

Significance
ATP13A2 is a lysosomal transporter that is genetically linked to an autosomal recessive variant of Parkinson’s disease and confers protection against α-synuclein toxicity in neurons. Here we show that an N-terminal hydrophobic domain of ATP13A2 specifically recognizes signaling lipids. Interactions with these signaling lipids enhance cytoprotection to mitochondrial stress. This study provides essential information for establishing the lysosomal function of ATP13A2 and suggests a therapeutic applicability in activating ATP13A2.
Keywords: mitochondria, lysosome, flippase, α-synuclein, P5-type ATPase
Abstract
ATP13A2 is a lysosomal P-type transport ATPase that has been implicated in Kufor–Rakeb syndrome and Parkinson’s disease (PD), providing protection against α-synuclein, Mn2+, and Zn2+ toxicity in various model systems. So far, the molecular function and regulation of ATP13A2 remains undetermined. Here, we demonstrate that ATP13A2 contains a unique N-terminal hydrophobic extension that lies on the cytosolic membrane surface of the lysosome, where it interacts with the lysosomal signaling lipids phosphatidic acid (PA) and phosphatidylinositol(3,5)bisphosphate [PI(3,5)P2]. We further demonstrate that ATP13A2 accumulates in an inactive autophosphorylated state and that PA and PI(3,5)P2 stimulate the autophosphorylation of ATP13A2. In a cellular model of PD, only catalytically active ATP13A2 offers cellular protection against rotenone-induced mitochondrial stress, which relies on the availability of PA and PI(3,5)P2. Thus, the N-terminal binding of PA and PI(3,5)P2 emerges as a key to unlock the activity of ATP13A2, which may offer a therapeutic strategy to activate ATP13A2 and thereby reduce α-synuclein toxicity or mitochondrial stress in PD or related disorders.
Neuronal fitness depends on optimal lysosomal function and efficient lysosomal delivery of proteins and organelles by autophagy for subsequent breakdown (1, 2). Kufor–Rakeb syndrome (KRS) is an autosomal recessive form of Parkinson’s disease (PD) associated with dementia, which is caused by mutations in ATP13A2/PARK9 (3). Mutations in or knockdown (KD) of ATP13A2 lead to lysosomal dysfunctions, including reduced lysosomal acidification, decreased degradation of lysosomal substrates (4), impaired autophagosomal flux (4, 5), and accumulation of fragmented mitochondria (5, 6). By contrast, overexpression (OE) of Ypk9p (i.e., the yeast ATP13A2 ortholog) protects yeast against toxicity of α-synuclein (7), which is the major protein in Lewy bodies, the abnormal protein aggregates that develop inside nerve cells in PD. This protective effect of ATP13A2 on α-synuclein toxicity is conserved in yeast, Caenorhabditis elegans, and rat neuronal cells (7). Because ATP13A2 imparts resistance to Mn2+ (7–9) and Zn2+ (10–12), it was proposed that ATP13A2 may function as a Mn2+ (7–9) and/or Zn2+ transporter (10–12).
ATP13A2 belongs to the P5 subfamily of the P-type ATPase superfamily, which comprises five subfamilies (P1–5) of membrane transporters. P-type ATPases hydrolyze ATP to actively transport inorganic ions across membranes or lipids between membrane leaflets (reviewed in ref. 13). During the transport cycle, a phospho-intermediate is formed on a conserved aspartate residue (14). The human P5-type ATPases are divided into two groups, P5A (ATP13A1) and P5B (ATP13A2–5), but their transport specificity has not been established (14–16).
P-type ATPases comprise a membrane-embedded core of six transmembrane (TM) helices (M1–6) that form the substrate binding site(s) and entrance/exit pathways for the transported substrate (13). Whereas four extra C-terminal TM helices (M7–10) are common, additional N-terminal TM helices are only observed in the P1B heavy metal pumps (17). Topology predictions indicate that ATP13A2 and other P5 members also contain additional N-terminal TM helices, which might serve a subclass-specific function (14, 15). Here, we demonstrate that the ATP13A2 N terminus is a critical regulatory element involved in lipid binding and ATP13A2 activation, providing protection to mitochondrial stress in a cellular PD model.
Results
The N Terminus of ATP13A2 Does Not Transverse the Membrane but Is Embedded in the Cytosolic Membrane Leaflet.
To decipher the molecular function of ATP13A2, we focused on the properties of the membrane domain—that is, the membrane-embedded section where substrate binding and transport take place. Most P-type ATPases contain 10 TM helices, but a hydrophobic segment in the ATP13A2 N terminus may be an additional TM helix (Ma, residues 45–67), which would result in an uneven number of helices with the N- and C-terminal ends exposed on different sides of the membrane (Fig. S1A) (14, 15). We examined this unusual topology of 11 TM helices in ATP13A2 via a fluorescence protease protection (FPP) assay (18). GFP-N-ATP13A2 and ATP13A2-C-GFP fusion proteins were transiently expressed in HeLa cells, and after permeabilization with digitonin, cells were treated with trypsin. Protection of the GFP tag from trypsin-mediated proteolysis indicates that the tag resides at the luminal side, whereas rapid loss of fluorescence reveals that the GFP tag is cytosolic and prone to trypsin cleavage. ERO-GFP (an ER-resident luminal protein) and cytosolic GFP were expressed as positive (protected; Fig. 1A) and negative (unprotected; Fig. 1B) controls, respectively. The degradation of GFP-N-ATP13A2 (Fig. 1C) and ATP13A2-C-GFP (Fig. 1D) fluorescence shows that the N and C termini both reside in the cytosol, implying that ATP13A2 contains an even number of TM helices.
Fig. S1.

Supportive information on the ATP13A2 topology. (A) Prediction of 11 TM (M) helices in hATP13A2 (TMHMM v2). M numbering is indicated above the figure. M1 corresponds to the first M helix present in all P-type ATPases. Ma is the predicted N-terminal M helix. (B) The predicted N-glycosylation site, 1028NRTV (1028N boxed), in L7–8, is highly conserved. (C) A conserved glycine (G59) is found at the center of Ma, which might introduce a kink in the Ma helix. (D and E) Cartoons of the N-terminal fragments N-Ma-M1-GFP and N-Ma-GFP. Numbers in circles correspond to the verified positions in Fig. 1 A–H of the FPP assay.
Fig. 1.

Revised topology of hATP13A2. (A–H) FPP assay. Controls ERO-GFP (A) and GFP (B), full-length ATP13A2 with N- (C) or C-terminal GFP (D), and GFP-labeled ATP13A2 fragments (E–H) (N-Ma-M1-GFP, residues 1–251, E; N-Ma-GFP, residues 1–187, F; GFP-N-Ma-M1, residues 1–251, G; and GFP-N-Ma, residues 1–187, H) were expressed in HeLa cells and subjected to FPP. Images were acquired after 0, 3, and 5 min of trypsin treatment. (I) Immunoblot analysis of mitochondrial/lysosomal fractions of COS-1 cells overexpressing ATP13A2 WT and N1028A [mutant in the predicted N-glycosylation site in loop 7–8 (L7–8)] treated with or without PNGase F. (J) Representative immunoblot shows that N-term-Ma mainly associates with membranes (M) of COS-1 cells compared with the cytosolic fraction (C), both in the absence (–) and presence (+) of 0.6 M KCl. (K) Revised topology of ATP13A2 with 10 M helices and a membrane-associated N terminus. Numbers in circles correspond to the verified positions in panels A–I (n = 3 for all experiments). (L) N-Ma-GFP displays a vesicular pattern in HeLa cells. White box in the Left panel is magnified in the Right panel.
To examine the unexpected discrepancy between our data and the topology prediction, we performed further FPP assays on N-terminal fragments of ATP13A2 containing Ma with or without M1. These fragments were fused to GFP at the C terminus [N-Ma-M1-GFP, residues 1–251 of ATP13A2 (Fig. 1E) and N-Ma-GFP, residues 1–187 of ATP13A2 (Fig. 1F)]. In contrast to N-Ma-GFP (Fig. 1F), the fluorescent signal of N-Ma-M1-GFP (Fig. 1E) is clearly protected, showing that M1 protrudes into the lumen (Fig. S1D), whereas Ma remains cytosolic (Fig. S1E). This refutes the assumption that Ma spans the membrane. We further verified that the position of the N terminus in the N-terminal fragments corresponds to the position in the full-length ATP13A2. Via FPP on N-terminal GFP fusion constructs of Ma and Ma-M1, we confirm that the N terminus of both the GFP-N-Ma-M1 (Fig. 1G) and GFP-N-Ma (Fig. 1H) constructs is cytosolic like in the full-length. We therefore conclude that Ma-M1 is an integral membrane protein with a cytosolic and luminal terminus, whereas the N- and C-terminal ends of Ma reside at the cytosolic side of the membrane. This indicates that the fragments are not merely targeted to the luminal compartment for degradation in the lysosome. Moreover, the topology of the N-terminal fragments explains well why the topology of the full-length ATP13A2 differs from the prediction.
Several observations indicate that the M1–10 topology prediction is correct: The ATP13A2 C terminus (Fig. 1D) and the N-terminal start of the M1 helix (Fig. 1F) are cytosolic, whereas loop L7–8 is luminal, as N1028 is glycosylated on a conserved and predicted NxS/T glycosylation motif (Fig. S1B). Removal of the sugar moieties by PNGaseF treatment of COS-1 lysosomal membrane fractions reduced the protein mass of the ATP13A2 WT but not of the N1028A mutant (Fig. 1I).
The ATP13A2 N Terminus Is Sufficient for Late Endo-/Lysosomal Targeting.
In transfected HeLa cells, we observed that not only full-length ATP13A2 (Fig. 2A) (3) but also the N-Ma-M1 fragment fused to mCherry (mCh-N-Ma-M1) colocalize with Rab7 (Fig. 2B), a late endo-/lysosomal marker. This indicates that the N terminus of ATP13A2 contains sufficient information to direct its targeting to the late endo-/lysosomes.
Fig. 2.

The ATP13A2 N terminus is sufficient for lysosomal targeting. (A–E) mCherry- or GFP-labeled ATP13A2, Ma, or Ma-M1 were transiently transfected in HeLa cells together with different mCherry- or GFP-labeled cellular markers and subjected to fluorescence microscopy. The late endo-/lysosomal distribution of WT ATP13A2 (A), Ma-M1 (B), and Ma (C) is depicted. For comparison, the distribution of SERCA2b (D) and SPCA1a (E) versus Ma is shown. Rab7, SERCA2b, and SPCA1a are, respectively, late endo-/lysosomal, endoplasmic reticulum, and Golgi/secretory pathway markers (representative images of three independent experiments).
Moreover, we demonstrated that the shorter N-terminal fragment with only the Ma region also partially associates with vesicles of the late endo-/lysosomal compartment (Figs. 1L and 2C). Indeed, mCh-N-Ma, a fusion construct of mCherry with the first 187 residues of hATP13A2, was found to colocalize in part with Rab7 as well as with SPCA1a, the Golgi/secretory pathway Ca2+/Mn2+ pump (19), but not with SERCA2b, the endoplasmic reticulum Ca2+ ATPase (Fig. 2 C–E). We also observed that the N-terminal fragment N-Ma-GFP is present in the membrane fraction of transfected COS-1 cells, even in the presence of 0.6 M KCl (Fig. 1J). Thus, together with the FPP data, we conclude that the hydrophobic N-terminal N-Ma fragment is associated with the cytosolic leaflet of the late endo-/lysosomal membranes. A conserved glycine, G59, at the center of the ATP13A2 Ma stretch might introduce a kink in the helix that promotes the return of Ma to the cytosol (Fig. S1C).
Based on our findings, we propose a topology for ATP13A2 (Fig. 1K), with a membrane-associated N terminus resembling the membrane floating docking platform in P1B-type ATPases, which functions in substrate delivery (17).
The ATP13A2 N Terminus Interacts with Phosphatidic Acid and Phosphatidylinositol(3,5)bisphosphate.
The hydrophobic nature of the ATP13A2 N terminus and the interaction of N-Ma-GFP with the cytosolic membrane leaflet of late endo-/lysosomal vesicles (Fig. 1L) suggest that the N terminus may interact with specific lipids. To test this, a recombinant N-terminal fragment of ATP13A2 (187 residues), fused to GST (GST-N-Ma), was used to screen in vitro for lipid interactions by means of a protein–lipid overlay assay. Of the 31 different lipids tested (including phospholipids, cholesterol, and sphingolipids; Fig. 3 A–C), GST-N-Ma interacted most strongly with phosphatidic acid (PA) (Fig. 3 A and B, GST-N-Ma) and phosphatidylinositol(3,5)bisphosphate [PI(3,5)P2] (Fig. 3B, GST-N-Ma), two lipids that are enriched in the late endo-/lysosomal membranes. GST-N-Ma did not bind to the lysosomal lipid bis(monoacylglycero)phosphate (Fig. S2C), and purified GST did not interact with lipids (Fig. 3 A–C, GST).
Fig. 3.

The N terminus of ATP13A2 interacts with PI(3,5)P2 and PA. (A–C) Lipid–protein overlay with GST-N-Ma on membrane lipid strips (A), PIP strips (B), and sphingolipid strips (C) spotted with 30 different lipids. Labels along the left refer to the leftmost column of spots in each panel, and labels to the right refer to the second column in each panel. CHL, cholesterol; CL, cardiolipin; CM, ceramide; DAG, diacylglycerol; GD3, disiaganglioside-GD3; GM1, monosialoganglioside-GM1; LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; MS, myriosine; PA, phosphatidic acid; PC, phosphatidylcholine; PCH, psychosine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PHS, phytosphingosine; PI, phosphatidylinositol; PS, phosphatidylserine; SM, sphingomyelin; SPC, sphingosylphosphorylcholine; SPN, sphingosine; S1P, sphingosine-1-phosphate; TG, triglyceride. (D–F) Liposome floatation assay. LUVs of different lipid compositions were subjected to a sucrose gradient. N-NBD-PE fluorescence was used as a control for vesicle floating in the sucrose fractions [0%, 25%, 30%, and pellet (R)] (D). LUVs were incubated with MultiPIPgrip (E) and GST-N-Ma and GST (F). Data are represented as average ± SD (n = 8). Immunoblot of PIP grip in the 0%, 25%, and 30% wt/vol sucrose fractions of LUVs with PC or PC with 5 mol% PI(3,5)P2 (E). Immunoblot of GST-N-Ma in 0%, 25%, and 30% wt/vol sucrose fractions of LUVs containing PC with or without 5 mol% PA or PI(3,5)P2 (F, Upper panel). Purified GST was used as a negative control (F, Lower panel). (G) Bar graph representing the flotation GST-N-Ma depicted in panel F. *Statistical difference between PC versus PC/PA or PC/PI(3,5)P2 (1 mark, P < 0.01; 3 marks, P < 0.001) (n = 8) (ANOVA with Bonferroni post hoc test).
Fig. S2.

N-terminal mutations in ATP13A2 prevent the PA and PI(3,5)P2 interaction. (A) Schematic representation of the position and (B) conservation of three putative lipid binding sites in the ATP13A2 N terminus. ATP13A2 splice variant 1 contains a five-amino-acid insert at position 154, as indicated. Truncated protein fragments of GST-N-Ma are depicted underneath. (C–K) Custom-made lipid strips exposing a serial dilution (100–50–25–12.5–6.25–3.13–1.56 pmol per spot) of the lipids PC, PA, and PI(3,5)P2. Strips were incubated with equal concentrations of GST-N-Ma (C), mutated GST-N-Ma [G59A (D), 65FRWKP-FAWAP (E), 74RLRLR-ALALA (F), or 155KRVLR-AAVLA (G)], truncated protein fragments [ΔN1 (H), ΔN2 (I), or ΔC (J)], or the GST-N-Ma protein fragment of splice variant 1 of ATP13A2 (K). Additionally, no interaction of N-GST-Ma with a serial dilution of bis(monoacylglycero)phosphate (BMP) was observed (C).
To determine the position of the PA and PI(3,5)P2 interaction sites in the ATP13A2 N terminus, the N terminus was mutated in the hydrophobic Ma region (G59A) and in three putative lipid binding regions that hold conserved positively charged residues (site 1, 65FRWKP→FAWAP, which partially overlaps with Ma; site 2, 74RLRLR→ALALA; and a more distant site 3, 155KRVLR→AAVLA) (Fig. S2 A and B). Lipid arrays were incubated with equal amounts of mutated or WT GST-N-Ma protein fragments (Fig. S2 C–K). Whereas the G59A substitution had no effect (Fig. S2D), the PA and PI(3,5)P2 interactions were clearly affected by alanine substitutions in sites 1–3 (Fig. S2 E–G). Using truncated protein fragments of GST-N-Ma (Fig. S2A), we further demonstrated that mainly site 3 is required for the PA interaction (Fig. S2 H–J). Indeed, the ΔC fragment no longer interacts with PA, whereas PA binding is not affected in ΔN1 and reduced in ΔN2. In contrast, removal of sites 1 and 2 in the ΔN2 fragment, but not of site 3 in the ΔC fragment, resulted in an impaired PI(3,5)P2 binding.
Of interest, splice variant 1 of ATP13A2 contains an additional insert of five amino acids next to lipid binding site 3 (i.e., after residue 154; Fig. S2 A and B). This insert did not affect the lipid specificity, but an increased binding affinity toward PA versus PI(3,5)P2 was observed (Fig. S2K), further confirming that site 3 is important for the PA interaction. We therefore conclude that PA and PI(3,5)P2 interactions depend on three distinct charged regions in the N terminus of ATP13A2, of which site 1 partially overlaps with Ma.
To assess if GST-N-Ma also recognizes PA and PI(3,5)P2 in vesicular membranes, we performed a liposome floatation assay. Liposomes (large unilamellar vesicles, LUVs) of the indicated compositions (including 1 mol% N-NBD-PE marker) were floated through a density gradient, carrying along with them bound protein. N-NBD-PE fluorescence was highest in the 0% sucrose fraction, where liposomes float (Fig. 3D). MultiPIP Grip protein, which interacts with phosphorylated phosphatidyl-inositol lipids, was used as a positive floating control (Fig. 3E). The GST-N-Ma protein, but not GST alone, floated in the same fraction as the liposomes only when 5 mol% PA or PI(3,5)P2 was incorporated (Fig. 3 F and G). These data independently confirm that GST-N-Ma interacts with PA or PI(3,5)P2, and importantly, it does so when they are membrane integrated.
ATP13A2 Undergoes Autophosphorylation and Accumulates in the E1P State.
During the transport cycle, P-type ATPases undergo autophosphorylation to couple ATP hydrolysis to substrate transport (Fig. 4A) (13). Catalytic cycling between autophosphorylation and dephosphorylation depends on sequence motifs that are also recognized in ATP13A2 (Fig. S3 A and B) (13). We therefore tested whether ATP13A2 forms a phospho-intermediate on the aspartyl residue in the 508DKTGT consensus motif for autophosphorylation. Microsomes of COS-1 cells overexpressing ATP13A2 were incubated with γ-32P–labeled ATP. ATP13A2 formed a phospho-intermediate (Fig. 4C) that reached steady-state conditions in 60 s. The phospho-enzyme was sensitive to hydroxylamine, a hallmark of aspartyl phosphorylation (Fig. 4D) (20). In addition, the protein levels of ATP13A2 WT and D508N (a catalytically inactive mutant) were comparable in transfected COS-1 cells and rose high above background levels (Fig. 4B), but the D508N mutant of ATP13A2 was unable to undergo autophosphorylation (Fig. 4C). Thus, ATP13A2 forms a covalent aspartyl-phosphate bond on residue D508, representing the first evidence, to our knowledge, that ATP13A2 undergoes autophosphorylation. As the highest expression and autophosphorylation of ATP13A2 was observed in COS-1 microsomal membrane fractions (Fig. 4E), these were used for further study.
Fig. 4.

ATP13A2 autophosphorylation is regulated by PA and PI(3,5)P2. (A) Post-Albers reaction scheme of P-type ATPases. (B) Immunoblot shows ATP13A2 levels (arrow) in COS-1 microsomal fractions overexpressing WT ATP13A2 or a catalytically inactive mutant (D508N) versus empty vector (e.v.) control. GAPDH levels are indicated as loading control (*). (C) Representative radiogram displaying radioactive phospho-enzyme levels (EP) of WT (arrow) but not the D508N mutant. (D) Radiogram shows that ATP13A2 EP is sensitive to hydroxylamine (HA). (E) Mitochondrial/lysosomal (M/L), microsomal (µ), and cytosolic (C) fractions of COS-1 cells overexpressing ATP13A2 were analyzed for ATP13A2 protein levels (WB) or autophosphorylation (EP). (F and G) The ATP13A2 phospho-enzyme was dephosphorylated by treatment with 5 mM cold ATP or ADP. MOPS reaction buffer was used as a negative control. Radiogram after 30 s treatment (F, arrow indicates ATP13A2) and time-dependent decrease in phospho-enzyme levels based on radiogram analysis at different time points (G). Nontransfected (NT) cells served as the negative control. (H) Autophosphorylation of WT ATP13A2 with or without 100 µM MnCl2 or ZnCl2. (I) Representative example of the effect of different lipids or combination of lipids on ATP13A2 phospho-enzyme (EP) formation. The Bottom panel in I displays the loading control (LC)—that is, Coomassie staining of the SDS/PAGE gel. (J) Bar graph representing average ± SEM of the data in I. *Statistical difference between nontreated versus lipid-treated and PI(3,5)P2 versus PA/PI(3,5)P2, respectively (2 marks, P < 0.01; 3 marks, P < 0.001) (n = 4) (ANOVA with Dunnetts) (n = 3).
Fig. S3.

Two predicted homology models of ATP13A2. (A) Homology models of Homo sapiens ATP13A2 splice variant 3 [Left, modeled by SWISS-Model, template Protein Data Bank (PDB) ID code 3RFU] and of Danio Rerio ATP13A2 (Right, PDB ID code A1A5E5, Protein Model Portal). Three cytosolic domains are recognized: the nucleotide-binding domain (N, wheat), phosphorylation domain (P, blue), and actuator domain (A, green); and a fourth domain, the membrane domain (M, pink), is also indicated. The lysine residue of the 649KGSPE sequence, which is important for nucleotide binding, the aspartate residue of the autophosphorylation sequence 508DKTGT, and the glutamate residue of the 341TGE sequence, which is required for dephosphorylation, is depicted, respectively, as a purple, red, and blue side chain. In the M domain, the 465PPALP recognition sequence for P5B ATPases (putative substrate-binding site) is shown as a green side chain. Sequence numbers correspond to the human ATP13A2 splice variant 2. (B) Conserved P-type ATPase fingerprint motifs in various organisms—that is, H. sapiens, Mus musculus, and D. rerio. In other P-type ATPases, M4 coordinates substrate binding and ATP hydrolysis via a tightly controlled allosteric mechanism. This involves the formation of a Mg2+ binding site for ATP binding at the phosphorylation site, which would depend on the sequence motifs 745TGDN and 863GDGAND. The strong conservation of these typical P-type sequence motifs indicates that ATP13A2 might couple ATP hydrolysis to substrate transport across the membrane or between membrane leaflets.
Because P5A-type ATPases autophosphorylate predominantly to the E1P state (20, 21), we tested whether this is also true for members of the P5B-type ATPases such as ATP13A2. In other P-type ATPases, only the E1P state is sensitive to ADP as a result of the reverse reaction with ADP (Fig. 4A) (20). We observed that phospho-enzyme levels rapidly decline with 5 mM ADP (Fig. 4 F and G), indicating that ATP13A2 accumulates in the E1P state. The slow apparent dephosphorylation of the phospho-enzyme in the presence of nonlabeled ATP (Fig. 4 F and G) further indicates that ATP13A2 is only slowly turning over in the forward reaction. We conclude that ATP13A2 undergoes autophosphorylation to the E1P state, where it may wait for activation.
Autophosphorylation Is Affected Not by Mn2+ or Zn2+ but by PA and PI(3,5)P2.
The catalytic turnover of P-type ATPases depends on the presence of the substrate to be transported (13). Because ATP13A2 may be a Mn2+ (7–9) or Zn2+ (10–12) pump, we tested whether Mn2+ or Zn2+ activate ATP13A2 turnover. However, the steady-state phosphorylation levels were unaffected by 100 µM MnCl2 or ZnCl2, indicating that ATP13A2 is unlikely to transport Mn2+ or Zn2+ (Fig. 4H).
Some P-type ATPases are regulated by phosphatidylinositol lipids, and furthermore, the P4 ATPases transport lipids from one membrane leaflet to the other (13). Here, we compared the effect of the N-terminal binding lipids PA and PI(3,5)P2 on ATP13A2 autophosphorylation with that of the nonbinding lipids phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS). The phospho-enzyme levels increased most strongly with PA or PI(3,5)P2. In the presence of combined PA and PI(3,5)P2, phospho-enzyme levels were significantly lower than in conditions with PI(3,5)P2 (Fig. 4 I and J), indicating that phospho-enzyme turnover is affected. This suggests that PA and PI(3,5)P2 are regulating ATP13A2 activity.
Lipid-Mediated ATP13A2 Activity Confers Protection in a Cellular Model of PD.
Stable human neuroblastoma SHSY5Y cells with altered ATP13A2 expression (KD; WT or D508N OE) versus control (firefly luciferase OE, FLUC) were generated by lentiviral transduction to assess the cellular implications of the ATP13A2 lipid interactions. The ATP13A2 WT and D508N OE levels were comparable (Fig. 5A), and only WT undergoes autophosphorylation (Fig. 5B). In the KD cell line ATP13A2, mRNA levels were reduced by 75.4 ± 8.9%. The cellular model was validated by confirming previous findings that ATP13A2 WT OE protects against whereas KD exacerbates the cellular toxicity toward Mn2+ and Zn2+ exposure (Fig. S4 A and B) (7–12).
Fig. 5.

PA and PI(3,5)P2 provide ATP13A2-mediated protection to mitochondrial stress in a cellular model of PD. Cellular models were generated in SHSY5Y cells of WT or kinetic dead (D508N) OE of ATP13A2 or sh-ATP13A2 (KD), in comparison with control (FLUC OE). The OE levels of ATP13A2 WT and D508N were compared via immunoblotting (A), and the autophosphorylated enzyme intermediate (EP) of ATP13A2 WT (B) was confirmed (µ, microsomal membranes; M/L, mitochondrial/lysosomal membrane fractions). (C and D) The effect of PA (FIPI, 100 nM) and PI(3,5)P2 (YM-201636, 200 nM) depletion on ATP13A2’s protective response against 24-h exposure to rotenone was assessed by MUH assay. C, control; F, PLD inhibitor FIPI; R, rotenone; Y, PIKfyve inhibitor YM-201636. Bar graphs represent average ± SD of relative cell viability normalized to FLUC control. *Statistical differences between R FLUC and R WT/D508N/KD, unless a bar indicates the statistical difference between R and R+Y/F (1 mark, P < 0.05; 2 marks, P < 0.01; 3 marks, P < 0.001) (n = 3) (ANOVA with Bonferroni post hoc test).
Fig. S4.

ATP13A2 protects SHSY5Y cells against Mn2+, Zn2+, and rotenone-induced cytotoxicity. To validate the protective role of ATP13A2 against 24 h exposure to zinc (A, 100 µM ZnCl2) or manganese (B, 1 µM MnCl2), SHSY5Y cells with ATP13A2 OE (WT-OE) and KD (sh-ATP13A2), in comparison with control (FLUC), were assessed by MUH-based analysis. PI-stained flow cytometry was performed to confirm the effect of FIPI (100 nM) and YM-201636 (200 nM), alone or in combination, on rotenone- (rot, 2.5 µM) induced cell death (C and D). Moreover, caspase-dependent apoptosis was demonstrated by pretreatment of WT-OE and sh-ATP13A2 cell lines with the caspase inhibitor zVAD (50 µM) before 24 h exposure to rotenone and assessed by PI-stained flow cytometry. Data are the mean of three independent experiments ± SD.
The implications of the ATP13A2 lipid interactions were evaluated in a well-established cellular PD disease model of mitochondrial complex I inhibition by rotenone to evoke mitochondrial stress (22, 23). Rotenone reduced cell viability in all cell lines (Fig. 5 C and D), which is mirrored by an increased caspase-dependent cell death (Fig. S4 C and D). Importantly, ATP13A2 WT OE, but not D508N OE, significantly protects SHSY5Y cells from mitochondrial stress, indicating that ATP13A2 activity is required for the protective effect (Fig. 5 C and D and Fig. S4C). In contrast, ATP13A2 KD increased cytotoxicity compared with the FLUC cells (Fig. 5 C and D).
Interfering with PI(3,5)P2 formation using YM-201636 (a selective PIKfyve lipid kinase inhibitor) blunted the protective effect observed in the WT OE cells close to the level of the control and D508N OE (Fig. 5C and Fig. S4C), indicating that ATP13A2 activity is impaired. We also tested the effect of 5-fluoro-2-indolyl des-chlorohalopemide (FIPI; a selective inhibitor of phospholipase D, PLD) in the presence of rotenone. Like YM-201636, FIPI blunted the protective effect of ATP13A2 against rotenone to the level of FLUC and D508N OE cells (Fig. 5D and Fig. S4C). In ATP13A2 KD cells, YM-201636 and FIPI did not influence the impact of rotenone (Fig. 5 C and D and Fig. S4C). In addition, treatment with either YM-201636 or FIPI did not induce cell death (Fig. S4C).
Together, our data show that in conditions in which PA or PI(3,5)P2 formation is inhibited, ATP13A2 no longer protects against mitochondrial stress, in line with an activating effect of the lipids on ATP13A2.
Discussion
The Unique Membrane-Associated N-Terminal Domain of ATP13A2.
ATP13A2 contains a membrane-associated N terminus that interacts with the endo-/lysosomal lipids PA and PI(3,5)P2, which regulate ATP13A2 activity in vitro and in a cellular model of PD.
We started by refuting the predicted topology of 11 TM helices (14, 15) based on the cytosolic position of the N and C termini of ATP13A2-GFP fusion constructs. Importantly, these GFP fusion constructs are properly expressed and folded, as they are targeted to the late endo-/lysosomes and undergo efficient autophosphorylation (Fig. S5 A–C), validating their use for a topology analysis. The position of the glycosylation site and C terminus further shows that the C terminal part of ATP13A2 is in line with the topology prediction and corresponds to the classical TM topology of other P-type ATPases (13). By analyzing N-terminal fragments of ATP13A2, we were able to unambiguously explain the discrepancy with the topology prediction. Via a combined approach of FPP, fluorescence microscopy, membrane fractionation and lipid interaction studies, we demonstrated that the N terminus is embedded in the cytosolic membrane leaflet of the late endo-/lysosomal membranes, where it specifically interacts with lipids. Of interest, the membrane-associated N terminus of ATP13A2 resembles the N-terminal membrane floating helix of the P1B-ATPase subfamily (17). Like in the heavy metal transporters, the N terminus of ATP13A2 might also serve as a substrate-delivery site or interaction site for chaperones or regulatory proteins (17).
Fig. S5.
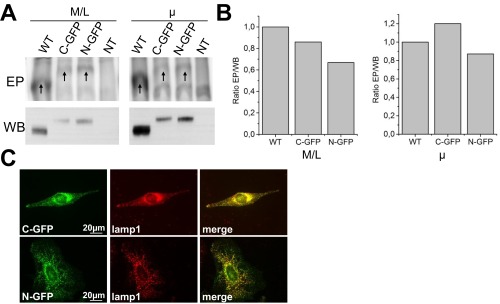
N- or C-terminal GFP-tagged ATP13A2 protein is active and targeted to the late endo-/lysosomes. (A) Mitochondrial/lysosomal (M/L) and microsomal (µ) fractions of COS-1 cells, transiently transfected with ATP13A2 WT, or GFP-tagged ATP13A2 (C- or N-terminal, respectively C-GFP and N-GFP), were analyzed for expression via immunoblot (WB) or for autophosphorylation on a radiogram (EP). As a control, nontransfected cells (NT) were included in the assay. Arrows indicate the phosphorylated enzyme. (B) The ratio of the autophosphorylation and expression levels (EP/WB) shows that a similar ratio of the ATP13A2-labeled protein undergoes autophosphorylation as the nontagged control, indicating that the GFP-labeled ATP13A2 proteins efficiently autophosphorylate. (C) N- or C-GFP–tagged ATP13A2 colocalizes with Lamp1 in transiently transfected HeLa cells. Lysosomes were visualized by means of a lamp1 antibody (Abcam, AB25630) and AlexaFluor 594.
PA and PI(3,5)P2 Connect ATP13A2 to Endo-/Lysosomal Pathways in Neurodegeneration.
Via protein-lipid overlays, floatation assays and site-directed mutagenesis, we demonstrate that PA and PI(3,5)P2 interact with the ATP13A2 N terminus involving positively charged residues that in part overlap with the hydrophobic Ma segment. Alternative splicing of the ATP13A2 transcript renders two splice variants that vary in the N terminus close to the lipid binding site 3, and we demonstrate that this relates to differences in the relative PA/PI(3,5)P2 sensitivity.
Importantly, the data indicate that the protective effect of ATP13A2 in a cellular PD model of rotenone-induced mitochondrial stress depends on the availability of PA and PI(3,5)P2. In fact, PA and PI(3,5)P2 connect ATP13A2 to important endo-/lysosomal vesicular trafficking pathways implicated in neurodegeneration. First, PI(3,5)P2 functions as an organelle tag in lysosomes and late endosomes and recruits or activates proteins involved in membrane trafficking (24). PI(3,5)P2 deficiency causes Charcot–Marie–Tooth disease (25), whereas in Vac14-deficient mice it results in massive neurodegeneration (26). PI(3,5)P2 regulates endo-/lysosome morphology, trafficking, acidification, and autophagy (reviewed in ref. 24). PI(3,5)P2 further controls membrane fusion and fission by activating the mucolipin transient receptor potential channel 1 (27). ATP13A2 might mediate some of the functions of PI(3,5)P2, as loss of ATP13A2 results in an increased lysosomal pH (4) and impaired autophagy (4, 5).
Secondly, signaling pools of PA are produced via hydrolysis of PC by PLD, the diacylglycerol kinase, or the lysophosphatidic acid acyl transferase pathways (28). As PA is a conical phospholipid, PA induces a negative membrane curvature and promotes membrane fission (29) and fusion (30). PA also stimulates the activity of the mTOR complex in autophagy (31). Interestingly, PLD1 inhibition disturbs α-synuclein clearance via autophagy, whereas OE of PLD1 reduces α-synuclein toxicity (32). As such, PA might regulate ATP13A2 activity during autophagy, as ATP13A2 confers protection against α-synuclein toxicity (7) and also mediates mitochondrial clearance (5).
ATP13A2 Autophosphorylation Is Stimulated by PA and PI(3,5)P2.
The autophosphorylation assay facilitated the first biochemical study of ATP13A2 and will be important to study disease mutants and uncover the transported substrate.
Although we recapitulate the cell-protective effects of ATP13A2 in conditions of Mn2+ and Zn2+ toxicity, our biochemical analysis indicates that in contrast to what was suggested in the literature (7–12), ATP13A2 most likely is not a Mn2+ or Zn2+ transporter, as the steady-state levels of autophosphorylated ATP13A2 remain unaffected by Mn2+ or Zn2+.
What could then be the transport function of ATP13A2? Like P5A ATPases (20, 21), ATP13A2 undergoes autophosphorylation and resides in the E1P state, suggesting this might be a general feature of P5-type ATPases. The accumulation in the E1P state and slow turnover of ATP13A2 indicate that ATP13A2 remains inactive, pending further activation. The ATP13A2 transport activity is required during mitochondrial stress, as ATP13A2, but not the catalytically dead form D508N, protects cells against rotenone toxicity. The protective effect of the active ATP13A2 further depends on the availability of PI(3,5)P2 and PA. Together with the stimulatory effect of both lipids on ATP13A2 autophosphorylation, it is likely that PA and PI(3,5)P2 are required for ATP13A2 activation, but presumably they are not the transported substrates of ATP13A2. Indeed, phosphatidylinositol lipids are locally formed and removed exclusively at the cytosolic leaflet of the membrane, whereas PA is a short living signaling molecule in cells that is rapidly converted into diacylglycerol.
Both PA (29) and PI(3,5)P2 (24) connect ATP13A2 to key endo-/lysosomal trafficking pathways, suggesting that ATP13A2 is implicated in vesicular transport. This is further supported by the observation that many of the ATP13A2 interacting proteins in human (33) or genetic modifiers of YPK9 in yeast (14) are implicated in vesicular transport. The previously reported effects of ATP13A2 on exosome release (10), Zn2+ (10–12) and Mn2+ (7–9) homeostasis and toxicity, mitochondrial clearance (5), and α-synuclein detoxification (7, 10) might all relate to a role in vesicular transport (14). Based on the phylogenetic relationship with the P4-type ATPases and the lipid effects on autophosphorylation, ATP13A2 may also be a lipid flippase in vesicular pathways (14). Moreover, ATP13A2 might be considered as a P1B/P4 hybrid pump, as it shares typical structural features with P1B-type ATPases and functional properties with P4-type ATPases.
In conclusion, the ATP13A2 N terminus interacts with the endo-/lysosomal lipids PA and PI(3,5)P2, which regulate ATP13A2 activity to provide protection during mitochondrial stress in a cellular model of PD. In a variety of model organisms, ATP13A2 protects against α-synuclein–induced toxicity (7). Targeting the N terminus may offer a modality to therapeutically activate the prosurvival characteristics of ATP13A2.
Materials and Methods
Full methods are described in detail in SI Materials and Methods.
DNA Constructs.
hATP13A2‐pDONR221 was provided by Susan Lindquist, Whitehead Institute for Biomedical Research, Cambridge Center, Cambridge, MA (7). The construct was transferred via Gateway ATP13A2 into vectors containing GST, GFP, or mCherry tags. Mutations were introduced via QuikChange Mutagenesis (Stratagene) and verified by sequencing (LGC Genomics).
Immunoblotting.
Immunoblotting was performed as in ref. 34. A3361 (Sigma) and SY3072 (homemade, epitope 119PSPQSQAEDGRSQAAVGAV) were used as ATP13A2 antibodies. Proteins were detected via enhanced chemiluminescence (Pierce) and a ChemiDoc imager (Bio-Rad).
Fluorescence Microscopy.
HeLa cells transiently transfected with ATP13A2 WT, D508N, or protein fragments (GFP‐ or mCherry‐tagged) were cultured in chamber slides and, after 48 h, subjected to fluorescence microscopy (Olympus IX81). For co‐localization, cells were co‐transfected with labeled SERCA2b, SPCA1a (19), and RAB7.
FPP.
HeLa cells transfected with ATP13A2-GFP constructs were subjected to FPP as described (18). The minimal effective digitonin (Sigma, D141) concentration (20–40 µM), optimal time of digitonin treatment (30–90 s), and minimal effective trypsin concentration (100–150 µg/mL) were determined for each experiment in cells expressing GFP or ERO-GFP.
Glycosylation Assay.
COS-1 lysosomal membranes overexpressing ATP13A2 WT or N1028A were treated with Peptide N-glycosidase F (PNGaseF, NewEngland Biolabs, manufacturer’s protocol). Differences in mass before and after treatment were analyzed via immunoblotting.
Lipid Overlay Assay.
Recombinant GST-N-Ma was affinity purified from BL21-AI Escherichia coli. The protein (1.36 nM final concentration) was applied on membrane strips with lipid spots (Echelon) according to the manufacturer’s protocol.
Liposome Floatation Assay.
The protein, LUVs, 0.2% wt/vol ovalbumin and buffer B (10 mM Tris⋅HCl pH 7.0, 150 mM NaCl) were mixed in a total volume of 80 µL (final lipid and protein concentration, 1.6 mM and 25.6 nM) and subjected to a sucrose gradient centrifugation (90 min, 186,000 × g, 4 °C). GST-labeled proteins were visualized via immunoblotting (34).
Autophosphorylation Assay.
The assay was performed as previously described (20). An additional washing step with 0.3 M hydroxylamine was conducted as indicated. To assess the effect of ADP or ATP on phospho-enzyme levels, 30 s after adding [γ-32P] ATP, the mixture was incubated with 5 mM ADP or 5 mM nonlabeled ATP before the reaction was quenched with stop solution at various time points. To assess the effect of lipids, COS-1 microsomes were supplemented with 5 mol% of additional lipid. The lipid/protein ratio in intracellular membranes was estimated at 200 nmol phospholipids per 1 mg protein (35).
Cell Viability Assay.
Stable SHSY5Y cells expressing FLUC (control), ATP13A2 WT, D508N, or sh-ATP13A2 were seeded in a 96-well plate (7,500 cells per well) and incubated with 100 µM ZnCl2 (Sigma), 1 µM MnCl2 (Sigma), 2.5 µM rotenone (Sigma) alone or combined with 200 nM YM-201636 (Calbiochem, DMSO), and/or 100 nM FIPI (Sigma, DMSO). After 24 h, cells were washed and incubated with 0.01 mg/mL MUH (4-methylumbelliferyl heptanoate, PBS; Sigma) for 30 min at 37 °C to assess cell viability. Fluorescence was measured on a FlexStation (Molecular Devices).
Statistical Analysis.
Data are presented as the average ± SD, and n indicates the number of independent experiments. Statistical analysis was conducted by ANOVA with a Bonferroni post hoc test.
SI Materials and Methods
Membrane Fractionation.
COS-1 cells were transiently transfected with GeneJuice (Novagen). At 48 h after transfection, cells were harvested and fractionated as described (34). The nuclear (1,000 × g, 10 min, 4 °C), mitochondrial‐lysosomal (12,000 × g, 20 min, 4 °C), and microsomal fractions (200,000 × g, 35 min, 4 °C) were collected. Fractions were suspended in 0.25 M sucrose with protease inhibitors (SigmaFast, Sigma). Protein concentration was determined using the Q‐bit fluorimetric method (Life Technologies).
To prepare membrane fractions of COS-1 cells overexpressing N-Ma-GFP, lysis was performed in 10 mM Tris⋅HCl pH 7.5 and 0.5 mM MgCl2 by 40× gentle pipetting. Membranes were collected by centrifugation (1,000 × g, 10 min, 4 °C and 100,000 × g, 35 min, 4 °C). The final pellet was dissolved in an equal volume as the supernatant.
Purification of GST-N-Ma.
Recombinant GST-N-Ma was affinity purified from BL21-AI cells transformed with a pDEST15 vector. Lysis of bacterial pellets, removal of inclusion bodies, and affinity purification were conducted as described by Life Technologies. GST-N-Ma was eluted in 50 mM Tris∙HCl pH 8.3, 150 mM NaCl, and 50 mM glutathion (reduced).
Lipid–Protein Overlay.
Membrane strips with lipid spots (Echelon) were blocked in buffer A (10 mM Tris∙HCl pH 7.0, 150 mM NaCl, 0.1% Tween20, 0.2% ovalbumin) for 1 h at room temperature (RT). Subsequently, the protein was added for 1 h (1.36 nM final concentration). After three washing steps of 5 min with washing buffer (10 mM Tris∙HCl pH 7.0, 150 mM NaCl, 0.1% Tween20), the membranes were probed for 1 h with anti-GST antibody (GE Healthcare, 27–4577–01) in buffer A. Strips were then washed and incubated with secondary antibody (Sigma, A7650) in buffer A for 1 h. After washing, proteins were detected with ECL substrate (GE Healthcare) and a ChemiDoc imager (Bio-Rad).
Preparation of LUVs.
Liposomes were composed of 99 mol% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC; Avanti Polar Lipids, APL) and 1 mol% 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2–1,3-benzoxadiazol-4-yl) (N-NBD-PE, APL) or 94 mol% DOPC, 1 mol% N-NBD-PE, and 5 mol% of either 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA, APL) or 1,2-dipalmitoylphosphatidylinositol(3,5)bisphosphate [PI(3,5)P2, Echelon]. The indicated amount of DOPC, N-NBD-PE, and DOPA in chloroform was first dried under a N2 flow and then in a desiccator. PI(3,5)P2 was dissolved in H2O-saturated butanol at 55 °C and added to the lipid film. After vortexing, the butanol was evaporated at 55 °C with N2 gas, and lipids were dried in the desiccator. Lipids were rehydrated in buffer B (10 mM Tris∙HCl pH 7.0, 150 mM NaCl) to a final lipid concentration of 2 mM and vortexed for 5 min. The multilamellar vesicles were converted into LUVs by passing the liposome suspension 41 times through a 0.2-µm polycarbonate filter with a miniextruder (APL).
Liposome Floatation Assay.
The protein, LUVs, 0.2% ovalbumin and buffer B were mixed in a total volume of 80 µL (final lipid and protein concentration, 1.6 mM and 25.6 nM). Then, 80 µL of buffer B containing 60% sucrose was added. A 150-µL sample was overlaid with 150 µL of buffer B containing 25% sucrose, followed by 150 µL of buffer B. Sucrose gradients were centrifuged for 90 min at 186,000 × g at 4 °C. Three 150-µL fractions were collected (top, 0%; middle, 25%; bottom, 30%), and the pellet was suspended in 150 µL of buffer B. Fluorescence of N-NBD-PE was detected on SDS/PAGE gels (Typhoon reader, excitation 488 nm, detection with 555BP), and proteins were visualized via immunoblotting (34).
Autophosphorylation Assay.
COS-1 membranes (40 μg) were added to EP reaction buffer (17 mM Hepes pH 6.5, 160 mM KCl, 2 mM MgCl2, 1 mM DTT, 5 mM NaN3) to a final volume of 95 μL. The reaction was initiated on ice by adding [γ-32P] ATP (2 μCi, 5.125 µM) and stopped after 60 s with 400 μL stop solution (20% trichloroacetic acid, 10 mM phosphoric acid). After precipitation on ice for 20 min, samples were centrifuged (20,000 × g, 30 min, 4 °C). The pellet was washed twice with 400 μL of ice-cold stop solution and dissolved in sample buffer before acidic electrophoresis as previously described (20). An additional washing step with 0.3 M hydroxylamine was conducted as indicated. To assess the effect of ADP or ATP on phospho-enzyme levels, 30 s after adding [γ-32P] ATP, the mixture was incubated with 5 mM ADP or 5 mM nonlabeled ATP before the reaction was quenched with stop solution at various time points.
Autophosphorylation Assay in the Presence of Lipids.
The lipid/protein ratio in intracellular membranes was estimated at 200 nmol phospholipids per 1 mg protein (35). The COS-1 microsomes were supplemented with 5 mol% of additional lipid [egg yolk l-α-PE, APL; brain l-α-PS, APL; DOPC, APL; DOPA, APL and PI(3,5)P2; Echelon]. First, lipid films were rehydrated by vortexing in EP reaction buffer supplemented with 20 μg n-dodecyl-β-d-maltopyranoside (DDM, 2:1 protein/DDM ratio) followed by 10 min of incubation at RT. Then, 40 µg of microsomes was added, and after 10 min, detergent was extracted with Biobeads (Bio-Rad) for 1 h. Microsomes were then subjected to the autophosphorylation assay.
Lentiviral Transduction of SHSY5Y Cells.
SHSY5Y cells were grown in DMEM culture medium containing 1% glutamine and penicillin/streptomycin (Sigma), supplemented with 10% FBS (HyClone). Then, 100,000 SHSY5Y cells per well were plated in a 24-well plate and transduced with lentiviral vectors coding for firefly luciferase (FLUC control), ATP13A2 WT, and D508N (36). KD lentiviral vectors were generated as described (37) using -ccctgacgatagggacatcaat- as the target sequence against ATP13A2. After lentiviral transduction, cells were selected with puromycin (2 µg/mL, Gibco) or blasticidin (9 µg/mL, Invitrogen) before confirmation by immunoblotting or real-time RT-PCR. All cells were maintained at 37 °C, 5% CO2 for a maximum of 20 passages.
Cell Death Assay.
Cell death was assessed by propidium iodide (PI; Sigma) stained flow cytometry (Attune Flow Cytometer, Life Technologies). Cells in 12-well plates (100,000 cells per well) were pretreated for 1 h with 50 µM zVAD-fmk (Bachem), exposed to the above compounds, collected and resuspended in 0.1 mg/mL PI (Sigma) for 10 min in a final volume of 500 µL PBS, and analyzed.
Acknowledgments
This work was funded by the Michael J. Fox Foundation, the Jake’s Ride Award of the Bachmann-Strauss Foundation, the Interuniversity Poles of Attraction of the Belgian Science Policy Office (P7/13), the KU Leuven (OT/13/091 and OT/14/120), the Flanders Research Foundation (G.0768.10 and G.0927.14), and the Danish National Research Foundation. T.G.P. is funded by the Research Centre “bioSYNergy” (University of Copenhagen Excellence Program for Interdisciplinary Research). T.H. is a research fellow of the Flanders Research Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508220112/-/DCSupplemental.
References
- 1.Usenovic M, Krainc D. Lysosomal dysfunction in neurodegeneration: The role of ATP13A2/PARK9. Autophagy. 2012;8(6):987–988. doi: 10.4161/auto.20256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dehay B, et al. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy. 2012;8(9):1389–1391. doi: 10.4161/auto.21011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramirez A, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38(10):1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 4.Dehay B, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci USA. 2012;109(24):9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012;45(3):962–972. doi: 10.1016/j.nbd.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grunewald A, et al. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. 2012;33(8):1843.e1–1843.7. doi: 10.1016/j.neurobiolaging.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 7.Gitler AD, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41(3):308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan J, et al. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J Biol Chem. 2011;286(34):29654–29662. doi: 10.1074/jbc.M111.233874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Covy JP, Waxman EA, Giasson BI. Characterization of cellular protective effects of ATP13A2/PARK9 expression and alterations resulting from pathogenic mutants. J Neurosci Res. 2012;90(12):2306–2316. doi: 10.1002/jnr.23112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong SM, et al. Parkinson’s Disease linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alphaSynuclein externalisation via exosomes. Hum Mol Genet. 2014;23(11):2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- 11.Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet. 2014;23(11):2802–2815. doi: 10.1093/hmg/ddt623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsunemi T, Krainc D. Zn²⁺ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23(11):2791–2801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmgren MG, Nissen P. P-type ATPases. Annu Rev Biophys. 2011;40:243–266. doi: 10.1146/annurev.biophys.093008.131331. [DOI] [PubMed] [Google Scholar]
- 14.van Veen S, et al. Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front Mol Neurosci. 2014;7:48. doi: 10.3389/fnmol.2014.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sørensen DM, Buch-Pedersen MJ, Palmgren MG. Structural divergence between the two subgroups of P5 ATPases. Biochim Biophys Acta. 2010;1797(6-7):846–855. doi: 10.1016/j.bbabio.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Møller AB, Asp T, Holm PB, Palmgren MG. Phylogenetic analysis of P5 P-type ATPases, a eukaryotic lineage of secretory pathway pumps. Mol Phylogenet Evol. 2008;46(2):619–634. doi: 10.1016/j.ympev.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 17.Gourdon P, et al. Crystal structure of a copper-transporting PIB-type ATPase. Nature. 2011;475(7354):59–64. doi: 10.1038/nature10191. [DOI] [PubMed] [Google Scholar]
- 18.Lorenz H, Hailey DW, Wunder C, Lippincott-Schwartz J. The fluorescence protease protection (FPP) assay to determine protein localization and membrane topology. Nat Protoc. 2006;1(1):276–279. doi: 10.1038/nprot.2006.42. [DOI] [PubMed] [Google Scholar]
- 19.Baron S, et al. The secretory pathway Ca(2+)-ATPase 1 is associated with cholesterol-rich microdomains of human colon adenocarcinoma cells. Biochim Biophys Acta. 2010;1798(8):1512–1521. doi: 10.1016/j.bbamem.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 20.Sørensen DM, et al. Ca2+ induces spontaneous dephosphorylation of a novel P5A-type ATPase. J Biol Chem. 2012;287(34):28336–28348. doi: 10.1074/jbc.M112.387191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corradi GR, de Tezanos Pinto F, Mazzitelli LR, Adamo HP. Shadows of an absent partner: ATP hydrolysis and phosphoenzyme turnover of the Spf1 (sensitivity to Pichia farinosa killer toxin) P5-ATPase. J Biol Chem. 2012;287(36):30477–30484. doi: 10.1074/jbc.M112.363465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu F, et al. Rotenone impairs autophagic flux and lysosomal functions in Parkinson’s disease. Neuroscience. 2015;284:900–911. doi: 10.1016/j.neuroscience.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Mader BJ, et al. Rotenone inhibits autophagic flux prior to inducing cell death. ACS Chem Neurosci. 2012;3(12):1063–1072. doi: 10.1021/cn300145z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho CY, Alghamdi TA, Botelho RJ. Phosphatidylinositol-3,5-bisphosphate: No longer the poor PIP2. Traffic. 2012;13(1):1–8. doi: 10.1111/j.1600-0854.2011.01246.x. [DOI] [PubMed] [Google Scholar]
- 25.Chow CY, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448(7149):68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, et al. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci USA. 2007;104(44):17518–17523. doi: 10.1073/pnas.0702275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong XP, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin JJ, Loewen CJ. Putting the pH into phosphatidic acid signaling. BMC Biol. 2011;9:85. doi: 10.1186/1741-7007-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weigert R, et al. CtBP/BARS induces fission of Golgi membranes by acylating lysophosphatidic acid. Nature. 1999;402(6760):429–433. doi: 10.1038/46587. [DOI] [PubMed] [Google Scholar]
- 30.Blackwood RA, et al. Phospholipase D activity facilitates Ca2+-induced aggregation and fusion of complex liposomes. Am J Physiol. 1997;272(4 Pt 1):C1279–C1285. doi: 10.1152/ajpcell.1997.272.4.C1279. [DOI] [PubMed] [Google Scholar]
- 31.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294(5548):1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 32.Bae EJ, et al. Phospholipase D1 regulates autophagic flux and clearance of α-synuclein aggregates. Cell Death Differ. 2014;21(7):1132–1141. doi: 10.1038/cdd.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Usenovic M, et al. Identification of novel ATP13A2 interactors and their role in α-synuclein misfolding and toxicity. Hum Mol Genet. 2012;21(17):3785–3794. doi: 10.1093/hmg/dds206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vandecaetsbeek I, et al. Structural basis for the high Ca2+ affinity of the ubiquitous SERCA2b Ca2+ pump. Proc Natl Acad Sci USA. 2009;106(44):18533–18538. doi: 10.1073/pnas.0906797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujiki Y, Fowler S, Shio H, Hubbard AL, Lazarow PB. Polypeptide and phospholipid composition of the membrane of rat liver peroxisomes: Comparison with endoplasmic reticulum and mitochondrial membranes. J Cell Biol. 1982;93(1):103–110. doi: 10.1083/jcb.93.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heeman B, et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci. 2011;124(Pt 7):1115–1125. doi: 10.1242/jcs.078303. [DOI] [PubMed] [Google Scholar]
- 37.Osório L, et al. Viral vectors expressing a single microRNA-based short-hairpin RNA result in potent gene silencing in vitro and in vivo. J Biotechnol. 2014;169:71–81. doi: 10.1016/j.jbiotec.2013.11.004. [DOI] [PubMed] [Google Scholar]