

Abstract
Background
Adenocarcinoma of the pancreas is one of the most aggressive cancer diseases affecting the human body. The oncogenic potential of this type of cancer is mainly characterized by its extreme growth rate triggered by the activation of signaling cascades. Modern oncological treatment strategies aim at efficiently modulating specific signaling and transcriptional pathways. Recently, anti-tumoral potential has been proven for several substances that are not primarily used in cancer treatment. In some tumor entities, for example, administration of glutamate antagonists inhibits cell proliferation, cell cycle arrest, and finally cell death.
To attain endogenic proof of NMDA receptor type expression in the pancreatic cancer cell lines PaTu8988t and Panc-1 and to investigate the impact of ketamine, s-ketamine, and the NMDA receptor antagonist MK 801 on proliferation, apoptosis, and necrosis in pancreatic carcinoma.
Methods
Cell proliferation was measured by means of the ELISA BrdU assay, and the apoptosis rate was analyzed by annexin V staining. Immunoblotting were also used.
Results
The NMDA receptor type R2a was expressed in both pancreatic carcinoma cell lines. Furthermore, ketamine, s-ketamine, and MK 801 significantly inhibited proliferation and apoptosis.
Conclusions
In this study, we showed the expression of the NMDA receptor type R2a in pancreatic cancer cells. The NMDA antagonists ketamine, s-ketamine, and MK 801 inhibited cell proliferation and cell death. Further clinical studies are warranted to identify the impact of these agents on the treatment of cancer patients.
Keywords: Ketamine, S-ketamine, MK 801, Proliferation, Apoptosis, Necrosis, NMDA, Pancreatic carcinoma, Cancer
Background
Malignant cancer diseases are one of the major causes of death worldwide. In 2008, malignant tumors resulted in about 7.6 million deaths (approximately 13 % of all causes of death), and this figure is expected to rise to over 13 million deaths per year in 2030 [1]. One type of malignant tumor that nearly always results in death because of its aggressive behavior is adenocarcinoma of the pancreas [2]. Due to the lack of characteristic early symptoms and suitable screening tests, 50 % of patients are already affected by infiltrated lymph nodes at the time of diagnosis. According to current knowledge, this tumor stage is no longer considered curable [3]. Most of these tumors are classified as surgically incurable and have a poor prognosis for treatment [4]. Thus, the 5-year survival rate of less than 5 % is particularly unfavorable [5]. Apart from surgery, alternative and curatively successful treatment options are rare because pancreatic carcinoma is extremely resistant to radiation treatment and chemotherapy [6]. Recently, however, anti-tumoral potential has been proven in several substances that are not primarily used in cancer treatment, and these findings may open up new therapeutic options in the future [7]. Glutamate antagonists, for example, inhibit cell proliferation [8–10] and induce cell cycle arrest [11] and cell death [12] in some tumor entities. MK 801 was used for this purpose in many preclinical studies [13], but the psychotropic effects of this glutamate antagonist significantly restrict its clinical application.
Ketamine, one of the most common N-methyl-D-aspartate (NMDA) receptor antagonists used in clinical practice, was first described in the literature in 1965 and approved as an anesthetic for clinical use in 1970 [14]. Ketamine is a racemic derivative consisting of the two enantiomers s(+)ketamine and r(−)ketamine. S(+)ketamine has a four times higher binding capacity at the phencyclidine binding site of the NDMA receptor than r(−)ketamine [15]. Additionally, ketamine interacts with opioid, monaminic, and muscarinic receptors as well as with voltage-gated Ca2+ channels and the AMPA receptor [16]. However, the effect of ketamine fundamentally differs from that of other anesthetic drugs. Because even subnarcotic doses of ketamine result in pronounced analgesia [17], this NMDA receptor antagonist is often used in preventive pain management but also in cancer pain therapy in patients with opiate-resistant pain [18].
Aim of this study was to scan NMDA receptor type expression in pancreatic cancer cells. A further objective was to investigate the effects of ketamine, s-ketamine, and the uncompetitive NMDA receptor antagonist MK 801 on cell proliferation, apoptosis, and necrosis in pancreatic carcinoma.
Materials and Methods
Cell lines
The human pancreatic adenocarcinoma cell lines PaTu8988t and Panc-1 were obtained from Professor Ellenrieder (Philipps University of Marburg, Germany). PaTu8988t and Panc-1 cells were maintained in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, Steinsheim, Germany) supplemented with 10 % fetal calf serum (FCS) (Sigma-Aldrich) and 5 % Myco Zap (Lonza Verviers SPRL, Verviers, Belgium). Cells were cultured at 37 °C in humidified CO2 atmosphere (5 %) and maintained in monolayer culture. Experiments were done with cells at ~70-80 % confluence.
Antibodies and reagents
Ketamine, s-ketamine, and MK 801 were purchased from Sigma-Aldrich. Final concentrations were achieved by diluting drugs in standard growth media. All solutions were prepared freshly prior to use. For immunoblotting, membranes were probed with antibodies against NMDA R1, NMDA R2a, NMDA R2b (BD Bioscience, Heidelberg, Germany), and ß-Actin (Sigma-Aldrich).
Subcellular fractionation and immunoblotting
Cells were washed twice with cold DPBS and collected by centrifugation at 4000 rpm at 4 °C for 10 min. Lysates were then resuspended in RIPAE-Buffer (5 mL Triton X100, 190 mg EDTA, 0.5 g SDS, 2.5 g Deoxycolid Acid, 500 mL DPBS, proteinase inhibitors) for 15 min and centrifuged at 13.000 rpm for 30 min. Supernatants were transferred to new cups and incubated on ice. 30 μg of total lysates were analyzed by SDS-PAGE and blotted onto nitrocellulose. Upon protein extraction and gel transfer, membranes were washed in TBS washing buffer and incubated with peroxidase-conjugated secondary antibodies. Immunoreactive proteins were visualized by means of an enhanced chemiluminescence detection system (Western Blotting Detection Reagent, GE Healthcare).
Cell proliferation
The quantification of cell proliferation was based on the measurement of BrdU incorporation during DNA synthesis. The test was done according to the manufactures protocol (Cell proliferation ELISA BrdU, Roche applied science, Mannheim Germany). In brief, cells were incubated with 100 μL of the test compounds for 48 h (0.1-1000 μM ketamine, s-ketamine, and MK 801). After 32 h incubation, cells were additionally treated with BrdU labeling solution for the last 16 h. The culture medium was removed, cells were fixed, and DNA was denatured. Afterwards, cells were incubated with Anti-BrdU-POD solution for 90 min, and antibody conjugates were removed in three washing cycles. Immune complexes were detected by means of TMB substrate for 15 min and quantified by measuring the absorbance at 405 nm and 490 nm. All tests were done in duplicates with eight wells per treatment group and repeated at least twice.
Apoptosis analysis
Apoptosis assays by annexin V staining were conducted according to the manufacturer’s instructions (BD Pharming). In brief, PaTu8988t and Panc-1 cells were incubated with test reagents (10 μM ketamine, 10 μM s-ketamine, and 10 μM MK 801). Staurosporine was used for positive control and standard growth medium for negative control. After 0 h, 3 h, 6 h, 16 h, or 24 h incubation time, supernatant was decanted from the cells to preserve floating cells. Adherent cells were rinsed with warm PBS (Sigma Aldrich) and harvested by standard trypsinization. Afterwards, harvested and floating cells were mixed, washed, and resuspended in binding buffer at a final concentration of 106 cells/ml. 100 μL of the cell suspension containing 105 cells was resuspended in 5 μL of FITC Annexin plus 5 μL of propidium iodide, followed by 15 min incubation at room temperature protected from light. Cells were washed and resuspended with 400 μL of binding buffer. Finally, cells were analyzed by flow cytometry using FACS Calibur (BD Bioscience) and Cellquest Pro software (BD Bioscience). All tests were done in duplicates and repeated twice.
Statistical analysis
Data are presented as mean ± SD. The non-parametric Mann–Whitney-U-Test was used for statistical evaluation of the data. P values < 0.05 were considered significant. IBM SPSS Statistics (Vs. 20; IBM New York, US) and Excel Vs. 2010 (Microsoft, Redmond, USA) packages were employed for statistical analysis.
Results
Endogenic expression of NMDA receptors in pancreatic cancer cells
The first aim was to obtain evidence for the actual expression of the NMDA receptor types R1, R2a, and R2b in pancreatic cancer cells (Fig. 1). Expression of the NMDA receptor types R1 and R2b could not be proven in any of the cell lines used (row 1 + 3). In contrast, the NMDA receptor type R2a was expressed in the pancreatic cancer cell lines PaTu8988t and Panc-1 (row 2). As control, endogenic expression of ß-Actin was shown (row 4).
Fig. 1.
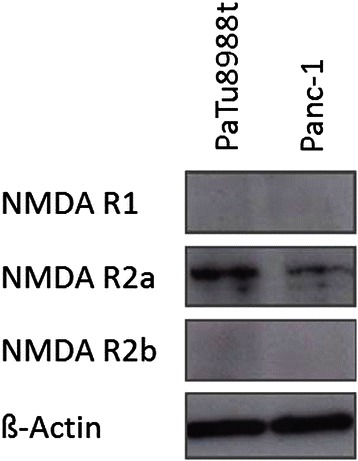
Immunblotting and proof of the endogenic expression of the NMDA receptor types R1, R2a, R2b, and ß-Actin in PaTu8988t and Panc-1 pancreatic cancer cells
Cell proliferation behavior
PaTu8988t and Panc-1 pancreatic cancer cells were stimulated with 0.1 μM, 1 μM, 10 μM, 100 μM, and 1000 μM ketamine (a), s-ketamine (b), or MK 801 (c) each for 48 h. As a result, proliferation was significantly inhibited in both cell lines at concentrations of 1000 μM ketamine, 1000 μM s-ketamine, and 1000 μM MK 801 (Fig. 2). 1 μM MK 801 yielded a slight but significant increase in proliferation by 5 % +/− 24 % in PaTu8988t cells compared to the untreated control group (Fig. 2c). A further increase in proliferation of 23 % +/− 15 % was found in Panc-1 pancreatic cancer cells at a concentration of 0.1 μM ketamine compared to the untreated control group (Fig. 2a).
Figs. 2.
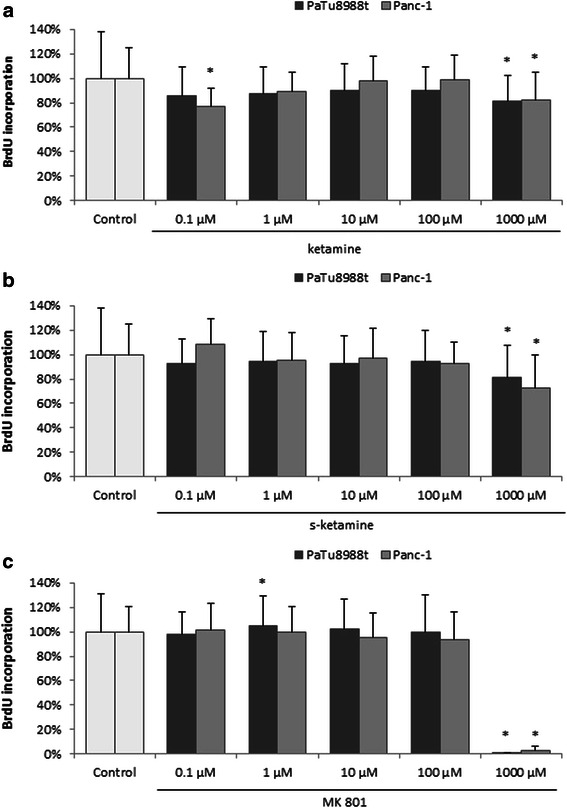
The effects of ketamine (a), s-ketamine (b), and MK 801 (c) on cell proliferation in PaTu8988t and Panc-1 pancreatic carcinoma cell lines in vitro. Cell proliferation was quantified by measuring BrdU incorporation. (*) indicate statistical significance at p < 0.05 compared to untreated control
Analysis of apoptosis and necrosis
The annexin V staining apoptosis assay was used to determine whether stimulation with ketamine, s-ketamine, or MK 801 incurred apoptosis or necrosis. Incubating s-ketamine in the pancreatic cancer cells PaTu8988t for 3 h (Fig. 3b) reduced the apoptotic cell fraction phase to 62 % +/− 27 % compared to the untreated samples. Ketamine and MK 801 did not induce any changes in cell death behavior.
Figs. 3 and 4.
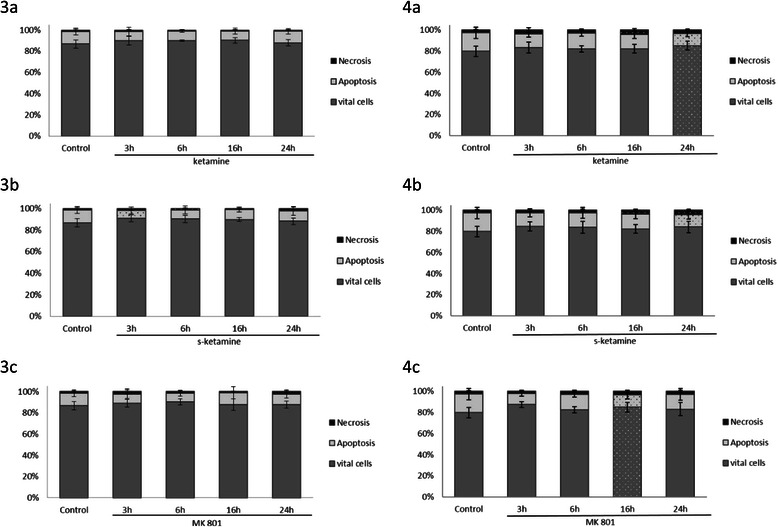
The effects of ketamine (a), s-ketamine (b), and MK 801 (c) on apoptosis in PaTu8988t (Figs. 3) and Panc-1 (Figs. 4) pancreatic carcinoma cell lines in vitro. For apoptosis analysis, cancer cells were stained with Annexin V. (*) indicate statistical significance at p < 0.05 compared to untreated control
In contrast to the untreated control samples in the cell line Panc-1, the apoptosis rate (Figs. 4) was significantly reduced after 24 h incubation with ketamine (a) (65 % +/− 17 %) and s-ketamine (b) (68 % +/− 24 %) as well as after 16 h incubation with MK 801 (c) (67 % +/− 23 %).
In contrast, the necrosis rate increased after 16 h stimulation with ketamine and, in vital cells, after 24 h incubation with ketamine (Fig. 4a).
Stimulation with s-ketamine (Fig. 4b) significantly increased necrosis after 6 h, 16 h, and 24 h, and stimulation with MK 801 (Fig. 4c) significantly increased the vital cells.
No other significant changes in the apoptosis rate were observed during the other timeframes. Staurosporine, an often employed method for inducing apoptosis, was used as a positive control for the testing procedure and induced significant apoptosis in the pancreatic cancer cells (Figure not shown).
Discussion
Three types of ionotropic glutamate receptors are known that are termed according to their prototypic receptor-agonist N-methyl-D-aspartate (NMDA), 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl) propanoic acid (AMPA), and kainate (KA) [19]. N-methyl-D-aspartate (NMDA), which belongs to the family of ionotropic glutamate receptors, is vital for the transmission of stimulating signals between nerve cells [20]. Physiologically, NMDA plays a major role in processes of synaptic plasticity and memory formation [21]. NMDA is also important during various-neurolopathological conditions, such as Alzheimer disease, Parkinson disease, multiple sclerosis, epilepsy, or depression [22]. For many years, NMDA receptor expression was associated with the central nervous system, but recent trials have shown that functional NMDA receptors may also be expressed in tumors[23]. The receptor type has already been successfully proven in various tumor cell lines, for instance, in neuronal tumors (astrocytoma, glioma, and neuroblastoma), rhabdomyosarcoma, medulloblastoma, thyroid carcinoma, lung carcinoma, colon carcinoma, and mamma carcinoma as well as in T-cell chronic lymphocytic leukemia, multiple myeloma [23], and carcinoma of the larynx [25].
In the present study, NMDA receptor type R2a expression could be detected in the pancreatic carcinoma cells PaTu8988t and Panc-1. However, the influence of the NMDA receptor on the oncogenic behavior of cancer cells has not yet been sufficiently investigated. Kalariti et al. assumed in 2005 that the NMDA receptor may be a critical factor in tumor development, tumor growth, and the metastasis of tumor cells [26]. Furthermore, NMDA receptor expression in prostate carcinoma was increased in comparison to healthy prostate tissue [27]. Watanabe et al. showed in 2008 that the NMDA receptor subunit R2a plays an important role in regulating proliferation by accelerating the cell cycle in cell lines of gastric cancer [28]. In other clinical studies, blocking of the NMDA receptor subunit R2a inhibited the proliferation of cancer cells of lung carcinoma and esophagus carcinoma [29, 30].
The next step in our trial was therefore to investigate the impact of ketamine, s-ketamine, and the noncompetitive NMDA receptor antagonist MK 801 on the proliferation, apoptosis, and necrosis of pancreatic cancer cells.
In their study including 31 patients, Idyall et al. investigated the clinical effects of intravenously injected ketamine and its pharmacokinetics. After the injection of 2 mg/kg of ketamine iv, plasma concentration was analyzed by means of gas–liquid chromatography. The average maintenance dose was 41 μg/kg/min, which corresponded to a plasma concentration of 9.3 μM [31].
As our aim was to investigate the direct effect of ketamine, s-ketamine and MK 801 on cancer cell lines, we used concentrations similar to clinically achievable plasma concentrations (0,1 μM – 1000 μM).
We found that incubation with high-dose ketamine, s-ketamine, and MK 801 significantly inhibited cell proliferation in all cell lines. Lee et al. described in 2009 that administration of s-ketamine induced mitochondrial apoptosis in hepatocellular carcinoma [12]. In breast cancer cells, however, the anti-apoptotic protein BCL-2 was up-regulated, and both cell invasion and the proliferation rate were increased [32]. In our study, all test substances also decreased the rate of apoptosis in pancreatic cancer cells. The main effect of ketamine, s-ketamine, and MK 801 is based on the noncompetitive blockade of the NMDA receptor complex. During this process, ketamine binds to the phencyclidine (PCP) binding site inside the NMDA channel, inhibiting the effectiveness of NMDA antagonists, such as glutamate, NMDA, or glycine [33].
Tumor progression including the proliferation and apoptosis of cells is regulated by various signaling cascades. As a first messenger, calcium is of vital importance in this respect [34]. NMDA receptor activation increases the intracellular calcium concentration and activates Ca2+-dependent systolic guanylate cyclase [35]. Calcium influx into cell cytosol results in the calcium-dependent activation of a secondary messenger, which may again activate proteins of various types − amongst others transcription factors − that significantly influence the further behavior of tumor cells [36].
Conclusions
In this study, we showed the expression of the NMDA receptor type R2a in pancreatic cancer cells. The NMDA antagonists ketamine, s-ketamine, and MK 801 inhibited cell proliferation and cell death.
The perioperative period is assumed to be a critical time with regard to cancer dissemination and metastasis [37], probably due to a fatal combination of several factors: surgical manipulation results in the release of cancer cells into circulation, growth factor levels are excessively enhanced during the wound healing process, and anticancer immune surveillance is impaired by perioperative immunosuppression [38].
Results of several preclinical and clinical studies have indicated that the selection of special anesthesia techniques, such as regional anesthesia, may affect cancer recurrence after surgery [39, 40] . Several well-established anesthetic and analgesic agents have been tested for their anticancer potency [41–45].
Modern treatment strategies of cancer diseases aim at efficaciously modulating specific signaling and transcriptional pathways (for instance, VEGF antibodies [45], tyrosine kinase inhibitors for treating chronic lymphatic leukemia [46], or EGFR antibodies for treating advanced colon carcinoma) [47]. The therapeutic potential of inhibiting Ca2+ channels or Ca2+-regulating enzymes in tumor cells has been debated for many years [48]. The basis and prerequisite of such new therapeutic approaches to a ‘targeted therapy’ necessitates comprehensive knowledge of the particular carcinogenesis. For this reason, further clinical studies are required to identify the underlying mechanisms. The identification and characterization of cellular receptors as well as their extracellular activators will help establish new therapeutic options in the treatment of aggressive pancreatic carcinoma.
Acknowledgements
We thank Renate Lange, Sigrid Bamberger, Regina Lindner, Gabriele Bollwein, Marion Schindler, and Ruth Spaeth for excellent technical assistance. We thank Monika Schoell for linguistic support.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have made substantial contributions to the conception, design, analysis, and the interpretation of this research article. They have been involved in the critical revision of the manuscript with regard to important intellectual content. All authors have given their final approval for the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Contributor Information
Manuela Malsy, Phone: +49 941 944 7801, Email: Manuela.Malsy@ukr.de.
Kristina Gebhardt, Email: Kristina.Gebhardt@ukr.de.
Michael Gruber, Email: Michael.Gruber@ukr.de.
Christoph Wiese, Email: Christoph.Wiese@ukr.de.
Bernhard Graf, Email: Bernhard.Graf@ukr.de.
Anika Bundscherer, Email: Anika.Bundscherer@ukr.de.
References
- 1.WHO. Cancer fact sheet No 297; 2012.
- 2.Oberstein P, Kenneth P. Pancreatic cancer: why is it so hard to treat? Therap Adv Gastroenterol. 2013;6:321–327. doi: 10.1177/1756283X13478680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vincent A, Herman J, Schulick R, Hruban R, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gnoni A, Licchetta A, Scarpa A, Azzariti A, Brunett A, Simone G, et al. Carcinogenesis of pancreatic adenocarcinoma: precursor lesion. Int J Mol Sci. 2013;14:19731–62. [DOI] [PMC free article] [PubMed]
- 5.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 6.Doi R, Imamura M, Hosotani R, Imaizumi T, Hatori T, Takasaki K, et al. Surgery versus radiochemotherapy for resectable locally invasive pancreatic cancer: final results of a randomized multi-institutional trial. Surg Today. 2008;38:1021–8. [DOI] [PubMed]
- 7.Egberts J, Cloosters V, Noack A, Scniewind B, Thon L, Klose S, et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008;5:1443–50. [DOI] [PubMed]
- 8.Parada-Turska J, Rzeski W, Majdan M, Kandefer-Szerszen M, Turski WA. Effect of glutamate receptor antagonists and antirheumatic drugs on proliferation of synoviocytes in vitro. Eur J Pharmacol. 2006;535:95–7. [DOI] [PubMed]
- 9.Jimi N, Segawa K, Minami K, Sata T, Shigematsu A. Inhibitory effect of the intravenous anesthetic, ketamine, on rat mesangial cell proliferation. Anesth Analg. 1997;84:190–195. doi: 10.1213/00000539-199701000-00034. [DOI] [PubMed] [Google Scholar]
- 10.Shiga Y, Minami K, Segawa K, Uezono Y, Shiraishi M, Sata T, et al. The inhibition of aortic smooth muscle cell proliferation by the intravenous anesthetic ketamine. Anesth Analg. 2004;99:1408–12. [DOI] [PubMed]
- 11.Rzeski W, Ikonomidou C, Turski L. Glutamate antagonists limit tumor growth. Biochem Pharmacol. 2002;64:1195–1200. doi: 10.1016/S0006-2952(02)01218-2. [DOI] [PubMed] [Google Scholar]
- 12.Lee S, Wu T, Yu P, Chen R. Apoptotic insults to human Hep G2 cells induced by S-(+)-ketamine occurs through activation of a Bax-mitochondra-caspase protease pathway. Br J Anaesth. 2009;102:80–89. doi: 10.1093/bja/aen322. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi F, Hirata Y, Akram H, Kamitori K, Dong Y, Sui L, et al. FOXO/TXNIP pathway is involved in the suppression of hepatocellular carcinoma growth by glutamate antagonist MK 801. BMC Cancer. 2013;13:468–79. [DOI] [PMC free article] [PubMed]
- 14.Persson J. Ketamine in pain management. CNS Neurosci Ther. 2013;19:396–402. doi: 10.1111/cns.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craven R. Ketamine. Anaesthesia. 2013;1:48–53. doi: 10.1111/j.1365-2044.2007.05298.x. [DOI] [PubMed] [Google Scholar]
- 16.Hirota K, Lambert DG. Ketamine:its mechanism(s) of action and unusual clinical uses. Br J Anaesth. 1996;77:441–444. doi: 10.1093/bja/77.4.441. [DOI] [PubMed] [Google Scholar]
- 17.Tawfic QA. A review of the use of ketamine in pain management. J Opioid Manag. 2013;9:379–388. doi: 10.5055/jom.2013.0180. [DOI] [PubMed] [Google Scholar]
- 18.Bredlau AL, Thakur R, Korones DN, Dworkin RH. Ketamine for Pain in Adults and Children with cancer: a systematic Review and Synthesis of the Literature. Pain Med. 2013;14:1505–1507. doi: 10.1111/pme.12182. [DOI] [PubMed] [Google Scholar]
- 19.Szczurowska E, Mares P. Glutamate, glutamate receptors and downstream signaling pathways. Int J Biol Sci. 2013;9:948–959. doi: 10.7150/ijbs.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cull-Candy S and Leszkiew D. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;255:re16. [DOI] [PubMed]
- 21.Warburton E, Barker G, Brown M. Investigations into the involvement of NMDA mechanisms in reconition memory. Neuropharmacology. 2013;74:41–47. doi: 10.1016/j.neuropharm.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mueller H, Meador-Woodruff J. NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder. Schizophr Res. 2004;71:361–370. doi: 10.1016/j.schres.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 23.Luksch H, Uckermann O, Stepulak A, Hendruschk S, Marzahn J, Bastian S, et al. Silencing of selected glutamate receptor subunits modulates cancer growth. Anticancer Res. 2011;10:3181–92. [PubMed]
- 24.Stepulak A, Luksch H, Gebhardt C, Uckermann O, Marzahn J, Sifringer M, et al. Expression of glutamate receptor subunits in human cancers. Histochem Cell Biol. 2009;132:435–45. [DOI] [PubMed]
- 25.Stepulak A, Luksch H, Uckermann O, Sifringer M, Rzeski W, Polberg K, et al. Glutamate Receptors in Laryngeal Cancer cells. Anticancer Res. 2011;31:565–73. [PubMed]
- 26.Kalariti N, Pissimissis N, Koutsilieris M. The glutamatergic system outside the CNS and in cancer biology. Expert Opin Investig. 2005;12:1487–1496. doi: 10.1517/13543784.14.12.1487. [DOI] [PubMed] [Google Scholar]
- 27.Abdul M, Hoosein N. N-methyl-D-aspartate receptor in human prostate cancer. J Membr Biol. 2005;205:125–128. doi: 10.1007/s00232-005-0777-0. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe K, Kanno T, Oshima T, Miwa H, Tashiro C, Nishizaki T. The NMDA receptor NR2A subunit regulates proliferation of MKN45 human gastric cancer cells. Biochem Biophys Res Commun. 2008;367:487–490. doi: 10.1016/j.bbrc.2007.12.167. [DOI] [PubMed] [Google Scholar]
- 29.Kim M, Yamashita K, Baek J, Park H, Carvalho A, Osada M, et al. N-Methyl-D-aspartae receptor type 2B is epigenetically inactivated and exhibits tumor-suppressive activity in human esophageal cancer. Cancer Res. 2006;66:3409–18. [DOI] [PubMed]
- 30.This is Number 30Stepulak A, Sifringer M, Rzeski W, Endesfelder S, Gratopp A, Pohl E, et al. NMDA antagonist inhibits the extracellular signal-regulated kinase pathway and supresses cancer growth. Proc Natl Acad Sci U S A. 2005;102:15605–10. [DOI] [PMC free article] [PubMed]
- 31.This is Number 31Idvall J, Ahlgren I, Aronsen K, Stenberg P. Ketamine infusions: Pharmacokonetics and clinical effects. Br J Anaesth. 1979;51:1167–73. [DOI] [PubMed]
- 32.He H, Chen J, Xie WP, Cao S, Hu HY, Yang LQ, et al. Ketamine used as an acesodyne in human breast cancer therapy causes an undesirable side effect, upregulating anti-apoptosis protein Bcl-2 expression. Genet Mol Res. 2014;12:1907–15. [DOI] [PubMed]
- 33.Kress HG. Mechanisms of action of ketamine. Anaesthesist. 1997;46:8–19. doi: 10.1007/PL00002469. [DOI] [PubMed] [Google Scholar]
- 34.Monteith G, Davis F, Roberts-Thomson S. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem. 2012;287:31666–31673. doi: 10.1074/jbc.R112.343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petroiano G, Osswald P. Anästhesie in Frage und Antwort. 3. Deutschand: Springerverlag; 2000. Ketamin; pp. 99–102. [Google Scholar]
- 36.Bengtson C, Bading H. Nuclear calcium signaling. Adv Exp Med Biol. 2012;970:377–405. doi: 10.1007/978-3-7091-0932-8_17. [DOI] [PubMed] [Google Scholar]
- 37.Tavare A, Perry N, Benzonana L, Takata M, Ma D. Cancer recurrence after surgery: direct and indirect effects of anesthetic agents. Int J Cancer. 2012;130:1237–1250. doi: 10.1002/ijc.26448. [DOI] [PubMed] [Google Scholar]
- 38.Goldfarb Y, Ben-Eliyahu S. Surgery as a risk factor for breast cancer recurrence and metastasis: mediating mechanisms and clinical prophylactic approaches. Breast Dis. 2006;26:99–114. doi: 10.3233/BD-2007-26109. [DOI] [PubMed] [Google Scholar]
- 39.Xuan W, Hankin J, Zhao H, Yao S and Ma D. The potential benefits of the use of regional anesthesia in cancer patients. Int J Cancer. 2014. [DOI] [PubMed]
- 40.Chen WK, Miao C. The effect of anesthetic technique on survival in human cancers: a meta-analysis of retrospective and prospective studies. PLoS One. 2013;8:56540. doi: 10.1371/journal.pone.0056540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Werdehausen R, Braun S, Essmann F, Schulze-Osthoff K, Walczak H, Lipfert P, et al. Lidocaine induces apoptosis via the mitochondrial pathway independently of death receptor signaling. Anesthesiology. 2007;107:136–43. [DOI] [PubMed]
- 42.Huang H, Benzonana L, Zhao H, Watts H, Perry N, Bevan C, et al. Prostate cancer cell malignancy via modulation of HIF-1 α pathway with isoflurane and propofol alone and in combination. Br J Cancer. 2014;111:1338–49. [DOI] [PMC free article] [PubMed]
- 43.Basu G, Pathangey L, Tinder T, Gendler S, Mukherjee P. Mechanisms underlying the growth inhibitory effects of the cyclo-oxygenase-2 inhibitor celecoxib in human breast cancer cells. Breast Cancer Res. 2005;7:422–435. doi: 10.1186/bcr1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kushida A, Inada T, Shingu K. Enhancement of antitumor immunity after propofol treatment in mice. Immunopharmacol Immunotoxicol. 2007;29:477–486. doi: 10.1080/08923970701675085. [DOI] [PubMed] [Google Scholar]
- 45.Bidart M, Berger F, Pelletier L. Anti-angiogenetic therapie: from theory to practice. Ann Biol Clin (Paris) 2013;71:527–535. doi: 10.1684/abc.2013.0883. [DOI] [PubMed] [Google Scholar]
- 46.Burger J, Montserrat E. Coming full circle: 70 years of chronic lymphocytic leukemia cell redistribution, from glucocorticoids to inhibitors of B-cell receptor signalling. Blood. 2013;121:1501–1509. doi: 10.1182/blood-2012-08-452607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Modest D, Hiddemann W, Heinemann V. Chemotherapy of metastatic colorectal cancer. Internist. 2014;55:37–42. doi: 10.1007/s00108-013-3314-8. [DOI] [PubMed] [Google Scholar]
- 48.Roderick H, Cook S. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer. 2008;8:361–375. doi: 10.1038/nrc2374. [DOI] [PubMed] [Google Scholar]