition experiments, 5 × 104 iDCs were infected with 1 × 108 GE of each virus (2 × 108 GE total) per well. Infected cells were washed 1X in PBS after overnight incubation, and supernatants collected and frozen at −80°C after 48 h. Viral RNA was isolated (QIAamp Viral RNA Mini Kit) and amplified by one-step RT-PCR (One-Step RT-PCR Kit) with primers specific to sites in NS1 conserved among Nicaraguan DENV-2 isolates (Forward 5′-GAATCCCCTTCAAAACTAGC-3′; Reverse 5′-ATGTCCGGCTGTGACCAAG-3′) to generate a 950-kb amplicon. Two μL of viral RNA was amplified with 4μL 5X Buffer, 1μL dNTPs, 1μL each primer (10μM stock), 0.8μL One-Step RT enzyme mix (Qiagen), and 1μL RNaseOUT (Invitrogen) in a final reaction volume of 20μL. Viral RNA was reverse transcribed for 30 min at 50°C and heat-inactivated for 15 min at 95°C, and cDNA was amplified by 30 cycles of 94°C for 45 sec, 54°C for 45 sec, and 72°C for 1.5 min, followed by a final extension at 72°C for 10 min. Amplification was confirmed by electrophoresis on 1% agarose, and amplicons were directly sequenced by adding 0.5μL PCR product to 0.5μL NS1 sequencing primer (5′-GAGGGCATTTGTGGAATCC-3′) in a final volume of 13μL, amplifying using the BigDye Terminator method, and sequencing on an ABI3730xl DNA analyzer (UC Berkeley DNA Sequencing Facility). ABI chromatograms were analyzed via the PolySNP PERL script (73), and the relative proportion of each virus in the dual infection was calculated by averaging the proportions of all valid SNPs. The relative fitness of each virus in each pairwise dual infection was calculated as previously described (72). Briefly, the proportion of each isolate at a given time-point after infection (fo) was divided by its initial proportion in the inoculum (io) to derive a relative fitness (ω) value (ω = fo/io).
Neutralization assays
Reporter viral particles (RVPs) (94) that represented the WHO reference viruses (DENV-1, WestPac-74; DENV-2, S16803; DENV-3, CH53489) were a kind gift of Molecular Integral, Inc. (Philadelphia, PA) (95). RVP neutralization assays were performed by incubating 25 μL of each RVP with 25 μL diluted patient serum [starting at a 1:10 dilution (serum + RVP) in RPMI/10% FBS plus P/S, pH 8.0] shaking at room temperature for 1 h at 100 rpm and adding 4 × 104 Raji/DC-SIGN cells (ATCC)/well in a total volume of 100 μL. After incubation for 48 h at 37°C, Green Fluorescent Protein (GFP)-positive cells were enumerated on an LSR Fortessa flow cytometer (BD Biosciences) and analyzed in FlowJo v.7.2.5 (TreeStar Software). Relative infection was determined using an RVP-only control. Human dengue-naïve serum (Red Cross) and anti-DENV polyvalent sera (WHO Reference Sera; NIBSC, UK) were used as negative and positive controls, respectively. NT50 values were calculated using nonlinear regression analysis with a variable slope (GraphPad 5.0; Prism). Samples were classified as seropositive for the serotype with the higher NT50 as compared to the other serotypes whenever the NT50 exceeded 40.
Statistical analysis
To analyze disease severity, we classified study participants as DF, DHF or DSS using traditional WHO criteria (1). Secondary outcomes of disease severity included low platelet count (≤50,000 cells/mL) (74). Viral clade was assigned as described above. All analyses were restricted to subjects with known gender, age, immune status, season of infection, infecting viral clade, and primary outcome. This resulted in a sample of 136 out of 154 participants from the Hospital study and 95 out of 107 participants from the Cohort study. Fisher’s exact test was used to compare proportions. Kruskal Wallis and Mann Whitney/Wilcoxon Rank-Sum tests were used for all nonparametric tests of >2 or 2 groups, respectively. Odds ratios for our primary and secondary outcomes were estimated using logistic regression in univariate and multivariate analyses adjusting for season of infection, immune status (previous DENV exposure), gender, and age.
To compare rates of DSS over the course of illness for each clade, the logrank test was used. For birth year analyses, subjects were classified as “DENV-3-era” if they were born before 1999 and “post-DENV-3-era” if they were born in 1999 or after. We adjusted for the changing representation of birth groups over time using the chi square test with a Rao-Scott correction. To further refine immunological background, children were sub-classified into groups based on birth year: (i) <1994 (“pre DENV-3”), (ii) 1994–1995 (“early DENV-3”) and (iii) 1996–1998 (“later DENV-3”), (iv) 1999–2001 (“DENV-2”), (v) 2002–2004 (“DENV-1”), and (vi) 2005–2007 (“recent DENV-2”). The chi square goodness of fit test was used to compare the observed and expected distribution of cases of dengue. The Rao-Scott adjustment was used to adjust for the year span of each birth group, as birth groups that spanned more years would be expected to have more cases of dengue. We constructed a full mixed model, by clade, of viremia on the log scale as a repeated measure, with disease classification as a fixed effect and hospital day of sample as a random effect. P-values ≤0.05 were considered to be significant, while P-values >0.05 but =0.1 were considered to be nonsignificant trends. All analyses were performed in SAS version 9.2 (SAS Institute Inc., Cary, NC).
Supplementary Material
Acknowledgments
We thank our Nicaraguan study team based at the Hospital Infantil Manuel de Jesús Rivera, the Centro de Salud Sócrates Flores Vivas, the Sustainable Sciences Institute, and the National Virology Laboratory in the Centro Nacional de Diagnóstico y Referencia, Ministry of Health in Managua for their dedication, excellent work to ensure high-quality medical attention and study performance, tireless data entry, top-notch laboratory work, and stellar database management and support, especially M. Amador, S. Arguello, W. Avilés, Y. Buitrago, J. R. Cisneros, D. Elizondo, C. Flores, N. Fitzpatrick, G. Gutierrez, S. Hammond, J. Herrera, G. Kuan, B. López, R. López, J. C. Matute, J. Medina, J. Mercado, J. C. Mercado, B. Moraga, M. Monterrey, A. Munguia, F. Narvaez, G. Navas, S. Ojeda, Z. Orozco, O. Ortega, L. Pérez, M. A. Pérez, M. Reyes, C. Romero, C. Rocha, C. Saborio, L. Saenz, N. Sanchez, S. Silva, K. Standish, M. J. Vargas, U. Vargas, and other study personnel. We also thank A. Gonzalez for his continued support over the years. Finally, we are indebted to the children who participated in the study and to their parents. We thank the Broad Institute’s Sequencing and Biological Samples Repository Platforms, E. Ryan, C. Malboeuf, L. Green, R. Erlich, J. Levin, R. Newman, L. Larson, F. Viloria, B. Weiner, and P. Charlebois for their assistance in generation of the genomic data, and U.C. Berkeley’s flow cytometry and DNA sequencing facilities for assistance in sample analyses. We are grateful to E. Holmes, M. Emerman, and K. Williams for comments on this work and manuscript and to P. Parameswaran, R. Gonzalez, and C. Quiner for experimental assistance. We thank B. Doranz and K. Mattia at Integral Molecular, Inc. for reporter viral particles used in this study, and L. Solomon for assistance with figure preparation.
Funding: This project has been funded with Federal funds from the National Institutes of Health, Department of Health and Human Services, from the National Institute of Medical Services under grant GM087405 (EH), and from the National Institute of Allergy and Infectious Diseases under grants AI65359 (AB) and AI62100 (EH, AB) and contracts HHSN272200900018C (BWB) and HHSN272200900006C (BWB); grant VE-1 (EH) from the Pediatric Dengue Vaccine Initiative; and a Helen Hay Whitney Postdoctoral Fellowship (MO). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/XXX/XXXraXX/DC1
Materials and Methods
fig. S1. National Dengue Surveillance data in Managua, Nicaragua.
fig. S2. Nicaraguan DENV-2 evolution.
fig. S3. Fitness of NI-1 and NI-2B isolates in vitro.
fig. S4. Patient viremia by clade and day post-onset of symptoms in DF and DHF/DSS cases.
fig. S5. Cases of severe dengue (DHF/DSS) by clade in “post-DENV-3-era” Hospital cases in later seasons.
fig. S6. Focus Reduction Neutralization Test (FRNT) analysis of Cohort sera using Nicaraguan DENV isolates.
table S1. Nonsynonymous and UTR mutations in DENV-2.
table S2. Occurrences of Nicaraguan clade-defining mutations in non-Nicaraguan complete genome sequences available in public databases.
table S3. Association of NI-1 viruses with more severe disease outcomes in later study years as compared to NI-2B viruses in univariate analysis, and adjusted for age, gender, and previous infection.
table S4. Primer sequences used for genotyping in this study.
Author contributions: Conceived and designed the experiments: MO, AB, AM, MRH, EH. Performed the experiments: MO, AM, YT, MZ, SS, NJL, MRH. Analyzed the data: MO, AB, AM, MZ, AN, AG, MRH, EH. Contributed materials/reagents/analysis tools: AB, BWB, MRH, EH. Wrote the paper: MO, AB, AM, MZ, AG, MRH, EH.
Competing Interests: The authors declare no competing financial or other conflicts of interest.
Accession numbers: The genomic sequences of DENV-2 isolates analyzed in this study are in GenBank under the following accession numbers: FJ850117, FJ850118, FJ850062, FJ850063, FJ850119, FJ850064, FJ850065, FJ850066, FJ873808, FJ850120, FJ850121, GQ199897, GQ199895, GQ199898, FJ850060, FJ850061, FJ744704, FJ744745, FJ744705, FJ744744, FJ898477, FJ898478, GQ868646, EU482629, FJ390390, EU482749, EU482750, EU482752, EU482753, EU482770, FJ226066.1, EU482680, EU482754, EU482755, EU482756, EU482758, FJ906956, EU482759, EU482760, EU482681, EU482761, EU482762, EU482763, EU482771, EU482444, EU482688, EU482695, EU482769, EU569699, FJ410291, FJ906961, FJ906962, FJ850053, FJ850054, EU482597, EU482598, EU482599, EU482632, EU482600, FJ898436, EU482633, FJ850115, FJ898435, EU482636, EU482637, EU596484, GQ199874, EU482631, EU482602, EU482639, FJ744741, FJ850067, EU482748, EU482757, EU482689, EU482693, EU482696, EU482603, EU482634, GQ199866, GQ199868, EU482620, FJ390391, FJ882593, FJ182014, FJ898432, FJ547090, EU482630, FJ205885, FJ744703, FJ744709, FJ744708, FJ810418, EU482751, FJ478455, EU482682, EU482772, EU482773, EU482683, EU482684, EU482685, EU482686, EU482687, EU482690, EU482691, EU482692, EU482694, EU482766, GQ199869, EU482621, EU569692, EU569693, FJ373300, EU596495, EU660404, FJ850116, EU482622, EU569694, EU596496, EU660405, EU596497, EU569695, FJ373301, EU569696, EU660406, EU569697, EU482623, EU596498, EU569698, FJ478459, EU596499, EU569700, EU569701, EU621672, EU569702, EU482624, EU596500, EU482625, EU482626, EU482627, EU482628, FJ639833, FJ639834, FJ639835, FJ850050, FJ639836, FJ639837, FJ744706, FJ744707, FJ906960, FJ850051, FJ898434, EU596483, EU482638, EU482601, FJ744743, FJ744742, GQ199896, FJ898479. Additional sequences used in the phylogenetic analysis can be found as GenBank: AF100467, AF100468, AF100465, AF100469, U87411, AF100462, AF022437, AF022434, AF100460, AF100459, AF100463, AF100464, AF100461, AF100466, M20558, AB122021, AB122020, AB122022, AY702035, AF208496, AY702034, AY702035, AY702036, AY702038, and AY702037.
REFERENCES AND NOTES
- 1.World Health Organization. Dengue Haemorrhagic Fever: Diagnosis, Treatment, Prevention and Control. 2. World Health Organization; Geneva: 1997. [Google Scholar]
- 2.Halstead SB. Dengue. Lancet. 2007;370:1644–1652. doi: 10.1016/S0140-6736(07)61687-0. [DOI] [PubMed] [Google Scholar]
- 3.Sangkawibha N, Rojanasuphot S, Ahandrik S, Viriyapongse S, Jatanasen S, Salitul V, Phanthumachinda B, Halstead SB. Risk factors in dengue shock syndrome: a prospective epidemiologic study in Rayong, Thailand. I. The 1980 outbreak. Am J Epidemiol. 1984;120:653–669. doi: 10.1093/oxfordjournals.aje.a113932. [DOI] [PubMed] [Google Scholar]
- 4.Thein S, Aung MM, Shwe TN, Aye M, Zaw A, Aye K, Aye KM, Aaskov J. Risk factors in dengue shock syndrome. Am J Trop Med Hyg. 1997;56:566–572. doi: 10.4269/ajtmh.1997.56.566. [DOI] [PubMed] [Google Scholar]
- 5.Burke DS, Nisalak A, Johnson DE, Scott RM. A prospective study of dengue infections in Bangkok. Am J Trop Med Hyg. 1988;38:172–180. doi: 10.4269/ajtmh.1988.38.172. [DOI] [PubMed] [Google Scholar]
- 6.Guzmán MG, Kouri G, Valdes L, Bravo J, Alvarez M, Vazques S, Delgado I, Halstead SB. Epidemiologic studies on Dengue in Santiago de Cuba, 1997. Am J Epidemiol. 2000;152:793–799. doi: 10.1093/aje/152.9.793. [DOI] [PubMed] [Google Scholar]
- 7.Alvarez M, Rodriguez-Roche R, Bernardo L, Vázquez S, Morier L, Gonzalez D, Castro O, Kouri G, Halstead SB, Guzman MG. Dengue hemorrhagic fever caused by sequential dengue 1–3 virus infections over a long time interval: Havana epidemic, 2001–2002. Am J Trop Med Hyg. 2006;75:1113–1117. [PubMed] [Google Scholar]
- 8.Hubert B, Halstead SB. Dengue 1 virus and dengue hemorrhagic fever, French Polynesia, 2001. Emerging Infect Dis. 2009;15:1265–1270. doi: 10.3201/eid1508.081500. [DOI] [PubMed] [Google Scholar]
- 9.Halstead SB. Pathogenesis of dengue: challenges to molecular biology. Science. 1988;239:476–481. doi: 10.1126/science.3277268. [DOI] [PubMed] [Google Scholar]
- 10.Sabin AB. Research on dengue during World War II. Am J Trop Med Hyg. 1952;1:30–50. doi: 10.4269/ajtmh.1952.1.30. [DOI] [PubMed] [Google Scholar]
- 11.Kochel TJ, Watts DM, Gozalo AS, Ewing DF, Porter KR, Russell KL. Cross-serotype neutralization of dengue virus in Aotus nancymae monkeys. J Infect Dis. 2005;191:1000–1004. doi: 10.1086/427511. [DOI] [PubMed] [Google Scholar]
- 12.Halstead SB, Suaya JA, Shepard DS. The burden of dengue infection. Lancet. 2007;369:1410–1411. doi: 10.1016/S0140-6736(07)60645-X. [DOI] [PubMed] [Google Scholar]
- 13.Guzman MG, Alvarez M, Rodriguez-Roche R, Bernardo L, Montes T, Vazquez S, Morier L, Alvarez A, Gould EA, Kouri G, Halstead SB. Neutralizing antibodies after infection with dengue 1 virus. Emerging Infect Dis. 2007;13:282–286. doi: 10.3201/eid1302.060539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boonnak K, Slike BM, Burgess TH, Mason RM, Wu SJ, Sun P, Porter K, Rudiman IF, Yuwono D, Puthavathana P, Marovich MA. Role of dendritic cells in antibody-dependent enhancement of dengue virus infection. J Virol. 2008;82:3939–3951. doi: 10.1128/JVI.02484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balsitis SJ, Williams KL, Lachica R, Flores D, Kyle JL, Mehlhop E, Johnson S, Diamond MS, Beatty PR, Harris E. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog. 2010;6:e1000790. doi: 10.1371/journal.ppat.1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halstead SB, O’Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J Exp Med. 1977;146:201–217. doi: 10.1084/jem.146.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zellweger RM, Prestwood TR, Shresta S. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe. 2010;7:128–139. doi: 10.1016/j.chom.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothman AL. Dengue: defining protective versus pathologic immunity. J Clin Invest. 2004;113:946–951. doi: 10.1172/JCI21512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurane I, Ennis F. Immunopathogenesis of dengue virus infection. In: Gubler DJ, Kuno G, editors. Dengue and Dengue Hemorrhagic Fever. CAB International; New York: 1997. pp. 273–290. [Google Scholar]
- 20.Rothman AL. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat Rev Immunol. 2011;11:532–543. doi: 10.1038/nri3014. [DOI] [PubMed] [Google Scholar]
- 21.Wearing HJ, Rohani P. Ecological and immunological determinants of dengue epidemics. Proc Natl Acad Sci USA. 2006;103:11802–11807. doi: 10.1073/pnas.0602960103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams B, Boots M. Modelling the relationship between antibody-dependent enhancement and immunological distance with application to dengue. J Theor Biol. 2006;242:337–346. doi: 10.1016/j.jtbi.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Nagao Y, Koelle K. Decreases in dengue transmission may act to increase the incidence of dengue hemorrhagic fever. Proc Natl Acad Sci USA. 2008;105:2238–2243. doi: 10.1073/pnas.0709029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Recker M, Blyuss KB, Simmons CP, Hien TT, Wills B, Farrar J, Gupta S. Immunological serotype interactions and their effect on the epidemiological pattern of dengue. Proc Biol Sci. 2009;276:2541–2548. doi: 10.1098/rspb.2009.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham RR, Juffrie M, Tan R, Hayes CG, Laksono I, Ma’roef C, Erlin, Sutaryo, Porter KR, Halstead SB. A prospective seroepidemiologic study on dengue in children four to nine years of age in Yogyakarta, Indonesia I. Studies in 1995–1996. Am J Trop Med Hyg. 1999;61:412–419. doi: 10.4269/ajtmh.1999.61.412. [DOI] [PubMed] [Google Scholar]
- 26.Guzmán MG, Kouri GP, Bravo J, Soler M, Vazquez S, Morier L. Dengue hemorrhagic fever in Cuba, 1981: a retrospective seroepidemiologic study. Am J Trop Med Hyg. 1990;42:179–184. doi: 10.4269/ajtmh.1990.42.179. [DOI] [PubMed] [Google Scholar]
- 27.Kliks SC, Nimmanitya S, Nisalak A, Burke DS. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am J Trop Med Hyg. 1988;38:411–419. doi: 10.4269/ajtmh.1988.38.411. [DOI] [PubMed] [Google Scholar]
- 28.Halstead SB. Neutralization and antibody-dependent enhancement of dengue viruses. Adv Virus Res. 2003;60:421–467. doi: 10.1016/s0065-3527(03)60011-4. [DOI] [PubMed] [Google Scholar]
- 29.Guzmán MG, Kourí G, Valdés L, Bravo J, Vázquez S, Halstead SB. Enhanced severity of secondary dengue-2 infections: death rates in 1981 and 1997 Cuban outbreaks. Rev Panam Salud Publica. 2002;11:223–227. doi: 10.1590/s1020-49892002000400003. [DOI] [PubMed] [Google Scholar]
- 30.Stephens HA, Klaythong R, Sirikong M, Vaughn DW, Green S, Kalayanarooj S, Endy TP, Libraty DH, Nisalak A, Innis BL, Rothman AL, Ennis FA, Chandanayingyong D. HLA-A and -B allele associations with secondary dengue virus infections correlate with disease severity and the infecting viral serotype in ethnic Thais. Tissue Antigens. 2002;60:309–318. doi: 10.1034/j.1399-0039.2002.600405.x. [DOI] [PubMed] [Google Scholar]
- 31.Falcon-Lezama JA, Ramos C, Zuniga J, Juarez-Palma L, Rangel-Flores H, Garcia-Trejo AR, Acunha-Alonzo V, Granados J, Vargas-Alarcon G. HLA class I and II polymorphisms in Mexican Mestizo patients with dengue fever. Acta Trop. 2009;112:193–197. doi: 10.1016/j.actatropica.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 32.Sakuntabhai A, Turbpaiboon C, Casadémont I, Chuansumrit A, Lowhnoo T, Kajaste-Rudnitski A, Kalayanarooj SM, Tangnararatchakit K, Tangthawornchaikul N, Vasanawathana S, Chaiyaratana W, Yenchitsomanus PT, Suriyaphol P, Avirutnan P, Chokephaibulkit K, Matsuda F, Yoksan S, Jacob Y, Lathrop GM, Malasit P, Desprès P, Julier C. A variant in the CD209 promoter is associated with severity of dengue disease. Nat Genet. 2005;37:507–513. doi: 10.1038/ng1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen TH, Nguyen TL, Lei HY, Lin YS, Le BL, Huang KJ, Lin CF, Do QH, Vu TQH, Lam TM, Yeh TM, Huang JH, Liu CC, Halstead SB. Association between sex, nutritional status, severity of dengue hemorrhagic fever, and immune status in infants with dengue hemorrhagic fever. Am J Trop Med Hyg. 2005;72:370–374. [PubMed] [Google Scholar]
- 34.Sierra B, Alegre R, Pérez AB, García G, Sturn-Ramirez K, Obasanjo O, Aguirre E, Alvarez M, Rodriguez-Roche R, Valdés L, Kanki P, Guzmán MG. HLA-A, -B, -C, and -DRB1 allele frequencies in Cuban individuals with antecedents of dengue 2 disease: advantages of the Cuban population for HLA studies of dengue virus infection. Hum Immunol. 2007;68:531–540. doi: 10.1016/j.humimm.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Halstead S, Streit T, Lafontant J, Putvatana R, Russell K, Sun W, Kanesa-thasan N, Hayes C, Watts D. Haiti: absence of dengue hemorrhagic fever despite hyperendemic dengue virus transmission. Am J Trop Med Hyg. 2001;65:180–183. doi: 10.4269/ajtmh.2001.65.180. [DOI] [PubMed] [Google Scholar]
- 36.Thisyakorn U, Nimmannitya S. Nutritional status of children with dengue hemorrhagic fever. Clin Infect Dis. 1993;16:295–297. doi: 10.1093/clind/16.2.295. [DOI] [PubMed] [Google Scholar]
- 37.Garcia G, Sierra B, Perez AB, Aguirre E, Rosado I, Gonzalez N, Izquierdo A, Pupo M, Danay Diaz DR, Sanchez L, Marcheco B, Hirayama K, Guzman MG. Asymptomatic dengue infection in a Cuban population confirms the protective role of the RR variant of the FcgammaRIIa polymorphism. Am J Trop Med Hyg. 2010;82:1153–1156. doi: 10.4269/ajtmh.2010.09-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes WJ, Rosen L. Fatal hemorrhagic disease and shock associated with primary dengue infection on a Pacific island. Am J Trop Med Hyg. 1974;23:495–506. doi: 10.4269/ajtmh.1974.23.495. [DOI] [PubMed] [Google Scholar]
- 39.Gubler DJ, Reed D, Rosen L, Hitchcock JCJ. Epidemiologic, clinical, and virologic observations on dengue in the Kingdom of Tonga. Am J Trop Med Hyg. 1978;27:581–189. doi: 10.4269/ajtmh.1978.27.581. [DOI] [PubMed] [Google Scholar]
- 40.Gubler DJ, Suharyono W, Lubis I, Eram S, Gunarso S. Epidemic dengue 3 in central Java, associated with low viremia in man. Am J Trop Med Hyg. 1981;30:1094–1099. doi: 10.4269/ajtmh.1981.30.1094. [DOI] [PubMed] [Google Scholar]
- 41.Gubler DJ, Trent DW. Emergence of epidemic dengue/dengue hemorrhagic fever as a public health problem in the Americas. Infect Agents Dis. 1993;2:383–393. [PubMed] [Google Scholar]
- 42.Kouri GP, Guzman MG, Bravo JR, Triana C. Dengue haemorrhagic fever/dengue shock syndrome: lessons from the Cuban epidemic, 1981. Bull WHO. 1989;67:375–380. [PMC free article] [PubMed] [Google Scholar]
- 43.Rico-Hesse R, Harrison L, Salas R, Tovar D, Nisalak A, Ramos C, Boshell J, de Mesa M, Nogueira R, da Rosa A. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology. 1997;230:244–251. doi: 10.1006/viro.1997.8504. [DOI] [PubMed] [Google Scholar]
- 44.Rico-Hesse R, Harrison LM, Nisalak A, Vaughn DW, Kalayanarooj S, Green S, Rothman AL, Ennis FA. Molecular evolution of dengue type 2 virus in Thailand. Am J Trop Med Hyg. 1998;58:96–101. doi: 10.4269/ajtmh.1998.58.96. [DOI] [PubMed] [Google Scholar]
- 45.Leitmeyer KC, Vaughn DW, Watts DM, Salas R, Villalobos I, Chacon D, Ramos C, Rico-Hesse R. Dengue virus structural differences that correlate with pathogenesis. J Virol. 1999;73:4738–4747. doi: 10.1128/jvi.73.6.4738-4747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bennett SN, Holmes EC, Chirivella M, Rodriguez DM, Beltran M, Vorndam V, Gubler DJ, McMillan WO. Selection-driven evolution of emergent dengue virus. Mol Biol Evol. 2003;20:1650–1658. doi: 10.1093/molbev/msg182. [DOI] [PubMed] [Google Scholar]
- 47.Bennett SN, Holmes EC, Chirivella M, Rodriguez DM, Beltran M, Vorndam V, Gubler DJ, McMillan WO. Molecular evolution of dengue 2 virus in Puerto Rico: positive selection in the viral envelope accompanies clade reintroduction. J Gen Virol. 2006;87:885–893. doi: 10.1099/vir.0.81309-0. [DOI] [PubMed] [Google Scholar]
- 48.Balmaseda A, Hammond SN, Pérez L, Tellez Y, Saborío SI, Mercado JC, Cuadra R, Rocha J, Pérez MA, Silva S, Rocha C, Harris E. Serotype-specific differences in clinical manifestations of dengue. Am J Trop Med Hyg. 2006;74:449–456. [PubMed] [Google Scholar]
- 49.Zhang C, Mammen MP, Chinnawirotpisan P, Klungthong C, Rodpradit P, Monkongdee P, Nimmannitya S, Kalayanarooj S, Holmes EC. Clade replacements in dengue virus serotypes 1 and 3 are associated with changing serotype prevalence. J Virol. 2005;79:15123–15130. doi: 10.1128/JVI.79.24.15123-15130.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McElroy KL, Santiago GA, Lennon NJ, Birren BW, Henn MR, Munoz-Jordan JL. Endurance, refuge, and reemergence of dengue virus type 2, Puerto Rico, 1986–2007. Emerg Infect Dis. 2011;17:64–71. doi: 10.3201/eid1701.100961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vu TT, Holmes EC, Duong V, Nguyen TQ, Tran TH, Quail M, Churcher C, Parkhill J, Cardosa J, Farrar J, Wills B, Lennon NJ, Birren BW, Buchy P, Henn MR, Simmons CP. Emergence of the Asian 1 genotype of dengue virus serotype 2 in Viet Nam: in vivo fitness advantage and lineage replacement in South-East Asia. PLoS Negl Trop Dis. 2010;4:e757. doi: 10.1371/journal.pntd.0000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duong V, Simmons C, Gavotte L, Viari A, Ong S, Chantha N, Lennon NJ, Birren BW, Vong S, Farrar JJ, Henn MR, Deubel V, Frutos R, Buchy P. Genetic diversity and lineage dynamic of dengue virus serotype 1 (DENV-1) in Cambodia. Infect Genet Evol. 2011 Jul 2; doi: 10.1016/j.meegid.2011.06.019. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 53.Rico-Hesse R. Dengue virus virulence and transmission determinants. Curr Top Microbiol Immunol. 2010;338:45–55. doi: 10.1007/978-3-642-02215-9_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watts DM, Porter KR, Putvatana P, Vasquez B, Calampa C, Hayes CG, Halstead SB. Failure of secondary infection with American genotype dengue 2 to cause dengue haemorrhagic fever. Lancet. 1999;354:1431–1434. doi: 10.1016/S0140-6736(99)04015-5. [DOI] [PubMed] [Google Scholar]
- 55.Kanakaratne N, Wahala WMPB, Messer WB, Tissera HA, Shahani A, Abeysinghe N, de-Silva AM, Gunasekera M. Severe dengue epidemics in Sri Lanka, 2003–2006. Emerg Infect Dis. 2009;15:192–199. doi: 10.3201/eid1502.080926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Domingo E, Escarmís C, Sevilla N, Moya A, Elena SF, Quer J, Novella IS, Holland JJ. Basic concepts in RNA virus evolution. FASEB J. 1996;10:859–864. doi: 10.1096/fasebj.10.8.8666162. [DOI] [PubMed] [Google Scholar]
- 57.Libraty DH, Young PR, Pickering D, Endy TP, Kalayanarooj S, Green S, Vaughn DW, Nisalak A, Ennis FA, Rothman AL. High circulating levels of the dengue virus nonstructural protein NS1 early in dengue illness correlate with the development of dengue hemorrhagic fever. J Infect Dis. 2002;186:1165–1168. doi: 10.1086/343813. [DOI] [PubMed] [Google Scholar]
- 58.Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181:2–9. doi: 10.1086/315215. [DOI] [PubMed] [Google Scholar]
- 59.Armstrong P, Rico-Hesse R. Differential susceptibility of Aedes aegypti to infection by the American and Southeast Asian genotypes of dengue type 2 virus. Vector Borne Zoonotic Dis. 2001;1:159–168. doi: 10.1089/153036601316977769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Armstrong PM, Rico-Hesse R. Efficiency of dengue serotype 2 virus strains to infect and disseminate in Aedes aegypti. Am J Trop Med Hyg. 2003;68:539–544. doi: 10.4269/ajtmh.2003.68.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cologna R, Rico-Hesse R. American genotype structures decrease dengue virus output from human monocytes and dendritic cells. J Virol. 2003;77:3929–3938. doi: 10.1128/JVI.77.7.3929-3938.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cologna R, Armstrong PM, Rico-Hesse R. Selection for virulent dengue viruses occurs in humans and mosquitoes. J Virol. 2005;79:853–859. doi: 10.1128/JVI.79.2.853-859.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mota J, Rico-Hesse R. Humanized mice show clinical signs of dengue fever according to infecting virus genotype. J Virol. 2009;83:8638–8645. doi: 10.1128/JVI.00581-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasilakis N, Shell EJ, Fokam EB, Mason PW, Hanley KA, Estes DM, Weaver SC. Potential of ancestral sylvatic dengue-2 viruses to re-emerge. Virology. 2007;358:402–412. doi: 10.1016/j.virol.2006.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kochel TJ, Watts DM, Halstead SB, Hayes CG, Espinoza A, Felices V, Caceda R, Bautista CT, Montoya Y, Douglas S, Russell KL. Effect of dengue-1 antibodies on American dengue-2 viral infection and dengue haemorrhagic fever. Lancet. 2002;360:310–312. doi: 10.1016/S0140-6736(02)09522-3. [DOI] [PubMed] [Google Scholar]
- 66.Balmaseda A, Sandoval E, Pérez L, Gutiérrez CM, Harris E. Application of molecular typing techniques in the 1998 dengue epidemic in Nicaragua. Am J Trop Med Hyg. 1999;61:893–897. doi: 10.4269/ajtmh.1999.61.893. [DOI] [PubMed] [Google Scholar]
- 67.Balmaseda A, Hammond SN, Tellez Y, Imhoff L, Rodriguez Y, Saborío SI, Mercado JC, Perez L, Videa E, Almanza E, Kuan G, Reyes M, Saenz L, Amador JJ, Harris E. High seroprevalence of antibodies against dengue virus in a prospective study of schoolchildren in Managua, Nicaragua. Trop Med Int Health. 2006;11:935–942. doi: 10.1111/j.1365-3156.2006.01641.x. [DOI] [PubMed] [Google Scholar]
- 68.Harris E, Videa E, Pérez L, Sandoval E, Téllez Y, Pérez ML, Cuadra R, Rocha J, Idiaquez W, Alonso RE, Delgado MA, Campo LA, Acevedo F, Gonzalez A, Amador JJ, Balmaseda A. Clinical, epidemiologic, and virologic features of dengue in the 1998 epidemic in Nicaragua. Am J Trop Med Hyg. 2000;63:5–11. doi: 10.4269/ajtmh.2000.63.5. [DOI] [PubMed] [Google Scholar]
- 69.Hammond SN, Balmaseda A, Pérez L, Tellez Y, Saborío SI, Mercado JC, Videa E, Rodriguez Y, Pérez MA, Cuadra R, Solano S, Rocha J, Idiaquez W, Gonzalez A, Harris E. Differences in dengue severity in infants, children, and adults in a 3-year hospital-based study in Nicaragua. Am J Trop Med Hyg. 2005;73:1063–1070. [PubMed] [Google Scholar]
- 70.Balmaseda A, Standish K, Mercado JC, Matute JC, Tellez Y, Saborío S, Hammond SN, Nuñez A, Avilés W, Henn MR, Holmes EC, Gordon A, Coloma J, Kuan G, Harris E. Trends in patterns of dengue transmission over 4 years in a pediatric cohort study in Nicaragua. J Infect Dis. 2010;201:5–14. doi: 10.1086/648592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guzmán MG, Vásquez S, Martínez E, Alvarez M, Rodríguez R, Kourí G, de los Reyes J, Acevedo F. Dengue en Nicaragua, 1994: reintroducción del serotipo 3 en las Américas. Bol Oficina Sanit Panam. 1996;121:102–110. [PubMed] [Google Scholar]
- 72.Quinones-Mateu ME, Ball SC, Marozsan AJ, Torre VS, Albright JL, Vanham G, van Der Groen G, Colebunders RL, Arts EJ. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J Virol. 2000;74:9222–9233. doi: 10.1128/jvi.74.19.9222-9233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hall GS, Little DP. Relative quantitation of virus population size in mixed genotype infections using sequencing chromatograms. J Virol Methods. 2007;146:22–28. doi: 10.1016/j.jviromet.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balmaseda A, Hammond SN, Pérez MA, Cuadra R, Solano S, Rocha J, Idiaquez W, Harris E. Short report: assessment of the World Health Organization scheme for classification of dengue severity in Nicaragua. Am J Trop Med Hyg. 2005;73:1059–1062. [PubMed] [Google Scholar]
- 75.Nisalak A, Endy TP, Nimmannitya S, Kalayanarooj S, Thisayakorn U, Scott RM, Burke DS, Hoke CH, Innis BL, Vaughn DW. Serotype-specific dengue virus circulation and dengue disease in Bangkok, Thailand from 1973 to 1999. Am J Trop Med Hyg. 2003;68:191–202. [PubMed] [Google Scholar]
- 76.Guzman MG, Kouri G. Dengue haemorrhagic fever integral hypothesis: confirming observations, 1987–2007. Trans R Soc Trop Med Hyg. 2008;102:522–523. doi: 10.1016/j.trstmh.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 77.Kapoor M, Zhang L, Ramachandra M, Kusukawa J, Ebner KE, Padmanabhan R. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J Biol Chem. 1995;270:19100–19106. doi: 10.1074/jbc.270.32.19100. [DOI] [PubMed] [Google Scholar]
- 78.Adams B, Holmes EC, Zhang C, Mammen MP, Nimmannitya S, Kalayanarooj S, Boots M. Cross-protective immunity can account for the alternating epidemic pattern of dengue virus serotypes circulating in Bangkok. Proc Natl Acad Sci USA. 2006;103:14234–14239. doi: 10.1073/pnas.0602768103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holmes EC, Grenfell BT. Discovering the phylodynamics of RNA viruses. PLoS Comput Biol. 2009;5:e1000505. doi: 10.1371/journal.pcbi.1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Avilés W, Ortega O, Kuan G, Coloma J, Harris E. Integration of information technologies in clinical studies in Nicaragua. PLoS Med. 2007;4:1578–1583. doi: 10.1371/journal.pmed.0040291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuan G, Gordon A, Aviles W, Ortega O, Hammond SN, Elizondo D, Nunez A, Coloma J, Balmaseda A, Harris E. The Nicaraguan pediatric dengue cohort study: study design, methods, use of information technology, and extension to other infectious diseases. Am J Epidemiol. 2009;170:120–129. doi: 10.1093/aje/kwp092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Standish K, Kuan G, Avilés W, Balmaseda A, Harris E. High dengue case capture rate in four years of a cohort study in nicaragua compared to national surveillance data. PLoS Negl Trop Dis. 2010;4:e633. doi: 10.1371/journal.pntd.0000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Colbert JA, Gordon A, Roxelin R, Silva S, Silva J, Rocha C, Harris E. Ultrasound measurement of gallbladder wall thickening as a diagnostic test and prognostic indicator for severe dengue in pediatric patients. Pediatr Infect Dis J. 2007;26:850–852. doi: 10.1097/INF.0b013e3180619692. [DOI] [PubMed] [Google Scholar]
- 84.Gubler DJ, Kuno G, Sather GE, Velez M, Oliver A. Mosquito cell cultures and specific monoclonal antibodies in surveillance for dengue viruses. Am J Trop Med Hyg. 1984;33:158–165. doi: 10.4269/ajtmh.1984.33.158. [DOI] [PubMed] [Google Scholar]
- 85.Lanciotti RS, Calisher CH, Gubler DJ, Chang GJ, Vorndam AV. Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction. J Clin Microbiol. 1992;30:545–551. doi: 10.1128/jcm.30.3.545-551.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vorndam AV, Kuno G. Laboratory diagnosis of dengue virus infections, in. In: Gubler DJ, Kuno G, editors. Dengue and Dengue Hemorrhagic Fever. CAB International; New York: 1997. pp. 313–333. [Google Scholar]
- 87.Balmaseda A, Guzmán MG, Hammond S, Robleto G, Flores C, Téllez Y, Videa E, Saborio S, Pérez L, Sandoval E, Rodriguez Y, Harris E. Diagnosis of dengue virus infection by detection of specific immunoglobulin M (IgM) and IgA antibodies in serum and saliva. Clin Diagn Lab Immunol. 2003;10:317–322. doi: 10.1128/CDLI.10.2.317-322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fernandez RJ, Vazquez S. Serological diagnosis of dengue by an ELISA inhibition method (EIM) Mem Inst Oswaldo Cruz. 1990;85:347–351. doi: 10.1590/s0074-02761990000300012. [DOI] [PubMed] [Google Scholar]
- 89.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 91.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 92.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 93.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 94.Ansarah-Sobrinho C, Nelson S, Jost CA, Whitehead SS, Pierson TC. Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology. 2008;381:67–74. doi: 10.1016/j.virol.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mattia K, Puffer B, Williams K, Gonzalez R, Murray M, Sluzas E, Pagano D, Ajith S, Bower M, Berduougo E, Harris E, Doranz B. Reporter virus particles for measuring neutralizing antibodies against each of the four dengue serotypes. PLoS One. 2011 doi: 10.1371/journal.pone.0027252. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.