

Significance
Human cytomegalovirus (HCMV) is an important human pathogen. Current anti-HCMV therapies suffer from toxicities, drug resistance, and/or pharmacokinetic limitations. A possible antiviral drug target is a two-subunit complex that orchestrates nuclear egress, an essential, unusual mechanism by which nucleocapsids move from the nucleus to the cytoplasm during viral replication. We solved the structure of the conserved core of one subunit of the complex, mapped the primary interaction interface with the other subunit, and tested the importance of specific residues for subunit interactions and viral replication. The combined biophysical and biological analyses presented here develop molecular understanding of nuclear egress and identify a groove that includes a large cavity on the subunit as an attractive target for yet to be identified inhibitors.
Keywords: herpesvirus, cytomegalovirus, drug target, protein–protein interactions, NMR
Abstract
Herpesviruses require a nuclear egress complex (NEC) for efficient transit of nucleocapsids from the nucleus to the cytoplasm. The NEC orchestrates multiple steps during herpesvirus nuclear egress, including disruption of nuclear lamina and particle budding through the inner nuclear membrane. In the important human pathogen human cytomegalovirus (HCMV), this complex consists of nuclear membrane protein UL50, and nucleoplasmic protein UL53, which is recruited to the nuclear membrane through its interaction with UL50. Here, we present an NMR-determined solution-state structure of the murine CMV homolog of UL50 (M50; residues 1–168) with a strikingly intricate protein fold that is matched by no other known protein folds in its entirety. Using NMR methods, we mapped the interaction of M50 with a highly conserved UL53-derived peptide, corresponding to a segment that is required for heterodimerization. The UL53 peptide binding site mapped onto an M50 surface groove, which harbors a large cavity. Point mutations of UL50 residues corresponding to surface residues in the characterized M50 heterodimerization interface substantially decreased UL50–UL53 binding in vitro, eliminated UL50–UL53 colocalization, prevented disruption of nuclear lamina, and halted productive virus replication in HCMV-infected cells. Our results provide detailed structural information on a key protein–protein interaction involved in nuclear egress and suggest that NEC subunit interactions can be an attractive drug target.
Herpesviruses encompass a large family of infectious agents, including important veterinary and human pathogens (1). Among the latter is human cytomegalovirus (HCMV), which can cause serious disease, particularly in immunocompromised individuals and newborns (2). Despite the importance of HCMV in these medically vulnerable populations, currently available treatment options suffer from issues with toxicities, drug resistance, and/or pharmacokinetics (2, 3), motivating the identification of new drug targets.
All herpesviruses of mammals, birds, and reptiles undergo a remarkable process known as nuclear egress as part of the viral lifecycle. It is generally accepted that, after assembly in the nucleus, the viral nucleocapsid undergoes envelopment to cross the inner nuclear membrane (INM) followed by deenvelopment to cross the outer nuclear membrane, resulting in release into the cytoplasm for continuation of the virion maturation process (4). Nuclear egress is orchestrated by a highly conserved, heterodimeric nuclear egress complex (NEC), which recruits one or more protein kinases to disrupt the nuclear lamina, permitting access of nucleocapsids to the INM, where the NEC induces budding of the nucleocapsid into the perinuclear space (5–13). In HCMV, the NEC is comprised of UL50, which is an INM protein, and UL53, which is a nucleoplasmic protein that is brought to the INM by its interaction with UL50. These two proteins and their murine CMV (MCMV) homologues, M50 and M53, are essential for replication and nuclear egress (8, 14–17) of their respective viruses. Although a process similar to herpesvirus nuclear egress was recently described for movement of ribonucleoprotein particles during Drosophila myogenesis (18), no host cell homolog of the NEC that would serve as mediator of this mechanism has yet been identified. Furthermore, no structural information currently exists for any NEC subunit across the Herpesviridae family.
To gain a better molecular understanding of herpesvirus nuclear egress, we used NMR methods to solve the structure of the conserved half of MCMV M50 and map residues on the surface of M50 that are involved in interactions with the other NEC subunit. We then tested the importance of several of these residues for heterodimerization of both the MCMV and HCMV NECs by looking at the effect of single-alanine mutations in both M50 and UL50 on binding affinity and replication of HCMV by looking at the effect of mutations in the context of NEC localization, nuclear lamina disruption, and virus production. Our results identified a subunit interaction interface with features that suggest that it could be an attractive antiviral drug target.
Results and Discussion
Solution-State NMR Structure of M50 (1–168).
MCMV M50 is a 316-aa protein with a predicted C-terminal transmembrane region (11) and an N-terminal one-half that is highly conserved among herpesvirus homologs (Figs. S1A and S2) (16, 19). The NMR structure of this N-terminal conserved core (M50; residues 1–168) was determined using ∼1,400 NOE-derived distance constraints, of which ∼350 were long-distance constraints. Most of the long-range constraints were derived from 3D time-shared 15N/13C-resolved nuclear Overhauser effect spectroscopy (NOESY) and 4D 13C-resolved heteronuclear multiple quantum coherence (HMQC)-NOESY-HMQC experiments performed on an isoleucine, leucine, valine (ILV)-labeled sample, where the terminal methyl groups of these residues were 13C-labeled and protonated in an otherwise deuterated background. In addition to NOEs, backbone dihedral constraints derived from the chemical shifts using TALOS+ (20) and orientation constraints derived from backbone amide residual dipolar couplings (RDCs) were used in the structure calculation. The structure was consequently refined in explicit water with 110 N-H RDCs. The RDC-refined and NOE-determined structures of M50 showed essentially indistinguishable secondary structure content and orientation of β-strands (Fig. S1C). The backbone rmsd for residues with secondary structure for the 15 lowest energy structures is 1.2 Å, and 98.8% of the residues in these 15 structures occupy the most favored and additionally allowed region in the Ramachandran plot. The complete statistics of the structure calculation as well as adherence to Ramachandran parameters are summarized in Table S1, and Fig. 1A displays an ensemble of the 15 lowest energy structures. Longitudinal and transverse relaxation times (T1 and T2) as well as hetero-NOE data (Fig. S1D) are consistent with a relatively rigid 18-kDa protein, with an average T2 of 50 ms.
Fig. S1.

Protein schematics and effect of RDC refinement. (A) Schematic of M50. The region demarcated in blue represents the residues expressed and used in the described NMR and ITC experiments. The M50 C-terminal transmembrane domain (TM) from residues 285–307 as predicted by TMHMM Server, version 2.0 (www.cbs.dtu.dk/services/TMHMM/) is demarcated in black. (B) Schematic of UL53. The region demarcated in orange represents the residues represented in the synthetic peptide used for the described NMR-based titration experiments. (C) The 15 lowest energy structures from the CYANA structure calculation of M50 (residues 1–168) in red aligned with the 15 lowest energy structures after the RDC and AMBER water refinements in blue. (D) T1, T2, and hetero-NOE values as derived from the associated NMR experiments (acquired at 291 K and plotted vs. residue number). These data show that M50 shows flexibility primarily in the loop regions of the protein but not in regions with defined secondary structure. Secondary structure elements (α-helices in blue and β-sheets in red) as determined by PROCHECK from the structure are depicted above the plots.
Fig. S2.

Clustal Omega (61) alignment of human herpesvirus homologs of M50. Colons indicate conservation between groups of strongly similar properties (scoring >0.5 in the Gonnet PAM 250 matrix). Periods indicate conservation between groups of weakly similar properties (scoring ≤0.5 in the Gonnet PAM 250 matrix). Residues with nonzero normalized ratios of peak intensities lower than 0.4 in titration experiments with 100% UL53 peptide and isotopically enriched M50 (1–168) are highlighted in red. Secondary structural elements (based on PROCHECK topology) from the M50 (1–168) lowest energy fold are blue in areas of α-helical character and red in areas of β-stranded character. TM, transmembrane domain. *Positions that have an identical residue.
Table S1.
NMR and refinement statistics for the M50 (1–168) structure
| M50 (1–168) | |
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | 1,384 |
| Intraresidue | 287 |
| Interresidue | |
| Short and medium range (1 ≤ |i − j| ≤ 5) | 715 |
| Long range (|i − j| > 5) | 357 |
| Hydrogen bonds | 99 |
| Total dihedral angle restraints | 282 |
| Φ | 141 |
| Ψ | 141 |
| RDC restraints | 110 |
| Alignment tensor* | |
| Aa (×10−4) | 7.715 ± 0.183 |
| Ar (×10−4) | 2.236 ± 0.190 |
| χ2 (×102) | 1.913 |
| Structure statistics | |
| Violations (mean and SD) | |
| Distance constraints (Å) | 0.08 ± 0.03 |
| Dihedral angle constraints (°) | 6.26 ± 3.53 |
| Maximum dihedral angle violation (°) | 30.36 |
| Maximum distance constraint violation (Å) | 0.27 |
| Ramachandran plot summary† (%) | |
| Most favored regions | 87.6 |
| Additionally allowed regions | 11.19 |
| Generously allowed regions | 0.4 |
| Disallowed regions | 0.0 |
| Average pairwise rmsd (Å) | |
| Heavy; ordered residues‡ | 1.8 |
| Backbone; ordered residues‡ | 1.2 |
| Backbone; selected residues§ | 1.0 |
Fitted with Module.
Calculated by PSVS using Procheck-NMR for 15 structures.
Calculated by PSVS for 15 structures (ordered residues: 5–32, 34–39, 41–43, 45–51, 54–99, 101–111, 113–124, 131–139, 145–153, and 158–168).
Ordered residues excluding the C-terminal α-helix 158–170 and flexible residues on the N terminus: 8–32, 34–39, 41–43, 45–51, 54–99, 101–111, 113–124, 131–139, and 145–153.
Fig. 1.
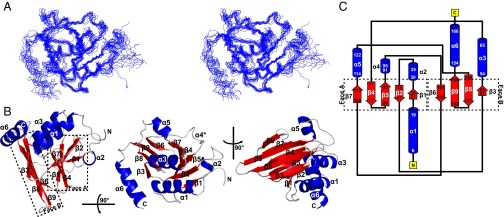
Solution-state structure of M50 (1–168) as determined by NMR. (A) The 15 lowest energy structures generated by the AMBER water refinement are shown overlaid using backbone atoms for residues 10–158 in side-by-side stereoview (Protein Data Bank ID code 5A3G). (B) Three viewpoints of a ribbon diagram of M50 (1–168) showing nine β-strands (red) and six α-helices (blue). Faces A and B of the β-taco are delineated in Left by the dotted outlines and labeled. *Helix-α4 is recognized as helical by PROCHECK (50) but not by default PyMOL (51) secondary structure identification. (C) Schematic of the topology of M50 (1–168).
The structure of M50 (1–168) is comprised of a spreading β-sandwich, with nine clearly defined β-strands interspersed by six α-helices. The distinct V shape of the β-sandwich and the positions of the helices (Fig. 1B) led to the structure being described by a colleague as a “β-taco.” Three of the α-helices are 10 aa or more in length, including one at the N terminus consistent with bioinformatics predictions (19), and the rest are short helices occurring between β-strands. Five of nine β-strands form one side (face A) of the fold, whereas the other four constitute the other side (face B). The opposing β-sheets are intricately woven together (Fig. 1C). β-strands 1 and 2 form an antiparallel sheet within face A, and β-strand 3 crosses over to face B and is returned back to face A as β-strand 4, which loops back into β-strand 5. β-strand 5 then traverses to face B through a 15-residue segment to form β-strand 6, which forms the edge of face B. β-strand 6 returns back to face A as β-strand 7 to complete the five antiparallel β-strands that make up face A. β-strand 7 then crosses over to face B through α-helix 5 to form the central two antiparallel β-strands 8 and 9, which complete the four β-strands of face B. β-strand 9 terminates in the final C-terminal α-helix 6. The strands ultimately cross five times between the two faces to form the β-taco fold.
Relationships to Other Proteins.
Motivated by the intricately interwoven topology of the M50 (1–168) structure, a number of databases were scanned to search for possible structural homologs. The CATHEDRAL Algorithm (based on the CATH database) (21), the Dali structural comparison database (22), FATCAT (23, 24), VAST (25), PDBeFold (26), and Skan (27, 28) (Table S2) returned hits that failed to encompass the overall M50 (1–168) fold. VAST and PDBeFold returned hits that approximated the dual β-sheet nature of the structure but with additional secondary structural differences. None of the databases returned identical top hits, and regions of structural similarity often encompassed less than one-half of the M50 (1–168) fold. Visual comparison of the overall protein topologies as well as attempts to align the structures revealed major differences, even within areas of structural similarity (Table S2). These results across multiple databases indicate that, although portions of the M50 (1–168) structural fold may be familiar from other known proteins, so far, the overall M50 (1–168) fold cannot be matched to any previously determined structure in the Protein Data Bank (PDB).
Table S2.
Analysis of top hits from the structural database comparisons
| Database and top hit | Score | RMSD (Å) |
| CATH Database (21) | ||
| YIGZ, a conserved hypothetical protein from E. coli k12 (CATH: 1VI7A02) | Sequential structure alignment program 77.8 | 5.7 |
| Residue range: 138–208; 71 residues | ||
| CATH domain: 1VI7A02 N:β1---α1-β2----β3----α2-β4:C | ||
| M50 (1–168) N:β5- -α1-β1-α2-β2-β3-α3-β4:C | ||
| Aligned secondary structures show additional elements in the M50 (1–168) fold in the region of structural similarity. The CATH domain has only a single β-sheet. Strand-β3 in M50 (1–168) is part of the opposite sheet that does not align with the sheet found in 1VI7A02, and α2 is an additional helix found in the M50 (1–168) fold that is not present in the CATH domain. Strand-β5 from the middle of face A of M50 (1–168) also makes the topologies inconsistent. | ||
| Dali Structural Comparison Database (22) | ||
| Collagenase from Clostridium histolyticum (2Y6I:A) | Z score: 3.1 | 3.2 |
| 71 Residues | ||
| 2Y6I chain A N:α1-β1----β2----α2-α3-β3-β4:C | ||
| M50 (1-168) N:α1-β1-α2-β2-β3-α3----β4-β5:C | ||
| Aligned secondary structures show additional elements in both 2Y6I:A and the M50 (1–168) fold in the region of structural similarity; 2Y6I:A has only a single β-sheet, whereas β3 from M50 (1–168) is part of a separate sheet. Helix-α2 from 2Y6I:A is much longer than the corresponding helix in the M50 (1-–68) fold, and α3 from 2Y6I:A is not even present in M50. | ||
| FATCAT (23, 24) | ||
| May6-15_pdb_90 (PDB May6-15 version, 90% nonredundant set with 35,746 structures) | ||
| Trafficking protein particle complex subunit 6A (2C0J:B) | Score: 122.89 | Opt: 3.10 |
| Residue range: 567–673; 101 residues | P value: 2.10e-03 | Chain: 3.13 |
| 2C0J chain B N:α1- -α2-α3-β1----β2-α3.5- -α4-β3-β4:C | ||
| M50 (1–168) -α6:C N:---α1-β1-α2-β2-β3-----α3-β4-β5- | ||
| Aligned secondary structures show additional elements in both 2C0J:B and the M50 (1–168) fold in the region of structural similarity. The initial helix of 2C0J:B aligns with the C-terminal helix of the M50 (1–168) fold, which is not consecutive with the remaining aligned structure; 2C0J:B has only a single β-sheet, whereas β3 from M50 (1–168) is part of a separate sheet. Helix-α2 from 2J3T:B is a significant helix not present in the M50 (1–168) fold. | ||
| scop175_90 (scop 1.75 version, 90% nonredundant set with 15,543 structures) | ||
| Multidrug efflux transporter AcrB E. coli (d1iwga3) | Score: 103.81 | Opt: 3.06 |
| 68 Residues | P value: 6.37e-03 | Chain: 3.24 |
| SCOP d1iwga3 N: β1------α1-β2----β3-β4-β5-α2----α3-β6:C | ||
| M50 (1–168) N:-β5-:C N:α1-β1-α2-------β2----β3-α3-β4:C | ||
| Aligned secondary structures show additional elements in both d1iwga3 and the M50 (1–168) fold in the region of structural similarity; d1iwga3 has only a single β-sheet and does not recapitulate the double β-sheet nature of the M50 (residues 1–168) fold. The major C-terminal helix of M50 (residues 1–168) is also not present in the region of structural similarity with d1iwga3. | ||
| VAST (25) | ||
| 5-Keto-4-deoxyuronate isomerase from E. coli (1XRU:B; residue range: 38–122; 63 residues) | 63 aligned residues | |
| 1XRU chain B N: β1----β2-β3-α1-β4------β5----β6-β7----β8:C | ||
| M50 (1–168) N:α1-β1-α2-β2-β3-α3-β4-β5- -β9- --β6-β7-α5-β8-:C | ||
| Aligned secondary structures show additional elements in the M50 (1–168) fold in the region of structural similarity. The only one of six helices of the M50 (1–168) fold is present in 1XRU:B; 1XRU:B does have two facing β-sheets, but the topologies are not identical, and strands in M50 (1–168) are either not present (β5) or present out of order (β9) in 1XRU:B. | ||
| PDBeFold (26) | ||
| No nonidentical matches with 70% similarity or higher; Pea lectin β-chain from Pisum sativum (1RIN:A) Residue range: 61–180; 71 residues | 40% of M50 (1–168) secondary structure was identified in the target protein | 3.94 |
| 46% of target chain secondary structure was identified in M50 (1–168) | ||
| 1RIN chain A N:-β8---α1-α2-β9-β10-β11-β12-β13:C | ||
| M50 (1–168) N:-β5- -α2- -β4-β7- -β8--β9-:C | ||
| Note that 1RIN:A showed a number of strands that were not identified as β-strands in PyMol or on the images present on the RCSB PDB website but strongly resembled them in parallel orientation. They have been taken as β-strands for the purpose of this analysis. | ||
| Aligned secondary structures show additional elements in both 1RIN:A and the M50 (1–168) fold in the region of structural similarity; 1RIN:A is lacking many of the major helices present in the M50 (1–168) fold, and the linear topologies of the two folds do not coincide in several areas. In addition, several of the β-strands in 1RIN:A and M50 (1–168) that align run in opposite directions from each other. | ||
| MarkUs Protein Function Annotation Server—Skan (28) Major NAD(P)H-flavin oxidoreductase from Aliivibrio fischeri (1VFR:A); 77 Residues | Protein Structural Distance score 0.72 | 3.99 |
| Structural Alignment Score (rmsd × 100/number aligned residues) 4.2 | ||
| 1VFR chain A N:α1-α2-β1-α3-α4-β2-α5-α6-α7-β3-α8-β4-α9:C | ||
| M50 (1–168) N: -β6- -β8- -β9- :C | ||
| Aligned secondary structures show additional elements in both 1VFR:A and the M50 (1–168) fold in the region of structural similarity; 1VFR:A has only a single β-sheet and does not recapitulate the double β-sheet nature of the M50 (residues 1–168) fold. Although both structures have a mixture of α-helices and β-sheets and roughly align, exact overlap between secondary structural elements is very poor. None of the major helices and most of the β-strands of the M50 fold are not represented by the elements in 1VFR:A. | ||
PDB, Protein Data Bank; RCSB, Research Collaboratory for Structural Bioinformatics.
To investigate whether any proteins of unknown structure might have a fold similar to that of M50 (1–168), we used threading with Phyre2 (29) in BackPhyre mode to screen several bacterial and eukaryotic sequence databases against the M50 (1–168) coordinates. These searches returned no confidence scores higher than ∼42% (for an Arabidopsis protein of unknown function) and fit no more than 43 amino acids (of 244 total) for a Caenorhabditis elegans protein (F18E9.8). In no case did the polypeptide segments of M50 that were fit form a compact domain. However, threading of viral protein sequences with Phyre2 predicted known homologs of M50, including HCMV UL50 (Fig. S3) (100% confidence score), to have structures highly similar to that of M50 (1–168). Threading of the sequence of the more distantly related HSV-1 homolog of M50 (UL34) onto the M50 structure returned an ∼98% confidence score (Fig. S3). Although lack of energy optimization in the Phyre2 calculations limits the likelihood that the actual structural homologies are quite so high, the striking scores support the suggestion that our structural understanding of M50 (1–168) also applies to its homologs in a variety of herpesviruses.
Fig. S3.

Stereo views of the PyMol (51) alignment of the Phyre2 (www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) (62) HCMV UL50 (purple) and HSV-1 UL34 threading results (red) with the M50 (1–168) lowest energy model (blue). Left and Center form a wall-eyed stereoview, and Center and Right form a cross-eyed stereoview. The free end of the structure depicted at the bottom is the N terminus. The orientation is a 90° clockwise rotation from Fig. 1B, Center.
Titration of a Conserved Peptide Identifies a Potential Binding Groove with a Large Cavity.
An important function of M50 and its homologs is to interact with their nucleoplasmic partners. To gain insight into this interaction, we hypothesized that the strong sequence conservation between both subunits of the HCMV and MCMV NECs would allow M50 to bind a peptide derived from a region of HCMV UL53 that was previously shown to be important for heterodimer formation (Figs. S1B and S4A) (30). We titrated this synthetic UL53 peptide into 15N-labeled M50 (1–168) and monitored the binding using a series of 1H-15N heteronuclear single-quantum coherence (HSQC) experiments. The titration resulted in line broadening and associated reductions of peak intensity of specific resonances in the M50 (1–168) spectrum, indicating that the UL53 peptide binding was in an intermediate exchange regime (Fig. 2 A and B). When the resonances that displayed substantial broadening are mapped onto the structure of M50 (1–168) (Fig. 2 C and D), the affected residues form a continuous grooved region that encompasses most of helix-α3 and -α6 and parts of helix-α5 and strand-β8 and -β9. A solvent-accessible cavity identification program (SCREEN) (28, 31) identified eight potential cavities on the M50 (1–168) structure. The number one ranked cavity and largest by surface area runs parallel to the C-terminal helix (α6) and is contained within the area identified by the titration experiment. These results suggested that the peptide was binding within this groove, forming the core of the NEC heterodimerization interface.
Fig. S4.

UL53 peptide and example ITC traces. (A) Clustal Omega alignment of the UL53 peptide (residues 58–85) and M53 (GenBank accession no. ACE95567.1) sequences shows a high degree of conservation between the HCMV and MCMV in the minimal conserved region previously identified (30, 61). Colons indicate conservation between groups of strongly similar properties (scoring >0.5 in the Gonnet PAM 250 matrix). Periods indicate conservation between groups of weakly similar properties (scoring ≤0.5 in the Gonnet PAM 250 matrix). Residues highlighted in cyan are those with aliphatic hydrophobic sidechains. Residues highlighted in green are those with aromatic hydrophobic sidechains. *Positions that have an identical residue. (B) ITC trace for M50 (residues 1–168) titrated into M53 (residues 50–292). (C) ITC trace for M50 (residues 1–168) V52A titrated into M53 (residues 50–292).
Fig. 2.

UL53 peptide binding site. (A) Heteronuclear single-quantum coherence spectrum of 15N M50 (1–168) with equimolar unlabeled UL53 peptide titrated into the sample. The expanded view shows Y57 as an example of a peak that shows chemical shift perturbation on the addition of the peptide and S82 (marked with an asterisk) as an example of a peak that shows no chemical shift perturbation. (B) Bar graph of the normalized ratio of peak intensities with equimolar unlabeled UL53 peptide present to peak intensities with no unlabeled UL53 peptide present for each residue. Areas highlighted by red boxes are residues that show marked reduction in peak intensity on addition of the peptide, and these areas correspond to the residues shown in red in C and D. (C) Ribbon representation of M50 (1–168), with residues showing a nonzero normalized ratio of peak intensity lower than 0.4 in titration experiments highlighted in red. (D) Surface representation of M50 (1–168), with residues showing a nonzero normalized ratio of peak intensity lower than 0.4 in titration experiments highlighted in red. Only a subset of residues has been labeled in C and D for clarity of presentation.
Importance of Residues in the Groove and Cavity for Heterodimerization.
To test whether residues within the groove and cavity are important for heterodimerization, we individually substituted nine different M50 (1–168) residues identified in the titration experiment with alanine. We also substituted seven of the corresponding residues and three others in HCMV UL50 (1–169) with alanine. Using isothermal titration calorimetry (ITC), we assayed the M50 mutants and the UL50 mutants along with the WT versions of these proteins for their ability to bind M53 (residues 103–333) or UL53 (residues 50–292), respectively. The substitutions resulted in a range of effects, with some causing reductions in binding affinity by a factor greater than 100 (Table 1 and Fig. S4 B and C). Substitutions that alter residues between positions 52 and 60 of either protein (at the bottom of the groove in Fig. 2 C and D) all decreased affinity by at least fivefold. This segment of UL50 and M50, particularly E56 and Y57, and the corresponding segment of their pseudorabies virus and Kaposi’s sarcoma herpes virus homologs have previously been shown to be important for interaction with the other NEC subunit in less quantitative coimmunoprecipitation and colocalization assays (16, 19, 32, 33). We also found that several substitutions that alter residues between positions 121 and 129 of M50 and 125 and 139 of UL50 (at the top of the groove and within the cavity in Fig. 2 C and D) also decreased binding, with substitution of a leucine (L129 in M50 and L130 in UL50) within the cavity having at least a 27-fold effect. Finally, substitution of a residue (N154A) within the loop connecting β9 to α6 in M50 also substantially decreased binding. Deletions of residues with the corresponding segments of the HSV-1 and Kaposi’s sarcoma herpes virus homologs decrease subunit interactions based on pull-down and colocalization experiments (33, 34). Our analysis provides a structural basis for these previous findings and identifies specific residues within a cavity of M50 and UL50 that are crucial for subunit interactions.
Table 1.
Effects of substitutions on subunit interactions and viral replication
| M50 (1–168) mutation | Kd (µM)* | Factor weaker than WT† | UL50 (1–169) mutation | Kd (µM) | Factor weaker than WT | HCMV replication phenotype‡ |
| WT | 0.97 ± 0.12 | 1 | WT | 0.29 | 1 | WT |
| V52A | NB | >100 | V52A | 1.4 ± 0.72 | 5 | 5- to 10-fold lower yield |
| E56A | N/A | N/A | E56A | 30–90 | >100 | Nonviable |
| Y57A | 4.1 ± 0.35 | 5 | Y57A | NB | >100 | N/A |
| S60A | 17 | 20 | S60A | N/A | N/A | N/A |
| Y61A | Insoluble | N/A | Y61A | Insoluble | N/A | N/A |
| E63A | 1.1 ± 0.33 | 4 | N/A | |||
| R65A | 0.55 ± 0.08 | 2 | N/A | |||
| I121A | 98 | >100 | I122A | N/A | N/A | |
| C124A | 76 | 87 | S125A | 2.2 | 7 | 5- to 10-fold lower yield |
| V128A | 0.89 | 1 | V129A | 0.32 | 1 | N/A |
| L129A | 60–85 | 68–100 | L130A | 7.9 | 27 | Nonviable |
| C133A | 0.67 ± 0.30 | 1 | N/A | |||
| D153A | 0.34 ± 0.14 | 0.4 | E154A | 0.51 ± 0.24 | 2 | N/A |
| N154A | 16 ± 2.5 | 16 | N155A | N/A | N/A | N/A |
N/A, not generated or tested (the M50 and UL50 Y61A mutant proteins were insoluble); NB, no detectable binding.
KD values ± SD were derived from ITC data for binding of M53 (residues 103–333) to WT and single point mutant M50 (residues 1–168) proteins and binding of UL53 (residues 50–292) to WT and single point mutant UL50 (residues 1–169) proteins.
Factors weaker than WT values were calculated by dividing the Kd value of the mutant by the Kd value of the WT protein.
Mutant BACs that produced no detectable spread or virus were considered nonviable. Factor reductions in yield of viable mutants were calculated by dividing the supernatant viral titer of the mutant virus by the supernatant viral titer of WT virus at day 7 postinfection (Fig. S5A).
Previous studies indicated that the interaction interface on M53 and UL53 includes a predicted amphipathic helical segment interrupted by a proline comprising residues 60–82 (Fig. S4A) and requires both hydrophobic residues and charged residues within this predicted helix (17, 30). We speculate that at least part of this helical segment fits within the groove in such a way as to allow burying of bulky hydrophobic sidechains present in the peptide (Fig. S4A). We found that replacement of certain large hydrophobic residues or neutral polar residues on M50 and UL50 with a smaller hydrophobic residue, alanine, resulted in reduced binding of a number of mutants to the nucleoplasmic partner (Table 1). Replacement of the negatively charged residue E56 also reduced binding. In contrast, replacement of a different negatively charged residue in M50, D153, with alanine resulted in a marginal increase in binding. Quantitative differences in binding affinity caused by substitutions of homologous residues (Table 1) show that there are variations in the exact mode of binding, even between closely related proteins, such as M50 and UL50; however, several of the residues that we have identified as important, such as E56 and L129 (L130 in UL50), are identical or similar across widely divergent herpesviruses (Fig. S2), which is consistent with similarities in binding mechanisms.
Importance During HCMV Replication.
To examine the importance of individual residues in the groove and cavity during HCMV replication, four substitutions in UL50, which have varying importance for heterodimerization in vitro, were introduced into a GFP-encoding BAC derived from HCMV strain AD169, pBADGFP (35), or modifications of this BAC, in which sequences encoding an FLAG tag have been fused to UL53 coding sequences (8). Electroporation of BACs containing WT UL50 and mutants V52A and S125A that had 5- to 10-fold reductions in UL53 binding in vitro (Table 1) into human foreskin fibroblasts (HFFs) resulted in a spreading infection, with increasing numbers of GFP-expressing cells showing cytopathic effect, and production of infectious virus. For V52A and S125A, after infection at a low multiplicity, the resulting mutant viruses displayed 5- to 10-fold reductions at 5–7 days postinfection, with S125A maintaining a reduced yield and V52A attaining a near-WT yield at 9 days postinfection (Table 1 and Fig. S5 A and B). In contrast, electroporation of BACs containing either a UL50 null mutation [as previously observed (8)], the E56A [as also observed recently (36)], or L130A substitutions that reduced binding to UL53 in vitro by 27- to >100-fold (Table 1) resulted in only individual cells expressing GFP and no detectable infectious virus. To determine if the effects on virus replication were caused by the introduced substitutions, we constructed rescued derivatives of the mutant BACs, in which each mutation was replaced with WT sequences. For the rescued derivatives of the E56A and L130A mutants (E56AR pBADGFP and L130AR pBADGFP, respectively), electroporation resulted in spread and cytopathic effect. The resulting viruses, and the viruses generated by electroporation of the rescued derivatives of V52A and S125A (V52AR and S125AR) all replicated indistinguishably from WT virus after infection at a low multiplicity (Fig. S5B). Thus, there was a strong positive correlation between the effects of substitutions on UL53 binding in vitro and their effects on viral replication in cells.
Fig. S5.

Substitution mutants, viral replication, UL53 distribution, and nuclear lamina disruption. (A) UL50 mutant viruses. V52A (□) and S125A (△) were compared with WT BADGFP virus (◆) after inoculation at a multiplicity of infection of 0.1 pfu per cell of 105 HFF cells per well in 24-well plates. For each data point in A and B, the supernatants from two wells were pooled on the indicated day postinfection, and titers were calculated by averaging plaque counts from duplicate titrations. Counts from duplicate titrations differed by less than a factor of two for all data points. Error bars are not shown. (B, Upper) Rescued derivatives of the UL50 null mutant 50NR (◆), the E56A mutant E56AR (△), and the L130 mutant L130AR (×). (B, Lower) The point mutants V52A (■) and S125A (▲) and their rescued derivatives V52AR (□) and S125AR (△), respectively, were compared with WT virus (BADGFP; □ in Upper and ○ in Lower) for replication after inoculation at a multiplicity of infection of 0.1 pfu per cell of 105 HFF cells per well in 24-well plates. (C) HFFs were electroporated with WT (i–iii) UL53-FLAG pBADGFP or (iv–vi) UL50 null UL53-FLAG pBADGFP or the E56A (vii–ix) UL53-FLAG pBADGFP or (x–xii) its rescued derivative E56AR UL53-FLAG pBADGFP. Cells were fixed on day 7 and stained with anti-FLAG antibody (red) or DAPI (blue), and virus-infected cells were visualized by confocal microscopy. (D) HFFs were (i–iii) mock-electroporated or electroporated with (iv–vi) WT, (vii–ix) UL50 null, (x–xii) E56A, or (xiii–xv) E56AR pBADGFP. The cells were fixed on day 7 and stained with antibody against lamin A/C (red), and the nucleus was stained with DAPI (blue). Virus-infected cells were observed for the appearance of the nuclear lamina using confocal microscopy. White arrows point to disruptions of nuclear lamina.
We then assessed the effects of the lethal substitutions E56A and L130A on the subcellular distribution of UL53 during infection by comparing the mutant viruses with their rescued derivatives. UL53 showed an almost exclusive localization to the nuclear rim in cells infected with the rescued derivatives, similar to what is seen with WT HCMV. However, in E56A and L130 mutant-infected cells, UL53 was distributed throughout the nucleus, similar to what is seen in cells infected with a UL50 null mutant. (Fig. 3A and Fig. S5C) (8). Furthermore, cells infected with the mutants L130A or E56A, displayed oval nuclei and almost intact nuclear lamina comparable with mock-infected cells or cells infected with a UL50 null mutant, whereas cells infected with the rescued derivatives cells showed deformation of nuclear shape, ruffling and thinning of the nuclear lamina, and generation of gaps in the lamina that are characteristic of WT HCMV infection (Fig. 3B and Fig. S5D). Thus, substitutions of individual residues in the groove and cavity that are lethal for HCMV prevent UL50–UL53 colocalization and the NEC-mediated disruption of the nuclear lamina that accompanies nuclear egress. These results taken together strongly suggest that HCMV NEC subunit interactions are essential for lamina disruption and replication.
Fig. 3.

Effects of substitutions on distribution of UL53 and disruption of nuclear lamina. HFFs were electroporated with (i–iv) the mutant L130A UL53-FLAG pBADGFP or (v–viii) the rescued derivative L130AR UL53-FLAG pBADGFP. Cells were fixed on day 7, stained with DAPI, and stained with either (A) anti-FLAG antibody (red) or (B) antibody against lamin A/C (green). Cells positive for GFP were visualized by confocal microscopy. In B, the red arrow points to an uninfected cell, and the white arrows point to gaps in nuclear lamina in cells infected with the rescued derivative.
Second Functional Surface.
Previous work with MCMV has identified dominant negative mutants of M50, which would be consistent with the protein being multifunctional. One of these functions is almost certainly its interaction with the nucleoplasmic subunit (M53), explaining the identified dominant negative phenotypes of insertions between E56 and Y57 and between D125 and K126 of M50 (37), because these residues are located within the core interaction interface described here. A different function may be defined by a dominant negative insertion between Y40 and S41 (37). These residues are found in a loop just after helix-α2. This loop is quite distal to the core interaction surface, and other insertions nearby (between K36 and N37 and between C43 and D44) permit M53 binding (16). Interestingly, substitutions of HSV-1 UL34 residues corresponding to the β1-strand of M50, which is in the vicinity of this loop, also result in a dominant negative phenotype without affecting subunit interactions as assessed by colocalization (38, 39). Thus, these dominant negative mutants seem to identify at least one additional functional surface of this NEC subunit.
Implications for Drug Discovery.
Our finding of lethal single-amino acid substitutions within the binding groove/cavity that disrupt NEC subunit interactions suggests that small molecules may be found that mimic this effect, thus ablating viral replication. This interaction interface consists primarily of hydrophobic and aromatic residues interrupted by a few charged amino acids, presenting an ideal landscape for drug design with complementing hydrophobic groups and hydrogen bond donors and acceptors to anchor a small-molecule drug. The complete conservation across human herpesviruses of residues, such as M50 E56, and the conservation of the hydrophobic character of others, such as M50 L129 (UL50 L130) (Fig. S2), which are important for binding and virus viability, raise the possibility that small molecules that bind these sites could have antiviral activity against a broad spectrum of herpesviruses. Such compounds might have considerable specificity given the apparent lack of cellular homologs of this subunit of the NEC.
Although nuclear egress was once thought to be limited to herpesviruses, recently, a broadly similar process has been identified for the export of ribonucleoprotein complexes in Drosophila (18, 40), suggesting that insight into herpesvirus nuclear egress may be more generally applicable than previously thought. Still, the unusual structure presented here of a crucial player in nuclear egress underscores the unusual nature of this process. Insights from this structure should apply to widely divergent herpesviruses. Moreover, a combined biophysical and genetic analysis of one of its key functions has identified a target that may permit the rational development of much needed, new therapeutics for diseases caused by HCMV and other herpesviruses.
Materials and Methods
Protein Expression and Purification.
Unlabeled and uniformly labeled (15N, 13C, and/or 2H) samples of C-terminal truncations of UL50, UL53, M50, and M53 as well as alanine mutants of UL50 and M50 were derived from constructs created and expressed using methods similar to those described in the work by Sam et al. (30). Isoleucine, leucine, valine-methyl–labeled samples were prepared using α-ketobutyrate and α-ketoisovalerate precursors as described (41, 42). More details are in SI Materials and Methods.
Structure and Interface Determination by NMR.
Standard NMR spectra for structure determination in addition to 3D 13C,15N time-shared NOESY (43) and 4D 13C-HMQC-NOESY-HMQC experiments (44) were acquired at 291.15 K with ∼150- to 300-µM M50 (1–168) protein samples in 25 mM NaPO4 (pH 6.5), 150 mM NaCl, and 1 mM DTT. All NMR data were processed using NMRPipe (45), and they were visualized and analyzed using CARA (46, 47) and CcpNmr Analysis (48). The structure calculation was run using CYANA. Dihedral angle constraints were extracted from the backbone chemical shifts using the program TALOS+ (20). The 50 lowest energy structures were selected for refinement using 15N-H RDC constraints in explicit water with AMBER through the WeNMR web interface (49). The core heterodimerization interface was mapped using transverse relaxation-optimized spectroscopy (TROSY)-based 15N-HSQC titration experiments with UL53 peptide (residues 58–85). More details are in SI Materials and Methods.
Structural Database Search.
The PDB coordinate file for the M50 (residues 1–168) fold was submitted to the CATHEDRAL Algorithm (based on the CATH database) (21), the Dali structural comparison database (22), FATCAT (23, 24), VAST (25), PDBeFold (26), and Skan, run through the MarkUs protein annotation server (27, 28), to search for structural homologs using default parameters. More details on top hits are in Table S2.
Structure Threading.
All threading calculations were done using the Phyre2 web portal (29). More details are in SI Materials and Methods.
ITC.
ITC experiments were carried out under conditions similar to those described previously in the work by Sam et al. (30). More details are in SI Materials and Methods.
Viruses.
pBADGFP-based BACs were electroporated into HFFs as described previously (8) to test for infectivity and when spread occurred, to generate infectious viruses (constructs are listed in Table S3, and constructions and primer sequences are listed in Table S4 and are described in SI Materials and Methods). HCMV replication after infection at a multiplicity of infection (MOI) of 0.1 was assessed as described previously (8). Titration was done by infecting 1 × 105 HFF cells per well (in a 24-well plate) with serial dilutions of harvested virus samples for 1 h, after which the inocula were replaced with media containing methylcellulose. After 14 days, the monolayers were stained with crystal violet, and plaques were counted using a dissecting microscope. Titers represent average values from duplicate samples.
Table S3.
Summary of HCMV AD169 BAC constructs
| Construct | Genetic background | Source reference | Change introduced |
| UL50Null | pBADGFP | 8 | |
| UL50NR | UL50 Null pBADGFP | 8 | |
| UL50Null 53-F | 53-F pBADGFP | 8 | |
| V52A BADGFP | pBADGFP | This study | UL50 mutation V52A |
| V52AR BADGFP | V52A BADGFP | This study | Restored the WT UL50 coding sequence |
| E56A BADGFP | pBADGFP | This study | UL50 mutation E56A |
| E56A 53-F BADGFP | 53-F pBADGFP | This study | UL50 mutation E56A in the UL53-FLAG BADGFP background |
| E56AR 53-F BADGFP | E56A 53-F pBADGFP | This study | Restored the WT UL50 coding sequence |
| S125A BADGFP | pBADGFP | This study | UL50 mutation S125A |
| S125AR BADGFP | S125A BADGFP | This study | Restored the WT UL50 coding sequence |
| L130A BADGFP | pBADGFP | This study | UL50 mutation L130A |
| L130AR BADGFP | pBADGFP | This study | Restored the WT UL50 coding sequence |
| L130A 53-F BADGFP | 53-F pBADGFP | This study | UL50 mutation L130A in the UL53-FLAG BADGFP background |
| L130AR 53-F BADGFP | L130AR 53-F pBADGFP | This study | Restored the WT UL50 coding sequence |
Table S4.
Primer sequences for HCMV AD169 BAC or BADGFP constructs
| Construct and primer | Sequence |
| V52A | |
| Forward | TCGGTGTGCGACGCCATGCTCAAGACAGACACGGCATATTGTGTCGAGTATCTACTCAGCTAGGGATAACAGGGTAATCG |
| Reverse | GCTCTCCCAGTAGCTGAGTAGATACTCGACACAATATGCCGTGTCTGTCTTGAGCATGGCGTCGCCAGTGTTACAACCAATTAAC |
| V52AR | |
| Forward | TCGGTGTGCGACGCCATGCTCAAGACAGACACGGTCTATTGTGTCGAGTATCTACTCAGCTAGGGATAACAGGGTAATCG |
| Reverse | GCTCTCCCAGTAGCTGAGTAGATACTCGACACAATAGACCGTGTCTGTCTTGAGCATGGCGTCGCCAGTGTTACAACCAATTAAC |
| E56A | |
| Forward | GCGACGCCATGCTCAAGACAGACACGGTCTATTGTGTCGCATATCTACTCAGCTACTGGGTAGGGATAACAGGGTAATCG |
| Reverse | GCACGTGGTCTGTGCGGCTCTCCCAGTAGCTGAGTAGATATGCGACACAATAGACCGTGTCTGCCAGTGTTACAACCAATTAACC |
| E56AR | |
| Forward | GCGACGCCATGCTCAAGACAGACACGGTCTATTGTGTCGAGTATCTACTCAGCTACTGGGTAGGGATAACAGGGTAATCG |
| Reverse | GCACGTGGTCTGTGCGGCTCTCCCAGTAGCTGAGTAGATActCGACACAATAGACCGTGTCTGCCAGTGTTACAACCAATTAACC |
| S125A | |
| Forward | TCGTCACGCTCAAGGACATCGAGGAGATCAAGCCCGCCGCCTACGGAGTGCTGACGAAGTGCTAGGGATAACAGGGTAATCG |
| Reverse | ATTTGCGCACCACGCACTTCGTCAGCACTCCGTAGGCGGCGGGCTTGATCTCCTCGATGTCCTTGCCAGTGTTACAACCAATTAAC |
| S125AR | |
| Forward | TCGTCACGCTCAAGGACATCGAGGAGATCAAGCCCTCGGCCTACGGAGTGCTGACGAAGTGCTAGGGATAACAGGGTAATCG |
| Reverse | ATTTGCGCACCACGCACTTCGTCAGCACTCCGTAGGCCGAGGGCTTGATCTCCTCGATGTCCTTGCCAGTGTTACAACCAATTAAC |
| L130A | |
| Forward | GGACATCGAGGAGATCAAGCCCTCGGCCTACGGAGTGGCCACGAAGTGCGTGGTGCGCAATAGGGATAACAGGGTAATCG |
| Reverse | CCGACGCCGAATTGGATTTGCGCACCACGCACTTCGTGGCCACTCCGTAGGCCGAGGGCTGCCAGTGTTACAACCAATTAAC |
| L130AR | |
| Forward | GGACATCGAGGAGATCAAGCCCTCGGCCTACGGAGTGCTGACGAAGTGCGTGGTGCGCAATAGGGATAACAGGGTAATCG |
| Reverse | CCGACGCCGAATTGGATTTGCGCACCACGCACTTCGTCAGCACTCCGTAGGCCGAGGGCTGCCAGTGTTACAACCAATTAAC |
Immunofluorescence.
HFFs were mock-electroporated or electroporated with pBADGFP constructs (WT, UL50 null, L130A, L130AR, E56A, and E56AR) and treated identically as described previously (8).
SI Materials and Methods
Plasmids.
As described in the work by Sam et al. (30), the sequences coding for UL50 residues 1–349 and UL53 residues 1–376 from HCMV strain AD169 and M50 residues 1–316 and M53 residues 1–333 from MCMV strain Smith were amplified by PCR using Platinum Pfx DNA Polymerase (Invitrogen) and cloned into an IMPACT-CN pTYB12 expression vector (New England Biolabs). The NdeI and EcoRI sites were used to construct an ORF that fused the N terminus of each target protein to the C terminus of the intein expression tag. Cleavage of the resulting fusion protein produced the target protein with three additional amino acids (AlaGlyHis) upstream of the N-terminal methionine residue. Truncations of UL50 (residues 1–169), M50 (residues 1–168), and M53 (residues 103–333) were engineered from the original constructs by introducing internal stop codons using the QuikChange protocol (Stratagene). A truncation of UL53 (residues 50–292) was engineered by PCR amplification from HCMV strain AD169 followed by cloning into a pGEX-6P-1 vector using the BamHI and EcoRI sites. Site-directed mutations to construct the alanine substitution mutants in Table 1 were introduced using QuikChange protocols (Stratagene). All engineered plasmids were sequenced using the Dana Farber Cancer Institute Molecular Biology Core Facility to confirm the introduction of the desired sequence changes and no others in the NEC subunit coding sequences.
Protein Expression and Purification.
Plasmids encoding C-terminal truncations of UL50, UL53, M50, and M53 as well as variants of UL50 and M50 with alanine substitutions were transformed into BL21-CodonPlus(DE3)-RP Escherichia coli (Stratagene). The 1-mL transformation culture was used to directly inoculate 30- to 50-mL starter cultures in LB with 50 µg/mL carbenicillin. For expression of unlabeled proteins, a starter culture was used to inoculate 4 to 6 L LB with 50 µg/mL carbenicillin. For expression of isotopically enriched proteins, a starter culture was used to inoculate 2 L M9 medium containing 50 µg/mL carbenicillin, 1 g 15N-NH4Cl, and 2 g 13C-glucose or 2H,13C-glucose. The M9 was made up in D2O for expression of perdeuterated proteins. When the OD at 600 nm of the culture reached ∼0.8, the culture was induced for protein overexpression with 0.3 mM isopropyl-β-d-thiogalactopyranoside for 16–24 h at 16 °C. Steps for purification and verification of the overexpressed protein were identical to those in the work by Sam et al. (30). Isoleucine, leucine, valine (ILV) methyl-labeled samples were prepared using α-ketobutyrate and α-ketoisovalerate precursors as described in the works by Gardner and Kay (41) and Rosen et al. (42).
Structure Determination by NMR.
Spectra were acquired at 291.15 K with ∼150- to 300-µM M50 (1–168) protein samples in 25 mM NaPO4 (pH 6.5), 150 mM NaCl, and 1 mM DTT. Traditional transverse relaxation-optimized spectroscopy (TROSY) -based backbone triple-resonance experiments (HNCA/HNCOCA, HNCO/HNCACO, and HNCACB) were conducted on an 15N,13C-perdeuterated sample to assign the backbone chemical shifts. Assignment of additional sidechain resonances used a combination of CCONH, HCCONH, and HCCH total correlation spectroscopy experiments carried out on an 15N,13C 60% deuterated sample with a mixing time of 18 ms used for the total correlation spectroscopy transfer. Distance constraints were obtained using 3D 15N- and 13C-dispersed nuclear Overhauser effect spectroscopy (NOESY) experiments on a uniformly labeled 15N,13C-labeled sample with a mixing time of 90 ms. 15N,13C,2H-ILV-labeled samples were used in 3D 13C,15N time-shared NOESY (43) and 4D 13C-heteronuclear multiple quantum coherence (HMQC)-NOESY-HMQC experiments (44) to provide additional constraints. A mixing time of 200 ms was used in both of these experiments. The 4D 13C-HMQC-NOESY-HMQC was recorded in a nonuniform fashion with 20% sampling of the grid points, and the full spectrum was reconstructed using an iterative soft thresholding (IST) algorithm (52). All NMR data were processed using NMRPipe (45), and they were visualized and analyzed using CARA (46, 47) and CcpNmr Analysis (48).
RDCs.
15NH RDCs used in the final step of the structure calculation after water refinement were obtained from the difference between the TROSY- and semi-TROSY(15N)–selected 3D HNCO spectra acquired on a triple-labeled isotropic and partially aligned M50 (residues 1–168) sample (53). The sample, buffer, and experimental conditions were similar to those for the backbone experiments described above. The 1H-15N resonances where further dispersed in the CO dimension for reliable assignment to alleviate problems caused by signal overlap; 10 mg/mL Pf1 filamentous phage was used to obtain partial alignment. The spectra were processed using NMRPipe (45) without using linear prediction.
Structure Calculation.
The NOESY cross-peaks in the 3D NOESY experiments were integrated using peakint (54). NOESY cross-peaks in the 4D NOESY experiment were integrated using the Make Distance Restraints module in CcpNmr Analysis with a reference distance of 3 Å (48). All other parameters were left at their default values. The volumes were converted to distance constraints using CALIBA (55). These constraints were used to run a CYANA structure calculation. Dihedral angle constrains were extracted from the backbone chemical shifts using the program TALOS+ (20). The 50 lowest energy structures were selected for refinement in explicit water with AMBER through the WeNMR web interface (49). Refinement was implemented with the AMBER99SB force field with the protein placed in a 10-Å box containing TIP3 water molecules along with RDC restraints. The RDC refined structure with explicit water was then fitted with Module, version 1.0 (56). The quality of the M50 structure was assessed and validated with the protein structure validation suite PSVS, version 1.5 (57) and Procheck-NMR (58).
Threading.
Threading calculations were performed using Phyre2 (29) on each of the HCMV UL50 (P16791; Uniprot) and HHV-1 UL34 (P10218; Uniprot) protein sequences fitting to the atomic coordinates from the M50 (1–168) structure. Phyre2 was run in one-to-one mode using the default settings of alignment method local with secondary structure scoring used and weighted 0.1. For UL34, Phyre2 found 20% sequence identity and aligned 147 residues, with a reported confidence score of ∼98%. For UL50, Phyre2 found 59% sequence identity and aligned 161 residues, with a confidence score of 100%.
Phyre2 using BackPhyre mode attempted to thread a library of eukaryotic and bacterial protein sequences onto the M50 (1–168) atomic coordinates.
Genomes Searched.
Arabidopsis thaliana
Bdellovibrio bacteriovorus
Caenorhabditis elegans WS220.66
Clostridium difficile
Drosophila melanogaster
Homo sapiens
Leishmania major strain Friedlin
Mus musculus
Mycobacterium tuberculosis CDC1551
Plasmodium falciparum
Saccharomyces cerevisiae
Sulfolobus solfataricus P2
Thermoplasma acidophilum
Agrobacterium tumefaciens C58 Cereon
Bacillus subtilis
Bartonella henselae Houston-1
Corynebacterium diphtheriae
Desulfitobacterium hafniense Y51
E coli K12
Lactobacillus casei ATCC 334
Neisseria meningitidis MC58
Pseudomonas aeruginosa
Staphylococcus aureus COL
Streptococcus pneumoniae D39
Streptomyces coelicolor
Synechococcus CC9311
Yersinia pestis CO92
NMR Titration Experiments.
A synthetic peptide of sequence RLTLHDLHDIFREHPELELKYLNMMKMA (UL53 residues 58–85) was purchased from the Tufts University Core Facility and used in a series of NMR titration experiments. The effect of the equimolar quantities of peptide on the resonances of M50 (residues 1–168) was observed using TROSY-based 15N-heteronuclear single-quantum coherence (HSQC) experiments on a 200 µM sample.
ITC.
ITC experiments were carried out under conditions identical to those described previously in the work by Sam et al. (30) using unlabeled recombinantly expressed UL50 (residues 1–169), M50 (residues 1–168), or single-alanine mutants of UL50 (1–169) or M50 (1–168) in the reservoir and UL53 (residues 50–292) or M53 (residues 103–333) in the syringe.
BAC Construction.
The BACs used in this study are summarized in Table S3. The UL50 null, its rescued derivative (UL50NR), and the UL50 null mutant expressing FLAG-tagged UL53 (UL50Null 53-F) have been previously described by Sharma et al. (8). BACs constructed for this study carrying UL50 mutation V52A, E56A, S125A, or L130A were generated using the PCR primers listed in Table S4 and the two-step Red recombination method by Tischer et al. (59, 60) in pBADGFP as described previously in the work by Sharma et al. (8). To construct the rescued BACs pBADGFP E56AR, L130AR, V52AR, and S125AR, WT sequences were restored to the respective mutant BACs using the relevant rescue PCR primers listed in Table S4 and the same methodology.
Viruses.
pBADGFP-based BACs were electroporated into HFFs as described previously (8) to test for infectivity and when spread occurred, to generate infectious viruses with the constructions that are described above. HCMV replication after infection at a multiplicity of infection of 0.1 was assessed as described previously (8). Briefly, 1 × 105 HFF cells were seeded per well in a 24-well plate and infected at a multiplicity of infection of 0.1 in duplicate for 1 h. The inocula were replaced with fresh medium postadsorption, and supernatant was harvested at various time points postinfection. Titration was done by infecting 1 × 105 HFF cells per well (in a 24-well plate) with serial dilutions of harvested virus samples for 1 h, after which the inocula were replaced with media containing methylcellulose. After 14 days, the monolayers were stained with crystal violet to identify multicell plaques, and these plaques were counted using a dissecting microscope. Titers represent average values from duplicate samples. Counts from duplicate titrations differed by less than a factor of two for all data points.
Acknowledgments
We thank Sven G. Hyberts for the hmsIST processing of the 4D NOESY; Remy Sounier and Jim Zhen-Yu Sun for assistance with the residual dipolar coupling refinement; Ekaterina Heldwein for the description of the fold as a β-taco; Natalia Reim, Jackie Havens, and Stephen Marvel-Coen for technical assistance; the Nikon Imaging Center at Harvard Medical School and its staff for assistance with acquisition and analysis of immunofluorescence data; and Rafael E. Luna for his comments on the manuscript. This study was funded by NIH Grants R01AI026077 (to J.M.H. and D.M.C.), P01GM047467 (to G.W.), P01CA68262 (to G.W.), and P41 EB002026 (to G.W.), and NIH National Institute of Diabetes and Digestive and Kidney Diseases Grant K01DK085198 (to H.A.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The coordinates for the M50 structures have been deposited in the Protein Data Bank, www.rcsb.org/pdb/home/home.do (PDB ID code 5A3G), and the NMR assignments for M50 have been deposited in BioMagResBank, www.bmrb.wisc.edu (accession no. BMRB-26580).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1511140112/-/DCSupplemental.
References
- 1.Pellett PE, Roizman B. Herpesviridae. In: Knipe DM, Howley PM, editors. Fields Virology. 6th Ed. Vol 2. Wolters Kluwer/Lippincott Williams & Wilkins Health; Philadelphia: 2013. pp. 1802–1822. [Google Scholar]
- 2. National Center for Immunization and Respiratory Diseases Division of Viral Diseases (2010) CDC: Cytomegalovirus (CMV) and Congenital CMV Infection: Overview (Centers for Disease Control, Atlanta, GA)
- 3.Boppana SB, Fowler KB. Persistence in the population: Epidemiology and transmisson. In: Arvin AM, Campadelli-Fiume G, Mocarski E, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge Univ Press; Cambridge, United Kingdom: 2007. pp. 795–813. [Google Scholar]
- 4.Mettenleiter TC. Budding events in herpesvirus morphogenesis. Virus Res. 2004;106(2):167–180. doi: 10.1016/j.virusres.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 5.Mettenleiter TC, Müller F, Granzow H, Klupp BG. The way out: What we know and do not know about herpesvirus nuclear egress. Cell Microbiol. 2013;15(2):170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 6.Klupp BG, et al. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci USA. 2007;104(17):7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J. Reconstitution of the Kaposi’s sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J Virol. 2012;86(1):594–598. doi: 10.1128/JVI.05988-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol. 2014;88(1):249–262. doi: 10.1128/JVI.02358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun. 2014;5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynolds AE, et al. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol. 2001;75(18):8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science. 2002;297(5582):854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- 12.Park R, Baines JD. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol. 2006;80(1):494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorenz M, et al. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem. 2015;290(11):6962–6974. doi: 10.1074/jbc.M114.627521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn W, et al. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci USA. 2003;100(24):14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu D, Silva MC, Shenk T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci USA. 2003;100(21):12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bubeck A, et al. Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J Virol. 2004;78(15):8026–8035. doi: 10.1128/JVI.78.15.8026-8035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lötzerich M, Ruzsics Z, Koszinowski UH. Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J Virol. 2006;80(1):73–84. doi: 10.1128/JVI.80.1.73-84.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Speese SD, et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell. 2012;149(4):832–846. doi: 10.1016/j.cell.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milbradt J, et al. Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J Biol Chem. 2012;287(28):24004–24016. doi: 10.1074/jbc.M111.331207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44(4):213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sillitoe I, et al. New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Res. 2013;41(Database issue):D490–D498. doi: 10.1093/nar/gks1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holm L, Rosenstrom P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye Y, Godzik A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics. 2003;19(Suppl 2):ii246–ii255. doi: 10.1093/bioinformatics/btg1086. [DOI] [PubMed] [Google Scholar]
- 24.Ye Y, Godzik A. FATCAT: A web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 2004;32(Web Server issue):W582–W585. doi: 10.1093/nar/gkh430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6(3):377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 26.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2288–2294. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 27.Petrey D, Honig B. GRASP2: Visualization, surface properties, and electrostatics of macromolecular structures and sequences. Methods Enzymol. 2003;374:492–509. doi: 10.1016/S0076-6879(03)74021-X. [DOI] [PubMed] [Google Scholar]
- 28.Petrey D, Fischer M, Honig B. Structural relationships among proteins with different global topologies and their implications for function annotation strategies. Proc Natl Acad Sci USA. 2009;106(41):17377–17382. doi: 10.1073/pnas.0907971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sam MD, Evans BT, Coen DM, Hogle JM. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J Virol. 2009;83(7):2996–3006. doi: 10.1128/JVI.02441-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nayal M, Honig B. On the nature of cavities on protein surfaces: Application to the identification of drug-binding sites. Proteins. 2006;63(4):892–906. doi: 10.1002/prot.20897. [DOI] [PubMed] [Google Scholar]
- 32.Paßvogel L, Trübe P, Schuster F, Klupp BG, Mettenleiter TC. Mapping of sequences in Pseudorabies virus pUL34 that are required for formation and function of the nuclear egress complex. J Virol. 2013;87(8):4475–4485. doi: 10.1128/JVI.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luitweiler EM, et al. Interactions of the Kaposi’s Sarcoma-associated herpesvirus nuclear egress complex: ORF69 is a potent factor for remodeling cellular membranes. J Virol. 2013;87(7):3915–3929. doi: 10.1128/JVI.03418-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang L, Baines JD. Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J Virol. 2005;79(6):3797–3806. doi: 10.1128/JVI.79.6.3797-3806.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strang BL, et al. A mutation deleting sequences encoding the amino terminus of human cytomegalovirus UL84 impairs interaction with UL44 and capsid localization. J Virol. 2012;86(20):11066–11077. doi: 10.1128/JVI.01379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milbradt J, et al. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol Cell Proteomics. 2014;13(8):2132–2146. doi: 10.1074/mcp.M113.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rupp B, et al. Random screening for dominant-negative mutants of the cytomegalovirus nuclear egress protein M50. J Virol. 2007;81(11):5508–5517. doi: 10.1128/JVI.02796-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roller RJ, Bjerke SL, Haugo AC, Hanson S. Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J Virol. 2010;84(8):3921–3934. doi: 10.1128/JVI.01638-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bjerke SL, et al. Effects of charged cluster mutations on the function of herpes simplex virus type 1 UL34 protein. J Virol. 2003;77(13):7601–7610. doi: 10.1128/JVI.77.13.7601-7610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jokhi V, et al. Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Reports. 2013;3(4):988–995. doi: 10.1016/j.celrep.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardner KH, Kay LE. Production and incorporation of 15N, 13C, 2H (1H-δ1 methyl) isoleucine into proteins for multidimensional NMR studies. J Am Chem Soc. 1997;119(32):7599–7600. [Google Scholar]
- 42.Rosen MK, et al. Selective methyl group protonation of perdeuterated proteins. J Mol Biol. 1996;263(5):627–636. doi: 10.1006/jmbi.1996.0603. [DOI] [PubMed] [Google Scholar]
- 43.Frueh DP, et al. Time-shared HSQC-NOESY for accurate distance constraints measured at high-field in (15)N-(13)C-ILV methyl labeled proteins. J Biomol NMR. 2009;45(3):311–318. doi: 10.1007/s10858-009-9372-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiller S, Ibraghimov I, Wagner G, Orekhov VY. Coupled decomposition of four-dimensional NOESY spectra. J Am Chem Soc. 2009;131(36):12970–12978. doi: 10.1021/ja902012x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 46.Keller RLJ. 2005. Optimizing the process of nuclear magnetic resonance spectrum analysis and computer aided resonance assignment. PhD thesis (Swiss Federal Institute of Technology Zürich, Steinerberg, Switzerland)
- 47.Keller RLJ. The Computer Aided Resonance Assignment Tutorial. CANTINA Verlag; Goldau, Switzerland: 2004. [Google Scholar]
- 48.Vranken WF, et al. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins. 2005;59(4):687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 49.Bertini I, Case DA, Ferella L, Giachetti A, Rosato A. A Grid-enabled web portal for NMR structure refinement with AMBER. Bioinformatics. 2011;27(17):2384–2390. doi: 10.1093/bioinformatics/btr415. [DOI] [PubMed] [Google Scholar]
- 50.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8(4):477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 51.Schrödinger LLC. 2011. The PyMOL Molecular Graphics System, Version 1.4.1.
- 52.Hyberts SG, Milbradt AG, Wagner AB, Arthanari H, Wagner G. Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J Biomol NMR. 2012;52(4):315–327. doi: 10.1007/s10858-012-9611-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kontaxis G, Clore GM, Bax A. Evaluation of cross-correlation effects and measurement of one-bond couplings in proteins with short transverse relaxation times. J Magn Reson. 2000;143(1):184–196. doi: 10.1006/jmre.1999.1979. [DOI] [PubMed] [Google Scholar]
- 54. Schäfer N (1992) Automatische Signalerkennung in heteronuklearen 3D und 4D NMR Spektren. Diploma thesis (Swiss Federal Institute of Technology Zürich, Steinerberg, Switzerland)
- 55.Güntert P, Braun W, Wüthrich K. Efficient computation of three-dimensional protein structures in solution from nuclear magnetic resonance data using the program DIANA and the supporting programs CALIBA, HABAS and GLOMSA. J Mol Biol. 1991;217(3):517–530. doi: 10.1016/0022-2836(91)90754-t. [DOI] [PubMed] [Google Scholar]
- 56.Dosset P, Hus JC, Marion D, Blackledge M. A novel interactive tool for rigid-body modeling of multi-domain macromolecules using residual dipolar couplings. J Biomol NMR. 2001;20(3):223–231. doi: 10.1023/a:1011206132740. [DOI] [PubMed] [Google Scholar]
- 57.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66(4):778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 58.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–291. [Google Scholar]
- 59.Tischer BK, Smith GA, Osterrieder N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol Biol. 2010;634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 60.Tischer BK, von Einem J, Kaufer B, Osterrieder N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques. 2006;40(2):191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 61.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: A case study using the Phyre server. Nat Protoc. 2009;4(3):363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]