

Significance
We demonstrate that hearts lacking the sarcomere protein cardiac myosin binding protein C (MYBPC) undergo altered development due to an extra round of postnatal cell division. Normal cardiac myocytes replicate rapidly during fetal life, undergo a final round of cell division shortly after birth, cease dividing, and increase in cell size during prepubescent life. MYBPC has an unexpected function—inhibition of myocyte cytokinesis. MYBPC-deficient myocytes undergo an additional round of cytokinesis, resulting in increased numbers of myocytes and a greater proportion of mononuclear myocytes in neonatal hearts. Our findings provide insights into the mechanisms of dilated cardiomyopathy caused by homozygous mutations that reduce MYBPC levels.
Keywords: myosin binding protein C, cardiac dilation, cardiac hypertrophy, cytokinesis, hyperplasia
Abstract
Homozygous cardiac myosin binding protein C-deficient (Mybpct/t) mice develop dramatic cardiac dilation shortly after birth; heart size increases almost twofold. We have investigated the mechanism of cardiac enlargement in these hearts. Throughout embryogenesis myocytes undergo cell division while maintaining the capacity to pump blood by rapidly disassembling and reforming myofibrillar components of the sarcomere throughout cell cycle progression. Shortly after birth, myocyte cell division ceases. Cardiac MYBPC is a thick filament protein that regulates sarcomere organization and rigidity. We demonstrate that many Mybpct/t myocytes undergo an additional round of cell division within 10 d postbirth compared with their wild-type counterparts, leading to increased numbers of mononuclear myocytes. Short-hairpin RNA knockdown of Mybpc3 mRNA in wild-type mice similarly extended the postnatal window of myocyte proliferation. However, adult Mybpct/t myocytes are unable to fully regenerate the myocardium after injury. MYBPC has unexpected inhibitory functions during postnatal myocyte cytokinesis and cell cycle progression. We suggest that human patients with homozygous MYBPC3-null mutations develop dilated cardiomyopathy, coupled with myocyte hyperplasia (increased cell number), as observed in Mybpct/t mice. Human patients, with heterozygous truncating MYBPC3 mutations, like mice with similar mutations, have hypertrophic cardiomyopathy. However, the mechanism leading to hypertrophic cardiomyopathy in heterozygous MYBPC3+/− individuals is myocyte hypertrophy (increased cell size), whereas the mechanism leading to cardiac dilation in homozygous Mybpc3−/− mice is primarily myocyte hyperplasia.
Dilated cardiomyopathy (DCM) leads to heart failure and is a leading cause of morbidity and mortality (1, 2). DCM is generally diagnosed as left ventricular (LV) dilation with associated reduction in cardiac contraction measured as impaired fractional shortening (3). Hearts from affected individuals frequently demonstrate myocyte elongation, myocyte death, and fibrosis, in addition to LV dilation. DCM results from a variety of environmental factors, such as viral infection and alcohol abuse, as well as from mutations in a number of genes including titin, lamin A/C, cardiac actin, cardiac myosin heavy chain, and phospholamban (reviewed in refs. 4–6). Whether all of these DCM-inducing factors activate the same or different cellular pathways to produce similar clinical features remains uncertain. The mechanisms by which mutations in the cardiac myosin binding protein C (MYBPC3) gene and other sarcomere protein genes lead to cardiac dilatation are under investigation.
MYBPC is a thick filament accessory protein component of the striated muscle sarcomere A band that constitutes 2–4% of the myofibril (discussed in ref. 7). Although there are four Mybpc genes in the mammalian genome, only cardiac Mybpc (Mybpc3) is expressed in embryonic, neonatal, and adult hearts (8, 9). Cardiac MYBPC interacts with at least four sarcomere components: myosin heavy chain, actin, myosin light chain 2, and titin (10–12). More than 400 cardiac MYBPC3 gene mutations have been identified in patients as a cause of hypertrophic cardiomyopathy (HCM), an autosomal dominant disorder resulting from defective sarcomeres (for reviews, see refs. 12, 13). Due to an ancient founder mutation, 4% of the population of India carries a truncating MYBPC3 mutation (14, 15). The majority of cardiac MYBPC3 mutations are predicted to encode truncated proteins that lack portions of either the carboxyl myosin and/or titin binding domains (7, 13). These truncating MYBPC3 mutations are thought to cause cardiac hypertrophy by inducing myocyte hypertrophy (increased cell size), rather than myocyte hyperplasia.
We and other researchers have created mice that carry a mutant cardiac Mybpc3 gene to create murine HCM models (16–18). Heterozygous mice, designated Mybpct/+, like humans bearing the same mutation, develop adult onset HCM. Homozygous MYBPC3 mutations are a much rarer cause of human DCM than autosomal dominant mutations in other sarcomere protein genes. However, homozygous Mybpct/t mice that express two mutant alleles and no wild-type cardiac Mybpc3 develop LV dilation by 3 d postbirth and have all of the features of DCM, including LV chamber dilation, albeit mildly impaired fractional shortening (16). Unlike most humans with DCM, homozygous mutant cardiac Mybpct/t mice have normal survival despite their cardiac disease. Other homozygous null cardiac Mybpc3 mice develop an identical phenotype (7, 17, 18). Hence, for the studies described here, we assume that the phenotype of the Mybpct/t mice is due to lack of MYBPC protein, rather than to small amounts of truncated protein. Recently, two groups have demonstrated that delivery of MYBPC to Mybpc3-null hearts restores cardiac function and morphology (19, 20). Here, we have begun to dissect the mechanism by which homozygous Mybpct/t hearts develop DCM.
Because Mybpct/t mice begin LV dilation within a few days postbirth (16), we hypothesized that this reflected abnormal development of neonatal myocytes. During fetal and early perinatal development in wild-type hearts, cardiomyocytes divide rapidly, producing hyperplastic cardiac growth (21). However, at 10 d postbirth, cardiomyocytes cease to divide and all subsequent increases in myocardial mass result from myocyte hypertrophy (22). Despite the importance of this phenomenon, little is known about the molecular basis for the transition from hyperplasic to hypertrophic-based myocardial growth. We hypothesized that abnormal cardiomyocyte growth, either hyperplastic or hypertrophic, in the perinatal period accounted for the LV dilation of Mybpct/t mouse hearts. To address this question, we have counted and measured cardiomyocytes from Mybpct/t and wild-type mice. We have also studied the consequences of reducing MYBPC levels by injecting Mybpc3-specific shRNA at birth. Neonatal cardiomyocytes lacking cardiac MYBPC, due to Mybpc3-specific shRNA knockdown, undergo an additional round of cytokinesis. We conclude that dramatic reductions in the amount of cardiac MYBPC leads to aberrant cell cycle regulation at the G1/S checkpoint, resulting in at least one extra round of myocyte division and DCM.
Results
Increased Numbers and Immaturity of Cardiac Myocytes in Mybpct/t Mice.
Cardiac tissues and myocytes from wild-type and Mybpct/t mice were studied from birth to postnatal day 35 (designated as P0–35). The hearts from P5 Mybpct/t mice had a significantly increased LV mass and LV/body weight ratio compared with wild-type mice (Fig. 1 A and B and Fig. S1A). Myocytes from P3 and P10 Mybpct/t mice were 20–30% wider than wild-type myocytes (Fig. S1 B and C), which we presume reflects myocyte immaturity (23) and/or abnormal spacing between parallel sarcomeres due to MYBPC deficiency (24). There was no significant difference in the length of myocytes from wild-type and Mybpct/t mice (Fig. S1D).
Fig. 1.

LV mass and myocyte nucleation in wild-type and Mybpct/t hearts. (A and B) Changes in LV mass (A) and LV weight/body weight ratios (B) of wild-type (filled circle) and Mybpct/t (open circle) mice from P0–20 (n = 5–10 neonates per time point). (C) Light micrographs of DAPI-stained isolated mononuclear (Upper panel) and binuclear (Lower panel) myocytes from 3-wk-old Mybpct/t mice visualized with bright field (BF) and fluorescent (DAPI) illumination. (Scale bar, 20 µm.) (D) Representative light micrographs of isolated LV myocytes from EdU-labeled Mybpct/t mice that are stained for nuclei (blue), troponin I (green), and EdU (red). A mononuclear myocyte (arrows) is indicated. (Scale bar, 20 µm.) (E) Distribution of mononuclear, binuclear, and multinuclear myocytes from 3-wk-old wild-type (gray), Mybpct/+(white box), and Mybpct/t (black) hearts. Data are presented from four mice per genotype, 200–300 cells per mouse (mean ± SD). All P values reflect comparison of wild-type and Mybpct/t hearts.
Fig. S1.
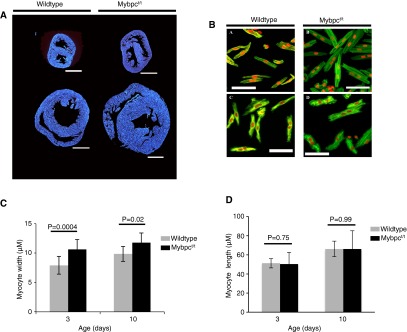
(A) DAPI-stained transverse sections of wild-type (Left) and Mybpct/t (Right) hearts from P3 (Upper) and P10 (Lower) mice. (Scale bar, 0.5 mm.) (B) Isolated ventricular myocytes from P3 wild-type (Top Left), P3 Mybpct/t (Top Right), P10 wild-type (Bottom Left), and P10 Mybpct/t (Bottom Right) fixed with 4% (wt/vol) paraformaldehyde and stained with propidium iodide (red) and phalloidin (green). (Scale bar, 50 µm.) (C) Width and (D) length of myocytes isolated from wild-type (gray) and Mybpct/t (black) in P3 and P10 mice (n = 13–18 cells, three animals per group).
As expected (22, 25, 26), nearly all wild-type adult LV myocytes were binuclear (Fig. 1 C–E). Analyses of younger mice confirmed this finding. At P21, ∼5% of wild-type and heterozygous Mybpct/+ LV myocytes were mononuclear and 90% were binuclear (Fig. 1E). However, significantly less Mybpct/t LV myocytes were binuclear (Fig. 1E; WT:Mybpct/+:Mybpct/t, 89.8%:89.7%:74.8%; P = 0.0003). Consistent with this observation was the observation that ∼threefold more Mybpct/t LV myocytes were mononuclear than either wild-type or heterozygous Mybpct/+ LV myocytes (Fig. 1E; WT:Mybpct/+:Mybpct/t, 5.8%:6.7%:18.3%; P < 4E-5). We defined myocyte numbers and nuclear morphology in hearts from 5-wk-old Mybpct/t and wild-type mice by immunohistochemical staining of 10 sections evenly distributed across the LV. Wheat germ agglutinin (WGA) was used to demarcate plasma membrane boundaries (Fig. 2 A and B), and nonmyocytes were excluded by size and absence of cardiac troponin I staining. In comparison with wild-type, Mybpct/t LV contained ∼40% more myocytes than wild-type LV (Fig. 2C; P = 0.006).
Fig. 2.

Increased numbers of myocytes in Mybpct/t compared with wild-type hearts. (A and B) Transverse heart sections stained with WGA at low (A; scale bar, 0.5 mm) and high (B; scale bar, 50 µm) magnification. (C) Quantification of myocytes from wild-type (gray) and Mybpct/t (black) mice. Data are presented from three mice per genotype, 10 sections per mouse (mean ± SD).
Previous studies have demonstrated that by P10 the majority of wild-type binuclear myocytes have exited the cell cycle (27). We hypothesized that the increased proportion of mononuclear to binuclear myocytes in Mybpct/t hearts reflected either premature cell cycle arrest before becoming binuclear or alternatively that mutant myocytes had continued cell cycle progression with cytokinesis—a delay in cell cycle exit that would account for greater myocyte number. To distinguish these models, we used three approaches to study cell cycle progression.
Mybpct/t Myocytes Have Delayed Cell Cycle Exit.
We injected the thymidine analog 5-ethynyl-2’-deoxyuridine (EdU) (28) into wild-type and Mybpct/t neonates at P1 through P5 and assessed DNA synthesis in isolated P14 myocytes (Fig. 1D). In comparison with wild type, there were significantly more EdU-positive mono- and binuclear myocytes from Mybpct/t mice (47.02 ± 2.50% vs. 29.35 ± 3.06%; P = 0.0003; four mice per genotype; 200–300 cells per mouse). To identify cells undergoing mitosis, we used an anti-histone H3 phosphorylation (pH3) antibody that detects pH3 of serine residue 10 during the mitotic (M) phase of the cell cycle (29). Immunohistochemistry analyses of pH3 at P3, P7, and P10 (Fig. 3 A and B) indicated almost twice as many myocytes undergoing mitosis in Mybpct/t than in wild-type hearts. Finally, we performed immunostaining for Aurora B kinase (Aurora B), which localizes to the central spindle during anaphase and in the midbody during cytokinesis (30). As cell cycle exit is arrested before cytokinesis during normal neonatal myocyte development, Aurora B kinase was rarely detected in the central spindle or midbody of wild-type myocytes. In contrast, P7 Mybpct/t myocytes had 15-fold more Aurora B staining, which was localized to the cytoplasmic bridge between daughter cells (Fig. 3 C and D). Based on the increase in EdU incorporation and pH3 and Aurora B immunostaining in Mybpct/t mice, we concluded that mutant myocytes have delayed cell cycle exit during neonatal life and undergo additional rounds of cell division. As a result, Mybpct/t mice had both more myocytes and more mononuclear myocytes than wild-type hearts.
Fig. 3.

pH3 and Aurora B expression in wild-type and Mybpct/t hearts. (A) Confocal micrographs of P7 wild-type and Mybpct/t LV sections stained with DAPI (blue), troponin I (green), and pH3 (red). (Scale bar, 20 µm.) (B) Quantification of pH3-positive myocytes in wild-type (gray) and Mybpct/t (black) hearts from P3, P7, and P10 mice. Data are presented from four mice per genotype, three sections per mouse (mean ± SD). (C) Confocal micrographs of cardiac sections from P7 wild-type and Mybpct/t mice stained with DAPI (blue), troponin I (green), and Aurora B (red). [Scale bar, (Upper) 20 µm and (Lower) 5 µm.] (D) Quantification of Aurora B located at the bridge of two daughter myocytes in wild-type (gray) and Mybpct/t (black) hearts from P7 mice. Data are presented from four mice per genotype, three sections per mouse (mean ± SD).
To further assess cell cycle regulation in neonatal (P10) Mypbct/t LV, we assessed expression of cell cycle-related genes by RNAseq (Table S1). One hundred and seven genes that are described as “cell division,” “cyclin-associated,” or “cell cycle” genes are expressed in the wild-type P10 LV. Expression of 70 cell cycle-related genes either increased or decreased (P < 0.001), whereas 38 of the cell cycle-related genes were expressed at similar levels in wild-type and Mypbct/t LV. Presumably these cell cycle-associated gene expression changes are related to changes in cell cycle exit found in neonatal Mypbct/t LV.
Table S1.
Cell cycle-associated gene expression at P10 in wild-type and Mypbct/t LV
| Gene | Chr | Start | End | Description | WT | Mybpct/t | Fold | P value |
| Differentially expressed cells | ||||||||
| Ccnyl1 | chr1 | 64737786 | 64772216 | Cyclin Y-like 1 | 9.9 | 5.4 | 1.84 | 4.E-08 |
| Ccnj | chr19 | 40905768 | 40923060 | Cyclin-J | 13.7 | 7.7 | 1.78 | 6.E-10 |
| Cdk13 | chr13 | 17807795 | 17896931 | Cell division protein kinase 13 | 24.3 | 14.5 | 1.68 | 9.E-14 |
| Bod1 | chr11 | 31565149 | 31571862 | Biorientation of chromosomes in cell division | 24.1 | 14.5 | 1.66 | 2.E-13 |
| Bod1l | chr5 | 42178777 | 42235554 | Biorientation of chromosomes in cell division | 52.3 | 32.5 | 1.61 | 5.E-24 |
| Cdk16 | chrX | 20265096 | 20277003 | Cell division protein kinase 16 | 158.2 | 98.7 | 1.60 | 2.E-68 |
| Ccni | chr5 | 93610958 | 93635521 | Cyclin-I | 99.6 | 64.1 | 1.56 | 6.E-39 |
| Cdk12 | chr11 | 98064618 | 98139845 | Cell division protein kinase 12 | 23.5 | 15.6 | 1.51 | 3.E-09 |
| Cntd1 | chr11 | 101140325 | 101150015 | Cyclin N-terminal domain-containing protein 1 | 7.7 | 5.1 | 1.50 | 7.E-04 |
| Cdc42ep3 | chr17 | 79733364 | 79754431 | Cdc42 effector protein 3 | 17.8 | 12.0 | 1.48 | 7.E-07 |
| Cdk2ap1 | chr5 | 124795447 | 124804637 | Cyclin-dependent kinase 2-associated protein 1 | 38.8 | 26.6 | 1.46 | 2.E-12 |
| Cdc73 | chr1 | 145454628 | 145549814 | Parafibromin | 8.9 | 6.1 | 1.45 | 7.E-04 |
| Cdc42bpb | chr12 | 112531182 | 112615929 | Serine/threonine-protein kinase MRCK beta | 95.7 | 67.3 | 1.42 | 3.E-25 |
| Dmtf1 | chr5 | 9118867 | 9161776 | Cyclin D binding myb-like transcription factor 1 | 11.9 | 8.4 | 1.41 | 3.E-04 |
| Cdc5l | chr17 | 45528838 | 45570656 | Cell division cycle 5-related protein | 26.7 | 20.6 | 1.30 | 3.E-05 |
| Cdc37l1 | chr19 | 29064983 | 29101169 | Hsp90 cochaperone Cdc37-like 1 | 55.7 | 43.1 | 1.29 | 4.E-09 |
| Cdkn1c | chr7 | 150644243 | 150646955 | Cyclin-dependent kinase inhibitor 1C | 28.8 | 22.5 | 1.28 | 4.E-05 |
| Ccny | chr18 | 9314041 | 9450148 | Cyclin-Y | 39.6 | 31.6 | 1.25 | 1.E-05 |
| Cdc42bpa | chr1 | 181891219 | 182095733 | Cell division cell 42 binding kinase alpha | 41.3 | 33.4 | 1.23 | 3.E-05 |
| Ccnd1 | chr7 | 152115835 | 152125830 | G1/S-specific cyclin-D1 | 84.1 | 68.5 | 1.23 | 5.E-09 |
| Ccar1 | chr10 | 62206675 | 62255116 | Cell division cycle and apoptosis regulator | 70.4 | 82.5 | 0.85 | 1.E-05 |
| Ccnk | chr12 | 109417947 | 109441569 | Cyclin-K | 37.6 | 45.3 | 0.83 | 1.E-04 |
| Cdc123 | chr2 | 5715339 | 5766006 | Cell division cycle protein 123 homolog | 40.1 | 49.7 | 0.81 | 6.E-06 |
| Ccnd2 | chr6 | 127075726 | 127101066 | G1/S-specific cyclin-D2 | 304.1 | 377.0 | 0.81 | 1.E-36 |
| Cdc23 | chr18 | 34790604 | 34811390 | Cell division cycle protein 23 homolog | 33.1 | 41.1 | 0.81 | 3.E-05 |
| Cdkn1b | chr6 | 134870418 | 134875543 | Cyclin-dependent kinase inhibitor 1B | 63.1 | 78.8 | 0.80 | 3.E-09 |
| Cdc42bpg | chr19 | 6306456 | 6325652 | Serine/threonine-protein kinase MRCK gamma | 19.1 | 24.2 | 0.79 | 6.E-04 |
| Cdk7 | chr13 | 101466979 | 101500897 | Cell division protein kinase 7 | 26.6 | 33.9 | 0.78 | 2.E-05 |
| Ccng2 | chr5 | 93696598 | 93705257 | Cyclin-G2 | 16.4 | 21.1 | 0.78 | 5.E-04 |
| Ccna2 | chr3 | 36463786 | 36470918 | Cyclin-A2 | 14.0 | 18.2 | 0.77 | 8.E-04 |
| Cdca4 | chr12 | 114058445 | 114067634 | Cell division cycle-associated protein 4 | 14.6 | 19.3 | 0.76 | 3.E-04 |
| Cdc34 | chr10 | 79144939 | 79151143 | Ubiquitin-conjugating enzyme E2 R1 | 54.1 | 72.8 | 0.74 | 7.E-14 |
| Cdc25b | chr2 | 131012686 | 131024233 | M-phase inducer phosphatase 2 | 8.0 | 11.6 | 0.70 | 4.E-04 |
| Ccnt1 | chr15 | 98373641 | 98398714 | Cyclin-T1 | 21.0 | 30.4 | 0.69 | 5.E-09 |
| Cdk4 | chr10 | 126500658 | 126504338 | Cell division protein kinase 4 | 59.0 | 85.7 | 0.69 | 2.E-23 |
| Cdk2ap2 | chr19 | 4097350 | 4099017 | Cyclin-dependent kinase 2-associated protein 2 | 14.8 | 21.5 | 0.69 | 7.E-07 |
| Cdc37 | chr9 | 20942984 | 20954350 | Hsp90 cochaperone Cdc37 | 108.4 | 161.9 | 0.67 | 2.E-48 |
| Cdc42ep1 | chr15 | 78673076 | 78681332 | Cdc42 effector protein 1 | 62.8 | 94.7 | 0.66 | 3.E-30 |
| Cdk2 | chr10 | 128134994 | 128142107 | Cell division protein kinase 2 | 20.0 | 31.3 | 0.64 | 2.E-12 |
| Cdc42ep4 | chr11 | 113588163 | 113613129 | Cdc42 effector protein 4 | 18.3 | 29.2 | 0.63 | 1.E-12 |
| Ptgis | chr2 | 167028695 | 167066037 | Prostacyclin synthase | 17.4 | 28.2 | 0.62 | 8.E-13 |
| Cdc27 | chr11 | 104363842 | 104411934 | Cell division cycle protein 27 homolog | 27.7 | 46.9 | 0.59 | 3.E-23 |
| Ccndbp1 | chr2 | 120834143 | 120842648 | Cyclin-D1–binding protein 1 | 21.0 | 36.4 | 0.58 | 1.E-19 |
| Cks1b | chr3 | 89219393 | 89222213 | Cyclin-dependent kinases regulatory subunit 1 | 17.7 | 31.0 | 0.57 | 1.E-17 |
| Rmnd1chr10+ | chr10 | 5914188 | 5943372 | Required for meiotic nuclear division protein 1 | 16.1 | 29.0 | 0.55 | 9.E-18 |
| Cdc16 | chr8 | 13757689 | 13781882 | SubName: Full, Cdc16 protein | 24.0 | 43.4 | 0.55 | 5.E-26 |
| Cdk6 | chr5 | 3343892 | 3522225 | Cell division protein kinase 6 | 6.6 | 11.9 | 0.55 | 3.E-08 |
| Ccne2 | chr4 | 11118500 | 11131926 | G1/S-specific cyclin-E2 | 5.2 | 9.5 | 0.55 | 6.E-07 |
| Cdkn2dchr9+ | chr9 | 123836859 | 123839607 | Cyclin-dependent kinase 4 inhibitor D | 14.0 | 26.4 | 0.53 | 3.E-18 |
| Cdkn2c | chr4 | 109333480 | 109339262 | Cyclin-dependent kinase 4 inhibitor C | 20.3 | 38.9 | 0.52 | 3.E-27 |
| Cdkn2dchr9- | chr9 | 21092907 | 21095653 | Cyclin-dependent kinase 4 inhibitor D | 14.9 | 29.0 | 0.52 | 4.E-21 |
| Cinp | chr12 | 112110819 | 112127355 | Cyclin-dependent kinase 2-interacting protein | 20.1 | 40.3 | 0.50 | 5.E-31 |
| Cdca8 | chr4 | 124595708 | 124614161 | Borealin | 7.7 | 16.4 | 0.47 | 6.E-15 |
| Cdk1 | chr10 | 68799382 | 68815660 | Cell division protein kinase 1 | 15.6 | 33.4 | 0.47 | 8.E-30 |
| Cdkn3 | chr14 | 47380215 | 47391200 | Cyclin-dependent kinase inhibitor 3 | 2.3 | 4.9 | 0.46 | 1.E-05 |
| Cdc42se1 | chr3 | 95032701 | 95040331 | CDC42 small effector protein 1 | 33.4 | 73.3 | 0.46 | 1.E-66 |
| Cks2 | chr13 | 51740600 | 51746031 | Cyclin-dependent kinase regulatory subunit 2 | 11.7 | 25.8 | 0.45 | 8.E-25 |
| Cdca5 | chr19 | 6085096 | 6091773 | Cell division cycle associated 5 | 2.4 | 5.5 | 0.43 | 8.E-07 |
| Mnd1 | chr3 | 83891855 | 83959708 | Meiotic nuclear division protein 1 homolog | 1.4 | 3.2 | 0.43 | 2.E-04 |
| Cdc42ep5 | chr7 | 4102861 | 4116301 | Cdc42 effector protein 5 | 12.1 | 30.2 | 0.40 | 3.E-35 |
| Cdca2 | chr14 | 68294410 | 68333898 | Cell division cycle-associated protein 2 | 3.9 | 9.8 | 0.39 | 1.E-12 |
| Cdca7l | chr12 | 119082333 | 119117179 | Cell division cycle-associated 7-like protein | 0.9 | 2.4 | 0.38 | 5.E-04 |
| Cdkl1 | chr12 | 70847834 | 70891694 | Cyclin-dependent kinase-like 1 | 3.2 | 9.4 | 0.34 | 1.E-14 |
| Cdc20 | chr4 | 118105505 | 118109948 | Cell division cycle protein 20 homolog | 10.5 | 31.7 | 0.33 | 4.E-48 |
| Cacybp | chr1 | 162133051 | 162142908 | Calcyclin-binding protein | 32.9 | 102.6 | 0.32 | 4.E-157 |
| Cdkn1a | chr17 | 29227930 | 29237667 | Cyclin-dependent kinase inhibitor 1 | 49.6 | 161.5 | 0.31 | 8.E-259 |
| Cdc6 | chr11 | 98769202 | 98785256 | Cell division control protein 6 homolog | 3.3 | 10.7 | 0.30 | 8.E-19 |
| Ccnb2 | chr9 | 70255495 | 70269361 | G2/M-specific cyclin-B2 | 13.1 | 43.8 | 0.30 | 2.E-73 |
| Cdca3 | chr6 | 124780193 | 124783719 | Cell division cycle-associated protein 3 | 9.9 | 33.3 | 0.30 | 2.E-56 |
| Cdc25c | chr18 | 34892650 | 34911187 | Cell division cycle 25 homolog C | 3.6 | 13.4 | 0.27 | 2.E-26 |
| Not differentially expressed cells | ||||||||
| Cdk18 | chr1 | 134010123 | 134036262 | Cell division protein kinase 18 | 10.5 | 8.0 | 1.30 | 8.E-03 |
| Cdk8chr5+ | chr5 | 147042825 | 147114450 | Cell division protein kinase 8 | 9.8 | 7.6 | 1.29 | 1.E-02 |
| Cdc14b | chr13 | 64290579 | 64376296 | Cell division cycle 14b | 15.0 | 11.7 | 1.28 | 3.E-03 |
| Ccnb1 | chr13 | 101548693 | 101556441 | G2/M-specific cyclin-B1 | 3.3 | 2.6 | 1.27 | 2.E-01 |
| Cdk8chr10+ | chr10 | 40069113 | 40203624 | Cell division protein kinase 8 | 15.0 | 12.2 | 1.23 | 1.E-02 |
| Cdk19 | chr10 | 40059369 | 40203624 | Cell division cycle 2-like 6 | 15.2 | 12.4 | 1.22 | 2.E-02 |
| mKIAA0432 | chr17 | 45544744 | 45570656 | Cell division cycle 5-like (Schizosaccharomyces pombe) | 19.5 | 16.4 | 1.19 | 2.E-02 |
| Scaper | chr9 | 55397689 | 55785922 | S-phase cyclin A-associated protein | 13.1 | 11.3 | 1.16 | 9.E-02 |
| Nucks1 | chr1 | 133807034 | 133832898 | Nuclear ubiquitous casein and cyclin-dependent | 50.2 | 44.1 | 1.14 | 3.E-03 |
| Cdc14a | chr3 | 115975470 | 116126950 | Dual specificity protein phosphatase CDC14A | 2.3 | 2.1 | 1.07 | 7.E-01 |
| Cdc42 | chr4 | 136875610 | 136913652 | Cell division control protein 42 homolog | 122.8 | 116.4 | 1.06 | 6.E-02 |
| Fam58b | chr11 | 78564007 | 78565231 | Cyclin-related protein FAM58A | 8.3 | 8.0 | 1.03 | 7.E-01 |
| Cdk17 | chr10 | 92623620 | 92729818 | Cell division protein kinase 17 | 26.4 | 25.9 | 1.02 | 7.E-01 |
| Cdc25a | chr9 | 109778082 | 109796394 | M-phase inducer phosphatase 1 | 8.0 | 7.9 | 1.02 | 8.E-01 |
| Ccnjl | chr11 | 43342285 | 43400499 | Cyclin-J–like protein | 3.6 | 3.5 | 1.01 | 9.E-01 |
| Ccnd3 | chr17 | 47641999 | 47736637 | G1/S-specific cyclin-D3 | 91.3 | 90.6 | 1.01 | 8.E-01 |
| Cdk5 | chr5 | 23924059 | 23929348 | Cell division protein kinase 5 | 18.7 | 18.7 | 1.00 | 1.E+00 |
| Ccng1 | chr11 | 40562053 | 40568788 | Cyclin-G1 | 191.9 | 193.1 | 0.99 | 8.E-01 |
| Ccnf | chr17 | 24360176 | 24388354 | Cyclin-F | 4.9 | 5.0 | 0.99 | 1.E+00 |
| Cdc42se2 | chr11 | 54530916 | 54601205 | CDC42 small effector protein 2 | 24.6 | 24.8 | 0.99 | 9.E-01 |
| Cdk11b | chr4 | 154998977 | 155024041 | Cell division cycle 2-like 1 | 66.5 | 67.8 | 0.98 | 6.E-01 |
| Cdk9 | chr2 | 32561301 | 32568304 | Cell division protein kinase 9 | 16.6 | 17.2 | 0.97 | 7.E-01 |
| Cdc26 | chr4 | 62042609 | 62069657 | Cell division cycle 26 | 18.4 | 19.0 | 0.96 | 6.E-01 |
| Cdc42ep2 | chr19 | 5916127 | 5924816 | Cdc42 effector protein 2 | 36.7 | 38.8 | 0.95 | 3.E-01 |
| Pigu | chr2 | 155103987 | 155183160 | Cell division cycle protein 91-like 1 | 22.6 | 24.5 | 0.92 | 2.E-01 |
| Cdc40 | chr10 | 40552698 | 40602949 | Pre-mRNA–processing factor 17 | 10.7 | 11.8 | 0.91 | 3.E-01 |
| Mrrf | chr2 | 35991917 | 36045803 | Ribosome-recycling factor, mitochondrial | 35.7 | 39.4 | 0.91 | 5.E-02 |
| Ccnh | chr13 | 85329081 | 85353328 | Cyclin-H | 17.3 | 19.5 | 0.89 | 1.E-01 |
| Inca1 | chr11 | 70501862 | 70513657 | Inhibitor of CDK interacting with cyclin A1 | 1.9 | 2.3 | 0.82 | 4.E-01 |
| Cdkl2 | chr5 | 92435100 | 92472044 | Cyclin-dependent kinase-like 2 | 2.4 | 3.0 | 0.81 | 3.E-01 |
| Cdk10 | chr8 | 125748740 | 125756156 | Cell division protein kinase 10 | 12.1 | 14.8 | 0.81 | 2.E-02 |
| Cdc7 | chr5 | 107393340 | 107413450 | Cell division cycle 7-related protein kinase | 3.0 | 3.8 | 0.78 | 2.E-01 |
| Rmnd1chr19+ | chr19 | 12979720 | 12981475 | Required for meiotic nuclear division protein 1 | 13.5 | 17.3 | 0.78 | 2.E-03 |
| Cdcrel-1 | chr16 | 18621903 | 18630031 | Cell division cycle 1 homolog; aka septin 5 | 8.2 | 10.7 | 0.76 | 1.E-02 |
| Cdk14 | chr5 | 4803384 | 5380251 | Cell division protein kinase 14 | 7.1 | 9.8 | 0.72 | 3.E-03 |
| Cdkl3 | chr11 | 51817722 | 51898169 | Cyclin-dependent kinase-like 3 | 6.2 | 8.8 | 0.70 | 3.E-03 |
| Cdc45 | chr16 | 18780539 | 18812065 | Cell division control protein 45 homolog | 3.7 | 5.5 | 0.67 | 9.E-03 |
| Cdca7 | chr2 | 72314275 | 72324947 | Cell division cycle-associated protein 7 | 3.2 | 5.3 | 0.60 | 1.E-03 |
Chr, chromosome; end, mm9 base pair position; fold, fold difference between WT and Mybpct/t gene expression at P10; P value, probability that the difference between Wt and Mybpct/t gene expression would occur by chance; start, mm9 base pair position. WT (wild-type) and Mybpct/t RNA expression of selected cell cycle genes is presented in reads per mapped reads.
Neonatal Depletion of Cardiac MYBPC Impairs Myocyte Maturation in Wild-Type Mice.
To confirm that altered myocyte maturation in Mybpct/t hearts was due to the myocyte-autonomous deficiency of MYBPC, we used the cardiotropic adeno-associated virus serotype 9 (AAV9) (31, 32) to deliver Mybpc3-specific shRNA to the neonatal heart. Two Mybpc3-specific shRNAs that targeted different regions of Mybpc3 RNA (Fig. S2A) were constructed with a colinear EGFP reporter gene downstream of the cardiac troponin T (cTnT) promoter, so as to exclude nonmyocyte expression (Fig. S2B).
Fig. S2.

AAV-mediated depletion of Mybpc3 in wild-type mice. (A) Location of 21-bp shRNA sequences targeting the Mypbc3 transcript. (B) Schematic representation of the AAV vector containing a colinear cTnT promoter and Mybpc3 shRNA-1 or shRNA-2.
AAV9-Mybpc3-shRNA was injected into the thoracic cavity of P1 wild-type neonates [5 × 1013 viral genomes (vg)/kg], and EGFP fluorescence was assessed to monitor shRNA expression. Cardiac EGFP fluorescence was detected 48 h after virus injection and continued for at least 5 mo (Fig. 4A) but was absent from all other organs (33). EGFP fluorescence was present in 60–80% of adult myocytes isolated from viral-transduced mice (Fig. 4B). Both of these shRNA constructs (denoted AAV9-Mybpc3-shRNA) had comparable efficiency in attenuating in vivo Mybpc3 RNA expression (Fig. 4C).
Fig. 4.

Depletion of Mybpc3 RNA by AAV-shRNA. (A) Hearts from three wild-type mouse hearts harvested 48 h after transduction with AAV9 lacking EGFP (center, arrow) or with AAV9-EFFP (two flanking center heart) visualized under fluorescent light. Right panel shows two hearts harvested 5 mo after AAV9-EFFP transduction. (Scale bar, 2 mm.) (B) Fluorescent microscopy of LV myocytes stained by DAPI (blue) and EGFP (green) isolated from wild-type mice transduced with AAV-LacZ shRNA (control, Upper panels) or AAV-Mybpc3 shRNA (Lower panels). DAPI was shown in red in merged images. (Scale bar, 50 µm.) (C) Quantitative real-time PCR analysis of LV Mybpc3 expression from mice transduced with AAV-Mybpc3 shRNA-1 or shRNA-2. Transcript levels were normalized to expression of the control AAV-LacZ shRNA. Data from three mice per group are presented (mean ± SD). (D) Distribution of mononuclear, binuclear, and multinuclear EGFP-expressing myocytes from 4-wk-old wild-type mice transduced (at P1) with AAV-LacZ shRNA (gray), AAV-Mybpc3 shRNA-1 (black), and AAV-Mybpc3 shRNA-2 (white). Data are presented from four mice per genotype, 200–300 cells per mouse (mean ± SD).
Four weeks after viral injection, EGFP-positive myocytes were isolated, and nuclear morphology was examined. Wild-type mice infected with a control shRNA (that targeted LacZ RNA; Materials and Methods) had ∼5% mono- and 90% binuclear myocytes (Fig. 4D). Among EGFP-positive myocytes, wild-type hearts injected with Mybpc3-specific shRNA had 5–6-fold more mononuclear myocytes than hearts injected with control shRNA (Fig. 4D). Mice infected with either of the two distinct Mybpc3-specific shRNA constructs showed similar results (Fig. 4D).
Myocyte Proliferation After Cardiac Injury in Mybpct/t Mice.
We considered whether Mybpct/t mice have better cardiac regenerative capacity after injury than wild-type mice, as myocyte division may play an important role in regeneration. To test this hypothesis, we performed sham or LV apical resections (26, 34), excising ∼15% of the total LV mass in P10 wild-type and Mybpct/t mice (Fig. 5 A–I). There is no difference in survival of the two genotypes after apical resection. Surviving neonates received a BrdU pulse 1 d later, and at P17 BrdU incorporation was assessed in hearts stained with cTnT-specific antibody, to exclude nonmyocytes, and with Ki67 antibody to detect cell cycle activity (Fig. 5 J and K). Sections from sham-operated Mybpct/t LV (47 ± 5 BrdU+;cTnT+ cells/mm2) had significantly more cTnT-positive, BrdU-labeled cells than sham-operated wild-type hearts (5 ± 0 BrdU+;cTnT+ cells/mm2; P = 0.005) or Ki67-labeled (18 ± 8 Ki67+;cTnT+ cells/mm2 vs. 3 ± 3 Ki67+;cTnT+ cells/mm2; P = 0.05; Fig. 5 J and K). Apical resection did not increase BrdU incorporation into cTnT-positive cells from either wild-type and Mybpct/t mice, and Ki67 staining was not increased in either wild-type or Mybpct/t LV (Fig. 5 J and K).
Fig. 5.

Cell proliferation in P10 wild-type and Mybpct/t mice after apical resection. Sections from sham-operated or LV apical resected hearts, 17 d postresection, from wild-type (A–C) or Mybpct/t (D–I) mice stained with hematoxylin/eosin (A, B, and D–F) or von Kossa (C and G–I). Dashed line indicates the resection plane. Note the marked calcium precipitate in apical resected Mybpct/t (H and I) but not wild-type (C) hearts. [Scale bar, (A, C–E, G, and H) 1 mm and (B, F, and I) 200 µm.] Quantification of BrdU-labeled, cTnT-positive myocytes (J, BrdU+;cTnT+) and Ki67-positive, cTnT-positive myocytes (K, Ki67+;cTnT+) isolated 17 d after sham (gray) or LV apical resection (black) of wild-type and Mybpct/t mice. Ten sections were counted from each of four mice per genotype, and cell counts are reported as stained cells per mm2 (mean ± SD).
Notably, dystrophic calcification, a marker of cell death and necrosis, was observed in apical resected regions in all Mybpct/t mice, but not in apical resected wild-type or sham-operated Mybpct/t mice (Fig. 5 B, H, and I). As cTnT-positive cells without nuclei surrounded these necrotic foci, we deduced that Mybpct/t myocytes were particularly sensitive to dystrophic calcification and death. The demise of Mybpct/t myocytes was unlikely to reflect inadequate vascular supply, as staining by the vascular endothelial marker CD31 was comparable in wild-type and Mybpct/t mice (Fig. S3). Dystrophic calcification also occurred in both apical and basal regions when apical resection was carried out in P1 Mybpct/t mice (Fig. S4). We suggest that Mybpct/t hearts were more susceptible to necrotic death after resection due to altered biomechanical properties that increased energy requirements due to enhanced actomyosin force (35) and/or elevated wall stress (24) in Mybpct/t myocytes. Mypbct/t mice appear to have reduced regenerative capacity compared with wild-type mice.
Fig. S3.

Cardiac, endothelial, and cell cycle markers expressed 7 d post-LV apical resection of P10 wild-type (A and B) and Mybpct/t mice (C and D). Heart sections were stained for DAPI or immunostained to detect cTnT (green) and Ki67 (red). An asterisk denotes location of dystrophic calcification in hematoxylin/eosin-stained Mybpct/t sections (Fig. S4). [Scale bars, (A and C) 200 μm; (B and D) 0.5 mm.]
Fig. S4.

Calcium precipitates in Mybpct/t mice after apical resection at P1. Sections from sham-operated or LV apical resected hearts, 7 d postresection, from wild-type (A and B) or Mybpct/t (C–H) mice stained with hematoxylin/eosin (A and C–E) or von Kossa (B and F–H). Note the marked calcium precipitate in apical resected Mybpct/t (G and H) but not wild-type (B) or sham (F) Mybpct/t hearts.
Discussion
MYBPC is critical for normal cardiac structure and function. Absence of MYBPC in the adult heart compromises the stiffness and rigidity of myofilaments (36, 37), reduces sarcomere packing density (24), and perturbs contractile performance (16), which might account for increased myocyte width in Mybpct/t mice and contribute to increased LV wall thickness (Fig. S1 C and D). We show that MYBPC also has unexpected roles in postnatal maturation of myocytes. Postnatal Mybpct/t myocytes appear morphologically immature, based on dimensions and a single nucleus, unlike postnatal wild-type myocytes. During neonatal development, Mybpct/t myocytes undergo additional rounds of division, resulting in increased myocyte numbers in Mybpct/t hearts (Fig. 6). Given its established role in myofilament lattice rigidity (1, 7, 12, 17, 18, 24), we conclude that MYBPC is critical for inhibiting postnatal myocyte cytokinesis, an essential step in myocyte maturation accompanying cell cycle exit.
Fig. 6.

A schematic of postnatal myocyte development in Mybpct/t and wild-type mice. In the perinatal period, wild-type myocytes develop dense mature myofibrillar structures as they undergo a final round of DNA replication without cytokinesis, resulting in 95% binuclear myocytes. Mybpct/t myocytes have reduced myofibrillar density and less rigid sarcomere, prolonged expression of cell cycle markers, resulting in more myocytes and larger proportions of mononuclear myocytes.
To progress through the cell cycle, myocytes must disassemble both the cytoskeleton and the contractile apparatus, the thick and thin myofilaments of the sarcomere. High-resolution confocal microscopy of immunostained neonatal myocytes has identified two phases of sarcomere dissolution. Before metaphase, proteins in the z disk and thin filament disassemble, whereas thick filament proteins, including myosin and MYBPC, maintain a mature, cross-striated pattern. Late in anaphase, thick filament proteins disassemble and remain dispersed until cytokinesis is complete (38).
Before cell cycle exit, neonatal mouse myocytes undergo karyokinesis without cytokinesis (39, 40). This phase in myocyte maturation is characterized by synchronized changes in the expression of cell cycle molecules; levels of activators (Cdk2, Cdk3, Cdk4, Cdk cofactors, and Ccnd1) are decreased, and levels of inhibitors (p21, p27KIP1, TSC2, p130, and Rb) are increased. Transgenic overexpression or genetic depletion in the heart of some cell cycle regulators (27, 41) has been demonstrated to partially overcome neonatal myocyte cell cycle arrest and enable additional rounds of cell division. Recent studies demonstrate that these reciprocal events are mediated in part by the transcriptional activator 14-3-3ε (42), the transcription factor Meis1 (43), and activation of the DNA damage response pathway induced by reactive oxygen species in neonatal mice (44).
MYBPC deficiency also impacted the regenerative capacity of the P10 mouse heart. Apical resection did not increase BrdU incorporation or Ki67 staining among cTnT-positive cells in Mybpct/t mice as it did in wild-type mice (Fig. 5 and Fig. S3). Moreover, as resected Mybpct/t hearts showed necrosis and dystrophic calcification, we suspect that MYBPC-deficient myofilaments are less resistant to sheer forces and perhaps more susceptible to depolymerization, as has been seen ex vivo (36, 45). These observations hint at the dichotomy inherent in a rigid sarcomere, a structure that can support the stress of contraction but that also impedes myocyte regeneration.
The findings presented here suggest two independent mechanisms by which MYBPC3 mutations can alter cardiac morphology. Individuals carrying heterozygous MYBPC3 mutations develop HCM (46), characterized by cardiac hypertrophy (increased heart mass and altered morphology). These changes in size and shape are due, at least in part, to the increased size of myocytes (hypertrophy) (47). By contrast, individuals homozygous for MYBPC3 mutations develop cardiac dilation, which we conclude is due to myocyte hyperplasia (increased numbers of myocytes). This myocyte hyperplasia is associated with increased numbers of mononuclear myocytes that may have reduced contractile function. Defining the relationship of homozygous MYBPC3 DCM to other more common forms of DCM will provide further insights into this clinically important pathophysiology.
Materials and Methods
Mouse Studies.
All mouse studies were performed with approved protocols in compliance with the Association for the Assessment and Accreditation of Laboratory Animal Care and Harvard Medical School. Mybpct/t (16) and wild-type mice (129SvEv background) were studied. The Mybpct/t alleles contain a PGK-neomycin resistance gene that disrupts exon 30 and is predicted to encode from a truncated peptide that terminates with amino acid residue 1,064 of the 1,270 residues in wild-type cardiac MYBPC. Protein chemical studies have demonstrated that this allele produces less than 10% of the normal amount of cardiac MYBPC.
Myocyte Isolation.
Ventricular myocytes were isolated from P1 to P10 mice using a modified collagenase dissociation protocol. After anesthetizing mice [2% (vol/vol) isoflurane inhalation], the thoracic cavity was opened and LV was perfused (2 mL/min) with prewarmed 37 °C buffer (126 mM NaCl, 4.4 mM KCl, 1 mM MgCl2, 4 mM NaHCO3, 30 mM 2,3-butanedione monoxime, 10 mM Hepes, 11 mM Glucose, 0.5 mM EDTA) with 0.09% Collagenase Type I (Worthington), 0.125% Trypsin, and 25 µM CaCl2 for 2 min. The perfused heart was then excised and placed in buffer with 100 µM CaCl2 and 2% BSA for 10 min at 37 °C. Ventricular tissue was minced, and myocytes were dispersed by gentle trituration of minced tissue through a wide bore disposable serologic pipette. The dispersed cells were filtered through a 100 µm nylon mesh and washed twice by centrifugation (50 × g for 3 min). The resulting cell pellet was suspended in 1 mL buffer with 100 µM CaCl2 and fixed by the addition of 1 mL 2% (wt/vol) paraformaldehyde in PBS and incubated on ice for 20 min. Fixed myocytes were washed twice in PBS by centrifugation (50 × g for 3 min) and stored at 4 °C.
For ventricular myocyte isolation from 3–4-wk-old mice, immediately after sacrifice, hearts were excised, the ascending aorta was quickly cannulated, and the hearts were perfused for 5 min with warm (37 °C) calcium Tyrode’s buffer (135 mM NaCl, 4 mM KCl, 0.33 mM NaH2PO4, 1.2 mM MgSO4, and 10 mM Hepes, pH 7.40), followed by 10–15 min of perfusion with collagenase B (0.56 mg/mL, Roche), collagenase D (0.48 mg/mL, Roche), and protease XIV (0.07 mg/ml, Sigma). The hearts were minced and filtered as described above. Cells were plated onto laminin-precoated coverslips (1 μg/cm2; Invitrogen) and cultured for 1 h in MEM (Sigma) containing 10 mM 2,3-butanedione monoxime to extract myocytes from other cardiac cells.
Immunohistochemistry.
Histochemical analyses were performed on heart sections fixed in 4% (wt/vol) paraformaldehyde overnight. Sections were treated with xylene (to remove paraffin), rehydrated, and permeabilized in 0.1% (vol/vol) Triton-X100 in PBS. Sections were incubated with primary antibodies applied at 1:200 dilution (unless otherwise indicated) in 0.1% (wt/vol) BSA in PBS overnight at 4 °C, and nonspecific antibody binding was blocked by 1.5% (vol/vol) FCS in PBS. Primary antibodies included the following: BrdU (rat anti-BrdU, Abcam ab6326, 1:200), Ki67 (rabbit anti-Ki67, Abcam ab15580, 1:200), cTnT (mouse anti-cTnT, Abcam ab8295, 1:200), cardiac troponin-I (rabbit anti-Tnni3, Abcam ab56357, 1:200), WGA (Invitrogen W32466, 1:400), pH3 (rabbit anti-pH3, Millipore 06–570, 1:250), and Aurora B (rabbit anti-Aurora B, Abcam ab2254, 1:250). Sections were washed in PBS and fluorophore-conjugated secondary antibody (Molecular Probes) diluted 1:200 in 1% FCS or donkey serum. Nuclei were counterstained with 0.3 µg/mL propidium iodide (Molecular Probes) or 2 µg/mL of 4’,6-diamidino-2-phenylindole (DAPI; Sigma). Myocytes were identified by visualizing sarcomeres with Nomarski (differential interference contrast) light microscope optics and cTnT expression. Fluorescent images of histological sections were captured using the Leica TCS NT confocal microscope system and subsequently analyzed and assembled using Image J and Adobe Photoshop software.
Myocyte Quantification.
WGA-stained heart sections were imaged using a Zeiss confocal microscope, and images were processed after color inversion, so that individual cells were highlighted as “particles.” The “Watershed” algorithm (Fiji; fiji.sc/) was used to resolve distinct particles that were close together. Particles were included based on the size of cardiomyocytes ranging from 150 to 3,000 pixels/cell.
AAV Production and Purification.
AAV vectors were packaged into AAV9 capsid by the triple transfection method using helper plasmids pAdΔF6 and plasmid pAAV2/9 (Penn Vector Core). We used 50 μg of plasmid DNA per 15-cm cell culture plate. Three days after transfection, AAV vectors were purified in Optiprep density gradient medium (d-1556, Sigma) by centrifugation and stored at −80 °C.
shRNA Vector Construction.
Two shRNA constructs specific for 21-base-pair sequences corresponding to cardiac Mypbc3 were constructed. The following targeted sequences were used: Mypbc3-specific shRNA-1, ccagagaaggcagaatctgaa; Mypbc3-specific shRNA-2, aagggtttgcctgcaacctgt; and shRNA control targeting LacZ, gactacacaaatcagcgattt.
RNA-Seq.
Hearts from mice were rapidly isolated, placed in room temperature PBS to evacuate blood, and then immersed in RNALater (Qiagen) at room temperature. We used 2 μg of total ventricular RNA to construct RNAseq sequencing libraries (48). At least 20 million 50-bp paired-end DNA reads were obtained from each library using the Illumina HiSeq2500. Data were processed as previously described (48).
Apical Resection.
LV apical resection procedures were performed as described (26). In brief, mice were anesthetized on ice to induce deep transient sedation, apnea, and asystole. The thoracic cavity was opened at the fourth intercostal space and the LV exposed. Approximately 15% of the ventricular apex was resected. Successful apical resection was assessed by visualization of the ventricular chamber immediately after resection. Following surgery, mice were rapidly warmed and monitored for viability. Sham-operated mice underwent identical procedures but without LV apex resection. We have independently verified that robust heart regeneration occurs in the neonatal mouse apical resection model (34).
EdU/BrdU Labeling.
To assess DNA synthesis in isolated myocytes, neonatal pups received an i.p. injection of EdU (Life Technologies, C10339, 5 mg/kg) for the first 5 d. To label cells with ongoing DNA synthesis, surviving mice subjected to sham or apical-resection surgery received a pulse (two s.c. injections 12 h apart) of BrdU (Sigma, 20 mg/mL solution in sterile PBS, 100 mg/kg dose) on the first day post-resection.
Statistical Analyses.
Significance was assessed by two-sample Student’s t test on selected groups, assuming two-tail heteroscedastic distributions. Multiple group comparison was assessed by ANOVA.
Acknowledgments
This work was supported in part by funding from NIH Grants 5R01HL080494, 5R01HL084553, and 1U01HL098166 (to C.E.S. and J.G.S.); NIH Grants HL117986 and AG040019 and the Leducq Transatlantic Network (to R.T.L.); NIH Grant F32HL117595 (to C.C.O.); NIH Grants R01HL085487 and R15HL124458 (to B.K.M.); and the Howard Hughes Medical Institute (C.E.S. and J.J.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1511004112/-/DCSupplemental.
References
- 1.Maron BJ, et al. American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 2.Go AS, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics—2014 update: A report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mann DL, Zipes DP, Libby P, Bonow RO, editors. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. Elsevier Mosby/Saunders; Philadelphia, PA: 2014. [Google Scholar]
- 4.Fatkin D, Seidman CE, Seidman JG. Genetics and disease of ventricular muscle. Cold Spring Harb Perspect Med. 2014;4(1):a021063. doi: 10.1101/cshperspect.a021063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123(1):19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mestroni L, Taylor MR. Genetics and genetic testing of dilated cardiomyopathy: A new perspective. Discov Med. 2013;15(80):43–49. [PMC free article] [PubMed] [Google Scholar]
- 7.Moss RL, Fitzsimons DP, Ralphe JC. Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ Res. 2015;116(1):183–192. doi: 10.1161/CIRCRESAHA.116.300561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gautel M, Fürst DO, Cocco A, Schiaffino S. Isoform transitions of the myosin binding protein C family in developing human and mouse muscles: Lack of isoform transcomplementation in cardiac muscle. Circ Res. 1998;82(1):124–129. doi: 10.1161/01.res.82.1.124. [DOI] [PubMed] [Google Scholar]
- 9.Yin Z, Ren J, Guo W. Sarcomeric protein isoform transitions in cardiac muscle: A journey to heart failure. Biochim Biophys Acta. 2015;1852(1):47–52. doi: 10.1016/j.bbadis.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfuhl M, Gautel M. Structure, interactions and function of the N-terminus of cardiac myosin binding protein C (MyBP-C): Who does what, with what, and to whom? J Muscle Res Cell Motil. 2012;33(1):83–94. doi: 10.1007/s10974-012-9291-z. [DOI] [PubMed] [Google Scholar]
- 11.Ackermann MA, Kontrogianni-Konstantopoulos A. Myosin binding protein-C: A regulator of actomyosin interaction in striated muscle. J Biomed Biotechnol. 2011;2011:636403. doi: 10.1155/2011/636403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flashman E, Redwood C, Moolman-Smook J, Watkins H. Cardiac myosin binding protein C: Its role in physiology and disease. Circ Res. 2004;94(10):1279–1289. doi: 10.1161/01.RES.0000127175.21818.C2. [DOI] [PubMed] [Google Scholar]
- 13.Sequeira V, Witjas-Paalberends ER, Kuster DW, van der Velden J. Cardiac myosin-binding protein C: Hypertrophic cardiomyopathy mutations and structure-function relationships. Pflugers Archiv. 2014;466(2):201–206. doi: 10.1007/s00424-013-1400-3. [DOI] [PubMed] [Google Scholar]
- 14.Kuster DW, Sadayappan S. MYBPC3’s alternate ending: Consequences and therapeutic implications of a highly prevalent 25 bp deletion mutation. Pflugers Archiv. 2014;466(2):207–213. doi: 10.1007/s00424-013-1417-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhandapany PS, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41(2):187–191. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McConnell BK, et al. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104(12):1771. doi: 10.1172/JCI7377C1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrier L, et al. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res. 2004;63(2):293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Harris SP, et al. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90(5):594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 19.Mearini G, et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun. 2014;5:5515. doi: 10.1038/ncomms6515. [DOI] [PubMed] [Google Scholar]
- 20.Merkulov S, Chen X, Chandler MP, Stelzer JE. In vivo cardiac myosin binding protein C gene transfer rescues myofilament contractile dysfunction in cardiac myosin binding protein C null mice. Circ Heart Fail. 2012;5(5):635–644. doi: 10.1161/CIRCHEARTFAILURE.112.968941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96(1):110–118. doi: 10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 22.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271(5 Pt 2):H2183–H2189. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 23.Snir M, et al. Assessment of the ultrastructural and proliferative properties of human embryonic stem cell-derived cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;285(6):H2355–H2363. doi: 10.1152/ajpheart.00020.2003. [DOI] [PubMed] [Google Scholar]
- 24.Palmer BM, et al. Effect of cardiac myosin binding protein-C on mechanoenergetics in mouse myocardium. Circ Res. 2004;94(12):1615–1622. doi: 10.1161/01.RES.0000132744.08754.f2. [DOI] [PubMed] [Google Scholar]
- 25.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87(2):521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porrello ER, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soonpaa MH, et al. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest. 1997;99(11):2644–2654. doi: 10.1172/JCI119453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradford JA, Clarke ST. Dual-pulse labeling using 5-ethynyl-2'-deoxyuridine (EdU) and 5-bromo-2'-deoxyuridine (BrdU) in flow cytometry. Curr Protoc Cytom. 2011;Chapter 7:Unit 7.38. doi: 10.1002/0471142956.cy0738s55. [DOI] [PubMed] [Google Scholar]
- 29.Gerdes J, et al. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133(4):1710–1715. [PubMed] [Google Scholar]
- 30.Poon RY. Aurora B: Hooking up with cyclin-dependent kinases. Cell Cycle. 2013;12(7):1019–1020. doi: 10.4161/cc.24307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: In vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18(1):43–52. doi: 10.1038/gt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther. 2005;5(3):285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- 33.Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342(6154):111–114. doi: 10.1126/science.1236921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryant DM, et al. A systematic analysis of neonatal mouse heart regeneration after apical resection. J Mol Cell Cardiol. 2015;79:315–318. doi: 10.1016/j.yjmcc.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michalek AJ, et al. Phosphorylation modulates the mechanical stability of the cardiac myosin-binding protein C motif. Biophys J. 2013;104(2):442–452. doi: 10.1016/j.bpj.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer BM, et al. Roles for cardiac MyBP-C in maintaining myofilament lattice rigidity and prolonging myosin cross-bridge lifetime. Biophys J. 2011;101(7):1661–1669. doi: 10.1016/j.bpj.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337(6099):1215–1218. doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahuja P, Perriard E, Perriard JC, Ehler E. Sequential myofibrillar breakdown accompanies mitotic division of mammalian cardiomyocytes. J Cell Sci. 2004;117(Pt 15):3295–3306. doi: 10.1242/jcs.01159. [DOI] [PubMed] [Google Scholar]
- 39.Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28(8):1737–1746. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- 40.Li F, Wang X, Gerdes AM. Formation of binucleated cardiac myocytes in rat heart: II. Cytoskeletal organisation. J Mol Cell Cardiol. 1997;29(6):1553–1565. doi: 10.1006/jmcc.1997.0403. [DOI] [PubMed] [Google Scholar]
- 41.Poolman RA, Li JM, Durand B, Brooks G. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res. 1999;85(2):117–127. doi: 10.1161/01.res.85.2.117. [DOI] [PubMed] [Google Scholar]
- 42.Kosaka Y, et al. 14-3-3ε plays a role in cardiac ventricular compaction by regulating the cardiomyocyte cell cycle. Mol Cell Biol. 2012;32(24):5089–5102. doi: 10.1128/MCB.00829-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahmoud AI, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497(7448):249–253. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puente BN, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157(3):565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulikovskaya I, McClellan GB, Levine R, Winegrad S. Multiple forms of cardiac myosin-binding protein C exist and can regulate thick filament stability. J Gen Physiol. 2007;129(5):419–428. doi: 10.1085/jgp.200609714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115(3):518–526. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marston S, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105(3):219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 48.Christodoulou DC, et al. 5’RNA-Seq identifies Fhl1 as a genetic modifier in cardiomyopathy. J Clin Invest. 2014;124(3):1364–1370. doi: 10.1172/JCI70108. [DOI] [PMC free article] [PubMed] [Google Scholar]