

Significance
Lung cancer is the leading cause of cancer death worldwide. About 10% harbor mutations in epidermal growth factor receptor (EGFR). Despite remarkable progress in treatment with EGFR inhibitors, only 5% of patients achieve tumor reduction >90%, even though all of those treated have EGFR mutations. Our study addressed this discrepant response by investigating the mechanism of innate drug resistance, i.e. resistance inherent in the tumor cells even before treatment begins and not acquired over its course. Because overcoming innate resistance will increase the primary response, our findings may provide an opportunity to develop new therapies to reduce the probability of emergent resistance to EGFR inhibitors.
Keywords: nonsmall cell lung cancer, EGFR inhibition, tyrosine kinase inhibitors, ERK1/2 paradoxical activation, innate drug resistance
Abstract
Nonsmall cell lung cancer (NSCLC) is the leading cause of cancer death worldwide. About 14% of NSCLCs harbor mutations in epidermal growth factor receptor (EGFR). Despite remarkable progress in treatment with tyrosine kinase inhibitors (TKIs), only 5% of patients achieve tumor reduction >90%. The limited primary responses are attributed partly to drug resistance inherent in the tumor cells before therapy begins. Recent reports showed that activation of receptor tyrosine kinases (RTKs) is an important determinant of this innate drug resistance. In contrast, we demonstrate that EGFR inhibition promotes innate drug resistance despite blockade of RTK activity in NSCLC cells. EGFR TKIs decrease both the mitogen-activated protein kinase (MAPK) and Akt protein kinase pathways for a short time, after which the Ras/MAPK pathway becomes reactivated. Akt inhibition selectively blocks the transcriptional activation of Ets-1, which inhibits its target gene, dual specificity phosphatase 6 (DUSP6), a negative regulator specific for ERK1/2. As a result, ERK1/2 is activated. Furthermore, elevated c-Src stimulates Ras GTP-loading and activates Raf and MEK kinases. These observations suggest that not only ERK1/2 but also Akt activity is essential to maintain Ets-1 in an active state. Therefore, despite high levels of ERK1/2, Ets-1 target genes including DUSP6 and cyclins D1, D3, and E2 remain suppressed by Akt inhibition. Reduction of DUSP6 in combination with elevated c-Src renews activation of the Ras/MAPK pathway, which enhances cell survival by accelerating Bim protein turnover. Thus, EGFR TKIs evoke innate drug resistance by preventing Akt activity and inactivating Ets-1 function in NSCLC cells.
Lung cancer is the leading cause of cancer death worldwide (1, 2). Of the two major histologic types, small cell and nonsmall cell (NSCLC), the latter is by far the more prevalent (∼85%). About 14% of NSCLCs harbor mutations in epidermal growth factor receptor (EGFR), a receptor tyrosine kinase (RTK). Despite remarkable progress in treating EGFR-mutated NSCLCs with EGFR tyrosine kinase inhibitors (TKIs), only 5% of patients achieve tumor reduction >90% (3). The remaining patients respond, but only partially, even though they too have EGFR mutations. We speculate that this limited primary response may be attributable to resistance inherent in the tumor cells even before treatment begins and not to resistance acquired over its course (4).
Recent studies demonstrated that RTK ligands secreted through paracrine, autocrine, and endocrine mechanisms in the tumor microenvironment are important determinants of therapeutic responses to anticancer kinase inhibitors (5–7). Indeed, hepatocyte growth factor (HGF), fibroblast growth factor (FGF), and neuregulin (NRG1) confer innate drug resistance to the highest number of cancer cell lines by activating RTKs and thus stimulating a prosurvival pathway, either the Ras/mitogen-activated protein kinase (MAPK)- or PI3K/Akt-signaling or both (5). Certainly, HGF-mediated activation of RTK Met is most suspected as the cause of innate resistance to anticancer agents (5–7). However, it is not clear to what extent activation of Met contributes to the innate drug resistance after EGFR inhibition in NSCLC cells.
In this study, we investigated the molecular machinery by which the Ras/MAPK pathway is activated after EGFR inhibition despite blockade of RTK activity in NSCLC cells.
Results
EGFR TKIs Paradoxically Activate ERK1/2 in EGFR-Mutated NSCLC Cells.
To investigate the biological response of NSCLC cells to EGFR inhibition, we used the cell lines HCC827, HCC2935, and H-1650, which express mutant forms of EGFR and wild-type HRAS, KRAS, NRAS, BRAF, PIK3CA, and PTEN. EGFR mutations include E746-A750del (HCC827 cells), E746-T751del and S752I (HCC2935 cells), and E746-A750del (H-1650 cells). These EGFR mutants were activated and phosphorylated in untreated cells (Figs. S1–S3, lane 1). To test whether we could recapitulate in cultured cells the clinical observations of the innate resistance to EGFR TKI, we treated HCC827 NSCLC cells with or without 1 μM gefitinib (Fig. 1A). A week later, 5% of the cells remained viable (Fig. 1A, Middle). Thereafter, the resistant cells commenced cell division and formed colonies in the presence of gefitinib (Fig. 1A, Right). These findings suggested that EGFR inhibition in the cultured cells mimics clinical observations of the innate resistance.
Fig. S1.

Gefitinib paradoxically activates ERK1/2 in EGFR-mutated NSCLC cells after 6–24 h of incubation. Western blot analysis. Exponentially growing HCC827 cells were treated with various concentrations of gefitinib and collected after 1 h (A), 6 h (B), 12 h (C), and 24 h (D) of treatment.
Fig. S3.

Gefitinib paradoxically activates ERK1/2 in EGFR-mutated NSCLC cells. (A–D) Western blot analysis. Exponentially growing HCC2935 cells (A and B) or H-1650 cells (C and D) were treated with gefitinib and collected after 1 h (A and C) and 6 h (B and D) of treatment.
Fig. 1.

Gefitinib paradoxically activates ERK1/2 in EGFR-mutated NSCLC cells. (A) Colony-forming assay on HCC827 cells with (Middle and Right) or without (Left) 1 μM gefitinib for 7 d (Left and Middle) or 28 d (Right). Cells were stained with 0.5% crystal violet containing 20% ethanol. (B and C) Western blot analysis. Exponentially growing HCC827 cells were treated with various concentrations of gefitinib and harvested at 1 h (B) and 12 h (C) after treatment. (D) Western blot analysis. HCC827 cells were treated with gefitinib for 12 h; 10 μM PD325901 was added, and the cells were cultured for 1 h before harvesting. (E) Active Ras pull-down assay. Total Ras expression was separately determined by Western blot analysis. (F) Western blot analysis. Exponentially growing HCC827 cells were infected with lentivirus expressing Myc-S17N K-Ras or an empty vector; 48 h after infection, cells were cultured with or without gefitinib for 12 h. (G) Phospho-ERK1/2–associated GST-Elk1 in vitro kinase assay (Top). Western blots of total cell lysates (Bottom). (H) Subcellular fractionation analysis. Arrowheads indicate full-length PARP (single arrowhead) and cleaved PARP (double arrowhead).
We next examined the biological behavior of the NSCLC cells immediately after EGFR inhibition. We found that the TKIs gefitinib (Iressa), erlotinib (Tarceva), and lapatinib (Tykerb) markedly reduced EGFR phosphorylation at Tyr1068 (Fig. 1 B and C and Figs. S1–S3) and Akt at Thr308 and Ser473 (Fig. 1B and Fig. S4). After 1 h of treatment, ERK1/2 phosphorylation was inhibited (Fig. 1B and Figs. S2A and S3 A and C), but surprisingly was reactivated after 6–24 h of gefitinib treatment (Fig. 1C and Figs. S1 B–D, S2 B and D, and S3 B and D). Similar results were seen in NSCLC cells bearing wild-type EGFR (H-1838 and H-2170; Fig. S5 A–D), although more gefitinib (≥0.8 μM) was required. ERK1/2 activation was nearly absent in the TKI-resistant cell line with T790M EGFR (H-1975; Fig. S5 E and F).
Fig. S4.

Gefitinib paradoxically activates the MAPK pathway in EGFR-mutated NSCLC cells. Western blot analysis was performed with antibodies specific for the indicated phosphorylation sites and total antibodies. Exponentially growing HCC827 cells were treated with different concentrations of gefitinib and collected after 12 h of treatment.
Fig. S2.

EGFR TKIs paradoxically activate ERK1/2 in EGFR-mutated NSCLC cells. (A–D) Western blot analysis. Exponentially growing HCC827 cells were treated with erlotinib (A and B) or lapatinib (C and D) at the indicated concentrations and collected after 1 h (A and C) and 6 h (B and D) of treatment.
Fig. S5.

Gefitinib paradoxically activates ERK1/2 in NSCLC cells bearing wild-type EGFR but not a drug-resistant mutant form after 6 h of incubation. (A–F) Western blot analysis. Exponentially growing H-1838 cells (A and B; EGFR wild-type EGFR), H-2170 cells (C and D; EGFR wild-type), and H-1975 cells (E and F; T790M + L858R TKI-resistance mutant form of EGFR) cells were treated with various concentration of gefitinib and collected after 1 h (A, C, and E) and 6 h (B, D, and F) h of treatment.
Gefitinib-induced ERK1/2 activation at 12 h was accompanied by MEK1/2 phosphorylation (Fig. 1C); 10 μM of a MEK inhibitor (PD325901) completely suppressed this activation (Fig. 1D). Thus, upstream MAPK signaling may be essential for ERK1/2 activation after EGFR inhibition. Indeed, in a pull-down assay with a GST-fusion protein containing the Ras-binding domain (RBD) of c-Raf, Ras remained activated after EGFR inhibition, as shown by SDS/PAGE and Western blot with a pan-Ras antibody (Fig. 1E).
To establish the importance of Ras activation in initiating the MAPK cascade after EGFR inhibition, we used a lentiviral gene expression system to make an S17N (asparagine substitution for serine 17) dominant-negative form of K-Ras. The dominant-negative effect reflects the mutant’s ability to sequester SOSs (upstream activators) and its inability to activate Rafs (downstream effectors) (8). Ectopic and endogenous expressions were distinguished by reduced mobility of the Myc epitope-tagged S17N K-Ras protein, which was also detected with an anti-Myc antibody (Fig. 1F). Ectopically expressed S17N K-Ras inhibited TKI-induced ERK1/2 activation, as shown by Western blot, suggesting that the ERK1/2 activation is dependent on Ras activity.
The activated ERK1/2 was evidently functional, as phosphorylation of GST-Elk1 was increased (Fig. 1G). Phosphorylated forms of ERK1/2 were predominantly localized in the nucleus rather than the cytoplasm (Fig. 1H).
RTKs Are Not Responsible for Ras/MAPK Activation Induced by EGFR Inhibitors.
Multiple RTKs are coactivated in NSCLCs and other types of solid tumors (9). To investigate whether sustained activation of Ras after EGFR inhibition is caused by phosphorylation and activation of RTKs other than EGFR, we used a phospho-RTK antibody array to assess the phosphorylation status of 28 major RTKs. In the absence of gefitinib, five RTKs were coactivated: EGFR, HER3, FGFR1, IGF1R, and Met (Fig. S6; numbers 1–5; Top). In cells treated with two concentrations of gefitinib for 12 h, phosphorylation of these five RTKs was reduced. No other RTKs were activated after EGFR inhibition.
Fig. S6.

Phosphorylation of EGFR, HER3, FGFR1, IGF1R, and Met was reduced in cells treated with gefitinib for 12 h. Phospho-RTK antibody arrays. The phosphorylation status of 28 major RTKs was simultaneously assessed.
Western blot analysis confirmed these observations (Fig. 2A and Fig. S7): gefitinib inhibited the activities of EGFR, HER3, FGFR1, IGF1R, and Met in a dose-dependent manner. These findings show that the EGFR mutation drives the activities of these RTKs in NSCLC cells and that EGFR inhibition collapses an extensive network of downstream signaling, consistent with a previous report (10). To confirm that targeted EGFR inhibition blocks the protein kinase activities of other coactivated RTKs in EGFR-mutated NSCLC cells, we also assessed the phosphorylation status of Shc, Gab1, and Gab2, which are phosphorylated by activated RTKs (11–13), and found gefitinib inhibition. Thus, the protein kinase activities of all RTKs were blocked (Fig. 2A and Fig. S7). Moreover, SHP2 was essentially inactivated at gefitinib doses ≥0.2 μM (Fig. 2A and Fig. S7). As SHP2 activation and association with Gab1 are critical for sustained ERK1/2 activation downstream of RTKs (14), RTKs are not responsible for sustained Ras activation after EGFR inhibition.
Fig. 2.

c-Src activates the EGFR/MAPK pathway in NSCLC cells and cooperates with loss of DUSP6 to activate ERK1/2 after EGFR inhibition. (A and B) Western blot analysis. Cells were treated with gefitinib at the indicated concentrations for 12 h. (C) Immunoprecipitation and immunoblotting. Protein was precipitated with mouse antibodies specific to control IgG or EGFR, followed by immunoblotting. (D) Subcellular fractionation analysis. (E) Western blot analysis and active Ras pull-down assay. HCC827 cells were treated with gefitinib for 12 h and further cultured with saracatinib for 1 h before harvesting. (F and G) HCC827 cells were treated with various concentrations of gefitinib for 12 h. (F) Quantitative PCR. Fold changes in gene expression were calculated as the ratio of expression in each sample treated with different concentrations of gefitinib to the level without it. (G) Western blot analysis. (H) Western blot analysis and active Ras pull-down assay. Exponentially growing HCC827 cells were infected with a lentivirus expressing Myc-tagged DUSP6 or empty vector lentivirus (mock infection); 48 h after infection, cells were cultured in the presence or absence of gefitinib for 12 h and harvested.
Fig. S7.

RTKs are not responsible for Ras/MAPK activation induced by EGFR inhibitors. Western blot analysis was performed using antibodies to indicated specific phosphorylation sites or total expression. Exponentially growing HCC827 cells were treated with different concentrations of gefitinib and collected after 12 h of treatment.
Activation of c-Src Is Sustained After EGFR Inhibition in NSCLC Cells.
EGFR mutations may contribute to aggressive human malignancies involving non-RTK c-Src (15). Indeed, overexpression of c-Src and EGFR in fibroblast cells causes synergistic increases in DNA synthesis, colony growth in soft agar, and tumor formation in nude mice (16). This synergy is dependent on c-Src–mediated phosphorylation of EGFR at Tyr845.
c-Src is activated in NSCLCs and promotes the survival of EGFR-mutated cells (17). TKI-sensitive mutations in EGFR’s catalytic domain reportedly confer the capacity to bind to c-Src (18). This binding is thought to activate c-Src by altering its conformation and subsequent phosphorylation status, potentially explaining the activation of c-Src in EGFR-mutated NSCLC cells (19). We evaluated EGFR phosphorylation at Tyr845 and Tyr1068 in HCC827 cells. We also tested the phosphorylation status of endogenous c-Src; in particular, we looked for phosphorylation residues that were active (Tyr419 in the activation loop of the kinase domain) or inactive (Tyr530 in the carboxyl terminus) (20). Finally, we examined mutated-EGFR–c-Src associations.
Gefitinib inhibited EGFR phosphorylation at Tyr845 and Tyr1068 (Fig. 2B). Tyr1068 autophosphorylation is necessary for Grb2-EGFR binding (21). Higher concentrations were required for inhibition at Tyr845 (≥1.6 μM) than at Tyr1068 (≥0.02 μM). Evidently, c-Src is active after EGFR inhibition. Consistent with this possibility, c-Src was stably phosphorylated at Tyr419 and not phosphorylated at Tyr530 after EGFR inhibition (Fig. 2B). After immunoprecipitation with EGFR antibodies (Fig. 2C), mutated EGFR associated directly with c-Src in the absence of gefitinib (lane 8), and EGFR inhibition did not alter this association (lanes 9–14). Because EGFR and c-Src were consistently localized in the plasma membrane, regardless of EGFR phosphorylation status (Fig. 2D), sustained c-Src activation likely reflects its association with mutated EGFR in the plasma membrane.
c-Src Activates the EGFR/MAPK Pathway After EGFR Inhibition in NSCLC Cells.
To link c-Src activity to sustained activation of the Ras/MAPK pathway after EGFR inhibition, we treated HCC827 cells with gefitinib for 12 h and cultured them in the presence or absence of the specific Src inhibitor saracatinib (10 μM) for 1 h before harvesting. Protein expression was analyzed by Western blot, and Ras-GTP levels were measured with a Ras pull-down assay (Fig. 2E). Saracatinib markedly reduced phosphorylation of c-Src at Thy419 (lanes 8–14), indicating that it inhibited c-Src kinase activity. It also decreased phosphorylation of EGFR and Shc in the absence of gefitinib (lane 8), resulting in inhibition of Ras-c-Raf-MEK1/2-ERK1/2 cell signaling. This finding suggests that c-Src promotes activation of EGFR and its downstream MAPK pathway in EGFR-mutated NSCLC cells, as reported (22). In the presence of gefitinib, loss of c-Src activity inhibited Ras, c-Raf, and MEK1/2 activities (lanes 9–14). However, ERK1/2 phosphorylation was reduced only modestly, suggesting that activation of the Ras/MAPK pathway after EGFR inhibition must be regulated at different levels.
EGFR Inhibition Severely Depletes DUSP6 Expression, Activating ERK1/2 by Interacting with Elevated c-Src in EGFR-Mutated NSCLC Cells.
MAPK signaling was once considered a linear and relatively simple receptor-to-nucleus pathway (23). However, we now know that negative feedback loops in the MAPK pathway form a complex network. Because protein phosphatases directly attenuate activated signal transduction pathways by dephosphorylating key kinase effectors, we analyzed expression profiles of the major 83 protein phosphatases from a gene expression data set of HCC827 cells treated with erlotinib for 12 h (GEO accession number GSE38310). EGFR TKI reduced levels of dual specificity phosphatase 6 (DUSP6), a negative regulator specific for ERK1/2 (23). We confirmed this finding with our own samples harvested after 12 h of treatment with gefitinib: mRNA levels of DUSP6 were reduced on quantitative PCR (Fig. 2F) and DUSP6 protein expression was markedly inhibited on Western blot (Fig. 2G). Moreover, DUSP6 levels remained inhibited despite a significant reduction of c-Src/Ras/Raf/MEK signaling (Fig. 2E).
To link EGFR inhibition directly to ERK1/2 activation by DUSP6 depletion, we determined whether ectopically expressed DUSP6 constitutively locks ERK1/2 in an inactive state after EGFR inhibition in HCC827 cells. Exponentially growing HCC827 cells were infected with a lentivirus expressing Myc-tagged DUSP6 or empty vector. After 48 h, cells were cultured in the presence or absence of various concentrations of gefitinib for 12 h and harvested; extracts were analyzed by Western blot. Ectopically expressed protein was distinguished from endogenous protein by its reduced mobility in the gel and was also detected by an antibody against the Myc epitope tag. Ectopically expressed DUSP6 completely inhibited gefitinib-induced ERK1/2 activation (Fig. 2H, lanes 9–14). However, Ras remained activated and MEK1/2 was phosphorylated after inhibition of both EGFR and ERK1/2 (Fig. 2H, lanes 9–14), highlighting an important role for c-Src in activating the Ras/Raf/MEK signaling pathway (Fig. 2E).
These findings indicate that c-Src activates the EGFR/MAPK pathway in NSCLC cells and cooperates with loss of DUSP6 to activate ERK1/2 after EGFR inhibition.
Gefitinib Activates ERK1/2 but Inhibits mRNA Expression of Cyclins D1, D3, and E2.
Cyclin D1 is a major transcriptional target of ERK1/2, and the MAPK signaling pathway is important for the G1–S cell cycle transition (24). We investigated expression of cyclin D1 and other G1 cell-cycle regulation molecules in cells in which ERK1/2 remained active after EGFR inhibition. We expected that levels of cyclin D1 and some G1 cyclins would increase in response to ERK1/2 activation. Surprisingly, after 12 h of treatment with gefitinib, cyclins D1, D3, E2, and p21 Cip1 were inhibited, but cyclin E1, CDK 2, 4, and 6, and p27 Kip1 were not, as shown by Western blot (Fig. 3A and Fig. S8).
Fig. 3.

Inhibition of Akt protein kinase after exposure to gefitinib is the primary cause of reduced expression of Ets-1, cyclins D1, D3, and E2, and DUSP6. (A) Western blot analysis of cyclin D1 and other G1 cell-cycle regulation molecules. (B) Quantitative PCR. Fold changes in gene expression were calculated as the ratio of expression in each sample treated with gefitinib to levels without it. (C) Schematic of −962 cyclin D1 promoter. (D) Western blot analysis of potential cyclin D1 regulation molecules. (E) Quantitative PCR of Ets-1. (F) Western blot. Ets-1 expression in HCC827 cells was depleted for 48 h with siRNA. Control: nontargeting siRNA. Mock: mock transfection. Arrowheads indicate Ets-1 and p42 and p44 DUSP6 proteins. (G) Western blot analysis. HCC827 cells were cultured in the presence or absence of different concentrations of gefitinib with or without PD325901 for 12 h. (H) Western blot analysis. Exponentially growing HCC827 cells were infected with a lentivirus expressing Myc-Myr-Akt1 or an empty vector; 48 h later, cells were cultured in the presence or absence of gefitinib for 12 h and harvested. The arrowhead shows cyclin D3-specific bands.
Fig. S8.

Gefitinib inhibits expression of cyclins D1, D3, and E2 but not cyclin E1, CDK 2, 4, and 6, and p27 Kip1. Western blot analysis of G1 cell-cycle regulation molecules.
To assess mRNA expression of cyclins D1, D3, E2, and p21 Cip1 after gefitinib exposure, we performed quantitative PCR (Fig. 3B). Gefitinib reduced mRNA levels of cyclins D1, D3, and E2, but not of p21 Cip1. Evidently, the reduced levels of p21 Cip1 protein likely reflect loss of cyclin–CDKs–p21 Cip1 complexes. Thus, although ERK1/2 was activated by EGFR inhibitors, its effect on cell proliferation was nullified by inhibition of its transcriptional target gene cyclins D1, D3, and E2.
EGFR Inhibition Specifically Decreases Ets-1 Protein, Which Negatively Regulates Expression of Cyclins D1, D3, E2, and DUSP6.
The –962 cyclin D1 promoter fragment has binding sites for the transcription factors AP-1 (c-Jun/c-Fos), Ets, TCF, and CREB (Fig. 3C) (25). Ras/MAPK-mediated activation of the cyclin D1 promoter requires Ets and CREB. As cyclin D1 transcription was inhibited despite ERK1/2 activation, we hypothesized that the cause was loss of Ets or CREB expression.
To test this hypothesis, we cultured HCC827 cells in the presence or absence of gefitinib for 12 h and analyzed cell extracts by Western blot with antibodies against c-Jun, Ets-1, Ets-2, TCF4, and CREB (Fig. 3D). We chose Ets-1 and Ets-2 because transactivation of their target genes is controlled by ERK1/2 (26, 27). In addition, HCC827 cells exclusively express TCF4 (Fig. S9). Gefitinib did not affect c-Jun, Ets-2, TCF4, or CREB (Fig. 3D), but quantitative PCR showed reduced Ets-1 mRNA (Fig. 3E). This gefitinib-induced decreased Ets-1 expression could explain why cyclin D1 transcription was inhibited despite elevated ERK1/2 activity after EGFR inhibition.
Fig. S9.
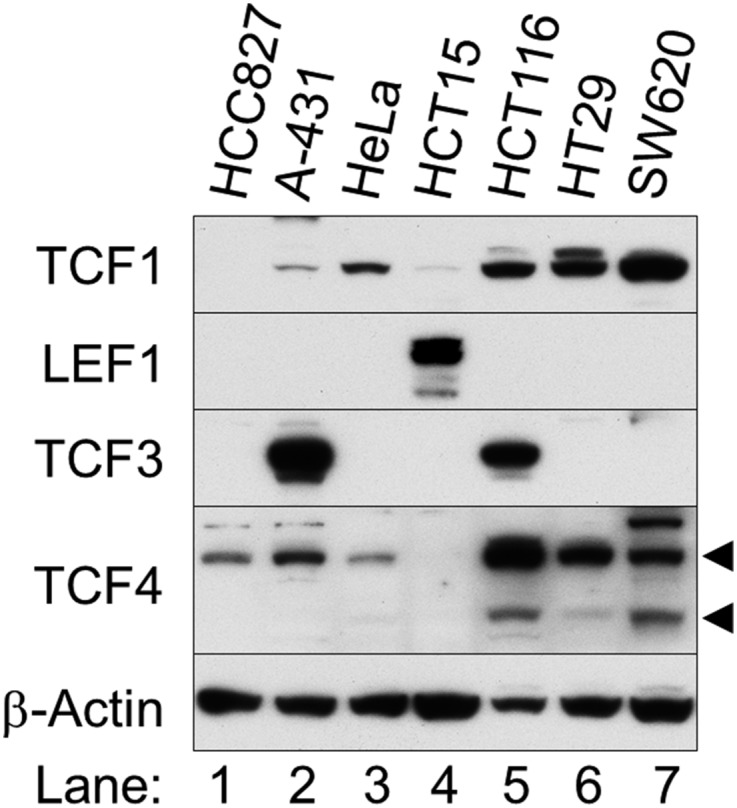
HCC827 cells exclusively express TCF4 among TCF/LEF family transcription factors. Western blot analysis is shown. Cell lysates were separately prepared from the indicated exponentially growing cell lines. Arrowheads indicate 58- and 79-kDa TCF4 splice variants.
To assess the role of Ets-1 in the expression of cyclins D1, D3, and E2, we used RNA interference (RNAi) to knock down Ets-1 expression. First, we measured Ets-1 protein in HCC827 cells transfected with control small interfering (si)RNA or Ets-1 siRNA oligonucleotides. Cells were harvested 48 h after transfection and analyzed by Western blot. Ets-1 siRNA, but not control siRNA, reduced Ets-1 levels (Fig. 3F). Next, we assessed the effects of Ets-1 depletion on DUSP6, cyclin D1, and other G1 cell-cycle regulators. We included DUSP6 because it is a downstream transcriptional target gene of Ets-1 (28, 29). As expected, expression of DUSP6, the cyclins, and p21 Cip1 was markedly decreased. Reduction of DUSP6 resulted in a significant increase of ERK1/2 phosphorylation without changing its total expression. As in gefitinib-treated cells, expression of cyclin E1 was not changed. These findings confirm that Ets-1 is essential for expression of DUSP6 and cyclins D1, D3, and E2 in HCC827 cells. They also verify that DUSP6 has a critical role in negative regulation of ERK1/2 kinase activity in the NSCLC cells.
Inhibition of Akt Protein Kinase After Exposure to Gefitinib Is the Primary Cause of Reduced Expression of Ets-1, Cyclins D1, D3, E2, and DUSP6.
To establish the relative importance of ERK1/2 activation and Akt reduction in Ets-1–mediated expression of cyclins D1, D3, E2, and DUSP6 after EGFR inhibition, we blocked ERK1/2 activity with a MEK inhibitor or enhanced Akt activity by introducing a constitutively active Akt1 in HCC827 cells.
HCC827 cells were treated with gefitinib and 10 μM PD325901 for 12 h and harvested for Western blot analysis (Fig. 3G). PD325901, a MEK inhibitor, inhibited both baseline and gefitinib-induced activation of ERK1/2 activity (lanes 8–14). In the absence of gefitinib, PD325901 attenuated expression of Ets-1, DUSP6, the cyclins, and p21 Cip1, despite phosphorylation and activation of Akt (lane 8). Addition of gefitinib inhibited phosphorylation of Akt and ERK1/2, but did not rescue expression of Ets-1, the cyclins, or DUSP6 (lanes 9–14). These observations suggest that ERK1/2 regulated expression of Ets-1, the cyclins, and DUSP6 proteins in these cells; however, gefitinib-induced ERK1/2 activation was not associated with reduced expression of Ets-1, the cyclins, and DUSP6.
When directed to membranes, Akt1 becomes constitutively active by the addition of a c-Src myristoylation (Myr) sequence (30). To examine this process, we generated an N-terminal Myr form of Akt1 in a lentiviral gene expression system. Ectopic and endogenous expression (Fig. 3H, lanes 8–14) were distinguished as described above. Ectopically expressed Myr-Akt1 was phosphorylated and activated regardless of EGFR inhibition, as shown by phospho-Akt blotting (lanes 9–14). Unexpectedly, Myr-Akt1 rescued expression of Ets-1, the three cyclins, p21 Cip1, and DUSP6. Although DUSP6 was increased less than the other proteins, gefitinib-induced ERK1/2 activation was inhibited. Thus, inhibition of Akt protein kinase after exposure to gefitinib is the primary cause of the reduction in the levels of Ets-1, DUSP6, and cyclins D1, D3, and E2.
Sustained Activation of ERK1/2 Enhances Cell Survival by Accelerating Bim Protein Turnover.
EGFR inhibition induces a BH3-only protein Bim, which is essential for apoptosis and caspase induction in EGFR-mutated NSCLC cells (31). EGFR TKIs generate all three splice variants of Bim: short (S), long (L), and extralong (EL), of which Bim EL is the most abundant. ERK1/2 phosphorylates Bim EL at Ser69, targeting it for degradation via the proteasome–ubiquitin pathway (32). Moreover, ERK1/2 activation can inhibit the binding of Bim to Bcl-2 family proteins such as Bcl-xL and Mcl-1 (33). Thus, ERK1/2 promotes cell survival by antagonizing Bim protein. To test this possibility in EGFR-inhibited cells, we treated HCC827 cells with different concentrations of gefitinib for 5 d. Cells were then further cultured with or without the addition of the MEK inhibitor PD325901 for 24 h before harvesting (Fig. 4A). Elevated ERK1/2 caused sustained phosphorylation of Bim EL at Ser69, resulting in rapid turnover of phosphorylated Bim EL (lanes 2–7). The other two splice variants of Bim S and L were barely detectable in these extracts. The MEK inhibitor reduced phosphorylation of ERK1/2 and prevented the ERK1/2-mediated phosphorylation of Bim protein, thereby suppressing degradation of nonphosphorylated Bim, restoring its expression, and facilitating caspase-3 activation (lanes 8–14).
Fig. 4.

Sustained activation of ERK1/2 enhances cell survival by accelerating Bim protein turnover. (A) Western blot analysis. HCC827 cells were cultured in the presence or absence of different concentrations of gefitinib for 5 d; medium was then changed and cells were further cultured with or without 10 μM PD325901 for 24 h before harvesting. Asterisk stands for nonspecific bands. (B) Schematic of the molecular mechanism of EGFR inhibition and innate drug resistance in EGFR-mutated NSCLC cells.
Discussion
HGF-Met signaling was previously identified as an important determinant of the innate drug resistance to EGFR TKIs in NSCLC cells (5). However, given that causes of acquired resistance to EGFR TKIs are multiple and complex (2), NSCLC cells may have more than one mechanism for innate resistance. In this paper, we investigated the molecular machinery by which the Ras/MAPK pathway is activated after EGFR inhibition, despite blockade of RTK activity in NSCLC cells (Fig. 4B). EGFR inhibitors decrease both the MAPK and Akt pathways for a short time, after which Ras and the MAPK pathway become reactivated. Akt inhibition selectively blocks the transcriptional activation of Ets-1, which inhibits its target gene, DUSP6. As a result, ERK1/2 is activated. Furthermore, elevated c-Src stimulates Ras GTP loading and activates Raf/MEK kinases. The outcome of persistent inactivation of Ets-1 is quiescence and survival of the NSCLC cells: quiescence is triggered by reduction of the Ets-1 target genes cyclins D1, D3, and E2, whereas elevated ERK1/2 accelerates Bim protein turnover, resulting in increased cell survival. Our findings may elucidate a new aspect of the innate resistance to EGFR TKIs in the absence of growth factors, without activation of RTKs.
To our knowledge, this is the first report of opposing effects of Akt on the MAPK pathway: transactivation of Ets-1 and inhibition of c-Raf. Akt-c-Raf crosstalk was previously reported to inhibit c-Raf kinase activity (34). However, we found that Akt activates Ets-1, an important downstream effector of ERK1/2. Conversely, EGFR inhibition prevents Akt activity, resulting in activation of c-Raf and two kinases downstream in the cascade, MEK1/2 and ERK1/2, but inhibits Ets-1 function. Thus, after EGFR inhibition, the Ras/MAPK pathway is simultaneously activated in three different ways: Ras activation by elevated c-Src, c-Raf stimulation by Akt inhibition, and ERK1/2 induction by DUSP6 depletion (Fig. 4B). Nonetheless, cyclin D1, a major transcriptional target of ERK1/2, is significantly inhibited after EGFR inhibition.
Our findings demonstrate that synthesis of Ets-1 and transactivation of its target genes requires ERK1/2 and Akt kinase activities (Fig. 4B). For this reason, although ERK1 and ERK2 were more active after EGFR inhibition, they could not transactivate Ets-1 target genes without Akt protein kinase. This insight reveals a previously unidentified point of convergence of MAPK and Akt signaling and may provide a strategy to treat cancers in which MAPK and Akt activities are abnormal. For example, B-Raf–mutated tumors are sensitive to MEK and B-Raf inhibitors, which attenuate cyclin D1 expression and cause G1 arrest (35, 36). Inhibition of both MEK and Akt or both B-Raf and Akt may result in more complete and lasting inhibition of Ets-1 target genes, including cyclin D1. Likewise, an essential role of Akt to maintain Ets-1 in an active state might shift the therapeutic approach to targeting the MAPK pathway. Despite the frequency of aberrant Ras/MAPK signaling in human cancer, the critical role of MEK1/2 in the pathway, and the availability of highly specific and potent MEK inhibitors, targeted chemotherapy for human malignancies has been disappointing, except for B-Raf–mutated melanoma (37). Because Ets-1 is the major effector regulating MAPK downstream target genes, our findings suggest an alternative approach to inhibiting the MAPK pathway—attenuating Akt activity. We believe that these insights will cause a shift in the therapeutic approach to targeting the MAPK pathway.
In NSCLC cells treated with EGFR inhibitors, persistent inactivation of Ets-1 induces the innate drug resistance. Defining the molecular mechanism for this process may provide insights into the role of Akt in regulation of Ets-1 and its target genes and might have important implications for targeted therapy for EGFR-driven cancers and the innate drug resistance to EGFR TKIs. We found a correlation between Ets-1 expression and its activity, as assessed by levels of its target genes, DUSP6 and cyclins D1, D3, and E2, with or without gefitinib. Moreover, EGFR inhibition reduced Ets-1 mRNA levels, the same mechanism as in the target genes. Thus, Ets-1 may autoregulate its transcript in NSCLC cells. In fact, consensus Ets-binding sites in the Ets-1 promoter regulatory region are necessary for its activation in cultured cells (38, 39). Therefore, once ERK1/2 and Akt activate Ets-1, positive feedback will exponentially increase its expression. Indeed, Ets-1 mRNA is increased in a K-Ras–transformed prostate epithelial cell line (40). Likewise, elevated Akt activity raises Ets-1 expression in prostate cancer (41).
Posttranslational modification of Ets family members is another mechanism for transactivation of Ets target genes (42). ERK1/2 phosphorylates Ets-1 at Thr38 and Ets-2 at Thr72, which increases their transactivational activity (26, 27). A recent study of macrophages in motheaten-viable mice showed that Thr72 of Ets-2 is phosphorylated and activated by Akt-mediated Jun-N-terminal kinase (43). Akt also induces transcriptional activity of an Ets family member, PU.1, by phosphorylating a residue in its transactivation domain (44). Therefore, transcription of Ets-1 might be enhanced by phosphorylation by Akt. However, Scansite motif analysis (45) showed that Ets-1’s potential Akt phosphorylation sites Thr73 and Ser282 are less stringent (within 2.672 and 2.233 percentiles, respectively) than its actual ERK1/2 phosphorylation residue Thr38 (within 0.744 percentile). Alternatively, Akt might phosphorylate two closely related transcriptional coactivating proteins to transactivate Ets-1 target genes, CREB binding protein (CREBBP) and p300, with which Ets-1 interacts (46). Moreover, Akt phosphorylates p300 at Ser1834, which is essential for its transcription from the promoter of intercellular adhesion molecule-1 (47), whose transcription is also activated by Ets-1 and Ets-2 (48, 49). Thus, Akt may activate the Ets-1 transcriptional machinery by phosphorylating its coactivator p300/CREBBP. Our protein motif analysis further supported this possibility. CREBBP has highly stringent potential Akt phosphorylation sites at Ser381, Ser1733, and Thr1833 (within 0.828, 0.538, and 0.235 percentile, respectively). All of these sites are in CREBBP’s CH1 and CH2/CH3 domains, which interact with Ets-1 (46). Nonetheless, more studies are warranted to define the mechanism of Akt-mediated transactivation of Ets-1 in NSCLC.
In this report, we demonstrate a new aspect of the innate drug resistance to EGFR TKIs without activation of RTKs. We investigated the mechanism by which the Ras/MAPK pathway is activated after EGFR inhibition despite blockade of RTK activity in NSCLC cells. We found that not only ERK1/2 but also Akt activity is essential to maintain Ets-1 in an active state. Therefore, despite high levels of ERK1/2, Ets-1 target genes including DUSP6 and cyclins D1, D3, and E2 remain suppressed in the absence of Akt activity after EGFR inhibition. Reduction of DUSP6 combines with c-Src to renew activation of the Ras/MAPK pathway, resulting in increased cell survival by accelerating Bim protein turnover. Because we found that addition of a MEK inhibitor enhances programmed cell death by rewiring apoptotic signaling, we may reduce the probability of emergent resistance to EGFR TKIs by the combined treatment of the TKI and MEK inhibitor, although further studies are required to address this question.
Materials and Methods
Chemicals, Cell Culture, DNA Plasmids, Small Interfering RNA, and Transfection.
The following were suspended in dimethyl sulfoxide: gefitinib, erlotinib, lapatinib, the MEK inhibitor PD325901, and the Src inhibitor saracatinib (all from LC laboratories). The following cell lines were from the American Type Culture Collection: HCC827, HCC2935, H-1650, H-1838, H-2170, H-1975, 293T, A-431, HeLa, HCT15, HCT116, HT29, and SW620. Cells were cultured in RPMI medium 1640 (Life Technologies) supplemented with 10% (vol/vol) FBS (Life Technologies) and were maintained in a 5% CO2 incubator at 37 °C.
The Myc-tagged S17N and G12V human K-Ras pcDNA3.1 expression vectors have been described (50). Human DUSP6 (Image Clone, Fisher Scientific), Myc-S17N human K-Ras, or Myr-mouse Akt1 (Upstate Biotechnology, Millipore) cDNA was subcloned into the pLenti-C-Myc-DDK lentiviral vector (OriGene). Ets-1 and control siRNA oligonucleotides were selected as recommended (SI03068632, FlexiTube siRNA, Qiagen; AllStars Negative Control, Qiagen). TransIT-LT1 (Mirus Bio) and Oligofectamine (Life Technologies) were used for transient DNA and siRNA transfections.
Lentiviral Gene Expression System.
Plasmid DNA containing DUSP6, S17N K-Ras, Myr-Akt1, or empty pLenti-C-Myc-DDK (control) was transfected into 293T cells together with lentiviral packaging plasmids pMDLg/pPRE, pRSV-Rev, and pMD2.G (provided by Didier Trono, Addgene, Cambridge, MA). After 16 h of incubation, the culture medium was replaced with fresh medium. The next day, cell culture supernatants containing viral particles expressing DUSP6, S17N K-Ras, Myr-Akt1, or empty lentiviral vector were collected and used with hexadimethrine bromide (polybrene, Sigma-Aldrich) to infect exponentially growing HCC827 cells. At 24 h after infection, cells were cultured in the presence or absence of gefitinib for 12 h and harvested.
Protein Preparation, Western Blot Analysis, and Phospho-RTK Antibody Array.
Total cellular protein was isolated with cell lysis buffer (Cell Signaling Technology). Nuclear-and-cytoplasmic or membrane-and-cytoplasmic protein fractions of cultured cells were obtained with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Protein Biology) or a Mem-PER Plus Membrane Protein Extraction Kit (Pierce Protein Biology). Equal amounts of protein were prepared by adding Reducing Red Loading Buffer (Cell Signaling Technology) and were resolved by SDS/PAGE (Novex Tris-Glycine Gels; Life Technologies).
Western blots were developed by enhanced chemiluminescence (ECL and SuperSignal West Pico, Pierce Protein Biology; Luminata Crescendo, Millipore; and ECL Prime, GE Healthcare Life Science), and detected by autoradiography film (Classic BX Autoradiography Film, MIDSCI). The following primary and secondary antibodies were used: phospho-Tyr1068 and Tyr845 EGFR (3777, 6963, Cell Signaling Technology), EGFR (1005, Santa Cruz Biotechnology; 4267, Cell Signaling Technology), phospho-ERK1/2 (9101, Cell Signaling Technology; E-4, Santa Cruz Biotechnology), ERK1/2 (9102, Cell Signaling Technology; K-23, Santa Cruz Biotechnology), β-actin HRP-conjugated (Sigma-Aldrich), phospho-Thr308 and Ser473 Akt (2965, 4060, Cell Signaling Technology), Akt (4691, Cell Signaling Technology), phospho-Ser299 A-Raf (4431, Cell Signaling Technology), A-Raf (4432, Cell Signaling Technology), phospho-Ser445 B-Raf (2696, Cell Signaling Technology), B-Raf (9434, Cell Signaling Technology), phospho-Ser259, Ser289/296/301, and Ser338 c-Raf (9421, 9431, 9427, Cell Signaling Technology), c-Raf (9422, Cell Signaling Technology; 610151, BD Transduction Laboratories), phospho-MEK1/2 (9121, Cell Signaling Technology), MEK1/2 (9126, Cell Signaling Technology), pan-(H, K, and N) Ras (Ras10, Pierce Protein Biology), PARP (9532, Cell Signaling Technology), β-tublin (2128, Cell Signaling Technology), phospho-Ser69 Bim (4585, Cell Signaling Technology), Bim (2933, Cell Signaling Technology), cleaved CASP3 (9664, Cell Signaling Technology), Myc-tag HRP conjugated (A-14, Santa Cruz Biotechnology), DUSP6 (3058, Cell Signaling Technology), phospho-Thy877 and Tyr1221/1222 HER2 (2241 and 2243, Cell Signaling Technology), HER2 (2165, Cell Signaling Technology), phospho-Thy1289 HER3 (4791, Cell Signaling Technology), HER3 (4754, Cell Signaling Technology), phospho-Tyr653/654 FGFR1 (3471, Cell Signaling Technology), FGFR1 (9740, Cell Signaling Technology), phospho-Tyr980, Tyr1131, Tyr1135, and Tyr1135/1136 IGF1Rβ (4568, 3024, 3021, and 3918; Cell Signaling Technology), IGF1Rβ (3018, Cell Signaling Technology), phospho-Tyr1003, Tyr1234/1235, and Tyr1349 Met (3135, 3077, and 3133; Cell Signaling Technology), Met (3127, Cell Signaling Technology), phospho-Thy239/240 Shc (S434, Cell Signaling Technology), Shc (610875, BD Transduction Laboratories), phospho-Tyr307 and Tyr627 Gab1 (3234 and 3233, Cell Signaling Technology), Gab1 (3232, Cell Signaling Technology), phospho-Ser159 and Tyr452 Gab2 (3884 and 3882, Cell Signaling Technology), Gab2 (3239, Cell Signaling Technology), phospho-Tyr580 SHP2 (5431, Cell Signaling Technology), SHP2 (3397, Cell Signaling Technology), phospho Tyr419 and Tyr530 c-Src (6943 and 2105, Cell Signaling Technology), nonphospho-Tyr419 and Tyr530 c-Src (2102 and 2107, Cell Signaling Technology), c-Src (2123, Cell Signaling Technology), cyclin D1 (A-12, Santa Cruz Biotechnology), cyclin D3 (610279, BD Transduction Laboratories; C-16, Santa Cruz Biotechnology), cyclin E1 (HE12, Calbiochem), cyclin E2 (4132, Cell Signaling Technology), p21 Cip1 (610233, BD Transduction Laboratories), p27 Kip1 (610241, BD Transduction Laboratories), c-Jun (9165, Cell Signaling Technology), Ets-1 (C-20, Santa Cruz Biotechnology; 6258, Cell Signaling Technology), Ets-2 (C-20, Santa Cruz Biotechnology), TCF1 (2203, Cell Signaling Technology), LEF1 (2230, Cell Signaling Technology), TCF3 (2883, Cell Signaling Technology), TCF4 (2569, Cell Signaling Technology), CREB (4820, Cell Signaling Technology), anti-mouse IgG HRP (RPN4201, GE Healthcare Life Science), and anti-rabbit IgG HRP (7074, Cell Signaling Technology).
A PathScan RTK Signaling Antibody Array Kit (7982, Cell Signaling Technology) was used according to the manufacturer’s instructions to assess the phosphorylation status of 28 major RTKs. Briefly, cell lysate was incubated on a slide, to which was added a biotinylated detection antibody mixture, streptavidin-conjugated horseradish peroxidase, and, finally, enhanced chemiluminescence reagents. Slide images were captured with autoradiography film.
Immunoprecipitation and Immunoblotting, Measurement of Ras-GTP Levels, and Phospho-ERK1/2–Associated GST-Elk1 in Vitro Kinase Assay.
Proteins were precipitated with agarose-conjugated antibodies to mouse normal IgG (sc-2343, Santa Cruz Biotechnology), and EGFR (528, Santa Cruz Biotechnology). Immunoprecipitates were subjected to SDS/PAGE and stained with antibodies against EGFR (1005, Santa Cruz Biotechnology), or c-Src (2123, Cell Signaling Technology). Ras-GTP levels were measured with the Active Ras Pull-Down and Detection Kit (Pierce; Cell Signaling Technology). The pulled-down Ras-GTP was detected by Western blot analysis with a pan-Ras antibody (H-Ras, K-Ras, and N-Ras).
A phospho-ERK1/2–associated GST-Elk1 in vitro kinase assay was performed according to the manufacturer’s instructions with a nonradioactive p44/42 MAP Kinase Assay Kit (9800, Cell Signaling Technology). Briefly, active p44/42-ERK1/2 kinase from cell extracts was precipitated by phospho-p44/42-ERK1/2 antibodies and analyzed with an in vitro kinase assay using GST-Elk-1 protein as a substrate. Elk-1 phosphorylation and input GST-Elk-1 protein were detected by Western blotting with antibodies against phospho-Ser383 Elk-1 (2B1, Cell Signaling Technology) and Elk-1 (9182, Cell Signaling Technology).
TaqMan Quantitative-PCR Assay.
Gene expression was analyzed in triplicate by TaqMan quantitative PCR (qPCR). mRNA was isolated with an SV Total RNA Isolation System (Promega). cDNA from 500 ng of total RNA was synthesized with an RT First Strand Kit (Life Technologies). cDNA (5 ng) was mixed with RT qPCR master mixes, and aliquots were placed with gene-specific primer sets. We used TaqMan assays from Life Technologies DUSP6 (Hs01044001_m1), cyclin D1 (Hs00765553_m1), cyclin D3 (Hs00426901_m1), cyclin E2 (Hs00180319_m1), p21 Cip1 (Hs00355782_m1), and Ets-1 (Hs00428287_m1). Expression levels normalized to endogenous GAPDH were determined by real-time PCR and analyzed at the UCSF Helen Diller Family Comprehensive Cancer Center Genome Analysis Core Facility.
Acknowledgments
The authors thank Kathryn Thompson, Jennifer Dang, Kirsten Copren, and the UCSF Helen Diller Comprehensive Cancer Center Genome Analysis Core Facility for their support of TaqMan quantitative PCR assays and Taku Tokuyasu for his valuable suggestions on GEO dataset analysis. This work was supported by grants (to O.T.) from the Joan and Irwin Jacobs Fund of the Jewish Community Foundation, Daiichi-Sankyo, and the American Head and Neck Society (Ballantyne Award).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1510733112/-/DCSupplemental.
References
- 1.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosell R, et al. Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 4.Lippert TH, Ruoff HJ, Volm M. Current status of methods to assess cancer drug resistance. Int J Med Sci. 2011;8(3):245–253. doi: 10.7150/ijms.8.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson TR, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kentsis A, et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med. 2012;18(7):1118–1122. doi: 10.1038/nm.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nassar N, Singh K, Garcia-Diaz M. Structure of the dominant negative S17N mutant of Ras. Biochemistry. 2010;49(9):1970–1974. doi: 10.1021/bi9020742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stommel JM, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 10.Guo A, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci USA. 2008;105(2):692–697. doi: 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Geer P, Wiley S, Gish GD, Pawson T. The Shc adaptor protein is highly phosphorylated at conserved, twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Curr Biol. 1996;6(11):1435–1444. doi: 10.1016/s0960-9822(96)00748-8. [DOI] [PubMed] [Google Scholar]
- 12.Weidner KM, et al. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature. 1996;384(6605):173–176. doi: 10.1038/384173a0. [DOI] [PubMed] [Google Scholar]
- 13.Holgado-Madruga M, Emlet DR, Moscatello DK, Godwin AK, Wong AJ. A Grb2-associated docking protein in EGF- and insulin-receptor signalling. Nature. 1996;379(6565):560–564. doi: 10.1038/379560a0. [DOI] [PubMed] [Google Scholar]
- 14.Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol. 2000;20(22):8513–8525. doi: 10.1128/mcb.20.22.8513-8525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci USA. 1999;96(4):1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biscardi JS, et al. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274(12):8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, et al. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am J Pathol. 2007;170(1):366–376. doi: 10.2353/ajpath.2007.060706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcotte R, Zhou L, Kim H, Roskelly CD, Muller WJ. c-Src associates with ErbB2 through an interaction between catalytic domains and confers enhanced transforming potential. Mol Cell Biol. 2009;29(21):5858–5871. doi: 10.1128/MCB.01731-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patwardhan P, Resh MD. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol Cell Biol. 2010;30(17):4094–4107. doi: 10.1128/MCB.00246-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aleshin A, Finn RS. SRC: A century of science brought to the clinic. Neoplasia. 2010;12(8):599–607. doi: 10.1593/neo.10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batzer AG, Rotin D, Ureña JM, Skolnik EY, Schlessinger J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol Cell Biol. 1994;14(8):5192–5201. doi: 10.1128/mcb.14.8.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donepudi M, Resh MD. c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell Signal. 2008;20(7):1359–1367. doi: 10.1016/j.cellsig.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013;280(2):489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003;3(3):233–245. doi: 10.1016/s1535-6108(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 25.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 26.Yang BS, et al. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol Cell Biol. 1996;16(2):538–547. doi: 10.1128/mcb.16.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slupsky CM, et al. Structure of the Ets-1 pointed domain and mitogen-activated protein kinase phosphorylation site. Proc Natl Acad Sci USA. 1998;95(21):12129–12134. doi: 10.1073/pnas.95.21.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekerot M, et al. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008;412(2):287–298. doi: 10.1042/BJ20071512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z, et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31(4):577–586. doi: 10.1093/carcin/bgq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996;271(36):21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- 31.Costa DB, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4(10):1669–1679, discussion 1680. doi: 10.1371/journal.pmed.0040315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luciano F, et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22(43):6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 33.Ewings KE, et al. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO J. 2007;26(12):2856–2867. doi: 10.1038/sj.emboj.7601723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286(5445):1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 35.Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joseph EW, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci USA. 2010;107(33):14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11(7):385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 38.Oka T, Rairkar A, Chen JH. Structural and functional analysis of the regulatory sequences of the ets-1 gene. Oncogene. 1991;6(11):2077–2083. [PubMed] [Google Scholar]
- 39.Majérus MA, Bibollet-Ruche F, Telliez JB, Wasylyk B, Bailleul B. Serum, AP-1 and Ets-1 stimulate the human ets-1 promoter. Nucleic Acids Res. 1992;20(11):2699–2703. doi: 10.1093/nar/20.11.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z, et al. Elevated expression of ETS-1 gene in a metastatic, tumorigenic human prostate epithelial cell line transformed by the v-Ki-ras oncogene. Int J Oncol. 1997;11(6):1179–1184. doi: 10.3892/ijo.11.6.1179. [DOI] [PubMed] [Google Scholar]
- 41.Smith AM, et al. ETS1 transcriptional activity is increased in advanced prostate cancer and promotes the castrate-resistant phenotype. Carcinogenesis. 2012;33(3):572–580. doi: 10.1093/carcin/bgs007. [DOI] [PubMed] [Google Scholar]
- 42.Tootle TL, Rebay I. Post-translational modifications influence transcription factor activity: A view from the ETS superfamily. BioEssays. 2005;27(3):285–298. doi: 10.1002/bies.20198. [DOI] [PubMed] [Google Scholar]
- 43.Smith JL, et al. ets-2 is a target for an akt (Protein kinase B)/jun N-terminal kinase signaling pathway in macrophages of motheaten-viable mutant mice. Mol Cell Biol. 2000;20(21):8026–8034. doi: 10.1128/mcb.20.21.8026-8034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rieske P, Pongubala JM. AKT induces transcriptional activity of PU.1 through phosphorylation-mediated modifications within its transactivation domain. J Biol Chem. 2001;276(11):8460–8468. doi: 10.1074/jbc.M007482200. [DOI] [PubMed] [Google Scholar]
- 45.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31(13):3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang C, Shapiro LH, Rivera M, Kumar A, Brindle PK. A role for CREB binding protein and p300 transcriptional coactivators in Ets-1 transactivation functions. Mol Cell Biol. 1998;18(4):2218–2229. doi: 10.1128/mcb.18.4.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol. 2005;25(15):6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Launoit Y, Audette M, Pelczar H, Plaza S, Baert JL. The transcription of the intercellular adhesion molecule-1 is regulated by Ets transcription factors. Oncogene. 1998;16(16):2065–2073. doi: 10.1038/sj.onc.1201726. [DOI] [PubMed] [Google Scholar]
- 49.Yockell-Lelièvre J, et al. Functional cooperation between Stat-1 and ets-1 to optimize icam-1 gene transcription. Biochem Cell Biol. 2009;87(6):905–918. doi: 10.1139/o09-055. [DOI] [PubMed] [Google Scholar]
- 50.Tetsu O, et al. Mutations in the c-Kit gene disrupt mitogen-activated protein kinase signaling during tumor development in adenoid cystic carcinoma of the salivary glands. Neoplasia. 2010;12(9):708–717. doi: 10.1593/neo.10356. [DOI] [PMC free article] [PubMed] [Google Scholar]