

Significance
Anti-trypanosomal drugs, used to tackle lethal human and animal diseases, target an unusual parasite DNA structure in a cellular compartment known as the mitochondrion. Using a high-throughput genetic approach to study drug resistance, we identified every component of a molecular rotor that couples ATP hydrolysis to proton transport across non-mitochondrial membranes. Surprisingly, this molecular machine was found to communicate with a related mitochondrial rotor and, when defective, rendered the mitochondrial DNA structure obsolete. Our findings reveal new potential mechanisms of multidrug resistance in trypanosomes. They also suggest that communication between these rotors in two separate cellular compartments could be conserved through evolution, reflecting an unanticipated and important aspect of environmental sensing and metabolic control in nucleated cells.
Keywords: brucei, mitochondrion, nagana, petite, samorin
Abstract
Kinetoplastid parasites cause lethal diseases in humans and animals. The kinetoplast itself contains the mitochondrial genome, comprising a huge, complex DNA network that is also an important drug target. Isometamidium, for example, is a key veterinary drug that accumulates in the kinetoplast in African trypanosomes. Kinetoplast independence and isometamidium resistance are observed where certain mutations in the F1-γ-subunit of the two-sector F1Fo-ATP synthase allow for Fo-independent generation of a mitochondrial membrane potential. To further explore kinetoplast biology and drug resistance, we screened a genome-scale RNA interference library in African trypanosomes for isometamidium resistance mechanisms. Our screen identified 14 V-ATPase subunits and all 4 adaptin-3 subunits, implicating acidic compartment defects in resistance; V-ATPase acidifies lysosomes and related organelles, whereas adaptin-3 is responsible for trafficking among these organelles. Independent strains with depleted V-ATPase or adaptin-3 subunits were isometamidium resistant, and chemical inhibition of the V-ATPase phenocopied this effect. While drug accumulation in the kinetoplast continued after V-ATPase subunit depletion, acriflavine-induced kinetoplast loss was specifically tolerated in these cells and in cells depleted for adaptin-3 or endoplasmic reticulum membrane complex subunits, also identified in our screen. Consistent with kinetoplast dispensability, V-ATPase defective cells were oligomycin resistant, suggesting ATP synthase uncoupling and bypass of the normal Fo-A6-subunit requirement; this subunit is the only kinetoplast-encoded product ultimately required for viability in bloodstream-form trypanosomes. Thus, we describe 30 genes and 3 protein complexes associated with kinetoplast-dependent growth. Mutations affecting these genes could explain natural cases of dyskinetoplasty and multidrug resistance. Our results also reveal potentially conserved communication between the compartmentalized two-sector rotary ATPases.
Kinetoplastid parasites, including African trypanosomes, South American trypanosomes, and Leishmania spp., cause a range of lethal and debilitating diseases in humans and animals, and there is an urgent need for new therapies (1). The kinetoplastids transmitted by tsetse flies, and the diseases they cause, are restricted to the African tsetse belt. These parasites include the major causes of nagana in livestock, Trypanosoma vivax and Trypanosoma congolense, and Trypanosoma brucei brucei, the animal parasite most often manipulated in the laboratory. Although Trypanosoma brucei gambiense, the most important cause of human infection, is not thought to circulate in cattle, Trypanosoma brucei rhodesiense is zoonotic, passing among humans and animal reservoirs. Trypanosoma equiperdum and Trypanosoma evansi are similar trypanosomes (2), but with defects in the kinetoplast, the large DNA network comprising the mitochondrial genome (3). These parasites cause dourine or surra disease in horses, camels, or water buffalo. These dyskinetoplastic parasites are not transmitted by tsetse flies because passage through the insect midgut requires kinetoplast-dependent mitochondrial elaboration (4). Instead, they are mechanically transmitted by other biting insects or sexually transmitted in Africa and outside Africa.
Several anti-trypanosomal drugs target the kinetoplast (5). Although this mitochondrial genome is essential in T. brucei, it is dispensable in T. equiperdum and T. evansi (6). As outlined below, studies on the mitochondrial F1Fo ATP synthase, also known as complex V, led to an explanation for this difference. ATP synthase is a large, multisubunit complex of the electron transport chain that couples the mitochondrial membrane potential, ΔΨmito, to proton transport and ATP synthesis in insect-stage T. brucei. In bloodstream-form T. brucei, which rely on glycolysis for energy production, the more minimal mitochondrion lacks cytochromes, Krebs cycle enzymes, and oxidative phosphorylation (4), and ATP synthase works in the other direction, hydrolyzing ATP to generate ΔΨmito (7–9); ΔΨmito is still required for mitochondrial protein import, redox homeostasis, and Fe-S cluster assembly. In this life cycle stage, the kinetoplast encodes a single essential protein, the Fo-A6-subunit of the membrane-sector of ATP synthase. A mutation in the nuclear genome-encoded γ-subunit in T. evansi or T. equiperdum can compensae for this A-subunit requirement and allows for kinetoplast loss (6). Indeed, these dyskinetoplastic trypanosomes have been referred to as petite mutants of T. brucei (10), sharing similarities with petite mutants of yeast (11). Thus, dyskinetoplastic trypanosomes use an alternative, Fo-independent mode of ΔΨmito generation involving the uncoupled F1 sector of ATP synthase and an ADP3-/ATP4- carrier (6, 12).
Isometamidium chloride (ISM; samorin, veridium, trypamidium) and diminazene aceturate (berenil) are the most important veterinary trypanocidal drugs. ISM, a phenanthridine related to ethidium bromide, is mainly used as a prophylactic against nagana disease in cattle, with millions of doses administered each year. This drug accumulates in the kinetoplast, and cells with mutations in the γ-subunit of ATP synthase are ISM resistant (5). Ethidium bromide has itself also been used against nagana. The diamidine, pentamidine, is another kinetoplast-targeting drug used against human trypanosomal diseases (13). Although kinetoplast biology is intimately linked to parasite biology and antiparasite therapy, our understanding of kinetoplast dependency, drug mode of action, and potential resistance mechanisms remains incomplete.
To further explore these areas, we screened a genome-scale RNA interference library in bloodstream-form T. brucei for ISM resistance mechanisms. Among 30 genes identified were those encoding possibly all 14 V-type H+ ATPase (V-ATPase) subunits, all 4 adaptin-3 subunits, and 5 endoplasmic-reticulum (ER) membrane complex subunits. Depletion of V-ATPase subunits, or the other complex subunits, was associated with probable ATP synthase uncoupling, kinetoplast-independent growth, and multidrug resistance. Our results reveal an unexpected link between V-ATPase function, mitochondrial ATP synthase function, and kinetoplast dependency and also provide candidate mechanisms to explain dyskinetoplasty and/or drug resistance in the field.
Results
An RNAi Screen Implicates Acidic Compartments in Sensitizing T. brucei to ISM.
ISM is a veterinary phenanthridine related to ethidium bromide, and this fluorescent, DNA-binding drug accumulates in the kinetoplast of T. brucei (Fig. 1A). Genome-scale RNA interference (RNAi) library screens previously revealed mechanisms of resistance to anti-trypanosomal compounds used in humans (14). As illustrated in the schematic (Fig. 1B), we ran a massive parallel RNAi screen (15) using ISM at a dose that is toxic to unperturbed cells (4.5 nM). During screening under tetracycline induction, each T. brucei cell produces dsRNA from a random genomic DNA fragment, or RNAi target, and RIT-seq (RNAi target sequencing) is then used to generate a read-out from the drug-resistant population that emerges. Preliminary analysis (Fig. S1) implicated genes encoding V-ATPase subunits, an adaptin-3 (AP-3) subunit, and putative ER membrane complex (EMC) subunits in drug resistance following knockdown. High-throughput RIT-seq confirmed and extended this hit list; 16.7 million sequence reads, or 88% of all reads, mapped to the reference genome (Fig. 1 C and D and Table S1). Among 30 loci identified were those encoding possibly all 14 V-ATPase subunits, all 4 AP-3 subunits, and 5 putative EMC subunits (Table S1). Another locus encodes a V-ATPase assembly factor. These hits and their functional clustering indicate genome-scale coverage and represent a remarkable level of cross-validation for each of the three complexes identified. The remaining six hits include genes encoding two proteins previously implicated in nifurtimox and/or pentamidine mode of action (14) and three small (11, 14, and 16 kDa) proteins each with two predicted transmembrane domains (Table S1). We speculate that some of these factors are associated with the V-ATPase, AP-3, or EMC complexes or otherwise contribute to their function. Example RIT-seq profiles for four V-ATPase subunit hits and all four AP-3 subunit hits are shown in Fig. 1D.
Fig. 1.

An RNAi screen implicates acidic compartment defects in isometamidium resistance. (A) The ISM structure is shown on the left, indicating the coupling of homidium (ethidium, pink background) with the p-aminophenyl-diazonium portion of diminazine (berenil, green background). T. brucei stained with ISM (red) and DAPI (blue) are shown on the right. K, kinetoplast; N, nucleus. ISM quenches the DAPI signal that would otherwise also stain the kinetoplast. (B) The schematic illustrates the genome-scale RNAi-library screen. RNAi was induced with tetracycline. RIT-seq, RNA-interference target sequencing. (C) The genome-wide RIT-seq map indicates hits from the RNAi screen; loci identified by >100 sequence reads that contain the RNAi vector barcode. V-ATPase, AP-3, and EMC subunits are indicated in green, orange, and black, respectively. See Table S1 for the full list. RPKM, reads per kilobase per million reads mapped. (D) The Artemis screen-shots show example hits in green and orange; flanking protein coding sequences are indicated as black bars. Red peaks, forward reads with RNAi-construct barcodes; blue peaks, reverse reads with RNAi-construct barcodes; gray peaks, all other reads.
Fig. S1.

Low-throughput RIT-seq. PCR products representing RNAi target fragments derived from the ISM RNAi screen were separated on an agarose gel. Sanger sequencing revealed targets representing genes encoding subunits of the protein complexes indicated.
Table S1.
Hits from the ISM RNAi screen
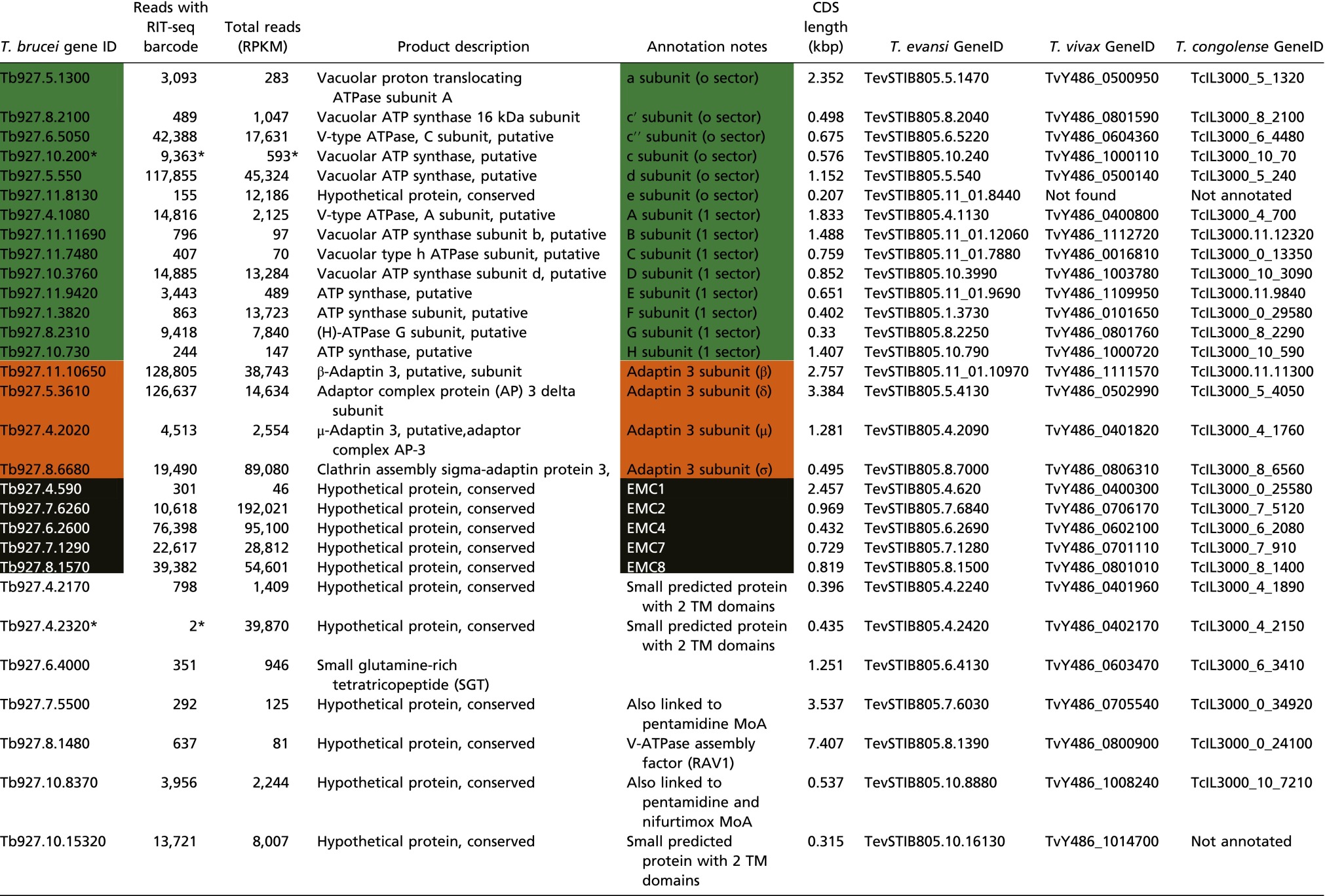 |
T. brucei CDSs with >100 reads containing the barcode found in the RNAi vector. MoA, mode of action; RPKM, reads per kilobase per million reads mapped. RIT-seq outputs: V-ATPase, green; AP-3, orange; EMC, black.
*Reads shown for Tb927.10.200 are within the Tb927.10.210 CDS but likely reflect knockdown Tb927.10.200 by targeting the untranslated region. Tb927.4.2320 was identified during a manual scan; the barcoded reads flank the CDS in this case. CDSs with reads simply because they are adjacent to hits (see Fig. 1D for examples) have been removed. Orthologous CDSs in Trypanosoma evansi, Trypanosoma vivax, and Trypanosoma congolense are also listed. Genes annotated as a second V-ATPase C subunit (Tb927.10.14040) and a putative EMC3 ortholog (Tb927.10.4760) did not register as hits.
Vacuolar ATPases are large, multisubunit rotary complexes that acidify intracellular organelles, contributing to macromolecular degradation and sorting, nutrient storage and mobilization, and pH and ion homeostasis (16). The Vo or membrane integral sector is responsible for proton transport from the cytoplasm to the lumen of intracellular compartments, comprising six subunits with a combined mass of ∼260 kDa in yeast and humans. The V1 sector is responsible for ATP hydrolysis and comprises eight subunits with a combined mass of ∼640 kDa in yeast and humans. Thus, we appear to have identified every T. brucei V-ATPase subunit in our screen (Table S1). In T. brucei, the V-ATPase is found in the lysosome and in acidocalcisomes (17); acidic, lysosome-related organelles found in cells from bacteria to humans and involved in phosphate and cation homeostasis. In the case of AP-3, we identified all four subunits. This adaptin mediates delivery of proteins to lysosome-related organelles in eukaryotes (18) and, in T. brucei, is localized to acidocalcisomes, Golgi, and late endosomes (19). We also identified five subunits of the conserved ER membrane complex or EMC, which plays a role in phospholipid transfer from the ER to mitochondria in yeast (20), but has not been characterized in T. brucei. Taken together, the outputs from our RNAi screen suggest an unexpected role for acidic compartments in sensitizing T. brucei to ISM.
V-ATPase Function Sensitizes T. brucei to ISM: Genetic and Chemical Validation.
We sought to validate the role of the V-ATPase in sensitizing T. brucei to ISM. We established a pair of independent V-ATPase subunit (Vo-c″: Tb927.6.5050) RNAi-based knockdown strains and demonstrated, as expected for an essential function, a severe growth defect associated with depletion of this V-ATPase subunit (Fig. 2A). It is likely that knockdown was incomplete in the cells selected for ISM resistance in our RNAi screen. Relatively low-efficiency knockdown is possible because the RNAi library comprises random-sheared DNA fragments that produce intermolecular dsRNA (21). We, therefore, used a low concentration of tetracycline (2 ng/mL) to produce intramolecular dsRNA and only a minor growth defect (Fig. 2A). To facilitate monitoring of V-ATPase depletion, we expressed an N-terminal MYC epitope-tagged Vo-c″ subunit from its native locus and observed effective V-ATPase subunit depletion using 2 ng/mL tetracycline (Fig. 2B); the Western blot was likely insufficiently sensitive to detect low-level expression in this particular case. We used these conditions to assess the impact of the V-ATPase on drug sensitivity. Both T. brucei strains with the V-ATPase subunit depleted displayed more than a 100-fold increase in the half-maximal effective concentration (EC50) for ISM, indicating dramatically increased resistance (Fig. 2C and Fig. S2A). These results were also confirmed by depletion of a second V-ATPase subunit (Vo-d: Tb927.5.550); once again, two independent strains displayed more than a 100-fold increased EC50 for ISM (Fig. S3 A–C).
Fig. 2.

V-ATPase function sensitizes cells to isometamidium. (A) Cumulative growth of V-ATPase subunit (Vo-c″: Tb927.6.5050) RNAi strains. Error bars indicate SD calculated from two independent strains. (B) Western blot analysis showing depletion of the V-ATPase c″ subunit that was MYC tagged at the native locus. The Coomassie panel serves as a loading control. Tetracycline was applied at 2 ng/mL. (C) Dose–response curves for ISM in V-ATPase RNAi strains. EC50 values are indicated. Error bars indicate SD from two experiments carried out in triplicate. No RNAi refers to the uninduced (−Tet) control. (D) Isobologram analysis with ISM and the V-ATPase inhibitor bafilomycin. The red dashed line indicates a simple additive relationship. Data points above this line indicate antagonism between the two compounds, which was calculated using data from three experiments. FIC50, 50% fractional inhibitory concentration.
Fig. S2.

EC50 analysis datasets derived using pairs of independent strains. (A) EC50 data for ISM before and after RNAi-based depletion of the V-ATPase, AP-3β, or EMC2 subunits. (B) EC50 data for acriflavine before and after RNAi-based depletion of the V-ATPase subunits. (C) EC50 data for oligomycin before and after RNAi-based depletion of the V-ATPase subunits. (D) EC50 data for sodium azide before and after RNAi-based depletion of the V-ATPase subunits. All error bars indicate SD derived from two independent strains.
Fig. S3.

A second V-ATPase subunit sensitizes cells to isometamidium. (A) Cumulative growth of V-ATPase RNAi strains for the Vo-d subunit (Tb927.5.550). Error bars indicate SD calculated from two independent strains. (B) Western blot analysis showing depletion of the MYC-tagged d-subunit. The Coomassie panel serves as a loading control. Tetracycline was applied at 2 ng/mL. (C) RNAi-based depletion of the d-subunit renders cells >100-fold resistant to ISM. Error bars indicate SD derived from two independent strains. (D) Flow cytometry analysis of ISM staining following d subunit RNAi. (E) Microscopy and flow cytometry analysis of Mitotracker staining following d-subunit RNAi. (Scale bars, 10 μm.) The magnified image shows the expected thread-like structure. (F) Cumulative growth of RNAi strains after exposure to acriflavine. Error bars indicate SD calculated from two independent strains.
We next used an orthogonal chemical approach to assess the role of V-ATPase function in sensitizing T. brucei to ISM. Bafilomycin is a specific inhibitor of the V-ATPase that binds to the c subunit of Vo (22). As predicted, and as demonstrated using isobologram analysis, bafilomycin strongly antagonizes ISM action (Fig. 2D). Thus, we obtained both genetic and chemical validation for identification of the V-ATPase in our RNAi screen and concluded that V-ATPase function sensitizes T. brucei to ISM.
AP-3 and the EMC Sensitize T. brucei to ISM.
We now sought to validate the roles of AP-3 and the EMC in sensitizing T. brucei to ISM. We established two independent RNAi-based knockdown strains for an AP-3 subunit (AP-3β: Tb927.11.10650) and two independent strains for an EMC subunit (EMC2: Tb927.7.6260) and assessed growth following induction with tetracycline. The AP-3 strains induced with 1 μg/mL tetracycline displayed a minor growth defect (Fig. 3A), whereas the EMC strains displayed a severe growth defect (Fig. 3B). As above, we used a low concentration of tetracycline (1 ng/mL), which induced only a minor growth defect (Fig. 3B), to assess the impact of the EMC on drug sensitivity. The T. brucei RNAi strains in which AP-3 (Fig. 3C and Fig. S2A) or EMC (Fig. 3D and Fig. S2A) subunits were targeted for knockdown were more than 100-fold resistant to ISM. Thus, we obtained genetic validation for identification of AP-3 and the EMC in our RNAi screen and conclude that these two additional complexes also sensitize T. brucei to ISM.
Fig. 3.

Adaptin-3 and the EMC sensitize cells to isometamidium. (A) Cumulative growth of AP-3 RNAi strains (AP-3β subunit: Tb927.11.10650). Error bars indicate SD calculated from two independent strains. (B) Cumulative growth of EMC RNAi strains (EMC2 subunit: Tb927.7.6260). Error bars as in A. (C) Dose–response curves for ISM in AP-3 RNAi strains. EC50 values are indicated. Error bars indicate SD calculated from two experiments carried out in triplicate. (D) Dose–response curves for ISM in EMC RNAi strains. Other details as in C. (E) Cells were stained with an acidocalcisome marker, α-VP1 (green), and counterstained with DAPI (blue). (Scale bars, 20 μm.) (C–E) RNAi strains were preinduced for 72 h in 2 ng/mL, 1 μg/mL, or 1 ng/mL tetracycline for the V-ATPase, AP-3, or EMC subunits, respectively.
Because our RNAi screen suggested a role for acidic compartments in sensitizing T. brucei to ISM, we examined the role of the above complexes in acidocalcisome biogenesis. Cells were depleted for a subunit of each complex and then stained with an antibody that recognizes VP1, an acidocalcisomal proton pyrophosphatase (23). Analysis of these cells confirmed a role for AP-3 in acidocalcisome biogenesis (19) but revealed no apparent perturbation in acidocalcisome biogenesis following RNAi against either V-ATPase or EMC subunits (Fig. 3E). These results indicate that V-ATPase and EMC depletion induce resistance to ISM without disrupting acidocalcisomes biogenesis.
ATP Synthase Uncoupling and Dyskinetoplasty in Cells with Acidic Compartment-Associated Defects.
Dyskinetoplastic trypanosomes (24), and T. brucei with ATP synthase γ-subunit mutations (5), display resistance to phenanthridines, including ISM and ethidium bromide, and nonintercalating diamidines, including pentamidine and berenil. These drugs selectively cleave kinetoplast DNA (25). To explore cross-resistance in our ISM-resistant strains, we determined EC50 values for these drugs. We observed cross-resistance in every case examined; to berenil following V-ATPase, AP-3, or EMC subunit depletion (Fig. S4A), to ethidium bromide following V-ATPase subunit depletion (Fig. S4B), and moderately to pentamidine following V-ATPase subunit depletion (Fig. S4C). Thus, disruption of acidic compartments renders T. brucei resistant to ISM, diminazine aceturate, ethidium bromide, and, to a lesser extent, pentamidine.
Fig. S4.

Acidic compartment defects induce cross-resistance to other kinetoplast-binding drugs. (A) RNAi-based depletion of the V-ATPase c″-, d-, AP-3β, or EMC2 subunits renders cells resistant to diminazene aceturate (berenil). (B) RNAi-based depletion of the V-ATPase c″ or d subunits renders cells resistant to ethidium bromide (EtBr). (Inset) Trypanosome stained with EtBr (red). N, nucleus; K, kinetoplast. (C) RNAi-based depletion of the V-ATPase c″ or d subunits renders cells moderately resistant to pentamidine. All error bars indicate SD derived from two independent strains.
We considered mechanisms by which V-ATPase, AP-3, or the EMC could sensitize cells to phenanthridines and diamidines. These complexes could facilitate endocytosis, as demonstrated for the human V-ATPase (26), or antitrypanosomal lytic action, as described for the T. brucei V-ATPase, which sensitizes cells to human serum trypanolytic apolipoprotein L1 (27). We exploited ISM fluorescence (Fig. 1A) to assess drug uptake following V-ATPase subunit depletion and repeatedly found no evidence for diminished accumulation relative to controls, using either microscopy or cytometry (Fig. 4A). Mitotracker staining also appeared to be unchanged in these cells, suggesting that ΔΨmito was not substantially perturbed (Fig. 4B). Drug uptake (Fig. S3D) and ΔΨmito (Fig. S3E) were also apparently unchanged following depletion of a second V-ATPase subunit.
Fig. 4.

ATP synthase uncoupling and dyskinetoplasty in cells with acidic compartment defects. (A) Flow cytometry and microscopy analysis of ISM staining following V-ATPase c″ subunit RNAi. The smaller panel indicates the ISM-induced shift in fluorescence. (B) Microscopy and flow cytometry analysis of staining with the cationic fluorophore, Mitotracker following V ATPase c″ subunit RNAi. The depolarizing agent, CCCP, prevents Mitotracker accumulation and serves as a control for loss of ΔΨmito. (Scale bars, 10 μm.) (C) Dose–response curves for acriflavine in V-ATPase c″ subunit RNAi strains. Error bars indicate SD from two experiments carried out in triplicate. (D) Proportions of cells that retain detectable DAPI-stained kinetoplasts following acriflavine exposure. The RNAi strains tested are indicated. Control cells are WT, untreated. Error bars represent SD calculated from two independent strains. (Inset) Two dyskinetoplastic trypanosomes. N, nucleus. (E) Cumulative growth of V-ATPase RNAi strains (Vo-c″ subunit) after exposure to acriflavine. Error bars indicate SD calculated from two independent strains. (F) Dose–response curves for oligomycin and sodium azide in V-ATPase RNAi strains (Vo-c″ subunit). Error bars as in C.
We next considered whether the V-ATPase, AP-3, or EMC complexes might sensitize cells to drugs by maintaining kinetoplast dependency. To test this hypothesis, we depleted complex subunits and exposed cells to acriflavine, an intercalating drug derived from acridine. Developed by Paul Ehrlich in the early 20th century and previously used against human trypanosomiasis, acriflavine induces kinetoplast loss (28) and was also used to generate the original petite mutant yeast colonies (29). T. brucei depleted for V-ATPase subunits displayed resistance to acriflavine (Fig. 4C and Fig. S2B), as did T. brucei depleted for AP-3 or EMC subunits, and, remarkably, populations lacking detectable kinetoplast-DNA were readily generated following acriflavine exposure (Fig. 4D). Kinetoplasts were undetectable in >90% of cells depleted for any one of the four subunits tested and were notably smaller in the remainder, whereas they were readily detectable in >99% of untreated WT cells. As expected, tetracycline removal following acriflavine exposure resulted in cell death (Fig. 4E and Fig. S3F). This observation indicated that cells remained kinetoplast independent while knockdown persisted and that kinetoplast dependency returned when V-ATPase subunit expression was restored.
The results above indicate that V-ATPase, AP-3, or EMC depletion allows T. brucei to tolerate dyskinetoplasty. ATP synthase γ-subunit mutations support dyskinetoplasty by compensating for loss of the kinetoplast-encoded Fo-A6 subunit (6); the mutations uncouple the F1 and Fo sectors, allowing the F1 ATPase to operate independently (12). Oligomycin inhibits the coupled ATPase by binding the Fo (the o indicates oligomycin sensitivity) sector (30). Indeed, the T. brucei F1 ATPase was shown to be oligomycin insensitive when separated from the Fo component (31), and oligomycin resistance was observed almost 40 y ago in dyskinetoplastic trypanosomes that lost tsetse transmissibility (32). We therefore derived EC50 values for oligomycin in pairs of independent strains depleted for two distinct V-ATPase subunits. V-ATPase depletion increased oligomycin resistance more than threefold in every case (Fig. 4F and Fig. S2C). In contrast, the EC50 for sodium azide, an inhibitor of the F1-ATPase (33), was unchanged following V-ATPase depletion (Fig. 4F and Fig. S2D). These results suggest that V-ATPase activity maintains F1Fo-ATPase sector coupling in T. brucei. We propose that reduced V-ATPase activity allows growth that is independent of both the Fo component of ATP synthase and the kinetoplast. Indeed, the kinetoplast is dispensable when V-ATPase, AP-3, or EMC activity is reduced, and these cells are resistant to kinetoplast-targeting drugs.
Discussion
We used an RNAi library screen for ISM resistance in trypanosomes to explore drug mode of action, drug resistance mechanisms, and kinetoplast biology. The screen yielded 30 hits, 23 of which represented three multisubunit protein complexes. Roles for these V-ATPase, AP-3, and EMC complexes were all validated in multidrug sensitization. The mechanism of drug resistance in subunit-depleted cells was not reduced drug uptake but was rather altered ATP synthase function that, in turn, rendered the kinetoplast obsolete. We thereby unearthed three complexes that, on depletion, appear to allow Fo-independent growth. These findings reveal an unexpected link between V-ATPase function, mitochondrial ATP synthase function, and kinetoplast dependency.
Dyskinetoplastic T. evansi and T. equiperdum are essentially “petite” mutants of T. brucei (10) with a γ-subunit mutation that uncouples the ATP synthase (12). This uncoupling renders the Fo component obsolete and allows a soluble F1 component to sustain electrogenic exchange and ΔΨmito, likely in concert with an ADP3−/ATP4− carrier (6). Because the Fo-A6 subunit is ultimately the only essential protein encoded by the kinetoplast in bloodstream-form cells, the kinetoplast is also rendered obsolete by these γ-subunit mutations. Our results indicate that depletion of V-ATPase, AP-3, or EMC functions have the same effect as the previously described γ-subunit mutations. It should be noted here that the obsolete kinetoplast is readily lost, when its replication is disrupted for example, but it can be retained in its obsolete form. Thus, the presence or absence of a kinetoplast does not necessarily correlate with drug resistance associated with γ-subunit mutations or V-ATPase, AP-3, or EMC depletion. As pointed out recently (5), this may have hampered past attempts to correlate dyskinetoplasty with drug resistance (34).
It is tempting to conclude that phenanthridines and diamidines, especially the intercalating drugs, kill trypanosomatids primarily by targeting the kinetoplast, but they may have additional targets. These drugs selectively cleave kinetoplast DNA (25), and ethidium bromide also targets nuclear DNA (35, 36). In addition, pentamidine disrupts ΔΨmito (37), and this may be the basis of pentamidine sensitivity, demonstrated by us and by others (5), in T. brucei with dispensable kinetoplasts. Ultimately, ISM and diminazene aceturate resistance are of major concern (38), and some resistant field isolates appear to depend on adaptations that remain to be identified (6). Our results now show that V-ATPase, AP-3, or EMC mutations that reduce activity without substantially reducing viability could potentially contribute to natural dyskinetoplasty and/or drug resistance. As in the case of γ-subunit mutations in T. evansi and T. equiperdum (6, 10), other mechanisms involving ATP synthase uncoupling would also be expected to block normal tsetse-mediated transmission. Failure to infect tsetse is expected because energy production through oxidative phosphorylation, which normally operates in tsetse fly stages, would no longer be possible. Thus, even if these resistance mechanisms arise frequently, the parasites that harbor them should concomitantly become tsetse transmission incompetent, subsequently requiring mechanical or sexual transmission.
The vacuolar ATPase and the mitochondrial ATPase are structurally and mechanistically related multisubunit rotary motors (39) that, in bloodstream-form T. brucei, both use ATP to generate proton gradients (8). Control of one by the other, as suggested by our results, may reflect unanticipated inter-ATPase communication. Indeed, V-ATPase function has been linked to Wnt (40) and Notch (41) signaling and to nutrient-responsive signaling through pH control (42). Nutrient-responsive signaling has also been linked to mitochondrial DNA damage (43). In this context, it is worth considering evidence for V-ATPase to mitochondria communication in other eukaryotes. In yeast, V-ATPase mutations have an impact on petite phenotypes (44, 45), and V-ATPase function has also been linked to cellular aging and rejuvenation by controlling mitochondrial function (46). In human cells, vesicular transport operates between lysosomes and mitochondria (47). Indeed, there is growing evidence supporting roles for lysosomes in signaling and energy metabolism beyond their established roles in degradation and recycling (48). Interactions between the ER and mitochondria are also important for lipid exchange, as well as Ca2+ and energy homeostasis (49). Consistent with these findings, our results indicate that adaptin-3 and the ER membrane complex also control ATP synthase function in T. brucei, and we suggest that these complexes mediate inter-ATPase communication as a means to coordinate organelle function. Thus, regulated V-ATPase coupling, as described in other eukaryotes (50), could be coordinated with ATP synthase coupling, and this could operate via pH, Ca2+, lipid, or amino acid signaling pathways.
Previously described petite mutants of T. brucei have γ-subunit mutations associated with mitochondrial ATP synthase uncoupling and kinetoplast-independent growth (6, 10, 12). We now describe 30 non-mitochondrial proteins and 3 multisubunit complexes that appear to have the same effect when depleted. Our results reveal an unanticipated link between vacuolar ATPase function and mitochondrial ATP synthase function, which we speculate may reflect a conserved sensing mechanism involved in metabolic control. Our results also provide candidate mechanisms to explain cases of dyskinetoplasty and/or drug resistance in a group of important parasites.
Materials and Methods
Trypanosomes.
Bloodstream-form T. brucei, Lister 427, MiTat 1.2, clone 221a, and derivatives, including 2T1 cells (51), were transfected using a Nucleofector (Lonza) with cytomix for stem-loop RNAi constructs or T-cell Nucleofector solution for native locus tagging constructs. Transformants were cloned by limiting dilution and selected with blasticidin (10 μg/mL) or hygromycin (2.5 μg/mL) for constructs integrated at the tagged rDNA locus (51). Cumulative growth curves were generated from cultures seeded at 105 cells/mL, counted on a hemocytometer every 24 h, and diluted as necessary. Where induction of stem-loop RNAi with 1 μg/mL tetracycline was lethal, growth was analyzed with titrated tetracycline. For assays, stem-loop RNAi strains were preinduced for 72 h in 2 ng/mL, 1 μg/mL, or 1 ng/mL tetracycline for the V-ATPase, AP-3, or EMC subunits, respectively. EC50 assays were carried out using alamarBlue as previously described (52). Isobologram analysis was carried out using a checkerboard setup as previously described (53). ISM (Merial) was a gift from Graham Skellern, University of Strathclyde, UK. Bafilomycin, diminazene aceturate, ethidium bromide, acriflavine, and sodium azide were from Sigma. Pentamidine (Sanofi SA) was a gift from Vanessa Yardley, London School of Hygiene & Tropical Medicine, UK.
RNAi Screening and Data Analysis.
Determinants of ISM resistance were identified using an RNAi library screen as previously described (15). Briefly, the RNAi screen was carried out with 4.5 nM ISM. Cultures were split and supplemented with fresh drug as required, and DNA was extracted from drug-resistant cells on day 7. RNAi target fragments were amplified by PCR using the LIB2f and LIB2r primers and individually Sanger sequenced. The pooled products were then subjected to high-throughput RIT-seq. The PCR products were fragmented to ∼300 bp, and sequence libraries were prepared using standard protocols and kits from Thermo Scientific Ion Torrent. The libraries were sequenced on the Ion Torrent Proton platform using the 200-bp sequencing kit. Reads were mapped to the T. brucei 927 reference genome (v6, tritrypdb.org) with Bowtie 2 (54) using the following parameters: very-sensitive-local–phred33. Alignment files were manipulated with SAMtools (55) and a custom script (15), and data were further assessed using the Artemis genome browser (56).
Plasmid Construction.
Gene-specific RNAi fragments of 400–600 bp were amplified using PCR primers designed using RNAit (57) and cloned into pRPaiSL for the generation of stem-loop dsRNA as the trigger for RNAi (51). For epitope tagging of V-ATPase subunits at native loci, N-terminal fragments were amplified and cloned into pNATTAGx (51). Oligonucleotide sequences are available on request.
Protein Analysis.
Protein samples were stored in the presence of a protease inhibitor mixture (Roche) and were not boiled. Whole-cell lysates were separated by SDS/PAGE and immuno-blotted using an enhanced chemi-luminescent kit (GE Healthcare) according to the manufacturer’s instructions. MYC was detected on Western blots using polyclonal rabbit α-MYC (Europa; 1:2,000).
Microscopy and Flow Cytometry-Based Assays.
Immunofluorescence microscopy was carried out according to standard protocols. MYC was detected for fluorescence microscopy using polyclonal rabbit α-MYC (Europa; 1:2,000). Acidicalcisomes were detected in PFA-fixed cells using rabbit α-VP1 (1:300) primary antibody (23). Cells were typically mounted in Vectashield (Vector Laboratories) containing the DNA counterstain DAPI. Images were captured using a Nikon Eclipse E600 epifluorescence microscope in conjunction with a Coolsnap FX (Photometrics) CCD camera and processed in Metamorph 5.0 (Photometrics). ISM uptake was assessed using microscopy or flow cytometry. Briefly, 5 mL of a culture at ∼1 × 106 cells/mL was incubated for 60 min in medium containing 500 nM ISM; 1 mL was transferred to a 1.5-mL tube and spun at 4,500 × g for 1 min. Supernatant was removed, and the cells were washed in 1 mL PBS, spun a second time, and resuspended in fresh PBS; 300 μL was used for flow cytometry and 500 μL for microscopy. One microliter 1 mM MitotrackerRed CMXROS (Invitrogen) was added to 10-mL cultures and incubated at 37 °C for 5 min. Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) was added to cells for 1 h before analysis. These cells were either fixed in a final concentration of 2% (vol/vol) PFA for examination by microscopy or in a final concentration of 70% (vol/vol) methanol overnight for examination by flow cytrometry. For flow cytrometry, 700 μL of glacial methanol was added to each tube, which was mixed gently, incubated on ice for 10 min, and stored at 4 °C overnight. Samples were then spun at 2,000 × g at 4 °C for 10 min, washed in 1 mL PBS, and resuspended in 500 μL on ice; analysis was carried out within 2 h. The fluorescence from 30,000 events was detected using settings for the FL-2, PE channel (R- Phycoerythrin) from the 575/25 optical filter. Data were gated and analyzed as histograms. Acriflavine was added to cells at 20 nM for a further 72 h after 72 h of tetracycline induction. DAPI staining was then used to assess the presence of kinetoplasts. Cells were stained with ethidium bromide at 5 μM in a 10-mL culture at ∼1 × 106 cells/mL for 60 min. Cells were then washed in medium and fixed in 2% (vol/vol) PFA for microscopy.
Acknowledgments
We thank Graham Skellern (University of Strathclyde) for ISM, James Morris (Clemson University) and Paul Englund (Johns Hopkins University) for the RNAi plasmid library, Bernard Dujon (Pasteur Institute) for the I-SceI gene, Vanessa Yardley (London School of Hygiene & Tropical Medicine) for pentamidine, Norbert Bakalara (INSERM) and Lucia Guther (Dundee) for αVP1, and Achim Schnaufer for advice on F1Fo uncoupling. This work was supported by Senior Investigator Award 100320/Z/12/Z (to D.H.), Project Grant 093010/Z/10/Z (to D.H.), Strategic Award 083481/Z/07/Z (to the Division of Biological Chemistry & Drug Discovery, University of Dundee), and Core Grant 085349 (to M.P.B.; as part of the Wellcome Trust Centre for Molecular Parasitology and Glasgow Polyomics), all from The Wellcome Trust.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The RIT-seq sequence data have been deposited in the European Nucleotide Archive, www.ebi.ac.uk/ena (accession no. PRJEB8850/ERP009891).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505411112/-/DCSupplemental.
References
- 1.Pedrique B, et al. The drug and vaccine landscape for neglected diseases (2000-11): A systematic assessment. Lancet Glob Health. 2013;1(6):e371–e379. doi: 10.1016/S2214-109X(13)70078-0. [DOI] [PubMed] [Google Scholar]
- 2.Carnes J, et al. Genome and phylogenetic analyses of Trypanosoma evansi reveal extensive similarity to T. brucei and multiple independent origins for dyskinetoplasty. PLoS Negl Trop Dis. 2015;9(1):e3404. doi: 10.1371/journal.pntd.0003404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen RE, Englund PT. Network news: The replication of kinetoplast DNA. Annu Rev Microbiol. 2012;66:473–491. doi: 10.1146/annurev-micro-092611-150057. [DOI] [PubMed] [Google Scholar]
- 4.Schnaufer A, Domingo GJ, Stuart K. Natural and induced dyskinetoplastic trypanosomatids: How to live without mitochondrial DNA. Int J Parasitol. 2002;32(9):1071–1084. doi: 10.1016/s0020-7519(02)00020-6. [DOI] [PubMed] [Google Scholar]
- 5.Gould MK, Schnaufer A. Independence from kinetoplast DNA maintenance and expression is associated with multidrug resistance in Trypanosoma brucei in vitro. Antimicrob Agents Chemother. 2014;58(5):2925–2928. doi: 10.1128/AAC.00122-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dean S, Gould MK, Dewar CE, Schnaufer AC. Single point mutations in ATP synthase compensate for mitochondrial genome loss in trypanosomes. Proc Natl Acad Sci USA. 2013;110(36):14741–14746. doi: 10.1073/pnas.1305404110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nolan DP, Voorheis HP. The mitochondrion in bloodstream forms of Trypanosoma brucei is energized by the electrogenic pumping of protons catalysed by the F1F0-ATPase. Eur J Biochem. 1992;209(1):207–216. doi: 10.1111/j.1432-1033.1992.tb17278.x. [DOI] [PubMed] [Google Scholar]
- 8.Schnaufer A, Clark-Walker GD, Steinberg AG, Stuart K. The F1-ATP synthase complex in bloodstream stage trypanosomes has an unusual and essential function. EMBO J. 2005;24(23):4029–4040. doi: 10.1038/sj.emboj.7600862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vercesi AE, Docampo R, Moreno SN. Energization-dependent Ca2+ accumulation in Trypanosoma brucei bloodstream and procyclic trypomastigotes mitochondria. Mol Biochem Parasitol. 1992;56(2):251–257. doi: 10.1016/0166-6851(92)90174-i. [DOI] [PubMed] [Google Scholar]
- 10.Lai DH, Hashimi H, Lun ZR, Ayala FJ, Lukes J. Adaptations of Trypanosoma brucei to gradual loss of kinetoplast DNA: Trypanosoma equiperdum and Trypanosoma evansi are petite mutants of T. brucei. Proc Natl Acad Sci USA. 2008;105(6):1999–2004. doi: 10.1073/pnas.0711799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen XJ, Clark-Walker GD. The petite mutation in yeasts: 50 years on. Int Rev Cytol. 2000;194:197–238. doi: 10.1016/s0074-7696(08)62397-9. [DOI] [PubMed] [Google Scholar]
- 12.Šubrtová K, Panicucci B, Zíková A. ATPaseTb2, a unique membrane-bound FoF1-ATPase component, is essential in bloodstream and dyskinetoplastic trypanosomes. PLoS Pathog. 2015;11(2):e1004660. doi: 10.1371/journal.ppat.1004660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mäser P, et al. Antiparasitic agents: New drugs on the horizon. Curr Opin Pharmacol. 2012;12(5):562–566. doi: 10.1016/j.coph.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Alsford S, et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature. 2012;482(7384):232–236. doi: 10.1038/nature10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glover L, et al. Genome-scale RNAi screens for high-throughput phenotyping in bloodstream-form African trypanosomes. Nat Protoc. 2015;10(1):106–133. doi: 10.1038/nprot.2015.005. [DOI] [PubMed] [Google Scholar]
- 16.Forgac M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8(11):917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 17.Huang G, et al. Proteomic analysis of the acidocalcisome, an organelle conserved from bacteria to human cells. PLoS Pathog. 2014;10(12):e1004555. doi: 10.1371/journal.ppat.1004555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dell’Angelica EC. AP-3-dependent trafficking and disease: The first decade. Curr Opin Cell Biol. 2009;21(4):552–559. doi: 10.1016/j.ceb.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Huang G, et al. Adaptor protein-3 (AP-3) complex mediates the biogenesis of acidocalcisomes and is essential for growth and virulence of Trypanosoma brucei. J Biol Chem. 2011;286(42):36619–36630. doi: 10.1074/jbc.M111.284661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lahiri S, et al. A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 2014;12(10):e1001969. doi: 10.1371/journal.pbio.1001969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durand-Dubief M, Kohl L, Bastin P. Efficiency and specificity of RNA interference generated by intra- and intermolecular double stranded RNA in Trypanosoma brucei. Mol Biochem Parasitol. 2003;129(1):11–21. doi: 10.1016/s0166-6851(03)00071-9. [DOI] [PubMed] [Google Scholar]
- 22.Bowman BJ, McCall ME, Baertsch R, Bowman EJ. A model for the proteolipid ring and bafilomycin/concanamycin-binding site in the vacuolar ATPase of Neurospora crassa. J Biol Chem. 2006;281(42):31885–31893. doi: 10.1074/jbc.M605532200. [DOI] [PubMed] [Google Scholar]
- 23.Lemercier G, et al. A vacuolar-type H+-pyrophosphatase governs maintenance of functional acidocalcisomes and growth of the insect and mammalian forms of Trypanosoma brucei. J Biol Chem. 2002;277(40):37369–37376. doi: 10.1074/jbc.M204744200. [DOI] [PubMed] [Google Scholar]
- 24.Agbe A, Yielding KL. Kinetoplasts play an important role in the drug responses of Trypanosoma brucei. J Parasitol. 1995;81(6):968–973. [PubMed] [Google Scholar]
- 25.Shapiro TA, Englund PT. Selective cleavage of kinetoplast DNA minicircles promoted by antitrypanosomal drugs. Proc Natl Acad Sci USA. 1990;87(3):950–954. doi: 10.1073/pnas.87.3.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kozik P, et al. A human genome-wide screen for regulators of clathrin-coated vesicle formation reveals an unexpected role for the V-ATPase. Nat Cell Biol. 2013;15(1):50–60. doi: 10.1038/ncb2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lecordier L, et al. Identification of Trypanosoma brucei components involved in trypanolysis by normal human serum. Mol Microbiol. 2014;94(3):625–636. doi: 10.1111/mmi.12783. [DOI] [PubMed] [Google Scholar]
- 28.Riou GF, Belnat P, Benard J. Complete loss of kinetoplast DNA sequences induced by ethidium bromide or by acriflavine in Trypanosoma equiperdum. J Biol Chem. 1980;255(11):5141–5144. [PubMed] [Google Scholar]
- 29.Avers CJ, Pfeffer CR, Rancourt MW. Acriflavine induction of different kinds of “petite” mitochondrial populations in Saccharomyces cerevisiae. J Bacteriol. 1965;90:481–494. doi: 10.1128/jb.90.2.481-494.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyer PD. The ATP synthase--a splendid molecular machine. Annu Rev Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 31.Williams N, Frank PH. The mitochondrial ATP synthase of Trypanosoma brucei: Isolation and characterization of the intact F1 moiety. Mol Biochem Parasitol. 1990;43(1):125–132. doi: 10.1016/0166-6851(90)90137-b. [DOI] [PubMed] [Google Scholar]
- 32.Opperdoes FR, Borst P, de Rijke D. Oligomycin sensitivity of the mitochondrial ATPase as a marker for fly transmissability and the presence of functional kinetoplast DNA in African trypanosomes. Comp Biochem Physiol B. 1976;55(1):25–30. doi: 10.1016/0305-0491(76)90167-x. [DOI] [PubMed] [Google Scholar]
- 33.Bowler MW, Montgomery MG, Leslie AG, Walker JE. How azide inhibits ATP hydrolysis by the F-ATPases. Proc Natl Acad Sci USA. 2006;103(23):8646–8649. doi: 10.1073/pnas.0602915103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaminsky R, Schmid C, Lun ZR. Susceptibility of dyskinetoplastic Trypanosoma evansi and T. equiperdum to isometamidium chloride. Parasitol Res. 1997;83(8):816–818. doi: 10.1007/s004360050346. [DOI] [PubMed] [Google Scholar]
- 35.Glover L, Horn D. Trypanosomal histone γH2A and the DNA damage response. Mol Biochem Parasitol. 2012;183(1):78–83. doi: 10.1016/j.molbiopara.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy Chowdhury A, et al. The killing of African trypanosomes by ethidium bromide. PLoS Pathog. 2010;6(12):e1001226. doi: 10.1371/journal.ppat.1001226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lanteri CA, Tidwell RR, Meshnick SR. The mitochondrion is a site of trypanocidal action of the aromatic diamidine DB75 in bloodstream forms of Trypanosoma brucei. Antimicrob Agents Chemother. 2008;52(3):875–882. doi: 10.1128/AAC.00642-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delespaux V, Geysen D, Van den Bossche P, Geerts S. Molecular tools for the rapid detection of drug resistance in animal trypanosomes. Trends Parasitol. 2008;24(5):236–242. doi: 10.1016/j.pt.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Stewart AG, Laming EM, Sobti M, Stock D. Rotary ATPases--dynamic molecular machines. Curr Opin Struct Biol. 2014;25:40–48. doi: 10.1016/j.sbi.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 40.Cruciat CM, et al. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010;327(5964):459–463. doi: 10.1126/science.1179802. [DOI] [PubMed] [Google Scholar]
- 41.Yan Y, Denef N, Schüpbach T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev Cell. 2009;17(3):387–402. doi: 10.1016/j.devcel.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dechant R, et al. Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. 2010;29(15):2515–2526. doi: 10.1038/emboj.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garipler G, Mutlu N, Lack NA, Dunn CD. Deletion of conserved protein phosphatases reverses defects associated with mitochondrial DNA damage in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2014;111(4):1473–1478. doi: 10.1073/pnas.1312399111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garipler G, Dunn CD. Defects associated with mitochondrial DNA damage can be mitigated by increased vacuolar pH in Saccharomyces cerevisiae. Genetics. 2013;194(1):285–290. doi: 10.1534/genetics.113.149708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merz S, Westermann B. Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol. 2009;10(9):R95. doi: 10.1186/gb-2009-10-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012;492(7428):261–265. doi: 10.1038/nature11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soubannier V, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22(2):135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 48.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kornmann B. The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol. 2013;25(4):443–448. doi: 10.1016/j.ceb.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 50.Parra KJ, Chan CY, Chen J. Saccharomyces cerevisiae vacuolar H+-ATPase regulation by disassembly and reassembly: One structure and multiple signals. Eukaryot Cell. 2014;13(6):706–714. doi: 10.1128/EC.00050-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alsford S, Horn D. Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol Biochem Parasitol. 2008;161(1):76–79. doi: 10.1016/j.molbiopara.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker N, Alsford S, Horn D. Genome-wide RNAi screens in African trypanosomes identify the nifurtimox activator NTR and the eflornithine transporter AAT6. Mol Biochem Parasitol. 2011;176(1):55–57. doi: 10.1016/j.molbiopara.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh PK, Tack BF, McCray PB, Jr, Welsh MJ. Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am J Physiol Lung Cell Mol Physiol. 2000;279(5):L799–L805. doi: 10.1152/ajplung.2000.279.5.L799. [DOI] [PubMed] [Google Scholar]
- 54.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, et al. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28(4):464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Redmond S, Vadivelu J, Field MC. RNAit: An automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol Biochem Parasitol. 2003;128(1):115–118. doi: 10.1016/s0166-6851(03)00045-8. [DOI] [PubMed] [Google Scholar]