

Abstract
Purpose
Adrenocortical carcinoma (ACC) is a rare pediatric malignancy. It occurs in excess among individuals with the Li-Fraumeni syndrome, which results primarily from germline mutations in the TP53 gene. Prior series exploring frequencies of germline TP53 mutation among children with ACC have been small, geographically limited, or subject to referral bias. The functional consequence of mutations has not been related to phenotype. We provide a genotype-phenotype analysis of TP53 mutations in pediatric ACC and propose a model for tissue-specific effects based on adrenocortical ontogeny.
Patients and Methods
Eighty-eight consecutive, unrelated children with ACC, unselected for family history, underwent germline TP53 sequencing. Rate and distribution of mutations were identified. Functional analysis was performed for novel TP53 variants. Correlation with the International Agency for Research on Cancer p53 database further delineated mutational distribution, association with family history, and risk for multiple primary malignancies (MPMs).
Results
Germline mutations were present in 50% of children. These mutations did not correspond to the conventional hotspot mutations. There was a wide range of mutant protein function. Patients bearing alleles encoding protein with higher functionality were less likely to have a strong family cancer history, whereas those with greater loss of function had MPMs and/or positive family history. In patients with MPMs, ACC was the most frequent initial malignancy. Finally, we demonstrated age-dependent rates of TP53 mutation positivity.
Conclusion
TP53 mutations are prevalent in children with ACC but decline with age. Mutations result in a broad spectrum of functional loss. Effect of individual mutations may predict carrier and familial disease penetrance with potentially broad implications for clinical surveillance and counseling.
INTRODUCTION
Adrenocortical carcinoma (ACC) is diagnosed at a rate of 0.3 to 0.4 per million children annually.1,2 Advancing age, large tumors, and metastatic disease predict poorer prognosis. For patients with metastatic disease, survival rates are extremely poor (5-year overall survival of approximately 20%),2 and treatments have significant toxicities. Although treatment guidelines are largely based on knowledge of disease and trials in adults, a recent Children's Oncology Group trial assessed the outcome of combination chemotherapy and retroperitoneal lymph node dissection. In addition, this study documented the rate of TP53 germline alterations among North American and Brazilian children.
Given the rarity of this diagnosis, reports describing demographics, morphology, and outcomes of children with ACC had been primarily reflective of small case series. A registry comprising 254 children with adrenocortical tumors (International Pediatric Adrenocortical Tumor Registry) contributed substantially to understanding the landscape of this disease.2 Even this series, however, was limited by geographic bias, because 79.5% of the patients came from a region in southern Brazil. It is now recognized that these patients carry a common genetic aberration, the R337H allele of the TP53 tumor-suppressor gene.3 This allele exhibits unique biochemical properties, including pH-dependent loss of function and apparent wild-type activity in vitro,4 in contrast to the loss of function observed with other common alleles.3,5 Thus, it is possible that data derived from this population may not be truly reflective of the natural history of pediatric ACC, despite no apparent differences in clinical presentation between patients in this cohort and patients in a subsequent study.6
A fundamental role for TP53 in ACC was first recognized when ACC was identified as a component tumor of the Li-Fraumeni syndrome (LFS).7 LFS results, in most cases, from germline TP53 mutations.8 Among all families with germline TP53 mutations, the six most common alleles (R175H, G245S, R248Q, R248W, R273H, and R282W) occur in 20% of pedigrees and are collectively referred to as hotspot mutations. Germline TP53 testing is advocated for anyone diagnosed with ACC, regardless of age or family cancer history, in recognition of an excess of ACC among families with LFS and a high rate of TP53 mutations among individuals with ACC.9
Small series have reported that 50% to 80% of children with ACC carry germline TP53 mutations. Wagner et al10 demonstrated that three of six children with ACC carried mutations. Subsequently, Varley et al11 identified mutations in 10 of 12 children with ACC. This series was enriched for two alleles, P152L (n = 5) and R158H (n = 3), a finding that is not replicated when examining the overall distribution of TP53 alleles in patients with ACC.12 Both series reported small numbers of children from geographically limited catchments. A more recent study found that 12 (80%) of 15 children with a diagnosis of ACC carried germline mutations.13 However, patients in this study were identified on the basis of referral to a TP53 reference laboratory; thus, enrichment for patients with a positive family history may have skewed these findings.
Striking among TP53 mutation carriers who are relatives of children with ACC has been the existence of apparent low-penetrance alleles. Several reports have identified unaffected carriers of TP53 mutations, including P152L,11 R175L,14 and R337H,3,4 where penetrance may be as low as 2.4%.15 Although these cancer rates are high for the general population, among carriers of TP53 mutations, for whom lifetime risk of cancer is between 70% and 100%,16,17 the cancer penetrance is relatively low. These findings suggest that affected individuals may have additional genetic or environmental exposures that promote tumorigenesis. Because TP53 mutation carriers have a high lifetime risk of developing multiple primary malignancies (MPMs),18 lifelong surveillance for subsequent malignancies decreases morbidity and improves short-term survival.19
Previous studies have suggested tissue specificity of mutations in certain TP53 domains.20,21 Olivier et al22 demonstrated that ACCs were preferentially associated with mutations located in loops opposing protein-DNA contact. Other series, including those describing the Brazilian R337H cohort and the Manchester pediatric cohort,11 have shown a paucity of nonadrenal tumors among pedigrees with certain TP53 alleles.3,11 Given the unique biochemical properties of the R337H mutation, a mechanism for tissue specificity based on perinatal remodeling within the adrenal cortex was proposed.4,20
We sought to characterize the TP53 mutations associated with childhood ACC and to determine whether, in a large cohort, the conventional hotspot alleles were represented or whether adrenal-specific hotspots existed. We undertook functional characterization of novel alleles and integrated these findings with characteristics of disease presentation, family history, and tissue specificity of ACC alleles into a framework for adrenocortical tumorigenesis based on a fetal-origin hypothesis.
PATIENTS AND METHODS
Patients
The Children's Oncology Group study ARAR0332 investigated incidence and functional characteristics of germline TP53 mutations in children with ACC. Fifty-five patients were eligible for analysis in this study. In addition, patients ≤ 20 years old referred to The Hospital for Sick Children with a histologically confirmed diagnosis of ACC were invited to submit blood samples for testing (n = 34) as per current recommendations.9 Four patients have been previously described.10,23
Samples were analyzed regardless of family history of cancers or identifiable genetic syndromes. This study was approved by the institutional research ethics board. All participants provided informed consent.
Sequencing of exons 2 to 11 and multiplex ligation-dependent probe amplification analysis for gene dosage were performed as described in the Appendix (online only). Plasmids expressing TP53 variants were generated using site-directed mutagenesis (Appendix and Appendix Table A1, online only), and functional analysis was performed as described in the Appendix.
Database Query
The International Agency for Research on Cancer (IARC) p53 database (http://p53.iarc.fr)12,24 is a relational database based on all published reports and personal communications regarding individuals with germline and somatic TP53 mutations. This study was based on Revision 15 of the database.
RESULTS
We identified 88 children with ACC derived from a broad geographic catchment, unselected for family history. Demographics (Table 1) overlap closely with those described previously.2,6
Table 1.
Descriptive Analysis of Patients Included in the Current Study
| Characteristic | No. of Patients |
|||
|---|---|---|---|---|
| Wild Type (n = 34) | R337H (n = 20) | Variant (n = 34) | Total (N = 88) | |
| Sex | ||||
| Male | 10 | 6 | 12 | 28 |
| Female | 24 | 14 | 22 | 60 |
| Stage | ||||
| I | 8 | 9 | 10 | 27 |
| II | 3 | 3 | 7 | 13 |
| III | 5 | 7 | 7 | 19 |
| IV | 9 | 1 | 4 | 14 |
| Not specified | 9 | 0 | 6 | 15 |
| Age at diagnosis, years | ||||
| < 1 | 4 | 1 | 3 | 8 |
| 1-1.99 | 8 | 7 | 7 | 22 |
| 2-3.99 | 6 | 8 | 9 | 23 |
| 4-11.99 | 4 | 3 | 11 | 18 |
| 12-20 | 12 | 1 | 4 | 17 |
| Family history | ||||
| Chompret | 13 | 9 | 22 | |
| Eeles | 0 | 8 | 8 | |
| Birch | 0 | 1 | 1 | |
| Classic | 0 | 1 | 1 | |
NOTE. All 20 patients with the R337H mutation were from southeastern Brazil. Overall distribution of patients was comparable to that described in the International Pediatric Adrenocortical Tumor Registry (IPACTR). The one exception to this in the current series is a significantly higher proportion of patients with stage III or IV disease in this cohort versus IPACTR (47% v 24%, respectively; χ2 = 10.93, P = .0015). We attribute this to recruitment from an active clinical trial, which could bias toward enrollment of patients with more advanced disease. Indeed, the patients in this cohort derived from the Children's Oncology Group trial were far more likely to have presented with stage III or IV disease than those referred based solely on a diagnosis of adrenocortical carcinoma or in the IPACTR registry (χ2 = 21.83, P < .001), whereas there was no difference between these groups with respect to age, sex, or TP53 mutation status.
After excluding carriers of the TP53R337H allele (see Appendix), 34 (50%) of 68 children carried TP53 mutations. The distribution of mutation type is similar to that found among all patients with ACC, as well as that found among all individuals with germline TP53-associated tumors (Fig 1). It is distinct from distributions among other tumor-suppressor genes such as Rb, APC, and PRKAR1a, which are more commonly subject to frameshift and nonsense substitutions.25,26
Fig 1.

Distribution of germline TP53 mutation type in (A) the current cohort, (B) all reported pedigrees with adrenocortical carcinoma (ACC) in the International Agency for Research on Cancer (IARC) database, and (C) all pedigrees (all tumor types) in the IARC database. COG, Children's Oncology Group.
Only two of 34 variants in our cohort corresponded to a TP53 hotspot mutation, versus 20% among LFS patients overall (χ2 = 4.008, P = .045). There was no clustering of mutations in this series. Three mutations were present in more than one unrelated individual (c.375G>A, n = 327–29; C229R, n = 3; and a deletion of exons 10 to 11, n = 2). This stands in contrast to a previous report wherein two alleles predominated.11 Although the prior study, in isolation, suggests a mutational selectivity for ACC (an adrenal hotspot), based on our current findings, it more likely represents geographic clustering of the mutation and may suggest a common ancestor of the patients, despite none having been identified by the authors.
The scarcity of conventional hotspot mutations in families with ACC was recapitulated when reviewing the IARC p53 database,12 wherein only 12 (16%) of 75 families affected by ACC carried a hotspot allele. This rate is substantially lower than among families with other LFS-component tumors, including brain tumors (15 [26%] of 57 families) and bone cancers (11 [35%] of 31 families), suggesting that ACC may result from a distinct spectrum of TP53 mutations. Indeed, tumor-specific TP53 mutation spectra have previously been described.21,22
There was no significant difference between groups with germline TP53 wild-type and TP53 variant alleles with respect to sex or tumor stage (Table 1), whereas diagnosis before age 12 years was more likely associated with germline TP53 mutation than diagnosis in adolescence (Fisher's exact test, P = .043).
Several polymorphisms have been associated with altering the TP53 phenotype. We found no association between polymorphisms in codon 72 (PEX4), intron 3 (PIN3), or MDM2SNP309 and age or stage at presentation30,31 (Appendix Table A2, online only).
ACC-Associated TP53 Alleles Demonstrate a Spectrum of Functional Activity
Although hotspot mutations result in near-complete loss of TP53 activity,12 many of the variants identified in this study lacked prior functional characterization in mammalian cells. To determine whether the paucity of hotspot mutations had biologic significance, functional analysis was performed.
TP53-variant proteins were expressed in TP53 functionally null cells to determine protein function based on transactivation of a TP53-dependent luciferase reporter. By virtue of their identification among children with an LFS-component tumor, we anticipated that these variants would result in significant diminution of TP53 function. Remarkably, several variants retained significant levels of wild-type function in three independent cell lines, H1299, SaOS-2, and HAC1532 (Fig 2 and Data Supplement). In addition, several mutants demonstrated dominant-negative activity (Data Supplement).
Fig 2.

(A) Functional activity of TP53 variants expressed in H1299 cells as determined by transactivation of TP53-responsive luciferase reporter (or a control reporter with point mutations in the response element which abolish TP53 binding [MutBS], Data Supplement). Expression of TP53 was confirmed by Western blot. C275X and Ins933a variants result in truncated proteins with faster migration. Vinculin staining demonstrates equal protein loading. All lanes except C275X and Ins933a demonstrate a full-length TP53 (53 kDa) and a smaller breakdown product. Loss-of-function alleles are associated with higher expression (presumably mediated by lack of negative feedback). (B) Data in panel A ranked based on activity and correlated with clinical presentation. Black rectangles indicate a family history consistent with Li-Fraumeni syndrome (LFS)/Li-Fraumeni–like syndrome (LFLS). White rectangles indicate absence of a significant family history in a complete pedigree containing three or more generations. Hatched rectangles indicate insufficient data to establish or exclude a family history of cancer predisposition. MPM, multiple primary malignancies; MR ACC, multiply recurrent adrenocortical carcinoma; mutBS, mutated TP53 binding site; WT, wild type.
As a subsequent assay of TP53 function, variants were introduced into TP53-null cells in vitro, and their ability to interfere with colony formation was determined (Figs 3A to 3C). As with luciferase assays, a spectrum of functionality was observed (Fig 3D).
Fig 3.

Colony reduction assays. Fourteen days after transfection, plates were stained with crystal violet and the remaining colony numbers were quantified manually. Representative plates of (A) vector, (B) wild-type (WT) TP53-transfected, or (C) TP53R273H-transfected cells. R273H is a hotspot mutation with minimal residual TP53 function. (D) Quantification of data for all alleles is shown. MutBS, mutated TP53 binding site.
Residual TP53 Activity May Underlie Low-Penetrance Alleles
Loss of TP53 activity is associated with a family history of multiple malignancies. We were struck by the paucity of pedigrees with a strong history of cancer in this cohort, despite recognized associations of ACC with LFS. A family pedigree of three or more generations was available for 32 children. No patients with ACC harboring wild-type TP53 had a significant family history of cancer. Of 19 patients with germline TP53 mutations, 11 (58%) met classic, Birch, or Eeles definitions of LFS or Li-Fraumeni–like syndrome (LFLS; summarized by Gonzalez et al13). Of the remaining eight children with TP53 variants and no family cancer history, four patients had de novo mutations (Y163C, H193P, C275X, and E285V), two patients had a mutation carrier parent unaffected by cancer at age 42 years (G334R) and 51 years (F134Y), and parental DNA was unavailable for two patients (C229R and N235D), although extended four-generation family histories for these two patients failed to reveal early or multiple malignancies.
We compared TP53 function among those alleles with and without criteria for LFS/LFLS. For this analysis, we derived activity from extant data based on transactivation of TP53 target genes in Saccharomyces cerevisiae.33 Median TP53 function across eight different TP53 target genes was determined and classified according to presence of familial cancer (Table 2). Patients meeting LFS/LFLS criteria or with de novo mutations had lower activity alleles than those without a significant cancer history. In this analysis, the R337H allele, with previously estimated adrenocortical tumor penetrance of 2.6% to 9.9%,15,34 demonstrates a predicted activity of 69% of wild type. Thus, penetrance of cancer seems to be linked, at least partially, to the magnitude of residual TP53 function. ACC may present despite near-normal TP53 function, whereas other LFS-related cancers are uncommon in such circumstances.
Table 2.
Mutational Activity Among Individuals With and Without a Family History Consistent With Li-Fraumeni Syndrome/Li-Fraumeni–Like Syndrome
| Pedigree No. | Allele | Inheritance | Median Activity (according to Kato et al33; %) |
|---|---|---|---|
| Family cancer history present | |||
| 1 | P152L | Maternal | 16.1 |
| 21 | R158H | Maternal | 16.05 |
| 2 | P219S | Maternal | 9.15 |
| 41 | S241Y | Paternal | 0 |
| 45 | R273S | Maternal | 10.3 |
| 6 | T125T (c.375G>A) | Maternal | 0 (predicted) |
| 14 | T125T (c.375G>A) | Unknown* | 0 (predicted) |
| 8 | del exon 10-11 | Maternal | 0 (predicted) |
| 42 | del exon 10-11 | Maternal | 0 (predicted) |
| 87 | N311fs (c.911insA) | Maternal | NA |
| Family cancer history absent | |||
| 7 | F134Y | Paternal | 61.5 |
| 3† | Y163C | De novo | 5.5 |
| 5† | H193P | De novo | 11.8 |
| 44 | C229R | Unknown* | 30 |
| 23 | N235D | Unknown* | 37.6 |
| 4† | C275X | De novo | 0 (predicted) |
| 58† | E285V | De novo | 8.9 |
| 12 | G334R | Maternal | 86.6 |
| 47 | R267Q | Maternal | 21.5 |
Abbreviation: NA, not applicable.
Unknown denotes that parental DNA was unavailable and pedigree analysis could not establish an obligate carrier.
Patients with de novo germline mutations.
ACC Is the Most Common Initial Tumor in Multiply Affected Individuals and Families
Harboring a germline TP53 mutation places individuals at high lifetime risk for MPMs. The cumulative probability of a second cancer at 30 years is estimated at 57%.18 Thus, we asked whether ACC was commonly an early or a late tumor among individuals with MPMs.
Given our small sample size (six patients with MPMs), we reviewed all instances of individuals with TP53 mutations and MPMs (including ACC as one of the malignancies) in both the IARC database and in our own series (Appendix Table A3, online only). Of 33 individuals, 26 (79%) presented with ACC as their first cancer, either singly or synchronously with other tumors.
Among all families that include a member with ACC, ACC again seems to be the earliest presenting tumor. Chompret et al35 identified 13 families with TP53 mutation, and ACC was present in at least one family member in four families. In all four families, the individual with ACC was the youngest to present with malignancy.35 In the IARC p53 database, 69 families contain at least one member with an ACC (excluding R337H families). Of these, the first cancer diagnosis within the family was an ACC in 53 (77%) of 69 families.
DISCUSSION
Li et al7,36 identified ACC as one of several rare tumors arising among families with autosomal dominant cancer predisposition resulting from germline TP53 mutations.8 The relevance of TP53 dysfunction to ACC pathogenesis was thus established.
Recent estimates have reported up to 80% prevalence of TP53 mutations among children with ACC.13,37 This has been extrapolated to suggest that ACC rarely occurs in the absence of a germline TP53 mutation.38 Our data suggest this to be an overestimate, with the true prevalence of TP53 mutation to be 50%. The difference may be explained by an inherent methodologic distinction with the former study, which was based on referrals to a central reference laboratory and thus likely enriched for pedigrees with a history of cancers. Although the current study was designed to overcome these biases, we cannot rule out a negative referral bias to tertiary care wherein TP53 testing was not offered to patients with benign (or indeterminate) histology and/or milder disease course.
The high rate of germline TP53 mutation seems almost exclusively limited to individuals diagnosed with ACC in childhood. Indeed, in the current series, the rate of germline positivity decreased from 58% in those diagnosed before age 12 years to 25% in those diagnosed between 12 and 20 years old (Table 1, Fig 4). This is consistent with our prior finding that only 11% of patients with TP53-associated ACC were older than age 12 years and only 3% were older than age 30.39 In two recent series, mutation rates among adults were 3.8% (four of 103 patients)40 and 5.8% (three of 52 patients).41 Figure 4 illustrates aggregate data from our series, as well as those of Herrmann et al40 and Raymond et al.41
Fig 4.
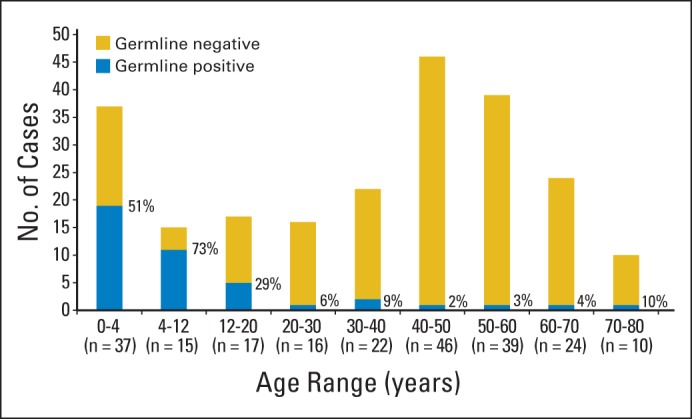
Prevalence of germline TP53 mutations based on age at diagnosis. Aggregate data from this study, as well as those of Herrmann et al40 and Raymond et al41 are included in this representation. Percentage of individuals with TP53 germline mutations is indicated.
Half of patients with TP53 variants lacked a family history of early or atypical cancers. This underscores the merit of the recommendation that all individuals with ACC be offered testing for TP539, regardless of family history, because a significant proportion of mutation carriers would otherwise have gone undetected. Nevertheless, we cannot anticipate the lifetime risk of developing cancer in those unaffected carriers of apparently milder mutations. Moreover, Figure 4 suggests that TP53 testing in patients with ACC older than age 40 years is of low yield and unlikely to have clinical ramifications, absent other primary malignancies or significant family history.
A particularly intriguing finding is the unanticipated wide spectrum of functional activity of TP53. We hypothesized that near-total loss of tumor-suppressor activity would be necessary for tumorigenesis. Instead, several alleles show retention of substantial function. We propose that the degree to which a TP53 mutation alters function impacts risk for MPMs and disease penetrance and may play a role in selective transformation of tissues based on their underlying biology. Alternatively, it is possible that total loss of one or more of the myriad functions of TP53 underlies these effects among those apparently milder alleles.
This wide variation in activity and the absence of observed family history or MPMs among those carrying milder mutants expand the concept of the gradient effect of TP5342, in which phenotype was argued to reflect TP53 activity. It would not be unexpected if tumors arising from individuals with milder mutations showed less genetic instability than those arising from patients with stronger loss-of-function alleles. A recent high-resolution genomic analysis of pediatric adrenocortical tumors identified fewer genomic alterations in tumors with TP53 wild type versus those with mutations.43 Analysis of germline DNA from six of these children revealed the absence of genomic aberrations, suggesting an early role for TP53 in genomic destabilization during tumorigenesis.
Carriers of TP53 mutations are at increased risk for MPMs. Mutation carriage necessitates lifelong surveillance, and screening of relatives is recommended. These data suggest that patients with milder TP53 alleles might be at decreased risk for MPMs. Also notable is the apparent association between milder alleles and a lower penetrance of cancer within families. Given the variable penetrance and tissue distribution of disease among individual TP53 alleles, we speculate that, ultimately, cancer surveillance for mutation carriers may be individualized based on specific alleles and/or modifier genetic or epigenetic variants. This will also have broad implications for genetic counseling and risk assessment in carrier relatives, although much work remains before this is realized.
We note distinct limitations to this analysis. An adequate family history was available for a minority of the individuals; thus, broad conclusions regarding penetrance cannot be drawn until more data are obtained. Moreover, the majority of patients were diagnosed within the past 10 years; therefore, conclusions regarding survival, recurrence, MPMs, and malignancies in mutation-positive siblings await longer follow-up, particularly in the context of a recent observation that carriers of TP53 mutations with partial transactivation develop cancer at a later age.12 Finally, we cannot refute the possibility that these alleles are not the dominant drivers in tumorigenesis, but may in fact be passengers or backseat drivers, modifying disease engendered by other driver mutations.
We and others have observed that ACC presents in a bimodal age distribution, with peaks in the first and fifth to sixth decade of life.39,44 Given the marked divergence in TP53 mutation prevalence in these two peaks, we propose that they evolve as a consequence of distinct molecular mechanisms. This is supported by differing genotype and expression profiles among pediatric and adult tumors.45,46 Whereas tumors arising in adults may result from gradual accumulation of genetic hits, we speculate that tumorigenesis in children reflects adrenal ontogeny and consequent susceptibility to perturbations in the physiologic transition from fetus to newborn.
The high proportion of children diagnosed with ACC within the first 2 years of life is consistent with an origin of disease in the fetal or neonatal period. The adrenal gland undergoes extensive postnatal remodeling as the fetal cortex undergoes a robust wave of apoptosis, decreasing from 70% to 3% of total adrenal volume.47–49 Within this context, even mild perturbations in proapoptotic signaling (via compromised TP53) could impair this remodeling. The apparent adrenal specificity of milder TP53 alleles would thus be conferred by the high apoptotic index, absent in other tissues. This massive apoptotic wave renders the gland vulnerable to mild perturbations in apoptotic signaling wherein failure of apoptosis in a single fetal cell may result in progression to the transformed state. Consequently tumorigenesis associated with seemingly mild TP53 mutations can be explained by a failure of physiologic apoptosis in fetal zone cells.
A fetal origin of disease is further supported by gene expression studies that demonstrate marked similarities between expression profiles of fetal adrenal cortex and ACCs, which are distinct from those of the definitive postnatal gland.45,50–52
We have demonstrated that half of children with ACC carry germline TP53 mutations and that the likelihood of mutation is highest in the first years of life, diminishing with age. The mutation carried in the germline (and its effects on protein function) is germane and likely influences the site of tumor formation, likelihood of subsequent malignancy, and disease penetrance among carriers. These data expand our understanding of the mutational spectrum underlying ACC tumorigenesis and suggest novel mechanisms whereby physiologic remodeling confers temporal and tissue selectivity to a tumorigenic process.
Supplementary Material
Acknowledgment
We thank Noa Alon for technical assistance and Uri Tabori and Rinnat Porat for valuable input during the course of this work and for critical evaluation of this article. We also thank all physicians and genetic counselors who referred patients to the ARAR0332 study and the SickKids Cancer Genetics Program. We are grateful to Stefanie Hahner, Victoria Raymond, Tobias Else, Thomas Giordano, and Gary Hamer for providing data regarding TP53 genotypes of adult patients' adrenocortical carcinoma. Finally, we thank Bill Rainey for providing the HAC15 cell line.
Appendix
METHODS
Patient Accrual
A total of 78 patients were enrolled onto Children's Oncology Group ARAR0332 (ClinicalTrials.gov identifier: NCT00304070). Consent for genetic testing was provided by 69 patients, and sufficient DNA was available for 56 patients. Two patients were common to both the Children's Oncology Group and the Toronto cohorts, and each patient is reported here only once.
TP53 Gene Sequencing and Dosage Analysis
Genomic DNA was extracted from peripheral-blood lymphocytes using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) per the manufacturer's directions. TP53 mutations were detected by BigDye direct Sanger sequencing of exons 2 to 11 and intron-exon boundaries of polymerase chain reaction products using an ABI automated sequencer (Applied Biosystems, Foster City, CA), as previously described (Tabori U, et al: Cancer Res 67:1415-1418, 2007). Limiting amounts of DNA were available for 10 of 21 patients from southeastern Brazil. For these patients, only exon 10 (containing codon 337) was sequenced. All carried the TP53R337H allele. Although we cannot rule out germline compound heterozygosity in these 10 patients (ie, a second TP53 variant outside of exon 10), it is exceedingly rare, with only case reports identified in the literature (Quesnel S, et al: Oncogene 18:3970-3978, 1999). Gene dosage was assessed using multiplex ligation-dependent probe amplification (MLPA) using the MRC-Holland SALSA MLPA PO56 TP53 probe set (MRC-Holland, Amsterdam, the Netherlands) according to the manufacturer's directions.
Among 88 unrelated probands, we identified 54 carriers of germline variants in the TP53 gene. Participants were recruited from sites in Canada and the United States (n = 67) and southern Brazil (n = 21). Of the 21 children from southern Brazil, all but one carried the R337H variant (the latter had no detectable TP53 variant). No carriers of this mutation were identified from centers outside of Brazil. This variant was demonstrated to arise from a founder mutation (Garritano S, et al: Hum Mutat 31:143-150, 2010; Pinto EM, et al: Arq Bras Endocrinol Metabol 48:647-650, 2004),43 and thus we do not consider these six samples as necessarily independent events. In addition, systematic screening for this variant in southern Brazil has led to increased mutation detection among this select population (Palmero EI, et al: Cancer Lett 261:21-25, 2008; Gomes MC, et al: Hered Cancer Clin Pract 10:3, 2012). Given the potential biases introduced, for the purposes of the prevalence estimates, we have excluded carriers of the R337H from our analyses. As such, we estimate the total prevalence of TP53 germline mutation in children with ACC to be 50% (34 of 68 patients).
The P47S variant of TP53 is generally thought to be a polymorphism without substantial effect on protein function (Sabra MM, et al: Thyroid 22:877-883, 2012). We validated its wild-type function in this study (Figs 2 and 3) and for the purposes of descriptive analyses have classified a patient with this allele as wild type. A silent germline mutation, c.375G>A (also referred to as T125T), has previously been shown to impair TP53 gene expression and segregate with cancer in families; thus, we consider it as a mutant TP53 for the purposes of descriptive analyses.27–29
Cell Culture
The human lung adenocarcinoma cell line, H1299, and osteosarcoma cell line, SaOS-2, were obtained from the American Type Culture Collection. Both H1299 (Lin DL, Chang C: J Biol Chem 271:14649-14652, 1996) and SaOS-2 (Chandar N, et al: Br J Cancer 65:208-214, 1992) are reported to be TP53-null cell lines. To confirm these reports, genomic DNA was extracted from confluent tissue culture by sodium dodecyl sulfate–proteinase K phenol-chloroform extraction. Genotype was confirmed by direct sequencing of the TP53 gene and by MLPA, as described earlier. H1299 and SaOS-2 were found to harbor deletions of exons 2 to 9 and 2 to 11, respectively. The genomic deletion in SaOS-2 cells extended telomeric to the TP53 gene on chromosome 17p and included deletion of the SHBG and ATP1B2 loci, whereas gene dosage of the more distal MPDU1 was normal. Cells were maintained as a monolayer in a humidified 5% carbon dioxide atmosphere at 37°C in Dulbeccos MEM (Wisent, Saint-Bruno, Quebec, Canada) with 10% Heat Inactivated Fetal Bovine Serum (Life Technologies, Carlsbad, CA) and penicillin-streptomycin (Wisent).
HAC15 cells, derived from the human adult adrenocortical carcinoma (ACC) H295R cell line, were the generous gift of Dr W. Rainey (Parmar J, et al: J Clin Endocrinol Metab 93:4542-4546, 2008). MLPA analysis, as described earlier, revealed normal gene dosage of exons 1 to 4, duplication of exons 5 to 7, heterozygous deletion of exon 8, homozygous deletion of exon 9, and a duplication of exons 10 to 11. Western blot analysis identified a single approximately 35-kDa immunoreactive protein, whereas functional analysis using TP53-responsive luciferase reporter (see later discussion in this Appendix and Data Supplement) showed no detectable TP53 activity (Data Supplement). HAC15 cells were propagated as described (Parmar J, et al: J Clin Endocrinol Metab 93:4542-4546, 2008).
HT1080 is a human fibrosarcoma cell line obtained from ATCC (Manassas, VA). Sanger sequencing of the entire TP53 coding region and MLPA analysis demonstrated wild-type sequence and gene dosage, respectively. Western blotting revealed an immunoreactive protein at approximately 53 kDa, and introduction of a TP53 response element–driven luciferase reporter demonstrated presence of endogenous TP53 function (Data Supplement). On the basis of these data, we consider HT1080 cells to express wild-type TP53. All cell lines were Mycoplasma negative.
Plasmid Construction
p53 was expressed in vitro using the pCDNA3.1-HA-wtp53 plasmid (generous gift of M. Irwin). Generation of single-nucleotide variants (SNVs), based on sequences identified among patients with ACC, was performed using polymerase chain reaction–based site-directed mutagenesis (QuikChange II or QuikChange Lightning; Stratagene, Santa Clara, CA) according to the manufacturer's directions. Mutagenic primer sequences are included in Appendix Table A1. The entire coding sequence of each plasmid was confirmed by direct sequencing of the entire insert.
C-terminal GFP fusion proteins were generated by site-directed mutagenesis of the GFP-p53 plasmid (Boyd SD, et al: Nat Cell Biol 2:563-568, 2000) (Addgene Plasmid No. 12091; Addgene, Cambridge, MA) using QuikChange Lightning, as described earlier, or by subcloning fragments from pCDNA-derived constructs. For generation of pGFP-p53-Ins933a (N311fs), a BamH1 restriction site was introduced, in-frame, immediately upstream of the predicted stop codon resulting from this frameshift mutation. Subsequent digestion with BamH1 to excise the nontranslated 3′ end of the cDNA and recircularization of the plasmid resulted in a C-terminal GFP fusion.
SNVs were selected for further functional analysis based on the following criteria: SNV was identified in the germline of an index patient with ACC; SNV was located within a coding exon of TP53 or at a splicing boundary; and function of the SNV had been characterized in ≤ one other publication (not including the comprehensive mutational analysis performed by Kato et al33 in Saccharomyces cerevisiae).
Cellular Transfection
Transfection was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. For Western blotting, colony reduction, and immunofluorescence, 2 × 105 cells were seeded on 10-cm2 plates 14 hours before transfection. For each plate, 2 μL of Lipofectamine diluted in serum-free medium (OptiMem; Life Technologies) were mixed with 1 μg of plasmid DNA in a total volume of 300 μL and incubated at room temperature for 20 minutes. The mixture was added to cells, and incubation was continued for 6 hours, followed by replacement with serum-containing DMEM and overnight incubation. For luciferase assays, 7 × 104 cells were seeded in 24-well plates and transfected as described earlier using 0.4 μL of Lipofectamine and 200 ng of plasmid DNA (178 ng of luciferase gene reporter plasmid [PG13 or MG15],32 2 ng of Renilla plasmid pR, and 20 ng of pDNA3.1, pCDNA3.1-HA-wtp53, or TP53 mutant constructs) diluted in 60 μL of OptiMem. PG13 is a luciferase gene reporter plasmid driven by a promoter containing 13 copies of the RGC TP53-responsive element. MG15 is a luciferase gene plasmid driven by 15 copies of a subtly mutated TP53 binding site and was used as a negative control to confirm the specificity of TP53-mediated transcription. Constitutive Renilla expression was used to normalize for transfection efficiency. TP53 activity was determined as luciferase activity normalized to Renilla activity, and data are expressed as activity relative to that of wild-type TP53.
Luciferase TP53 Transcriptional Activation Assay
Luciferase assays were performed using a dual-luciferase kit (Promega, Madison, WI) as per the manufacturer's directions. Briefly, 70,000 cells were transfected with a mixture of 178 ng of TP53 reporter (PG13 or MG1532), 2 ng of Renilla, and 20 ng of pCDNA-p53–derived plasmid. PG13 (wild-type binding site) carries 13 tandem repeats of a TP53-response element upstream of a luciferase transcript, whereas MG15 (mutated binding site) carries 15 repeats of the same response element with point mutations that abolish TP53 binding. Lysates were obtained 30 hours after transfection and were analyzed on a Berthold Lumat LB 9507 luminometer (Berthold Technologies, Oak Ridge, TN). All transfections were performed in triplicate. Results are typical of ≥ two independent replicates.
Dominant Negative Activity of TP53 Variants
The luciferase transcriptional activation assay was also used to ascertain dominant negative activity of TP53 variants using two different assays. First, wild-type and variant TP53 constructs were cotransfected into TP53-null H1299 cells with fixed amount of wild-type and increasing TP53 variant construct totaling 20 ng of total DNA. Ability of the variant to interfere with wild-type TP53 activity was subsequently assayed, as described earlier.
In a parallel approach, constructs carrying TP53 variants were transfected into HT1080 fibrosarcoma cells (TP53 wild type), and ability to interfere with endogenous TP53 transactivation of the PG13 reporter was determined by luciferase assay. Semiquantitative analysis of TP53 expression using Western blot of cell lysates demonstrated that transfection of 2 ng of TP53 plasmid resulted in transgene expression levels equivalent to endogenous TP53 (data not shown).
Intracellular pH Measurement
Cells maintained at 2% or 5% carbon dioxide atmosphere were treated with 10 μmol/L of SNARF-1-AM (Molecular Probes; Life Technologies) for 1 hour at 37°C. Intracellular pH (pHi) values were obtained by determining the ratio of the fluorescence emissions of the SNARF-1-AM compound at 575 nm and 635 nm. To calibrate the fluorescence values according to the pHi, control samples were incubated in high-[K+] buffers (20 mmol/L HEPES, 130 mmol/L KCl, 20 mmol/L NaCl, 5 mmol/L glucose, 1 mmol/L CaCl2, 1 mmol/L KH2PO4, and 0.5 mmol/L MgSO4) of different but known ionic strength (ranging from pH 6.5 to 8.5) containing 10 μmol/L of nigericin, a proton ionophore that promotes the equilibration between extracellular pH and pHi. In parallel, cells transiently transfected with the p53-R337H mutant maintained either at 5% or 2% carbon dioxide atmosphere were subjected to gene reporter assays to analyze for changes in p53 transcriptional activity under different pHis.
Western Blotting
Cell lysates were prepared as previously described (Durbin AD, et al: J Clin Invest 119:1558-1570, 2009) and electrophoresed on a 12% acrylamide gel, electroblotted onto polyvinylidene difluoride membrane, and blocked overnight in Odyssey blocking buffer (LI-COR, Lincoln, NE). The membrane was incubated for 1 hour at room temperature with mouse anti-p53 DO-1 (1:2,000; Calbiochem, San Diego, CA) and mouse anti-Vinculin (1:5,000; Millipore, Billerica, MA). Secondary antibodies included IR800 goat antimouse and IR680 goat anti-rabbit (1:5,000; LI-COR). Membranes were imaged using a LI-COR Odyssey infrared scanner.
Colony Reduction Assay
Transfections were performed as described earlier. All transfections were performed in triplicate. Six hours after transfection, cells were dissociated and plated at a density of 5 × 103 cells per 35-mm dish. Twenty-four hours after transfection, selective antibiotic G418 (Geneticin; Life Technologies) was added to transfectants at a concentration of 750 μg/mL. Cells were incubated for 14 days at 37°C, 5% carbon dioxide, with one change of media. Cells were then fixed in 80% methanol and 3.7% formaldehyde and stained with 0.25% crystal violet before visualization. Colonies were counted manually. Data were reported as percent reduction relative to vector-transfected controls. Percent reduction was calculated as follows: (colony count in experimental plate – mean colony count of vector-transfected plate) ÷ mean colony count of vector-transfected plate.
Meta-Analysis of Allelic Function in Different Tumor Types
A search of the International Agency for Research on Cancer (IARC) database (revision 15) by tumor morphology identified all TP53 germline missense mutations associated with the development of ACC, rhabdomyosarcoma, osteosarcoma, and choroid plexus carcinoma. Transactivation activity of each allele was determined as the median activity as measured across eight different TP53 regulated genes, based on data generated by Kato et al,33 as described, and available at http://p53.iarc.fr. Data were then plotted according to Tukey's method with GraphPad Prism version 5.03 for Windows (GraphPad Software, La Jolla, CA). Boxes represent the 25th and 75th percentiles, with the median reflected as a horizontal line within the box (medians for rhabdomyosarcoma and osteosarcoma are 0.00 and 0.04, respectively, and thus overlap the x-axis). Whisker length represents 1.5× the interquartile distance.
Chronologic Ranking of Tumor Sequence
The IARC p53 database (revision 15) was interrogated for all instances of ACC in TP53 mutation carriers. Individuals meeting these criteria with additional malignancies were identified and characterized. In addition, for each individual, other mutation-carrying members of the family were identified, and the ages of diagnosis with tumors were collated and analyzed as described in the text. Tumors were regarded as synchronous if they were diagnosed within 6 months of each other.
RESULTS
TP53 Pathway Polymorphisms
Polymorphisms in the TP53 gene and its target, the ubiquitin ligase, MDM2, have been implicated in altering age of presentation and disease severity for multiple tumor types. Genotypes of TP53 for a common polymorphism in exon 4 (P72R, rs1042522) (Bonafe M, et al: Clin Cancer Res 9:4860-4864, 2003; Dumont P, et al: Nat Genet 33:357-365, 2003; Salvioli S, et al: Cell Cycle 4:1264-1271, 2005; Ignaszak-Szczepaniak M, et al: Oncol Rep 16:65-71, 2006), a 16-bp duplication in intron 3 (Gemignani F, et al: Oncogene 23:1954-1956, 2004; Marcel V, et al: J Med Genet 46:766-772, 2009),31 and a polymorphism in the MDM2 promoter SNP309 (rs2279744) (Marcel V, et al: J Med Genet 46:766-772, 2009; Bond GL, et al: Cell 119:591-602, 2004; Ruijs MW, et al: Eur J Hum Genet 15:110-114, 2007)30 are described in Appendix Table A2. We did not identify a strong effect of any of these polymorphisms in this analysis.
Subcellular Localization Is Unaffected by Most Alleles
Mutations that alter subcellular localization (ie, by interfering with nuclear accumulation) may affect the ability of TP53 variants to direct transcription of target genes. When overexpressed in vitro, all but two variants appropriately localized to the nucleus, suggesting that loss of function resulted primarily from effects on protein function, rather than altered localization (Data Supplement).
pH-Dependent Loss of Function of TP53R337H
The TP53R337H allele has previously been reported to exert pH-dependent loss of function.4 We confirmed this in vitro, altering pHi by manipulating ambient carbon dioxide tension (Data Supplement).
Distribution of Alleles Associated With Other Li-Fraumeni Syndrome Tumors Distinguishes ACC and Choroid Plexus Carcinoma From Other Tumors
To determine whether this representation of variants bearing substantial residual activity was reflective of the larger population of children with ACC, we again referred to the published literature. By cross-referencing the genotype of all reported children with TP53-associated ACC from the IARC database12 with a comprehensive, yeast-based, functional analysis of all possible TP53 mutations,33 we derived the distribution of predicted activity for TP53 alleles associated with several Li-Fraumeni syndrome–component tumors. The distribution of TP53 activity of alleles associated with different tumor types is shown in the Data Supplement. ACC-associated variants included a greater proportion of alleles with retained activity than variants associated with other Li-Fraumeni syndrome–component tumors including rhabdomyosarcoma and osteosarcoma. Individuals diagnosed with choroid plexus carcinoma were also associated with greater retention of TP53 activity, an observation that may reflect common clinical behavior between the two tumors and/or shared embryonic lineage.
Table A1.
Primer Pairs Used for Site-Directed Mutagenesis in Generation of Adrenocortical Carcinoma–Associated TP53 Mutations
| Variant | Codon | Forward Primer | Reverse Primer |
|---|---|---|---|
| P72R | 72 | ggctgctccccgcgtggcccctg | caggggccacgcggggagcagcc |
| F134Y | 134 | gccctcaacaagatgtattgccaactggccaag | cttggccagttggcaatacatcttgttgagggc |
| R158L | 158 | cacccgcgtcctcgccatggcca | tggccatggcgaggacgcgggtg |
| R158H | 158 | cacccgcgtccacgccatggcca | tggccatggcgtggacgcgggtg |
| Y163C | 163 | cgccatggccatctgcaagcagtcacagc | gctgtgactgcttgcagatggccatggcg |
| R175L | 175 | gaggttgtgaggcactgcccccaccat | atggtgggggcagtgcctcacaacctc |
| R175H | 175 | gaggttgtgaggcactgcccccaccat | atggtgggggcagtgcctcacaacctc |
| H193P | 193 | ggcccctcctcagcctcttatccgagtgg | ccactcggataagaggctgaggaggggcc |
| F212I | 212 | ttggatgacagaaacactattcgacatagtgtggtgg | ccaccacactatgtcgaatagtgtttctgtcatccaa |
| P219S | 219 | catagtgtggtggtgtcctatgagccgcctg | caggcggctcataggacaccaccacactatg |
| C229R | 229 | gaggttggctctgaccgtaccaccatccact | agtggatggtggtacggtcagagccaacctc |
| S241Y | 241 | tacaactacatgtgtaacagttactgcatgggcgg | ccgcccatgcagtaactgttacacatgtagttgta |
| R248W | 248 | gggcggcatgaactggaggcccatcc | ggatgggcctccagttcatgccgccc |
| R248L | 248 | ggcggcatgaacctgaggcccatcctc | gaggatgggcctcaggttcatgccgcc |
| I254T | 254 | ggcccatcctcaccaccatcacactggaaga | tcttccagtgtgatggtggtgaggatgggcc |
| R267Q | 267 | tggtaatctactgggacagaacagctttgaggtgc | gcacctcaaagctgttctgtcccagtagattacca |
| R273H | 273 | gaacagctttgaggtgcatgtttgtgcctgtcctg | caggacaggcacaaacatgcacctcaaagctgttc |
| C275X | 275 | ctttgaggtgcgtgtttgaggtgctgggagagaccggc | gccggtctctcccagcacctcaaacacgcacctcaaag |
| E285V | 285 | gaccggcgcacagtggaagagaatctcc | ggagattctcttccactgtgcgccggtc |
| Ins933 | 311 | gcgagcactgcccaacaaacaccagctcc | ggagctggtgtttgttgggcagtgctcgc |
| R337H | 337 | cgtgggcgtgagcacttcgagatgttccgag | ctcggaacatctcgaagtgctcacgcccacg |
| R337H | 337 | ccgtgggcgtgagcacttcgagatgttcc | ggaacatctcgaagtgctcacgcccacgg |
| C275X-GFP | G275 | gctttgaggtgcgtgttcgggatccaccg | cggtggatcccgaacacgcacctcaaagc |
| Ins933a | G311 | gatccgtgggcgcgggatccaccg | cggtggatcccgcgcccacggatc |
| Ins933a-GFP | G-933-b | gatccgtgggcgtgggatccagcgcttcgagatg | catctcgaagcgctggatcccacgcccacggatc |
Table A2.
TP53 and MDM2 SNP Genotypes
| Patient No. | Age (years) | Sex | p53 Genotype | PEX4 | PIN3 | MDM2SNP309 |
|---|---|---|---|---|---|---|
| 1 | 2.4 | M | P152L | R/R | A1/A1 | G/G |
| 2 | 5 | F | P219S | R/R | A1/A1 | T/T |
| 3 | 2.7 | F | Y163C | P/R | A1/A1 | G/T |
| 4 | 3.7 | M | C275ter | R/R | A1/A1 | T/T |
| 5 | 1.1 | F | H193P | P/R | A1/A1 | G/T |
| 6 | 3 | M | T125T c375G>A (r.spl) | R/R | A1/A1 | T/T |
| 7 | 14.3 | F | F134Y | P/P | A2/A2 | G/T |
| 8 | 17.8 | F | del exon 10-11 | R/R | A1/A1 | T/T |
| 9 | 5 | F | R158L | R/R | A1/A1 | G/T |
| 10 | 14 | M | WT | R/R | A1/A1 | T/T |
| 11 | 0.5 | F | C229R | R/R | A1/A1 | T/T |
| 12 | 1.8 | F | G334R | R/R | A1/A1 | G/T |
| 13 | 2 | M | E180K | R/R | A1/A1 | G/T |
| 14 | 0.5 | F | T125T c375G>A (r.spl) | P/R | A1/A1 | G/T |
| 15 | 11 | M | G245C | R/R | A1/A1 | T/T |
| 16 | 3 | F | G245S | P/R | A1/A1 | T/T |
| 17 | 1 | F | R337H | R/R | A1/A1 | G/T |
| 18 | 1 | F | T125T c375G>A (r.spl) | P/R | A1/A2 | G/G |
| 19 | 15 | F | Q52fs | P/R | A1/A1 | G/G |
| 20 | 1.4 | F | WT | R/R | A1/A1 | G/T |
| 21 | 2.3 | F | R158H | P/R | A1/A2 | G/T |
| 26 | 2.2 | F | WT | R/R | A1/A2 | T/T |
| 27 | 17 | F | WT | P/R | A1/A1 | G/T |
| 28 | 13 | F | WT | R/R | A1/A1 | T/T |
| 29 | F | WT | R/R | A1/A1 | G/T | |
| 30 | 5 | M | WT | R/R | A1/A1 | G/T |
| 31 | 0.5 | F | WT | P/P | A1/A2 | G/T |
| 32 | 3 | M | WT | R/R | A1/A1 | T/T |
| 33 | 0.5 | F | WT | R/R | A1/A2 | T/T |
| 34 | 15 | F | WT | R/R | A1/A1 | G/T |
| 35 | 12 | M | WT | P/R | A1/A2 | T/T |
| 37 | 0.6 | F | WT | P/R | A1/A1 | G/T |
| 38 | 2 | F | I254T | P/R | A1/A2 | T/T |
| 40 | 3.6 | F | WT | R/R | A1/A1 | G/T |
| 41 | 7.9 | F | S241Y | P/R | A1/A1 | G/T |
| 42 | 1.4 | F | del exon 10-11 | R/R | A1/A1 | G/T |
| 43 | M | WT | R/R | A1/A1 | G/T | |
| 44 | F | C229R | R/R | A1/A1 | T/T | |
| 45 | 6.2 | M | R273S | P/R | A1/A2 | G/T |
| 46 | 1.6 | F | WT | R/R | A1/A1 | G/T |
| 47 | 4 | F | R267Q | P/R | A1/A1 | G/G |
| 48 | 3 | M | WT | P/P | A1/A2 | G/T |
| 49 | 0.5 | M | R248L | P/R | A1/A1 | G/T |
| 50 | 15 | M | P47S | P/P | A1/A1 | T/T |
| 51 | 17 | M | WT | R/R | A1/A1 | G/T |
| 52 | 8 | F | WT | P/R | A1/A1 | G/T |
| 53 | 1 | F | R213P | P/R | A1/A2 | T/T |
| 54 | 1 | F | WT | R/R | A1/A1 | T/T |
| 55 | 13 | F | WT | P/R | A1/A1 | G/T |
| 56 | 20 | F | WT | R/R | A1/A1 | T/T |
| 57 | 1 | F | WT | R/R | A1/A1 | G/G |
| 58 | 0.5 | M | E285V | R/R | A1/A1 | G/T |
| 59 | 7.4 | F | R273H | P/R | A1/A2 | G/T |
| 61 | 14 | F | WT | P/R | A1/A1 | G/T |
| 62 | 1 | F | R337H | P/R | A1/A1 | T/T |
| 64 | 3 | F | del exon 2-10 | P/P | A1/A1 | G/T |
| 65 | 1 | F | R337H | P/R | A1/A1 | G/T |
| 66 | 3 | F | R337H | R/R | A1/A1 | G/G |
| 67 | 3 | F | R337H | R/R | A1/A1 | G/T |
| 68 | 4 | F | R337H | R/R | A1/A1 | G/G |
| 70 | 12 | F | WT | P/P | A1/A2 | G/T |
| 71 | 13 | F | C229R | R/R | A1/A1 | G/T |
| 72 | F | WT | P/R | A1/A2 | G/T | |
| 73 | 1.3 | F | WT | R/R | A1/A1 | T/T |
NOTE. See text for details.
Abbreviations: F, female; M, male; SNP, single-nucleotide polymorphism; WT, wild type.
Table A3.
Individuals Identified in the Current Study and IARC Database With More Than One Primary Malignancy
| Mutation | Sex | First Tumor |
Second Tumor |
Third Tumor |
Activity (according to Kato et al33; %) | IARC Reference | |||
|---|---|---|---|---|---|---|---|---|---|
| Type | Age (years) | Type | Age (years) | Type | Age (years) | ||||
| G108_L111delinsIQ | M | ACC | 1 | Rhabdomyosarcoma | 15 | NA | 2 | ||
| T125T | F | ACC | NR | Cancer NOS | 35 | 0 | 41 | ||
| T125T* | F | ACC | 35 | Breast cancer | 35 | 0 | 221 | ||
| K132Q | F | ACC | 1 | Osteosarcoma | 11 | 7.1 | 191 | ||
| A138P | F | ACC | 2 | Rhabdomyosarcoma | 3 | 8.1 | 56 | ||
| Q144L | F | ACC | 1 | Osteosarcoma | 26 | Bowen's disease (SCC) | 31 | 54.6 | 105 |
| P151S | F | ACC | 1 | Fibrosarcoma | 4 | Rhabdomyosarcoma | 5 | 5.2 | 13 |
| P152L | F | ACC | 2 | Chondrosarcoma | 11 | Osteosarcoma | 12 | 16.1 | 68 |
| P152L | F | ACC | 1 | Osteochondroma | 13 | Leiomyosarcoma | 26 | 16.1 | 68 |
| R158H | F | ACC | 1 | Spindle cell sarcoma (breast) | 14 | 19.4 | 68 | ||
| Y163C | F | ACC | 2 | Astrocytoma anaplastic | 13 | 5.6 | NA | ||
| H179Y | F | Fibrosarcoma | 2 | ACC | 6 | Osteosarcoma | 14 | 22 | 170 |
| H193P* | F | ACC | 1 | CPC | 1 | 11.8 | 177 | ||
| H214R | F | Breast cancer | 35 | Brain cancer | 40 | ACC | 41 | 8.3 | 223 |
| Y236del | F | Anaplastic astrocytoma | 22 | ACC | 27 | 0 (predicted) | 21 | ||
| S241Y | F | CPC | 4 | ACC | 7 | 0 | NA | ||
| R248W | F | ACC | 1 | Cancer NOS | 29 | 0 | 2 | ||
| R248Q | F | ACC | 2 | PNET (brain) | 5 | 0 | 42 | ||
| R248W* | F | ACC | 1 | Ganglioneuroblastoma | 1 | 0 | 69 | ||
| R248W | M | ACC | 4 | CPP | 6 | 0 | 73 | ||
| R248W* | F | ACC | 1 | Neuroblastoma | 1 | 0 | 172 | ||
| V272M | F | Cervical cancer | 29 | ACC | 69 | 17.2 | NA | ||
| R273L* | F | ACC | 1 | Rhabdomyosarcoma | 1 | 0 | 62 | ||
| R273H* | M | ACC | 1 | Rhabdomyosarcoma | 1 | 0 | 146 | ||
| R273S | M | ACC | 6 | PNET (brain) | 11 | 10.3 | 177 | ||
| R273H | F | Rhabdomyosarcoma | 1 | ACC | 7 | Osteosarcoma | 7 | 0 | 128 |
| R282W | F | ACC | 2 | Osteosarcoma | 5 | 0 | 178 | ||
| E285Q | F | ACC | 1 | Osteosarcoma (periosteal) | 11 | 118.6 | 93 | ||
| E285V* | M | ACC | 1 | CPC | 1 | 8.9 | 181 | ||
| R337H | F | ACC | 7 | Renal carcinoma | 8 | 69 | 138 | ||
| R337P* | M | ACC | 5 | ALL | 5 | 8.1 | 219 | ||
| R337H | F | Leukemia | 7 | ACC | 10 | Sarcoma NOS | 10 | 69 | 196 |
| R342X | M | ACC | 2 | Medulloblastoma | 5 | NA | 178 | ||
NOTE. References for individual mutations are indexed on the IARC database, and the IARC citation is provided in the far right column.
Abbreviations: ACC, adrenocortical carcinoma; ALL, acute lymphoblastic leukemia; CPC, choroid plexus carcinoma; CPP, choroid plexus papilloma; F, female; IARC, International Agency for Research on Cancer; M, male; NA, not applicable; NOS, not otherwise specified; NR, not reported; PNET, primitive neuroectodermal tumor; SCC, squamous cell carcinoma.
Patients with synchronous tumors.
Footnotes
Supported in part by Grant No. MOP106639 from the Canadian Institutes of Health Research (D.M.) and by National Institutes of Health/National Cancer Institute Cancer Center Support CORE Grant No. CA21765 and the American Lebanese Syrian Associated Charities of St Jude Children's Research Hospital (R.C.R. and G.P.Z.). J.D.W. was funded by the Canadian Pediatric Endocrine Group and the Lawson Wilkins Pediatric Endocrine Society and was a postdoctoral fellow of the Canadian Child Health Clinician Scientist Program.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Financial support: David Malkin
Administrative support: Ana Novokmet
Provision of study materials or patients: Ana Novokmet, Raul C. Ribeiro, Carlos Rodriguez-Galindo, Gerard P. Zambetti, David Malkin
Collection and assembly of data: Jonathan D. Wasserman, Ana Novokmet
Data analysis and interpretation: Jonathan D. Wasserman, Carlos Rodriguez-Galindo, David Malkin
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Prevalence and Functional Consequence of TP53 Mutations in Pediatric Adrenocortical Carcinoma: A Children's Oncology Group Study
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Jonathan D. Wasserman
No relationship to disclose
Ana Novokmet
No relationship to disclose
Claudia Eichler-Jonsson
No relationship to disclose
Raul C. Ribeiro
No relationship to disclose
Carlos Rodriguez-Galindo
No relationship to disclose
Gerard P. Zambetti
No relationship to disclose
David Malkin
No relationship to disclose
REFERENCES
- 1.Parkin DM. Vol. II. Lyon, France: International Agency for Research on Cancer; 1998. International Agency for Research on Cancer: International Incidence of Childhood Cancer. [Google Scholar]
- 2.Michalkiewicz E, Sandrini R, Figueiredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004;22:838–845. doi: 10.1200/JCO.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 3.Ribeiro RC, Sandrini F, Figueiredo B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001;98:9330–9335. doi: 10.1073/pnas.161479898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiGiammarino EL, Lee AS, Cadwell C, et al. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat Struct Biol. 2002;9:12–16. doi: 10.1038/nsb730. [DOI] [PubMed] [Google Scholar]
- 5.Lomax ME, Barnes DM, Hupp TR, et al. Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene. 1998;17:643–649. doi: 10.1038/sj.onc.1201974. [DOI] [PubMed] [Google Scholar]
- 6.Ribeiro RC, Pinto EM, Zambetti GP, et al. The International Pediatric Adrenocortical Tumor Registry initiative: Contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol Cell Endocrinol. 2012;351:37–43. doi: 10.1016/j.mce.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Li FP, Fraumeni JF, Jr, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 8.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 9.Tinat J, Bougeard G, Baert-Desurmont S, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol. 2009;27:e108–109. doi: 10.1200/JCO.2009.22.7967. [DOI] [PubMed] [Google Scholar]
- 10.Wagner J, Portwine C, Rabin K, et al. High frequency of germline p53 mutations in childhood adrenocortical cancer. J Natl Cancer Inst. 1994;86:1707–1710. doi: 10.1093/jnci/86.22.1707. [DOI] [PubMed] [Google Scholar]
- 11.Varley JM, McGown G, Thorncroft M, et al. Are there low-penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet. 1999;65:995–1006. doi: 10.1086/302575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li-Fraumeni syndrome: Clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- 14.West AN, Ribeiro RC, Jenkins J, et al. Identification of a novel germ line variant hotspot mutant p53–R175L in pediatric adrenal cortical carcinoma. Cancer Res. 2006;66:5056–5062. doi: 10.1158/0008-5472.CAN-05-4580. [DOI] [PubMed] [Google Scholar]
- 15.Custódio G, Parise GA, Kiesel Filho N, et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol. 2013;31:2619–2626. doi: 10.1200/JCO.2012.46.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu CC, Shete S, Amos CI, et al. Joint effects of germ-line p53 mutation and sex on cancer risk in Li-Fraumeni syndrome. Cancer Res. 2006;66:8287–8292. doi: 10.1158/0008-5472.CAN-05-4247. [DOI] [PubMed] [Google Scholar]
- 17.Hwang SJ, Lozano G, Amos CI, et al. Germline p53 mutations in a cohort with childhood sarcoma: Sex differences in cancer risk. Am J Hum Genet. 2003;72:975–983. doi: 10.1086/374567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hisada M, Garber JE, Fung CY, et al. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90:606–611. doi: 10.1093/jnci/90.8.606. [DOI] [PubMed] [Google Scholar]
- 19.Villani A, Tabori U, Schiffman J, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: A prospective observational study. Lancet Oncol. 2011;12:559–567. doi: 10.1016/S1470-2045(11)70119-X. [DOI] [PubMed] [Google Scholar]
- 20.Hainaut P. Tumor-specific mutations in p53: The acid test. Nat Med. 2002;8:21–23. doi: 10.1038/nm0102-21. [DOI] [PubMed] [Google Scholar]
- 21.Glazko GV, Koonin EV, Rogozin IB. Mutation hotspots in the p53 gene in tumors of different origin: Correlation with evolutionary conservation and signs of positive selection. Biochim Biophys Acta. 2004;1679:95–106. doi: 10.1016/j.bbaexp.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Olivier M, Goldgar DE, Sodha N, et al. Li-Fraumeni and related syndromes: Correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643–6650. [PubMed] [Google Scholar]
- 23.Russell-Swetek A, West AN, Mintern JE, et al. Identification of a novel TP53 germline mutation E285V in a rare case of paediatric adrenocortical carcinoma and choroid plexus carcinoma. J Med Genet. 2008;45:603–606. doi: 10.1136/jmg.2008.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernandez-Boussard T, Rodriguez-Tome P, Montesano R, et al. IARC p53 mutation database: A relational database to compile and analyze p53 mutations in human tumors and cell lines—International Agency for Research on Cancer. Hum Mutat. 1999;14:1–8. doi: 10.1002/(SICI)1098-1004(1999)14:1<1::AID-HUMU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 25.Guimaraes DP, Hainaut P. TP53: A key gene in human cancer. Biochimie. 2002;84:83–93. doi: 10.1016/s0300-9084(01)01356-6. [DOI] [PubMed] [Google Scholar]
- 26.Horvath A, Bertherat J, Groussin L, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): An update. Hum Mutat. 2010;31:369–379. doi: 10.1002/humu.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warneford SG, Witton LJ, Townsend ML, et al. Germ-line splicing mutation of the p53 gene in a cancer-prone family. Cell Growth Differ. 1992;3:839–846. [PubMed] [Google Scholar]
- 28.Varley JM, Chapman P, McGown G, et al. Genetic and functional studies of a germline TP53 splicing mutation in a Li-Fraumeni-like family. Oncogene. 1998;16:3291–3298. doi: 10.1038/sj.onc.1201878. [DOI] [PubMed] [Google Scholar]
- 29.Soudon J, Caron de Fromentel C, Bernard O, et al. Inactivation of the p53 gene expression by a splice donor site mutation in a human T-cell leukemia cell line. Leukemia. 1991;5:917–920. [PubMed] [Google Scholar]
- 30.Bougeard G, Baert-Desurmont S, Tournier I, et al. Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li-Fraumeni syndrome. J Med Genet. 2006;43:531–533. doi: 10.1136/jmg.2005.037952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costa S, Pinto D, Pereira D, et al. Importance of TP53 codon 72 and intron 3 duplication 16bp polymorphisms in prediction of susceptibility on breast cancer. BMC Cancer. 2008;8:32. doi: 10.1186/1471-2407-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 33.Kato S, Han SY, Liu W, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Figueiredo BC, Sandrini R, Zambetti GP, et al. Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J Med Genet. 2006;43:91–96. doi: 10.1136/jmg.2004.030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chompret A, Brugières L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer. 2000;82:1932–1937. doi: 10.1054/bjoc.2000.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li FP, Fraumeni JF., Jr Soft-tissue sarcomas, breast cancer, and other neoplasms: A familial syndrome? Ann Intern Med. 1969;71:747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 37.Lindor NM, McMaster ML, Lindor CJ, et al. Concise handbook of familial cancer susceptibility syndromes–second edition. J Natl Cancer Inst Monogr. 2008;38:1–93. doi: 10.1093/jncimonographs/lgn001. [DOI] [PubMed] [Google Scholar]
- 38.Choong SS, Latiff ZA, Mohamed M, et al. Childhood adrenocortical carcinoma as a sentinel cancer for detecting families with germline TP53 mutations. Clin Genet. 2012;82:564–568. doi: 10.1111/j.1399-0004.2012.01841.x. [DOI] [PubMed] [Google Scholar]
- 39.Wasserman JD, Zambetti GP, Malkin D. Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Mol Cell Endocrinol. 2012;351:101–110. doi: 10.1016/j.mce.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herrmann LJ, Heinze B, Fassnacht M, et al. TP53 germline mutations in adult patients with adrenocortical carcinoma. J Clin Endocrinol Metab. 2012;97:E476–E485. doi: 10.1210/jc.2011-1982. [DOI] [PubMed] [Google Scholar]
- 41.Raymond VM, Else T, Everett JN, et al. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98:E119–E125. doi: 10.1210/jc.2012-2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zambetti GP. The p53 mutation “gradient effect” and its clinical implications. J Cell Physiol. 2007;213:370–373. doi: 10.1002/jcp.21217. [DOI] [PubMed] [Google Scholar]
- 43.Letouzé E, Rosati R, Komechen H, et al. SNP array profiling of childhood adrenocortical tumors reveals distinct pathways of tumorigenesis and highlights candidate driver genes. J Clin Endocrinol Metab. 2012;97:E1284–E1293. doi: 10.1210/jc.2012-1184. [DOI] [PubMed] [Google Scholar]
- 44.Wooten MD, King DK. Adrenal cortical carcinoma: Epidemiology and treatment with mitotane and a review of the literature. Cancer. 1993;72:3145–3155. doi: 10.1002/1097-0142(19931201)72:11<3145::aid-cncr2820721105>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 45.West AN, Neale GA, Pounds S, et al. Gene expression profiling of childhood adrenocortical tumors. Cancer Res. 2007;67:600–608. doi: 10.1158/0008-5472.CAN-06-3767. [DOI] [PubMed] [Google Scholar]
- 46.Faria AM, Almeida MQ. Differences in the molecular mechanisms of adrenocortical tumorigenesis between children and adults. Mol Cell Endocrinol. 2012;351:52–57. doi: 10.1016/j.mce.2011.09.040. [DOI] [PubMed] [Google Scholar]
- 47.Bocian-Sobkowska J, Woźniak W, Malendowicz LK. Postnatal involution of the human adrenal fetal zone: Stereologic description and apoptosis. Endocr Res. 1998;24:969–973. doi: 10.3109/07435809809032718. [DOI] [PubMed] [Google Scholar]
- 48.Coulter CL. Fetal adrenal development: Insight gained from adrenal tumors. Trends Endocrinol Metab. 2005;16:235–242. doi: 10.1016/j.tem.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 49.Mesiano S, Jaffe RB. Developmental and functional biology of the primate fetal adrenal cortex. Endocr Rev. 1997;18:378–403. doi: 10.1210/edrv.18.3.0304. [DOI] [PubMed] [Google Scholar]
- 50.Coulter CL. Functional biology of the primate fetal adrenal gland: Advances in technology provide new insight. Clin Exp Pharmacol Physiol. 2004;31:475–484. doi: 10.1111/j.1440-1681.2004.04031.x. [DOI] [PubMed] [Google Scholar]
- 51.Rainey WE, Carr BR, Wang ZN, et al. Gene profiling of human fetal and adult adrenals. J Endocrinol. 2001;171:209–215. doi: 10.1677/joe.0.1710209. [DOI] [PubMed] [Google Scholar]
- 52.Rainey WE, Parker CR, Jr, Rehman K, et al. The adrenal genetic puzzle: How do the fetal and adult pieces differ? Endocr Res. 2002;28:611–622. doi: 10.1081/erc-120016974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.