

Significance
A rapidly growing number of cryptic natural product biosynthetic gene clusters have been identified in bacterial DNA sequencing datasets. The metabolites encoded by most of these gene clusters remain uncharacterized because they are not readily activated using monoculture fermentation methods. The development of generic gene cluster activation strategies is needed to access molecules encoded by this rapidly growing collection of sequenced gene clusters. The promoter engineering platform outlined here provides a simple, cost-effective, and potentially scalable tool for the characterization of molecules encoded by gene clusters found in sequenced microbial (meta)genomes. We believe that this gene cluster activation platform will accelerate the discovery of biomedically relevant metabolites using (meta)genomics-driven natural products discovery methods.
Keywords: promoter engineering, indolotryptoline, environmental DNA
Abstract
Large-scale sequencing of prokaryotic (meta)genomic DNA suggests that most bacterial natural product gene clusters are not expressed under common laboratory culture conditions. Silent gene clusters represent a promising resource for natural product discovery and the development of a new generation of therapeutics. Unfortunately, the characterization of molecules encoded by these clusters is hampered owing to our inability to express these gene clusters in the laboratory. To address this bottleneck, we have developed a promoter-engineering platform to transcriptionally activate silent gene clusters in a model heterologous host. Our approach uses yeast homologous recombination, an auxotrophy complementation-based yeast selection system and sequence orthogonal promoter cassettes to exchange all native promoters in silent gene clusters with constitutively active promoters. As part of this platform, we constructed and validated a set of bidirectional promoter cassettes consisting of orthogonal promoter sequences, Streptomyces ribosome binding sites, and yeast selectable marker genes. Using these tools we demonstrate the ability to simultaneously insert multiple promoter cassettes into a gene cluster, thereby expediting the reengineering process. We apply this method to model active and silent gene clusters (rebeccamycin and tetarimycin) and to the silent, cryptic pseudogene-containing, environmental DNA-derived Lzr gene cluster. Complete promoter refactoring and targeted gene exchange in this “dead” cluster led to the discovery of potent indolotryptoline antiproliferative agents, lazarimides A and B. This potentially scalable and cost-effective promoter reengineering platform should streamline the discovery of natural products from silent natural product biosynthetic gene clusters.
Bacteria-based natural product discovery programs have traditionally relied on the random screening of culture broth extracts to identify novel natural products. Recent advances in DNA sequencing technologies have made it possible to envision sequence-first natural product discovery programs, where the scanning of DNA sequence data is used to identify gene clusters predicted to encode for novel metabolites (1, 2). A major limitation of this approach has been that gene clusters identified in DNA sequence data are often silent under common laboratory culture conditions and therefore the molecules they encode remain inaccessible (3). A growing body of evidence suggests that silent gene clusters are transcriptionally inactive and that activation of silent gene clusters can be achieved by methods that activate transcription (4–6). Although a number of strategies have been explored to activate silent gene clusters (7), no universal solution to this problem has yet arisen. Here we describe a potentially generic approach for inducing molecule production from silent natural products biosynthetic gene clusters through the multiplexed exchange of native promoters for constitutive synthetic promoters upstream of the biosynthetic operons in a gene cluster.
The development of a simple and cost-effective method for the multiplexed exchange of native promoters with experimentally optimized synthetic promoters has the potential to speed the discovery of new natural products from silent gene clusters found in (meta)genomic DNA sequencing efforts. One commonly proposed method for generically activating silent gene clusters is the resynthesis of clusters with codon optimization and incorporation of model regulated promoters (8, 9). Unfortunately, de novo synthesis of large natural product gene clusters remains technically challenging and expensive. In some previously reported gene cluster activation studies each individual gene in a biosynthetic gene cluster has been placed under the control of synthetic promoter using overlapping DNA sequences (10, 11). We speculated that because bacterial genes organized into operons are naturally coregulated it should be possible to simplify the problem of transcriptionally activating silent gene clusters to the activation of operons in gene clusters. This view ignores codon optimization and the detailed balancing of gene expression levels throughout a gene cluster with the belief that they are not prerequisites to accessing molecules from silent gene clusters. These assumptions are supported by recent reports where promoter reengineering of biosynthetic gene clusters has resulted in the successful production of new natural products from previously silent gene clusters (10–14).
Here we combine the construction of a collection of selectable synthetic promoter cassettes with transformation-associated recombination (TAR) in Saccharomyces cerevisiae to establish a simple and potentially scalable method for activating silent natural product biosynthetic gene clusters through multiplexed promoter exchange (Fig. 1A). Each synthetic promoter cassette is designed to contain a unique gene that complements an auxotrophic yeast strain (i.e., “auxotrophic marker”), pairs of sequence orthogonal actinomycetes constitutive promoters, and ribosome binding sites (RBSs). These cassettes enable the promoter engineering of gene clusters in yeast, followed by molecule production in Streptomyces. This method retains native operon structures, allowing for transcriptional refactoring with minimal synthetic nucleic acid input, thereby streamlining the promoter engineering process and rendering the procedure technically and economically accessible. The utility of this gene cluster activation approach is shown using model gene clusters that encode for either the indolocarbazole rebeccamycin (Reb cluster) or the aromatic polyketide tetarimycin (Tam cluster) (6, 13, 15). We then apply the method to the refactoring of a silent, and we believe naturally dead, environmental DNA (eDNA)-derived gene cluster that we predicted would encode for a novel indolotryptoline-based metabolite. This work resulted in the isolation of lazarimides A and B, which are alkaloids belonging to a rare family of indolotryptoline vacuolar ATPase inhibitors (16). The application of this method to the targeted activation of gene clusters related to other biomedically relevant metabolites or new gene clusters representing new molecular families should provide a simple potentially scalable approach for either improving known bioactive natural products or discovering new families of bioactive natural products.
Fig. 1.
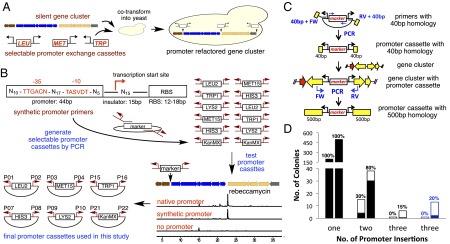
Construction of bidirectional promoter cassettes and multiplexed promoter exchange strategy. (A) Overview of promoter exchange strategy. (B) The bidirectional promoter cassettes were generated by PCR amplification of six yeast selectable makers with primers containing promoter/insulator/RBS combinations (SI Appendix). The generated promoter cassettes were cloned into TOPO vector 2.1 for their future maintenance. These bidirectional promoter cassettes were tested on the Reb gene cluster, and those that produced rebeccamycin were used for promoter engineering (SI Appendix). (C) The method to generate the 500-bp homology arms is outlined. A promoter cassette is inserted into a gene cluster using 40-bp homology arms and the region around this insertion is PCR-amplified to generate 500-bp homology arms. (D) The number of colonies generated from the insertion of one, two, or three promoter cassettes with 40-bp or 500-bp homology arms were compared using the Reb gene cluster (cluster A) and an unrelated eDNA-derived type II polyketide synthase gene cluster (cluster B). A representative collection of yeast colonies (>10) from each experiment was examined by PCR to determine the frequency with which all promoter cassettes were correctly inserted. The solid portion of the bars represents the fraction of colonies with correctly inserted promoter cassettes.
Results and Discussion
Design of Promoter Cassettes for Yeast Homologous Recombination.
Actinomycetes produce the majority of characterized biomedically relevant natural products (17), and as a result we believe that they are likely to be the most appropriate hosts for most heterologous expression studies focused on the discovery of novel bioactive metabolites. A key feature of our promoter exchange strategy was therefore the construction of a set of constitutively active actinomycetes promoters that could be used to drive transcription without the risk of interpromoter recombination. To this end we designed a set of sequence orthogonal promoter cassettes that are based on promoters identified by Seghezzi et al. (18) in a screen for active promoters from a library of sequences containing a random 17-bp spacer between consensus −10 (TTGACN) and −35 (TASVDT) sequences recognized by the housekeeping sigma factor σ70. The exact sequences from this study could not be used for promoter reengineering because each contains an identical RBS and spacer region, which would make them prone to interpromoter recombination if more than one were introduced into a gene cluster. To solve this problem, we added random 15-bp insulator sequences and unique natural Streptomyces RBSs (19) to 24 promoters identified by Seghezzi et al. (18). The spacer and RBS sequences were matched to give an overall GC content of ∼65% for each promoter element (SI Appendix). Pairs of promoter/spacer/RBS sequences were synthesized as primers (SI Appendix) containing 20-bp sequences homologous to the ends of yeast selectable marker genes. Five yeast auxotrophic genes (LEU2, MET15, TRP1, HIS3, and LYS2 encoding genes involved in l-leucine, l-methionine, l-tryptophan, l-histidine, and l-lysine biosynthesis, respectively) (20) and one antibiotic resistance gene (KanMX, aminoglycoside G418) (21) were chosen as selectable markers. Primer pairs were used to amplify each selectable marker to give a set of selectable promoter-exchange cassettes (Fig. 1B).
Each promoter cassette was tested in a promoter exchange experiment using the rebeccamycin (Reb) gene cluster (13). All gene clusters used in this study were cloned from soil metagenomes using Proteobacterial specific cosmid vectors. To permit promoter engineering in yeast and heterologous expression in actinomycetes hosts, clones carrying gene clusters were retrofitted with a yeast origin of replication (CEN/ARS), a yeast selectable marker (URA3), an origin of transfer (oriT) for intergeneric conjugation, the Streptomyces ΦC31 integrase, and the Streptomyces apramycin resistance gene (22). This results in an Escherichia coli:yeast:Streptomyces shuttle vector that permits the rapid movement of gene clusters between all three organisms.
To test our selectable promoter-replacement cassettes using the Reb gene cluster, each bidirectional promoter exchange cassette was amplified with primers designed to contain 40-bp sequences homologous to the sequences upstream of the Reb biosynthetic genes rebG and rebO, which flank the bidirectional promoter region in the Reb gene cluster (Fig. 1B). Each cassette was then cotransformed along with the Reb gene cluster containing cosmid into S. cerevisiae (BY4727 Δdnl4), and transformation reactions were plated on cassette-specific selective media to identify promoter exchanged recombinants. Promoter-replaced Reb constructs isolated from yeast were transformed into E. coli S17 and conjugated into Streptomyces albus where they were examined for the ability to confer the production of rebeccamycin to S. albus (SI Appendix).
S. cerevisiae strain BY4727 contains complete deletions of LEU2, MET15, HIS3, TRP1, LYS2, and URA3, making it useful for selection with all selectable markers used in our promoter cassettes (20). To avoid the nonhomologous end joining (NHEJ) of promoter cassettes, the Dnl4 gene, a DNA ligase IV known to be involved in NHEJ (23), was knocked-out in BY4727 (SI Appendix), and this strain was used as a standard strain for all promoter reengineering experiments.
Of the 12 bidirectional promoter cassettes we tested, eight led to the production of rebeccamycin (1). Six productive promoter/marker gene cassettes, one for each marker gene, were selected for use as promoter replacement tools in this study (Fig. 1B and SI Appendix). For each of the six active cassettes, we also constructed a corresponding set of cassettes with promoters containing 16-bp spacing between the −10 and −35 sequences (SI Appendix), which is reported to generically reduce promoter strength (24). All 12 DNAs were TOPO-cloned to provide a renewable source of each cassette for use in promoter exchange experiments (Fig. 1B).
Simultaneous Insertion of Multiple Promoter Cassettes.
Each round of recombination-mediated promoter exchange takes 3–5 d for selection and recovery of yeast colonies. To help simplify the replacement of multiple promoters in a gene cluster, we explored the feasibility of multiplexed promoter engineering in a single TAR reaction. The productivity of a multiplexed promoter exchange reaction likely depends on the method of transformation and the efficiency of homologous recombination, and therefore we explored the influence of both on promoter exchange reactions.
Both spheroplast- and LiAc-based transformation have been described as effective methods for introducing DNAs into yeast (25–27). We compared the efficiencies of both methods in single promoter replacement experiments and consistently found that LiAc transformation yielded 7 to 10 times more colonies than spheroplast transformation. Although spheroplast transformation has been reported to be more efficient for introducing large DNAs into yeast (26), LiAc transformation seems be more efficient for promoter engineering experiments, which require cotransformation of both large (a gene cluster) and small (promoter cassettes) DNAs.
Recombination efficiency is known to depend on the length of sequence overlap between two DNAs (28). With this in mind, we tested both 40- and 500-bp homology arms in multiplexed promoter exchange experiments. To generate promoter cassettes with 500-bp homology arms, promoter cassettes were first inserted in parallel into the target cluster using 40-bp homology arms. Promoter cassettes containing 500-bp homology arms were then PCR-amplified from these single promoter insertion constructs (Fig. 1C). In single insertion experiments, we consistently observed three times more colonies with promoter cassettes containing 500-bp homology arms than with cassettes containing 40-bp homology arms (Fig. 1D and SI Appendix). In multiplexed promoter exchange reactions, we observed successively fewer recombinants and fewer correct recombinants as we tried to exchange a larger number of promoters (Fig. 1D and SI Appendix).
In general, we find that LiAc cotransformation of a biosynthetic gene cluster and promoter cassettes flanked by 40-bp homology sequences can easily achieve simultaneous insertion of two promoter cassettes. Correct simultaneous insertion of three promoters was only achieved with cassettes containing longer (≥500 bp) homology arms. The longer homology arm promoter cassettes used in multiplexed promoter exchange experiments can be obtained by PCR using single promoter constructs created in parallel with 40-bp homology arms as a template (Fig. 1C). Although the production of longer homology arms using this two-step process requires an additional set of primers, the ability to multiplex the second recombination reaction should significantly simplify the reengineering of complex gene clusters.
Gene Cluster-Wide Promoter Exchange and Natural Product Production Using Model Systems.
With the construction of a set of constitutive bidirectional promoter cassettes and the ability to efficiently introduce these cassettes into gene clusters we sought to evaluate their utility for activating gene clusters using a multiplexed promoter exchange strategy. We initially explored this using well-characterized natural product biosynthetic gene clusters known to encode for either the tryptophan dimer rebeccamycin (Reb) or the aromatic polyketide tetarimycin (Tam). The Reb cluster is natively transcriptionally active (13), whereas the Tam cluster is known to be transcriptionally silent (6).
Reb cluster.
The Reb gene cluster used in this study was originally isolated from an Arizona soil metagenome and found to natively produce rebeccamycin when introduced into S. albus (13). The Reb gene cluster is predicted to contain three promoters: two promoters oriented in opposite directions between the glycosyltransferase rebG and oxidase rebO genes that were used to test our promoter cassettes and a third promoter upstream of the transcriptional regulator rebR gene (Fig. 2A). The bidirectional promoter site between the rebG and rebO genes was replaced with a TRP1 bidirectional promoter cassette and the unidirectional promoter in the upstream region of the rebR gene was replaced with a MET15 cassette in which only one promoter was incorporated into the amplicon used for recombination (i.e., a unidirectional promoter cassette) (SI Appendix). This construct was then moved from yeast through E. coli into S. albus for heterologous expression studies. HPLC analysis of organic extracts from cultures of S. albus transformed with the wild-type Reb gene cluster or with the promoter-exchanged Reb gene cluster showed no significant difference in the production of rebeccamycin, indicating that our promoter reengineering tools should be able to induce molecule production from transcriptionally silent natural product biosynthetic gene clusters.
Fig. 2.

Refactoring of the Reb (rebeccamycin, GenBank accession no. KF551872) and Tam (tetarimycin, GenBank accession no. JX843821) gene clusters. (A) Both promoter regions in the Reb cluster were replaced with synthetic promoters. HPLC analysis of extracts from S. albus cultures containing either the refactored (i) or wild-type cluster (ii) indicates that these cultures produce comparable levels of rebeccamycin. (B) The Tam gene cluster encodes tetarimycin A (2) but is silent in S. albus (iii). The three bidirectional (P1–P3) and one unidirectional (P4) promoter region in the Tam gene cluster was exchanged with synthetic bidirectional promoter cassettes. HPLC analysis of extracts from S. albus cultures either transformed with the fully promoter refactored gene cluster (i) or the Tam gene clusters activated through expression of the tamI SARP gene (ii) indicates that these cultures produce comparable levels of tetarimycin A (2).
Tam cluster.
The Tam gene cluster is an eDNA-derived type II (aromatic) polyketide synthase biosynthetic gene cluster that encodes for the antibiotics tetarimycin A (2) and B. In S. albus, this gene cluster is transcriptionally silent unless tamI, the gene cluster-specific SARP family positive regulator, is artificially up-regulated (6). The Tam gene cluster is predicted to contain six biosynthetic operons driven by four promoter regions (SI Appendix). As a second proof-of-concept experiment, we replaced all four promoter regions with synthetic promoter cassettes using two rounds of TAR (SI Appendix). In the first yeast transformation, LEU2-, MET15-, TRP1-, and HIS3-based promoter cassettes were inserted in parallel into the Tam gene cluster using 40-bp homology arms (SI Appendix). Promoter cassettes with 500-bp homology arms were then amplified from each reengineered gene cluster. In the second round of TAR, LEU2-, MET15-, and TRP1-based promoter cassettes with 500-bp homology arms were simultaneously inserted into the Tam gene cluster harboring the HIS3 promoter cassette. The successful insertion of all four promoter cassettes into the Tam cluster was confirmed by genotyping refactored gene clusters using PCR. The promoter reengineered Tam cluster was transformed into E. coli S17.1 and conjugated into S. albus for heterologous expression studies. Liquid chromatography–mass spectrometry analysis of culture broth extracts from S. albus transformed with either the promoter-refactored Tam cluster or the wild-type cluster activated through expression of the SARP regulatory element using the ermE* promoter showed essentially identical levels of tetarimycin production (Fig. 2 B, i), indicating that the complete promoter refactoring was able to replicate native levels of metabolite production by this gene cluster.
Resuscitation of a Dead Indolotryptoline Gene Cluster.
Silent gene clusters in need of activation increasingly appear in (meta)genomic DNA sequencing datasets. Here we use our promoter engineering method to activate a previously uncharacterized silent, and we believe naturally dead, eDNA-derived indolotryptoline gene cluster to produce a previously unknown indolotryptoline metabolite with potent human cell cytotoxicity.
Discovery of the Lzr gene cluster.
Tryptophan dimers (or bisindoles) are a structurally and functionally diverse class of natural products (29). The best studied of these are the indolocarbazoles staurosporine and rebeccamycin, which are kinase and topoisomerase inhibitors, respectively (30, 31). One very potent, but to date rarely encountered, and therefore underexplored, family of tryptophan dimers is the indolotryptolines. Indolotryptolines contain a core tricyclic tryptoline ring fused to an indole. The two naturally occurring indolotryptolines that have been characterized in fermentation based natural product discovery programs, are cladoniamide (3) and BE-54017 (4) (32, 33). Both exhibit potent human cell cytotoxicity and have recently been shown to function by inhibiting the vacuolar ATPase (16, 34). Vacuolar ATPases are responsible for pumping protons across the plasma membrane and acidifying an array of intracellular organelles (35). They have gained attention as potential therapeutic agents owing to the importance of intracellular pH gradients in a number of diseases (36, 37).
In an effort to expand the observed natural diversity of indolotryptolines and potentially improve their therapeutic prospects, we PCR-screened eDNA cosmid libraries using degenerate primers targeting genes that encode for tryptophan dimerization enzymes (32, 38). This led to the discovery of the Lzr gene cluster, which closely resembles the cladoniamide and BE-54017 gene clusters; however, it is predicted to encode tailoring enzymes (e.g., an extra halogenase and a cytochrome P450 oxidase) that are not used in the biosynthesis of any known indolotryptoline, suggesting that it should encode for a novel indolotryptoline congener.
The Lzr gene cluster was recovered from a previously archived Arizona desert soil eDNA library (39) on two overlapping eDNA cosmid clones (AZ25-292 and AZ25-153). The full-length Lzr gene cluster was reassembled from these two cosmids using TAR and a pTARa-based pathway-specific E. coli:yeast:Streptomyces shuttle capture vector to yield the bacterial artificial chromosome (BAC) BAC-AZ25-292/153. This BAC was transferred into S. albus for heterologous expression studies, but unfortunately this strain failed to produce any detectable clone-specific metabolite under all of the culture conditions we tested, indicating that the Lzr gene cluster is silent in S. albus. We used this silent cryptic gene cluster as a third model system for testing our promoter replacement tools.
Refactoring of the silent eDNA-derived Lzr gene cluster.
The outer edges of the Lzr gene cluster were defined based on comparisons to the BE-54017 and cladoniamide gene clusters and a BLAST analysis of genes surrounding the core indolotryptoline biosynthesis genes (Fig. 3A). The biosynthesis of indolotryptolines is well-characterized, making it possible to predict the function of most genes in the Lzr gene cluster (32). The four key stages of indolotryptoline biosynthesis involve dimerization of oxo-tryptophan to form a chromopyrrolic acid, oxidative aryl–aryl coupling to form an indolocarbazole, “flipping” of one of the indole rings to form an indolotryptoline, and tailoring to generate the final product. The Lzr gene cluster is predicted to contain seven transcriptional units controlled by three bidirectional (P1, P2, and P3) and one unidirectional (P4) promoter regions (Fig. 3 B, i). This cluster is conveniently organized such that successive activation of the three bidirectional promoter regions (P1, P2, and P3) is predicted to drive the expression of genes required to achieve the first, second, and third stages in indolotryptoline biosynthesis, respectively (Fig. 3C).
Fig. 3.
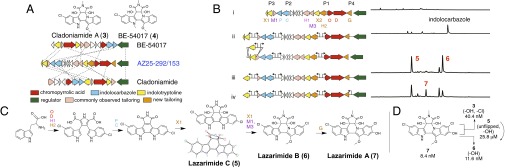
Activation of the Lzr gene cluster. (A) Comparison of the Lzr gene cluster with other indolotryptoline gene clusters. (B) Sequential replacement of promoters and the lzrXI pseudogene in the Lzr gene cluster and HPLC analysis of extracts from cultures of S. albus transformed with refactored gene clusters. (C) The proposed biosynthetic scheme for the lazarimides. The structures of lazarimides A (7), B (6), and C (5) were elucidated by NMR. The structure and absolute configuration of 5 were confirmed by X-ray crystallography (SI Appendix). (D) IC50 values for indolotryptolines against HCT-116 cancer cells.
In a series of single cassette insertions we replaced each bidirectional Lzr promoter region with a synthetic promoter cassette. As expected, P1 and P1+P2 replacement constructs produced chromopyrrolic and indolocarbazole intermediates, respectively (SI Appendix). The P1+P2+P3-replaced gene cluster, however, produced an indolocarbazole intermediate instead of the expected indolotryptoline intermediate (Fig. 3 B, ii). A close examination of lzrX1, the gene predicted to encode the oxidative enzyme that installs the C4c/C7a diol (32, 40), suggested the presence of a single base deletion that leads to a truncated and likely nonfunctional lzrX1 gene (i.e., pseudogene). Hence, the Lzr gene cluster seems to be not only silent but also dead owing to the disruption of the lzrX1 gene.
Although natural mutation rates for most environmental bacteria are not known, even if these rates are relatively low it would not be surprising if many secondary metabolite gene clusters, which are expected to be required only during specific potentially rare environmental events, accumulate mutations in key biosynthetic genes. Ideally, a gene cluster activation tool should therefore not only be able to awaken silent gene clusters through the replacement of promoters but also have the flexibility to “resuscitate” dead gene clusters through the exchange of pseudogenes with functional homologs found in closely related gene clusters. In an effort to resuscitate the Lzr cluster, we extended our promoter exchange method to allow for simultaneous insertion of both synthetic promoters and new genes into the gene cluster of interest (SI Appendix). In this case the abeX1 gene from the BE-54017 gene cluster (32), a full-length homolog of the lzrX1 pseudogene, and a promoter selection cassette were independently PCR-amplified to produce amplicons with 20-bp overlaps. A second round of PCR was then carried out to link the resulting amplicons into a single cassette containing 40-bp Lzr cluster-specific homology arms, two promoters, the full-length abeX1 oxidative gene, and the LYS2 marker gene (Fig. 3B and SI Appendix). This cassette was then used in a standard TAR promoter exchange reaction to replace both the disrupted lzrX1 gene and the P3 promoter region. Upon introduction of this cassette into the P3 site, the new P1+P2+P3 reengineered gene cluster was found to confer to S. albus the ability to produce new indolocarbazole [lazarimide C (5)]- and indolotryptoline [lazarimide B (6)]-based metabolites (Fig. 3 B, iii).
To complete the refactoring of the Lzr gene cluster, we replaced P4 with a unidirectional synthetic promoter cassette. Heterologous expression studies with this fully reengineered Lzr gene cluster showed the presence of one additional major metabolite (7) not seen in cultures of S. albus transformed with any previous reengineered constructs (Fig. 3 B, iv). Compound 7, which we have given the trivial name lazarimide A, was purified from large-scale cultures of S. albus transformed with the completely reengineered Lzr gene cluster, and its structure was solved using high-resolution electrospray ionization mass spectrometry and 1D and 2D NMR (SI Appendix). The general structure of the lazarimide series of metabolites was further confirmed with a crystal structure of the lazarimide intermediate lazarimide C (5) (Fig. 3C).
Lazarimide A (7) differs from cladoniamide and BE-54017 by both its halogenation pattern and the oxidation of the flipped indole moiety. The biosynthesis of lazarimides can be rationalized based on the predicted function of each gene in the Lzr gene cluster (Fig. 3C). Key novel features of the proposed biosynthetic scheme include the action of the predicted oxygenase lzrG and two predicted halogenases lzrH1 and lzrH2. The cytochrome P450, lzrG, is predicted to carry out the unique indolotryptoline core hydroxylation seen on Lazarimide A (7). lzrH1 and lzrH2 are predicted to be tryptophan-5 and tryptophan-6 halogenases, respectively. This difference in regiospecificity affords the unique halogenation pattern seen in lazarimides A (7), B (6), and C (5).
Because known indolotryptolines are potent human cell line toxins, previously unknown compounds 5–7, as well as cladoniamide A (3) as a control, were tested for cytotoxicity against HCT-116 human colon carcinoma cancer cells. The IC50s observed for compounds 3 and 5–7 were 40.4 nM, 25.8 μM, 11.6 nM, and 8.4 nM, respectively (Fig. 3D). In this series of closely related natural products, lazarimide A (7) exhibits the most potent cytotoxicity against human cells. Activation of the Lzr gene cluster demonstrates the utility of our platform toward characterizing new biomedically relevant metabolites through the activation not only of silent but also of naturally dead biosynthetic gene clusters.
Conclusions
The functional characterization of cryptic biosynthetic gene clusters identified in (meta)genome sequencing efforts remains a significant challenge because most of these clusters are silent under common laboratory culture conditions. Here, we demonstrate a potentially general and scalable yeast homologous recombination-based promoter reengineering platform for activating silent gene clusters through replacement of native promoters with constitutively active synthetic promoters. We initially use our promoter-engineering platform to refactor known clusters resulting in molecule production efficiencies that are comparable to those seen for the naturally active clusters. We then demonstrate the successful application of this method to the expression of the dead eDNA-derived Lzr gene cluster in a heterologous host, leading to the characterization of a potent, previously unknown, indolotryptoline-based natural product.
This methodology can be expanded to gene clusters that require replacements of more than the 12 promoters we provide through either (i) the use of additional promoter selection cassettes containing other auxotrophic markers and yeast strains with additional auxotrophies or (ii) the use of multiple integration sites in the Streptomyces host that would allow introduction of partially refactored pathways and permit reuse of existing promoter cassettes. The simple and potentially scalable gene cluster activation method we have developed should greatly facilitate the isolation of bioactive natural products from silent gene clusters identified in both strain-based and metagenome-based next-generation sequencing campaigns.
Materials and Methods
Construction of Promoter Exchange Cassettes.
The forward and reverse primers used to amplify yeast selectable markers were designed to contain unique promoter (44 bp)/spacer (15 bp)/RBS (12–18 bp) combinations (SI Appendix) and 20-bp sequences identical to the distal end of yeast marker genes. LEU2, MET15, TRP1, HIS3, LYS2, and KanMX selectable makers were amplified from plasmids pRS405, pRS401, pRS404, pRS403, pR317, and pFA6, respectively, using the primer sets listed in SI Appendix. PCR products were TA-cloned into TOPO vector pCR2.1 (Life Technologies) for stable maintenance of each selection cassette. Cloned promoter cassettes were sequenced using M13F(−21) and M13R primers to confirm each sequence. Primer sets used for the promoter test experiment with the Reb gene cluster are listed SI Appendix.
Promoter Exchanges.
For each promoter engineering experiment, amplicons were generated from the TOPO-cloned promoter cassettes using primers containing 40-bp sequences matching the targeted insertion site. Specific primers used in the reengineering of different gene clusters are listed in SI Appendix. Promoter cassettes were amplified using AccuPrime Taq High Fidelity DNA polymerase (Invitrogen). A standard 50-μL PCR contained 1 μL of template (10 ng/μL), 2.5 μL of each primer (5 μM), 25 μL of buffer G (Epicentre), 18.5 μL of water, and 0.5 μL of polymerase. The following gradient amplification protocol was used for LEU2 and LYS2 promoter cassettes: initial denaturation (95 °C, 5 min), 36 cycles of denaturation (95 °C, 30 s), annealing (gradient 40–60 °C across 12 wells, 30 s) and extension (72 °C, 5 min), and final extension (72 °C, 5 min). MET15, TRP1, HIS3, and KanMX promoter cassettes were amplified using the following single annealing temperature protocol: initial denaturation (95 °C, 5 min), 36 cycles of denaturation (95 °C, 30 s), annealing (55 °C, 40 s) and extension (72 °C, 3 min), and final extension (72 °C, 5 min). The resulting PCR products were column-purified (QIAprep spin columns; Qiagen) and either the cosmid or BAC clone harboring the target gene clusters were cotransformed into S. cerevisiae (BY4727 Δdnl4) using the LiAc/ss carrier DNA/PEG yeast transformation protocol published by Gietz and Schiestl (25). Briefly, yeast was grown overnight in 50 mL of YPD media containing G418 (200 μg/mL) at 30 °C. In the morning, 2 mL of the overnight culture was reinoculated into 50 mL of fresh YPD media containing G418 (200 μg/mL) and grown for ∼4 h (OD600 = 2.0). This culture was harvested by centrifugation (10 min, 3,200 × g), washed twice with sterile 4 °C water, and resuspended in 1 mL of sterile 4 °C water. For each transformation 100 μL of washed cells was transferred to a microfuge tube. The cells were collected by centrifugation (30 s, 18,000 × g) and resuspended in a transformation mix containing 36 μL of 1 M LiAc solution, 50 μL of 2 mg/mL carrier DNA (Salmon sperm DNA) solution, 240 μL of 50% (wt/vol) PEG 3350 solution, and 34 μL of Tris-EDTA containing 4 μg of cosmid or BAC vector and 4 μg of promoter cassettes. This transformation mix was incubated at 42 °C for 40 min. Cells were then collected by centrifugation (30 s, 18,000 × g), resuspended in 100 μL of water, and plated on the appropriate synthetic composite dropout media agar plates. Agar plates were incubated at 30 °C until colonies appeared. Colonies were checked by PCR for correct promoter insertion using primer pairs where one primer targeted the cassette and the second primer targeted the gene cluster (SI Appendix). DNA was isolated from PCR-positive yeast clones, transferred into E. coli S17.1, and then moved to S. albus by intergeneric conjugation for expression studies (SI Appendix).
Supplementary Material
Acknowledgments
We thank Emil Lobkovsky for his assistance with X-ray crystallography. This work was supported by NIH Grant GM077516.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. KR052816).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1507606112/-/DCSupplemental.
References
- 1.Blin K, et al. 2013. AntiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41(Web Server issue):W204–212.
- 2.Doroghazi JR, et al. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol. 2014;10(11):963–968. doi: 10.1038/nchembio.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen PR, Chavarria KL, Fenical W, Moore BS, Ziemert N. Challenges and triumphs to genomics-based natural product discovery. J Ind Microbiol Biotechnol. 2014;41(2):203–209. doi: 10.1007/s10295-013-1353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laureti L, et al. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc Natl Acad Sci USA. 2011;108(15):6258–6263. doi: 10.1073/pnas.1019077108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olano C, et al. Activation and identification of five clusters for secondary metabolites in Streptomyces albus J1074. Microb Biotechnol. 2014;7(3):242–256. doi: 10.1111/1751-7915.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kallifidas D, Kang HS, Brady SF. Tetarimycin A, an MRSA-active antibiotic identified through induced expression of environmental DNA gene clusters. J Am Chem Soc. 2012;134(48):19552–19555. doi: 10.1021/ja3093828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scherlach K, Hertweck C. Triggering cryptic natural product biosynthesis in microorganisms. Org Biomol Chem. 2009;7(9):1753–1760. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 8.Elena C, Ravasi P, Castelli ME, Peirú S, Menzella HG. Expression of codon optimized genes in microbial systems: Current industrial applications and perspectives. Front Microbiol. 2014;5:21. doi: 10.3389/fmicb.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medema MH, Breitling R, Takano E. Synthetic biology in Streptomyces bacteria. Methods Enzymol. 2011;497:485–502. doi: 10.1016/B978-0-12-385075-1.00021-4. [DOI] [PubMed] [Google Scholar]
- 10.Luo Y, et al. Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat Commun. 2013;4:2894. doi: 10.1038/ncomms3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shao Z, et al. Refactoring the silent spectinabilin gene cluster using a plug-and-play scaffold. ACS Synth Biol. 2013;2(11):662–669. doi: 10.1021/sb400058n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Challis GL. Mining microbial genomes for new natural products and biosynthetic pathways. Microbiology. 2008;154(Pt 6):1555–1569. doi: 10.1099/mic.0.2008/018523-0. [DOI] [PubMed] [Google Scholar]
- 13.Chang FY, Ternei MA, Calle PY, Brady SF. Discovery and synthetic refactoring of tryptophan dimer gene clusters from the environment. J Am Chem Soc. 2013;135(47):17906–17912. doi: 10.1021/ja408683p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamanaka K, et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc Natl Acad Sci USA. 2014;111(5):1957–1962. doi: 10.1073/pnas.1319584111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sánchez C, et al. The biosynthetic gene cluster for the antitumor rebeccamycin: Characterization and generation of indolocarbazole derivatives. Chem Biol. 2002;9(4):519–531. doi: 10.1016/s1074-5521(02)00126-6. [DOI] [PubMed] [Google Scholar]
- 16.Chang F-Y, Kawashima SA, Brady SF. Mutations in the proteolipid subunits of the vacuolar H+-ATPase provide resistance to indolotryptoline natural products. Biochemistry. 2014;53(45):7123–7131. doi: 10.1021/bi501078j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nett M, Ikeda H, Moore BS. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep. 2009;26(11):1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seghezzi N, Amar P, Koebmann B, Jensen PR, Virolle M-J. The construction of a library of synthetic promoters revealed some specific features of strong Streptomyces promoters. Appl Microbiol Biotechnol. 2011;90(2):615–623. doi: 10.1007/s00253-010-3018-0. [DOI] [PubMed] [Google Scholar]
- 19.Strohl WR. Compilation and analysis of DNA sequences associated with apparent streptomycete promoters. Nucleic Acids Res. 1992;20(5):961–974. doi: 10.1093/nar/20.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brachmann CB, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14(2):115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 21.Wach A, Brachat A, Pöhlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10(13):1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 22.Kim JH, et al. Cloning large natural product gene clusters from the environment: Piecing environmental DNA gene clusters back together with TAR. Biopolymers. 2010;93(9):833–844. doi: 10.1002/bip.21450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson TE, Grawunder U, Lieber MR. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997;388(6641):495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- 24.Aoyama T, et al. Essential structure of E. coli promoter: Effect of spacer length between the two consensus sequences on promoter function. Nucleic Acids Res. 1983;11(17):5855–5864. doi: 10.1093/nar/11.17.5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2(1):31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 26.Gietz RD, Woods RA. 2001. Genetic transformation of yeast. Biotechniques 30(4):816–820, 822–816, 828 passim.
- 27.Kallifidas D, Brady SF. Reassembly of functionally intact environmental DNA-derived biosynthetic gene clusters. Methods Enzymol. 2012;517:225–239. doi: 10.1016/B978-0-12-404634-4.00011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manivasakam P, Weber SC, McElver J, Schiestl RH. Micro-homology mediated PCR targeting in Saccharomyces cerevisiae. Nucleic Acids Res. 1995;23(14):2799–2800. doi: 10.1093/nar/23.14.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan KS, Drennan CL. Divergent pathways in the biosynthesis of bisindole natural products. Chem Biol. 2009;16(4):351–364. doi: 10.1016/j.chembiol.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreau P, et al. Syntheses and biological activities of rebeccamycin analogues. Introduction of a halogenoacetyl substituent. J Med Chem. 1999;42(4):584–592. doi: 10.1021/jm980396d. [DOI] [PubMed] [Google Scholar]
- 31.Tamaoki T, et al. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem Biophys Res Commun. 1986;135(2):397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 32.Chang FY, Brady SF. Cloning and characterization of an environmental DNA-derived gene cluster that encodes the biosynthesis of the antitumor substance BE-54017. J Am Chem Soc. 2011;133(26):9996–9999. doi: 10.1021/ja2022653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams DE, et al. Cladoniamides A-G, tryptophan-derived alkaloids produced in culture by Streptomyces uncialis. Org Lett. 2008;10(16):3501–3504. doi: 10.1021/ol801274c. [DOI] [PubMed] [Google Scholar]
- 34.Kimura T, et al. Synthesis and assignment of the absolute configuration of indenotryptoline bisindole alkaloid BE-54017. Org Lett. 2012;14(17):4418–4421. doi: 10.1021/ol3019314. [DOI] [PubMed] [Google Scholar]
- 35.Jefferies KC, Cipriano DJ, Forgac M. Function, structure and regulation of the vacuolar (H+)-ATPases. Arch Biochem Biophys. 2008;476(1):33–42. doi: 10.1016/j.abb.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pérez-Sayáns M, Somoza-Martín JM, Barros-Angueira F, Rey JMG, García-García A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat Rev. 2009;35(8):707–713. doi: 10.1016/j.ctrv.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10(10):767–777. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 38.Chang FY, Brady SF. Discovery of indolotryptoline antiproliferative agents by homology-guided metagenomic screening. Proc Natl Acad Sci USA. 2013;110(7):2478–2483. doi: 10.1073/pnas.1218073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Owen JG, et al. Multiplexed metagenome mining using short DNA sequence tags facilitates targeted discovery of epoxyketone proteasome inhibitors. Proc Natl Acad Sci USA. 2015;112(14):4221–4226. doi: 10.1073/pnas.1501124112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du Y-L, Williams DE, Patrick BO, Andersen RJ, Ryan KS. Reconstruction of cladoniamide biosynthesis reveals nonenzymatic routes to bisindole diversity. ACS Chem Biol. 2014;9(12):2748–2754. doi: 10.1021/cb500728h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.