

Abstract
Loss of basal forebrain cholinergic innervation of the hippocampus and severe neuronal loss within the hippocampal CA1 region are early hallmarks of Alzheimer’s disease, and are strongly correlated with cognitive status. Various therapeutic approaches involve attempts to enhance neurotransmission or to provide some level of neuroprotection for remaining cells. An alternative approach may involve the generation of new cells to replace those lost in AD. Indeed, a simple shift in the balance between cell generation and cell loss may slow disease progression and possibly even reverse existing cognitive deficits. One potential neurogenic regulator might be acetylcholine, itself, which has been shown to play a critical role in hippocampal development. Here, we report the effects of various cholinergic compounds on indices of hippocampal neurogenesis, demonstrating a significant induction following pharmacological activation of muscarinic M1 receptors, located on hippocampal progenitors in the adult brain. This is the first report that a small-molecule agonist may induce neurogenesis in the hippocampal CA1 region. Furthermore, such treatment reversed deficits in markers of neurogenesis and spatial working memory triggered by cholinergic denervation in a rodent model. This study suggests the use of small molecule, receptor agonists may represent a novel means to trigger the restoration of specific neuronal populations lost to a variety of neurodegenerative disorders, such as Parkinson’s, Alzheimer’s, Huntington’s and Amyotrophic Lateral Sclerosis.
Keywords: Alzheimer’s disease, Acetylcholine, Neurogenesis, Hippocampus
1. Introduction
The discovery of ongoing neurogenesis in the adult brain has opened up new avenues for cell replacement strategies targeting endogenous progenitor cells as a novel tissue resource. While various growth factors are well-known for their regulatory role during neurodevelopment, it is becoming increasingly apparent that neurotransmitters also regulate the generation and fate of neural progenitors (Cameron et al., 1998; Nguyen et al., 2001) both during embryogenesis (Barker et al., 1998; Miranda-Contreras et al., 1999; Coronas et al., 2004), and into adulthood (Duman et al., 2001; Powrozek et al., 2004; Van Kampen et al., 2004; Encincas et al., 2006; Kotani et al., 2006; Borta and Hoglinger, 2007). Acetylcholine (ACh) is one such neurotransmitter (Hohmann, 2003; Mohapel et al., 2005).
In the adult brain, new neurons are continuously generated throughout life in the hippocampal dentate gyrus (DG) (Eriksson et al., 1998). Newborn cells in this region are contacted by cholinergic projections (Kotani et al., 2006) and express cholinergic receptors of both the nicotinic (Kaneko et al., 2006) and muscarinic subtypes (Mohapel et al., 2005; Kaneko et al., 2006). Removal of this cholinergic innervation, through depletion of forebrain ACh, impairs neurogenesis in the adult DG (Cooper-Kuhn et al., 2004; Mohapel et al., 2005; Van der Borght et al., 2005). Such deficits in hippocampal neurogenesis have been observed in several animal models of Alzheimer’s disease (AD) (Haughey et al., 2002; Feng et al., 2001; Wang et al., 2004; Donovan et al., 2006), a disease marked by significant degeneration of basal forebrain cholinergic neurons (Whitehouse et al., 1982; Coyle et al., 1983). In AD patients, a reduction in neural progenitors has been reported, with a strong association between neurogenesis and the degree of cholinergic loss (Ziabreva et al., 2006).
In light of the pivotal role the hippocampus plays in learning and memory (Morris et al., 1982; Squire, 1982; Jacobs and Schenk, 2003), and the strong association between neurogenesis and hippocampal function (Shors et al., 2001; Madsen et al., 2003; Rola et al., 2004), measures designed to enhance hippocampal neurogenesis could have therapeutic value in AD. We have previously reported significant elevations in neurogenesis following chronic exposure to dopaminergic agonists (Van Kampen and Robertson, 2005), which was capable of reversing cellular and locomotor deficits when applied to a rodent model of Parkinson’s disease (PD) (Van Kampen and Eckman, 2006). In this report, we examine whether a similar approach could be applied to the cholinergic system for possible therapeutic application in AD. Specifically, we sought to determine whether cholinergic agonist treatment would regulate neurogenesis and restore cellular and behavioural deficits associated with cholinergic cell loss.
2. Materials and methods
2.1. Animals
All studies used 250 g female Sprague–Dawley rats (Harlan). Animals were housed in a temperature-controlled environment with a 12 h light/dark cycle and ad libitum access to standard rat chow and water. All animal experimentation was conducted in accordance with the NIH guidelines for the care and use of laboratory animals and were approved by the Mayo Foundation Institutional Animal Care and Use Committee (IACUC).
2.2. Drug delivery
For intracerebroventricular delivery, animals were anaesthetized using isoflurane (1%) and placed in a Kopf stereotaxic frame. Stainless steel indwelling cannulae were placed into the left lateral ventricle (A.P. 0.00, M.L. +1.30, D.V. −3.50). The cannula (30Ga; Plastics ONE, Raonoke, Virginia) was fixed to the skull using dental acrylic and jeweler’s screws. Each cannula was attached, by 50 PE polyethylene tubing, to an osmotic minipump (Alza, 2002, 0.5 μl/h, 2 weeks; Alza, 2004, 0.25 μl/h, 4 weeks), which was placed under the skin at the base of the neck. Each pump was filled with either the cholinesterase inhibitor physostigmine (10, 50 μg/h), the nicotinic receptor agonist nicotine (0.5, 5 μg/h), the muscarinic agonist, oxotremorine (1, 5, 30 μg/h), the nonselective cholinergic agonist, carbachol (1, 5, 30 μg/h) (Sigma, St. Louis, MO) or their vehicle, 0.9% saline. For antagonist studies, pumps were filled with either saline (0.9%), or oxotremorine (30 μg/h), along with one of various doses of the M1 receptor-selective antagonist, pirenzepine (0, 1, 5, 20 μg/h) or the nicotinic receptor antagonist, mecamylamine (0, 1, 5, 20 μg/h).
2.3. Bromodeoxyuridine administration
During the drug treatments, all animals received daily injections of bromo-deoxyuridine (BrdU) (50 mg/kg, i.p.) (Sigma, St. Louis, MO), to label proliferating cells.
2.4. Immunolesioning
The immunotoxin, 192IgG saporin, was used to create selective basal forebrain cholinergic lesions. The immunotoxin, 192IgG saporin (SAP), consists of a monoclonal antibody to the low affinity/p75 nerve growth factor receptor, 192IgG, that is coupled to the ribosome-inactivating protein, saporin (Wiley, 1992). The p75 NGFr is highly expressed in basal forebrain cholinergic neurons relative to other neurons or regions, allowing for a selective neurotoxic lesion. SAP has been used to create such selective basal forebrain cholinergic lesions with the aim of clarifying the role of cholinergic dysfunction in the deficits associated with AD (Wiley et al., 1995; Rossner, 1997).
Animals received acute intracerebroventricular infusion of the immunotoxin, 192IgG saporin (5 μg/5 μl). Thus, animals were anaesthetized using isoflurane (1%) and placed in a Kopf stereotaxic frame. The immunotoxin, 192IgG saporin was injected unilaterally into the left lateral ventricle (A.P. 0.00, M.L. +1.30, D.V. −3.50) (5 μg/5 μl) at a rate of 1 μl/min via an infusion cannula connected by polyethylene tubing (50 PE) to a 50 μl Hamilton syringe driven by a Harvard pump. Following infusion, the toxin was permitted to diffuse away from the cannula for 2 min before withdrawal.
2.5. Radial arm maze
Following 4 weeks of treatment, animals were placed on a food-restricted diet. Animals had free access to food 4 h at the same time each day, beginning immediately following the scheduled completion of each RAM trial. Following one week of food-restriction, animals underwent an adaptation phase using the radial arm maze (RAM). Each animal was placed on the center platform and permitted to explore for 5 min. During this phase, the reward, Cheetos®, was scattered throughout the RAM. Then, the reward was then placed half-way down the arms chosen to be baited. Finally, the reward was placed at the end of the baited arms. Following 3 days of the adaptation phase, each animal was given one trial per day, five trials per week, for a total of 25 trials. The RAM consisted of 8 arms, of which 5 were baited with the reward. The baited and unbaited arms were chosen randomly for each rat and remained constant throughout the experiment. At the beginning of each trial, the animal was placed on the central platform and permitted to move throughout the maze until either all five rewards had been taken or 10 min had elapsed. Entries into unbaited arms and re-entries into previously visited arms were recorded as reference memory and working memory errors, respectively. For analysis, data were divided into 5 blocks of 5 trials each, and the scores averaged over each block. Drug treatment was terminated 3 days prior to the final block of testing.
2.6. Immunohistochemistry
Animals were sacrificed by transcardial perfusion with 4% paraformaldehyde. Brains were removed and postfixed for 24 h in 4% paraformaldehyde followed by cryoprotection in 30% sucrose. Symmetrical 40 μm-thick sections were cut on a freezing microtome and stored in a Millonigs solution. Every twelfth section was processed for immunohistochemistry. Free-floating sections were pretreated with 50% formamide/280 mM, incubated in 2 M HCl at 37 °C for 30 min, and rinsed in 0.1 M boric acid (pH 8.5) at room temperature for 10 min. Sections were incubated in 1% H2O2 in phosphate buffered saline for 15 min, in blocking solution (3% goat or donkey serum/0.3% Triton X-100/Tris-buffered saline [TBS]) for 1 h at room temperature, followed by the appropriate antibody at 4 °C overnight. Anti-BrdU antibodies were mouse monoclonal anti-BrdU (1:250, Chemicon) or sheep polyclonal anti-BrdU (1:10,000, RDI). The other primary antibodies were monoclonal mouse anti-glial fibrillary acidic protein (GFAP) (1:1000, Chemicon), monoclonal mouse anti-Tuj1 (1:1000, Chemicon), monoclonal mouse anti-NeuN (1:1000; Chemicon), polyclonal goat anti-choline acetyltransferase (ChAT) (1:3000, Chemicon), and proliferating cell nuclear antigen (PCNA) (1:1000, Santa Cruz). For fluorescent visualization, sections were incubated with the respective secondary antibody conjugated to either Alexa 488, Alexa 594, Alexa 633, or Alexa 350 (Molecular Probes). In between steps, sections were washed for 3 × 10 min in TBS. Sections were mounted on unsubbed glass slides and coverslipped in Fluoromount. Fluorescence signals were detected with a Zeiss axiophot laser scanning confocal microscope LSM 510 Meta at excitation/emission wavelengths of 535/565 nm, 470/505 nm, and 585/615 nm.
2.7. Pyknotic cells
Separate sets of hippocampus sections were counterstained with Cresyl Violet acetate, dehydrated and coverslipped with Permount (Fisher Scientific). Pyknotic cells were counted on these sections as a morphological measure of cell death on every 12th section. Cells were considered pyknotic if they lacked a nuclear membrane, had pale or absent cytoplasm and darkly stained condensed chromatin.
2.8. Cell counts
Unbiased stereological cell counts were obtained using a computer-assisted image analysis system, Zeiss AIMM (Axiovision Interactive Measurement Module). Immunopositive cells were counted in every 12th section through the dentate gyrus and CA1 region of the hippocampus using a 20× objective (sampling frame area, 90,000 μm2) containing an optical grid. The total number of immunopositive cells was counted in each region of each section and volume measurements of the selected region were used to estimate the number of cells per mm3. A double-labeled BrdU/phenotypic marker-positive cell was defined as having the strongest intensity of both immunolabels within the same or neighbouring 1.5-μm-thick optical section through the cell in a consecutive Z-series of at least 10 sections, at 200× magnification and with a resolution of 1024 × 1024 pixels. Cells not present in their entirety were excluded. All double-labeling was confirmed by rotating the image along each axis to ensure signals were localized within the same cell rather than separate cells in close apposition. Slides were blind-coded to eliminate experimenter bias.
2.9. Statistical analysis
Data were analyzed using a multivariant analysis of variance. Where significant F-values were obtained, planned pair-wise comparisons were made using Newman–Keuls. Differences were considered statistically significant when p < 0.05.
3. Results
3.1. Pharmacological activation of muscarinic M1 receptors enhances hippocampal cell proliferation
First, we sought to determine the effects of various cholinergic compounds on the induction of neurogenesis in intact animals. Following 10 days of intracerebroventricular infusion, we observed dose-dependent elevations in immunolabeling for both BrdU and the endogenous proliferation marker, PCNA, in response to the muscarinic agonist, oxotremorine, and inverse dose-dependent effects in response to the nonselective cholinergic agonist, carbachol in the subgranular zone of the DG (BrdU: F7,47 = 34.46, p < 0.0001; PCNA: F7,47 = 35.51, p < 0.0001) (Fig. 1a). Maximal induction (~458%) was observed following the highest dose of oxotremorine (30 μg/h), while only lower doses of carbachol appeared to elevate cell proliferation in the DG (~450%). Physostigmine, triggered a modest, yet significant, elevation (~102%) in BrdU labeling. However, no changes in PCNA immunolabeling were found, suggesting that this cholinesterase inhibitor may influence neurogenesis by enhancing the survival of newly-generated cells rather than through direct effects on cell proliferation. Conversely, and as an additional indication of specificity, infusion with the nicotinic receptor agonist, nicotine, resulted in significant reductions in both BrdU and PCNA immunolabeling in the DG compared to saline-treated controls. As the hippocampal cornus ammonis 1 (CA1) region undergoes a severe neuronal loss in AD (Ball, 1977; Davies et al., 1992; Fukutani et al., 2000), one closely associated with disease severity (Bobinski et al., 1998; Rössler et al., 2002; von Gunten et al., 2006), we also examined effects on cell proliferation in the CA1 region. Baseline levels of cell proliferation were lower than that observed in the DG but were similarly affected by pharmacological activation (BrdU: F7,47 = 34.46, p < 0.0001; PCNA: F7,47 = 33.48, p < 0.0001) (Fig. 1b).
Fig. 1.
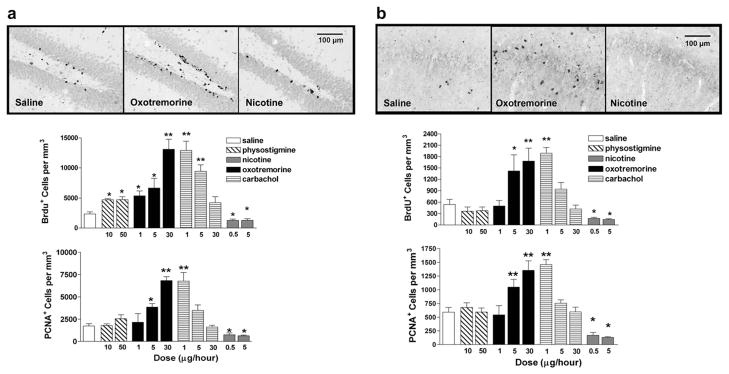
Cholinergic drugs differentially regulate indices of cell proliferation in the adult hippocampus. a, Both BrdU+ and PCNA+ cell counts were significantly reduced following chronic administration of nicotine in the DG, while the muscarinic agonists, oxotremorine and carbachol induced a dose-dependent and inversely dose-dependent elevation in immunolabeling, respectively, for both markers of cell proliferation. Slight elevations in BrdU labeling were also observed in response to the AChE inhibitor, phystostigmine. Representative photomicrographs depict BrdU immunolabeling in the DG following saline, nicotine or oxotremorine treatment. b, Similar elevations in both indices of cell proliferation were also evident in the CA1 region following treatment with oxotremorine or carbachol. By contrast, nicotine significantly reduced BrdU+ and PCNA+ cells in the CA1 region. Representative photomicrographs depict BrdU immunolabeling in the CA1 region following saline, nicotine or oxotremorine treatment. Each bar represents the mean (±S.E. M.) (n = 6) number of immunopositive cells per mm3. ** sig. diff. from saline-treated controls, p < 0.001; *p < 0.05. Scalebar = 100 μm.
Muscarinic M1 receptors are the most predominant receptor subtype in the hippocampus, showing prominent localization to the soma and dendrites of CA1 pyramidal neurons and the granule cells of the DG (Levey et al., 1995) and have recently been found localized on newborn cells in the hippocampus (Mohapel et al., 2005). Here, oxotremorine-induced elevations in both hippocampal BrdU and PCNA immunolabeling were blocked by co-infusion of the muscarinic M1 receptor antagonist, pirenzepine, with no effect on baseline levels. There was no significant effect of the nicotinic antagonist, mecamylamine, indicating that this positive influence on cytogenesis is likely specifically mediated through the M1 receptor (Fig. 2a) [BrdU: (DG: F1,80 = 558.50, p < 0.0001, AGONIST main effect; F1,80 = 71.39, p < 0.0001, ANTAGONIST main effect; F1,80 = 47.61, p < 0.0001, AGONIST × ANTAGONIST interaction effect) CA1: (F1,80 = 432.01, p < 0.0001, AGONIST main effect; F1,80 = 33.24, p < 0.0001, ANTAGONIST main effect; F1,80 = 37.41, p < 0.0001, AGONIST × ANTAGONIST interaction effect)] [PCNA: (F1,80 = 287.41, p < 0.0001, AGONIST main effect; F1,80 = 39.80, p < 0.0001, ANTAGONIST main effect; F1,80 = 32.95, p < 0.0001, AGONIST × ANTAGONIST interaction effect) CA1: (F1,80 = 329.54, p < 0.0001, AGONIST main effect; F1,80 = 32.97, p < 0.0001, ANTAGONIST main effect; F1,80 = 27.46, p < 0.0001, AGONIST × ANTAGONIST interaction effect)]. Consistent with the induction of cell proliferation being mediated through muscarinic receptor activation, co-administration of pirenzepine also blocked elevations in BrdU labeling triggered by low dose carbachol (1 μg/h) (Fig. 2b) (DG: F1,80 = 219.45, p < 0.0001, AGONIST main effect; F1,80 = 39.77, p < 0.0001, ANTAGONIST main effect; F1,80 = 27.46, p < 0.0001, AGONIST × ANTAGONIST interaction effect CA1: F1,80 = 224.50, p < 0.0001, AGONIST main effect; F1,80 = 21.43, p < 0.0001, ANTAGONIST main effect; F1,80 = 24.76, p < 0.0001, AGONIST × ANTAGONIST interaction effect). While a low dose of carbachol nearly tripled the number of BrdU-labeled cells in the hippocampus, higher doses were ineffective. However, when co-administered with the nicotinic receptor antagonist, mecamylamine (5, 20 μg/h), the number of BrdU-labeled cells significantly increased (Fig. 2b) (DG: F1,80 = 32.07, p < 0.0001, AGONIST main effect; F1,80 = 47.85, p < 0.0001, ANTAGONIST main effect; F1,80 = 39.53, p < 0.0001, AGONIST × ANTAGONIST interaction effect CA1: F1,80 = 347.22, p < 0.0001, AGONIST main effect; F1,80 = 41.32, p < 0.0001, ANTAGONIST main effect; F1,80 = 24.02, p < 0.0001, AGONIST × ANTAGONIST interaction effect). Thus, the nonselective agonist, carbachol, may have mixed effects on cell proliferation, with nicotinic-mediated suppression masking muscarinic-mediated activation at higher doses.
Fig. 2.

Agonist-induced regulation of cell proliferation is blocked by receptor-selective antagonists indicating positive regulation mediated by the muscarinic M1 receptor. a, BrdU+ cell counts in the DG and CA1 regions of the hippocampus following oxotremorine. Chronic intraventricular administration of the M1 receptor antagonist, pirenzepine, but not the nicotinic antagonist, mecamylamine, dose-dependently blocked oxotremorine-induced elevations in BrdU immunolabeling in both the DG and CA1. b, Chronic intraventricular administration of the M1 receptor antagonist, pirenzepine, also dose-dependently blocked low dose carbachol-induced elevations in BrdU immunolabeling. By contrast, the nicotinic receptor antagonist, mecamylamine, had no effect. As described earlier, a higher dose of carbachol (30 μg/h) failed to affect cell proliferation. Co-administration with mecamylamine, however, dose-dependently elevated BrdU immunolabeling while co-administration with pirenzepine significantly reduced BrdU immunolabeling. Each bar represents the mean (±S.E.M.) (n = 6) number of BrdU+ cells per mm3. ** sig. diff. from saline-treated controls, p < 0.001, *p < 0.05; ++ sig. diff. from oxotremorine alone, p < 0.001, +p < 0.05.
3.2. Pharmacological activation of muscarinic receptors reverses deficits in hippocampal neurogenesis following cholinergic denervation
The loss of cholinergic neurons is an early and invariant feature in patients with Alzheimer’s disease (Whitehouse et al., 1982; Coyle et al., 1983). Having identified oxotremorine as modulator of cytogenesis in both the DG and CA1 region of the hippocampus in normal, healthy rats, we sought to determine if this could be extended into a disease model. Infusion of the immunotoxin, 192IgG saporin (SAP), results in selective basal forebrain cholinergic cell loss and this model has been utilized extensively to study the role of cholinergic dysfunction in AD (Wiley, 1992; Rossner, 1997). As expected, infusion of SAP resulted in a pronounced loss of ChAT fiber density in the hippocampus and basal forebrain cholinergic neurons (over 80%) (F1,11 = 119.48, p < 0.0001) (Fig. 3). Consistent with earlier reports (Cooper-Kuhn et al., 2004; Mohapel et al., 2005), SAP-lesioned animals had significantly fewer BrdU+ and PCNA+ cells in both the DG (BrdU; F1,23 = 113.26, p < 0.0001, LESION main effect) (PCNA: F1,23 = 137.14, p < 0.0001, LESION main effect) and CA1 region (BrdU; F1,23 = 30.29, p < 0.0001, LESION main effect) (PCNA: F1,23 = 17.39, p < 0.0001, LESION main effect), when compared to sham-operated control animals (Fig. 4a). Following oxotremorine treatment, both BrdU+ and PCNA+ cell counts in the DG and in the CA1 region of the hippocampus were significantly elevated, effectively reversing lesion-induced impairments in cell proliferation [DG: (BrdU; F1,23 = 183.49, p < 0.0001, TREATMENT main effect) (PCNA: F1,23 = 127.34, p < 0.0001, TREATMENT main effect)] [CA1: (BrdU; F1,23 = 60.87, p < 0.0001, TREATMENT main effect) (PCNA: F1,23 = 27.48, p < 0.0001, TREATMENT main effect)]. We also examined cell death, as determined by the number of pyknotic cells, revealed using cresyl violet (Fig. 4a). While the number of pyknotic figures was elevated in the DG following denervation, oxotremorine failed to reduce these numbers (DG: F1,23 = 49.06, p < 0.0001, LESION main effect; F1,23 = 2.25, p = 0.1489, TREATMENT main effect) (CA1: F1,23 = 0.52, p = 0.48, LESION main effect; F1,23 = 1.75, p = 0.2006, TREATMENT main effect), suggesting the observed elevations in BrdU+ cells are not due to a decrease in apoptotic cell death.
Fig. 3.

Loss of Basal Forebrain Cholinergic Neurons Following Immunolesioning. Representative photomicrographs depicting choline acetyltransferase immunolabeling in the a,b dentate gyrus, c,d CA1 region and e,f medial septum of the adult rat brain following administration of a,c,e saline, or b,d,f 192IgG saporin. g, ChAT+ cell counts were significantly reduced in the medial septum four and eight weeks following saporin exposure. Each bar represents the mean (±S.E.M.) (n = 6) number of ChAT+ cells **per mm3. ** sig. diff. from saline-treated controls, p < 0.001. Scalebar = (a–d) 50 μm, (e,f) 100 μm.
Fig. 4.
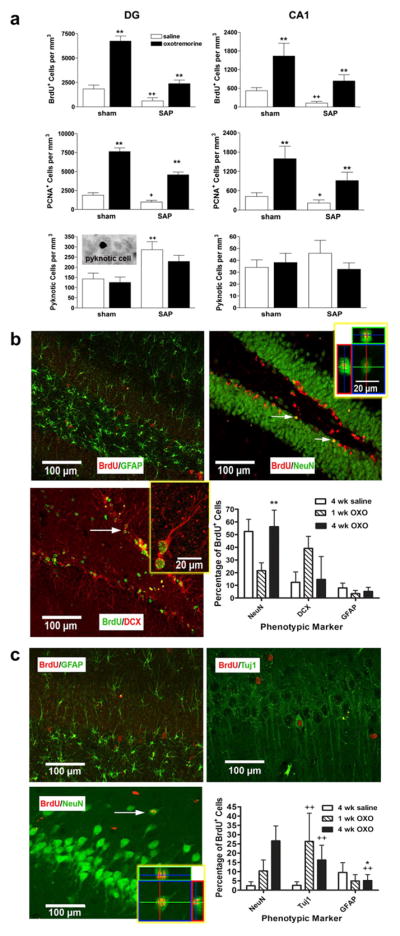
Muscarinic receptor activation restores deficits in cell proliferation following immunolesioning with the majority of newly-generated cells expressing a neuronal phenotype. a, BrdU+ and PCNA+ cell counts were significantly reduced in both regions of the hippocampus following cholinergic depletion using 192IgG saporin. The muscarinic agonist, oxotremorine, induced a significant elevation in cell proliferation in sham animals and reversed the deficits in cell proliferation observed following immunolesioning without affecting apoptosis, as indicated by pyknotic cell counts. Each bar represents the mean (±S.E.M.) (n = 6) number of immunopositive cells per mm3. ** sig. diff. from saline-treated controls, p < 0.001; ++sig. diff. from sham controls, p < 0.001, +p < 0.05. Inset depicts an example of a pyknotic cell in the hippocampus. b,c, Representative fluorescent photomicrographs taken from the hippocampus of animals treated with oxotremorine for 4 weeks. b, In the DG, the majority of BrdU+ cells were also labeled with either the mature neuronal marker, NeuN, or doublecortin (DCX), a marker of immature migrating neurons. Significantly more BrdU+ cells expressed NeuN after 4 weeks as compared to 1 week, suggesting a time-dependent maturation. Few cells were co-labeled with the glial marker, GFAP. c, In the CA1 region, approximately 44% of BrdU+ cells were also labeled with NeuN or the immature neuronal marker, betaIII tubulin (Tuj1). Again, significantly more BrdU+ cells expressed the mature neuronal marker following 4 weeks, as compared to 1 week. Oxotremorine treatment significantly elevated the percentage of BrdU+ expressing a neuronal marker. Few cells were co-labeled with GFAP. Each bar represents the mean (±S.E.M.) (n = 6) percentage of BrdU+ cells also labeled for a phenotypic marker. ** sig. diff. from 1 week treatment, p < 0.001, *p < 0.05; ++ sig. diff. from saline-treated control, p < 0.001. Insets depict enlarged views, rotated along the x and y axes, scalebar = 20 μm. Arrows indicate representative examples of a double-labeled cell. Scalebar = 100 μm.
In order to examine the effect of 4-week treatment with oxotremorine on differentiation to neurons, we performed multiple immunolabeling experiments examining the number of BrdU+ cells co-labeled with antibodies against the immature neuronal markers, Tuj1 or doublecortin (DCX), the mature neuronal marker, NeuN, or the glial marker, GFAP. While oxotremorine treatment triggered a robust increase in the number of newly-generated cells, it did not significantly alter the phenotype adopted by these cells in the DG, with the majority going on to express neuronal markers. Following 4 weeks of oxotremorine treatment, approximately 56% of the BrdU+ cells co-labeled for NeuN and 15% co-labeled for DCX in the DG (Fig. 4b). A subset of animals, examined after only one week of treatment, were found to have significantly fewer BrdU+ cells co-labeled for the mature neuronal marker (DG: F1,23 = 21.67, p = 0.0002, TIME main effect) (CA1: F1,23 = 6.83, p = 0.0166, TIME main effect), consistent with a time-dependent maturation of newly-generated neurons, rather than nonselective uptake of BrdU by pre-existing mature neurons undergoing apoptosis. In the CA1 region, only a very small percentage of BrdU+ cells expressed neuronal markers (~2%) in control animals. However, following 4 weeks of oxotremorine treatment, we found a significant increase in the percentage of BrdU+ cells co-labeled for NeuN (~27%) (F1,23 = 17.68, p = 0.0004, TREATMENT main effect; F1,23 = 5.61, p = 0.0281, TREATMENT × TIME interaction effect) (Fig. 4c), suggesting that oxotremorine treatment promotes a neuronal phenotype in the CA1. While the CA1 region is not noted for neuronal turnover, these findings are consistent with previous reports of new neurons following transient global ischemia (Bueters et al., 2008), growth factor infusion (Nakatomi et al., 2002) and even exposure to isoflurane (Elsersy et al., 2004).
3.3. Pharmacological activation of muscarinic receptors reverses deficits in spatial memory following cholinergic denervation
Cholinergic denervation by SAP immunolesioning has been shown to trigger deficits in spatial working memory (Dornan et al., 1996; Wrenn and Wiley,1998), including impairments in radial arm maze performance (Shen et al., 1996; Walsh et al., 1996; Lehmann et al., 2002). In order to determine whether agonist-induced restoration of neurogenesis was accompanied by improvements in spatial learning, we examined the number of reference and working memory errors committed during 20 trials in the RAM. Consistent with previous reports, animals tended to make fewer errors over time, with the exception of SAP immunolesioned animals. While no significant differences in reference memory errors were observed among the various groups (F1,23 = 0.85, p = 0.3678, LESION main effect; F1,23 = 3.43, p = 0.179, TREATMENT main effect; F4,96 = 1.90, p = 0.1182, BLOCK main effect) (Fig. 5a), SAP immunolesioned animals displayed significantly more working memory errors than sham controls during the last two blocks of trials (Fig. 5b) (F1,23 = 178.06, p < 0.0001, LESION main effect; F1,23 = 66.59, p < 0.0001, TREATMENT main effect; F4,96 = 39.51, p < 0.0001, BLOCK main effect; F4,96 = 10.52, p < 0.0001, LESION × BLOCK interaction effect). However, following chronic intraventricular administration of oxotremorine, these errors were significantly reduced (F4,96 = 8.64, p < 0.0001, LESION × TREATMENT × BLOCK interaction effect), making their performance in the RAM indistinguishable from sham controls. In order to rule out any nonspecific effects of motor activity on performance, we examined spontaneous locomotor activity in an open field one day after all RAM testing was complete. There was no significant effect of either SAP or oxotremorine on spontaneous locomotor activity (F1,23 = 0.99, p = 0.3322, LESION main effect; F1,23 = 1.93, p = 0.1804, TREATMENT main effect) (Fig. 5c). Thus, recovery of hippocampal neurogenesis following oxotremorine treatment was accompanied by an improvement in spatial working memory.
Fig. 5.
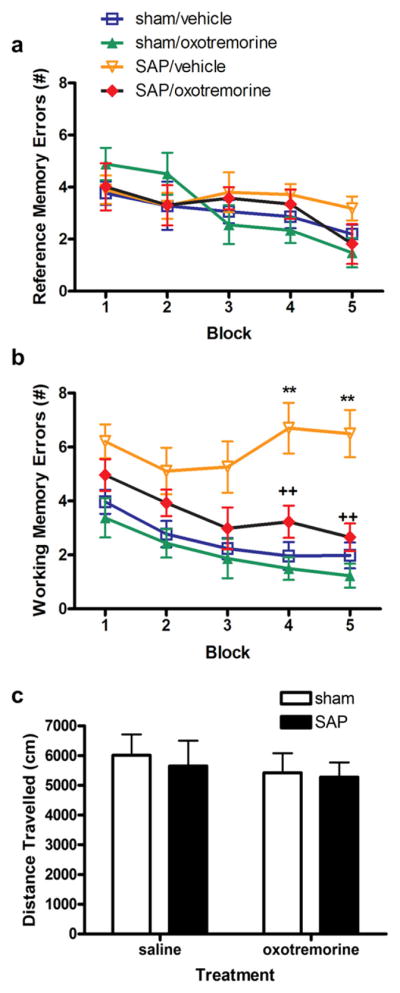
Muscarinic receptor activation reverses impairment in spatial working memory following immunolesioning. a, None of the groups differed in the number of reference errors committed. Reference memory errors were defined as entries into arms that were never baited. Each point represents the mean (±S.E.M.) (n = 6) number of reference memory errors. b, Significantly more working memory errors were committed by immunolesioned animals in the last two blocks of trials. These deficits in spatial working memory were significantly attenuated in animals treated with oxotremorine. Oxotremorine treatment was terminated 3 days prior to the final block of trials. In this final block, spatial working memory performance remained elevated. Working memory errors were defined as entries into previously visited arms. Each point represents the mean (±S.E.M.) (n = 6) number of working memory errors. ** sig. diff. from sham control, p < 0.001. c, No changes in locomotor activity were observed in any of the groups tested. Each bar represents the mean (±S.E.M.) (n = 6) total distance traveled (cm) in 1 h.
4. Discussion
Here, we have shown that selective activation of muscarinic receptors is capable of triggering an induction of cell proliferation not only in the DG, but also the CA1 region of the hippocampus, an area of severe neuronal loss in AD (Davies et al., 1992; Bobinski et al., 1998). As a complement to the BrdU labeling, we also examined immunolabeling for PCNA, an endogenous marker of cell proliferation. Since PCNA is an endogenous marker, antibodies only label cells proliferating at the time of death, in contrast to BrdU, which is taken within the cell and maintained through migration and differentiation. Thus, the increase in PCNA labeling within the hippocampus suggests that oxotremorine-induced increases in BrdU-positive cell counts are likely to reflect an effect on cell proliferation rather than an indirect effect on cell survival (Carvey et al., 2001; Du et al., 2005). Furthermore, impairments in cell proliferation, in a model of basal forebrain cholinergic cell loss, are counteracted by chronic muscarinic activation. This restoration of cell proliferation is accompanied by a time-dependent increase in the number of newly-generated cells expressing neuronal markers and by a reversal of cognitive deficits characteristic of this model (Dornan et al., 1996; Wrenn and Wiley, 1998).
Muscarinic M1 receptors are G-protein-coupled receptors and are the most abundant receptor subtype in the hippocampus where they show prominent localization to the soma and dendrites of CA1 pyramidal neurons and the granule cells of the DG (Levey et al., 1995) as well as hippocampal progenitors of the adult brain (Mohapel et al., 2005). There is accumulating evidence that G-protein-coupled receptors and their signaling molecules are important for growth stimulation (Rozengurt,1986; Gutkind,1998). Extracellular signal-regulated kinases, originally noted for their activation by growth factors, are known to also be activated by G-protein-coupled receptors including muscarinic receptors. The muscarinic receptor-mediated cell proliferation reported here may involve the activation of multiple downstream signaling pathways (Larocca and Almazan, 1997; Ma et al., 2000; Li et al., 2001; Zhao et al., 2003), including the Ras pathway which, in turn, promotes the phosphorylation of the mitogen-activated protein kinases (MAPK), thought to be important in cell proliferation (Crespo et al., 1995; Marshall, 1995). Other signaling pathways including PI3-K-Akt, PKC, c-Src and Ca2+, may also be involved, either alone or through some complex interaction (Ma et al., 2004). Reductions in hippocampal M1 receptor expression (Wang et al., 1992) and disturbances in G-protein coupling (Wang and Freidman, 1994) in the brains of AD patients may play a significant role in the disease mechanism (Caccamo et al., 2006) and resulting cognitive impairments (Rossi et al., 2005; Caccamo et al., 2006).
The opposing influences of muscarinic and nicotinic receptor activation on cell proliferation observed in the current study highlight the importance of selective receptor stimulation. Indeed, when the nonselective cholinergic agonist, carbachol, was administered, an inverse dose–response was observed. With a higher binding affinity to muscarinic acetylcholine receptors (Abood et al., 1993), lower doses of carbachol would provide more selective muscarinic activation, while higher doses would result in a relative increase in nicotinic receptor activation. Thus, in light of the negative effects nicotinic receptor activation was shown to have on cell proliferation, their increased activation by higher doses of carbachol would counteract muscarinic receptor-mediated elevations. This was confirmed by co-administration of receptor-selective antagonists, which revealed M1 muscarinic receptor-mediated induction of hippocampal cell proliferation following low doses of carbachol and nicotinic receptor-mediated suppression of cell proliferation at higher doses. Thus, the nonselective cholinergic agonist, carbachol, has differential effects on hippocampal cell proliferation that appear to be dependent upon the dose and, consequently, the cholinergic receptor type involved.
This oppositional dynamic may also underlie the relatively low effectiveness of the cholinesterase inhibitor, physostigmine, observed in these studies. By blocking acetylcholinesterase activity, physostigmine elevates available synaptic levels of ACh, increasing activation of muscarinic receptors yet failing to provide the receptor selectivity that appears to be necessary for effective induction of hippocampal cell proliferation. Thus, the minimal effects observed with physostigmine may be due to the absence of selective muscarinic receptor activation. This paradigm is similar to that seen with dopamine, as dopamine D1-like and D2-like receptor activation has been reported to produce mutually opposing effects on cell proliferation (Ohtani et al., 2003; Van Kampen et al., 2004). These two receptor subtypes may act, in essence, as a ‘gas’ and ‘brake’ in the regulation of cell genesis.
The hippocampus is believed to be critically involved in learning and memory (Scoville and Milner, 1957; Squire, 1982; Moscovitch et al., 2005), particularly spatial learning (Morris et al., 1982; Jacobs and Schenk, 2003) and it has been suggested that the hippocampal changes observed in AD may underlie some of the cognitive impairments characteristic of this disease. The primary site of hippocampal pathology in AD is the pyramidal cell layer of Ammon’s horn, especially the CA1 region, which exhibits the most severe neuronal loss (Davies et al., 1992; Fukutani et al., 2000; Kril et al., 2004), one closely correlated with the degree of cognitive impairment (Bobinski et al., 1998; von Gunten et al., 2006). In rodents, neuronal numbers in the CA1 region are similarly correlated with cognitive function (Stepanichev et al., 2004; Roberge et al., 2008), with activity in this region associated with RAM memory tasks (Vann et al., 2000; Touzani et al., 2003) and CA1 lesions resulting in impaired spatial working memory, as assessed by RAM performance (Dillon et al., 2008). Here, cholinergic denervation, modeling that which occurs in AD, reduced markers of hippocampal neurogenesis and resulted in cognitive deficits, both of which were effectively reversed following chronic muscarinic receptor activation. Furthermore, improvements in spatial working memory were observed long after agonist treatment was terminated, suggesting the cognitive recovery was not merely an acute pharmacological effect of the drug. The induction of neurogenesis in a brain region severely affected in AD and correlated with cognitive function, may be of therapeutic benefit in AD patients.
Classical neurotransmitters appear to play an important role in the development of the CNS and some vestige of this growth regulatory function may persist in the adult brain. In previous work, we have generated new dopaminergic neurons through the apparent activation of a specific dopamine receptor subtype, the D3 receptor (Van Kampen et al., 2004; Van Kampen and Robertson, 2005; Van Kampen and Eckman, 2006), and applied this to a model of PD as a novel cell replacement strategy (Van Kampen and Eckman, 2006). Similar to what we report here concerning cholinergic compounds, we found that selective receptor activation was critical, with evidence for other dopaminergic receptor subtypes having either no effect or even an inhibitory effect on cell proliferation. These principles may have application for disorders involving the loss of other cell types in other regions. For example, striatal neurons are regulated during neurodevelopment by NMDA receptor activity (Luk et al., 2003; Gandhi et al., 2008) and cell proliferation can be enhanced by mGluR5 receptor activation (Gandhi et al., 2008), making it a potential target for a similar cell replacement strategy in Huntington’s disease. Domain-specific proliferative effects mediated by specific receptor subtypes provides an important mechanism allowing generation of the correct complement of neuronal subtypes to re-populate areas affected in neurodegenerative disorders. The concept of selective receptor activation regulating the generation of specific populations of cells is novel and may alter how we view the brains endogenous capacity for self-repair, given the appropriate cues.
References
- Abood LG, Saraswati M, Lerner-Marmarosh N, Hashmi M. Affinity ligands and related agents for brain muscarinic and nicotinic cholinergic receptors. Biochemical Pharmacology. 1993;45:2143–2148. doi: 10.1016/0006-2952(93)90028-u. [DOI] [PubMed] [Google Scholar]
- Ball MJ. Neuronal loss, neurofibrillary tangles and granulovacuolar degeneration in the hippocampus with ageing and dementia. Acta Neuropathologica. 1977;37:111–118. doi: 10.1007/BF00692056. [DOI] [PubMed] [Google Scholar]
- Barker JL, Behar T, Li YX, Liu QY, Ma W, Maric D, Maric I, Schaffner AE, Serafini R, Smith SV, Somogyi R, Vautrin JY, Wen XL, Xian H. GABAergic cells and signals in CNS development. Perspectives on Developmental Neurobiology. 1998;5:305–322. [PubMed] [Google Scholar]
- Bobinski M, de Leon MJ, Tarnawski M, Wegiel J, Bobinski M, Reisberg B, Miller DC, Wisniewski HM. Neuronal and volume loss in CA1 of the hippocampal formation uniquely predicts duration and severity of Alzheimer disease. Brain Research. 1998;805:267–269. doi: 10.1016/s0006-8993(98)00759-8. [DOI] [PubMed] [Google Scholar]
- Borta A, Hoglinger GU. Dopamine and adult neurogenesis. Journal of Neurochemistry. 2007;100:587–595. doi: 10.1111/j.1471-4159.2006.04241.x. [DOI] [PubMed] [Google Scholar]
- Bueters T, von Euler M, Bendel O, von Euler G. Degeneration of newly formed CA1 neurons following global ischemia. Experimental Neurology. 2008;209:114–124. doi: 10.1016/j.expneurol.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Billings LM, Green KN, Marintez-Coria H, Fisher A, LaFeria FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Hazel TG, McKay RD. Regulation of neurogenesis by growth factors and neurotransmitters. Journal of Neurobiology. 1998;36:287–306. [PubMed] [Google Scholar]
- Carvey PM, McGuire SO, Ling ZD. Neuroprotective effects of D3 dopamine receptor agonists. Parkinsonism and Related Disorders. 2001;7:213–223. doi: 10.1016/s1353-8020(00)00061-4. [DOI] [PubMed] [Google Scholar]
- Cooper-Kuhn CM, Winkler J, Kuhn HG. Decreased neurogenesis after cholinergic forebrain lesion in the adult rat. Journal of Neuroscience Research. 2004;77:155–165. doi: 10.1002/jnr.20116. [DOI] [PubMed] [Google Scholar]
- Coronas V, Bantubungi K, Fombonne J, Krantic S, Schiffmann SN, Roger M. Dopamine D3 receptor stimulation promotes the proliferation of cells derived from the post-natal subventricular zone. Journal of Neurochemistry. 2004;91:1292–1301. doi: 10.1111/j.1471-4159.2004.02823.x. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219:1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- Crespo C, Xu N, Simonds WF, Gukind JS. Ras-dependent activation of MAP-kinases pathway mediated by G protein βγ subunits. Nature. 1995;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- Davies DC, Horwood N, Isaacs SL, Mann DM. The effect of age and Alzheimer’s disease on pyramidal neuron density in the individual fields of the hippocampal formation. Acta Neuropathologica. 1992;83:510–517. doi: 10.1007/BF00310028. [DOI] [PubMed] [Google Scholar]
- Dillon GM, Qu X, Marcus JN, Dodart JC. Excitotoxic lesions restricted to the dorsal CA1 field of the hippocampus impair spatial memory and extinction learning in C57/BL6 mice. Neurobiology of Learning and Memory. 2008;90:426–433. doi: 10.1016/j.nlm.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. Journal of Comparative Neurology. 2006;495:70–83. doi: 10.1002/cne.20840. [DOI] [PubMed] [Google Scholar]
- Dornan WA, McCampbell AR, Tinkler GP, Hickman LJ, Bannon AW, Decker MW, Gunther KL. Comparison of site-specific injections into the basal forebrain on water maze and radial arm maze performance in the male rat after immunolesioning with 192IgG saporin. Behavioral Brain Research. 1996;82:93–101. doi: 10.1016/s0166-4328(97)81112-6. [DOI] [PubMed] [Google Scholar]
- Du F, Li R, Huang Y, Li X, Le W. Dopamine D3 receptor-preferring agonists induce neurotrophic effects on mesencephalic dopamine neurons. European Journal of Neuroscience. 2005;22:2422–2430. doi: 10.1111/j.1460-9568.2005.04438.x. [DOI] [PubMed] [Google Scholar]
- Duman RS, Nakagawa S, Malberg J. Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology. 2001;5:836–844. doi: 10.1016/S0893-133X(01)00358-X. [DOI] [PubMed] [Google Scholar]
- Elsersy H, Sheng H, Lynch JR, Moldovan M, Pearlstein RD, Warner DS. Effects of isoflurane versus fentanyl-nitrous oxide anesthesia on long-term outcome from severe forebrain ischemia in the rat. Anesthesiology. 2004;100:1160–1166. doi: 10.1097/00000542-200405000-00018. [DOI] [PubMed] [Google Scholar]
- Encincas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8233–8238. doi: 10.1073/pnas.0601992103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nature Medicine. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Miller MW, Ware CB, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Fukutani Y, Cairns NJ, Shiozawa M, Sasaki K, Sudo S, Isaki K, Lantos PL. Neuronal loss and neurofibrillary degeneration in the hippocampal cortex in late-onset sporadic Alzheimer’s disease. Psychiatry and Clinical Neurosciences. 2000;54:523–529. doi: 10.1046/j.1440-1819.2000.00747.x. [DOI] [PubMed] [Google Scholar]
- Gandhi R, Luk KC, Rymar VV, Sadikot AF. Group 1 mGluR5 metabo-tropic glutamate receptors regulate proliferation of neuronal progenitors in specific forebrain developmental domains. Journal of Neurochemistry. 2008;104:155–172. doi: 10.1111/j.1471-4159.2007.04955.x. [DOI] [PubMed] [Google Scholar]
- Gutkind JS. Cell growth control by G protein-coupled receptors: from signal transduction to signal integration. Oncogene. 1998;17:1331–1342. doi: 10.1038/sj.onc.1202186. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. Journal of Neurochemistry. 2002;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- Hohmann CF. A morphogenetic role for acetylcholine in mouse cerebral neocortex. Neuroscience and Biobehavioral Reviews. 2003;27:351–363. doi: 10.1016/s0149-7634(03)00066-6. [DOI] [PubMed] [Google Scholar]
- Jacobs LF, Schenk F. Unpacking the cognitive map: the parallel map theory of hippocampal function. Psychological Reviews. 2003;110:285–315. doi: 10.1037/0033-295x.110.2.285. [DOI] [PubMed] [Google Scholar]
- Kaneko N, Okano H, Sawamoto K. Role of the cholinergic system in regulating survival of newborn neurons in the adult mouse dentate gyrus and olfactory bulb. Genes to Cells. 2006;11:1145–1159. doi: 10.1111/j.1365-2443.2006.01010.x. [DOI] [PubMed] [Google Scholar]
- Kotani S, Yamauchi T, Teramoto T, Ogura H. Pharmacological evidence of cholinergic involvement in adult hippocampal neurogenesis in rats. Neuroscience. 2006;142:505–514. doi: 10.1016/j.neuroscience.2006.06.035. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Hodges J, Halliday G. Relationship between hippocampal volume and CA1 neuron loss in brains of humans with and without Alzheimer’s disease. Neuroscience Letters. 2004;361:9–12. doi: 10.1016/j.neulet.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Larocca J, Almazan G. Acetylcholine agonists stimulate mitogen-activated protein kinase in oligodendrocyte progenitors by muscarinic receptors. Journal of Neuroscience Research. 1997;50:743–754. doi: 10.1002/(SICI)1097-4547(19971201)50:5<743::AID-JNR11>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Lehmann O, Bertrand F, Jeltsch H, Morer M, Lazarus C, Will B, Cassel JC. 5,7-DHT-induced hippocampal 5-HT depletion attenuates behavioural deficits produced by 192 IgG-saporin lesions of septal cholinergic neurons in the rat. European Journal of Neuroscience. 2002;15:1991–2006. doi: 10.1046/j.1460-9568.2002.02037.x. [DOI] [PubMed] [Google Scholar]
- Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. Expression of m1–m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. Journal of Neuroscience. 1995;15:4077–4092. doi: 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Ma W, Zhang L, Barker JL, Stenger DA, Pant HC. Activation of phosphatidylinositol-3 kinase (PI-3K) and extracellular regulated kinases (Erk1/2) is involved in muscarinic receptor-mediated synthesis in neural progenitor cells. Journal of Neuroscience. 2001;21:1569–1579. doi: 10.1523/JNEUROSCI.21-05-01569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kennedy TE, Sadikot AF. Glutamate promotes proliferation of striatal neuronal progenitors by an NMDA receptor-mediated mechanism. Journal of Neuroscience. 2003;23:2239–2250. doi: 10.1523/JNEUROSCI.23-06-02239.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Maric D, Li BS, Andreadis JD, Grant GM, Liu QY, Schaffer KM, Chang YH, Zhang L, Pancrazio J, Pant HC, Stenger DA, Barker JL. Acetylcholine stimulates cortical precursor cell proliferation via muscarinic receptor activation and MAP kinase phosphorylation. European Journal of Neuroscience. 2000;12:1227–1240. doi: 10.1046/j.1460-9568.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- Ma W, Li BS, Zhang L, Pant HC. Signaling cascades implicated in muscarinic regulation of proliferation of neural stem and progenitor cells. Drug News Perspectives. 2004;17:258–266. doi: 10.1358/dnp.2004.17.4.829053. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Kristjansen PEG, Bolwig TG, Wortwein G. Arrested neuronal proliferation and impaired hippocampal function following fractionated brain irradiation in the adult rat. Neuroscience. 2003;119:635–642. doi: 10.1016/s0306-4522(03)00199-4. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Miranda-Contreras L, Benitez-Diaz PR, Mendoza-Briceno RV, Delgado-Saez MC, Palacios-Pru EL. Levels of amino acid neurotransmitters during mouse cerebellar neurogenesis and in histotypic cerebellar cultures. Developmental Neuroscience. 1999;21:147–158. doi: 10.1159/000017377. [DOI] [PubMed] [Google Scholar]
- Mohapel P, Leanza G, Kokaia M, Lindvall O. Forebrain acetylcholine regulates adult hippocampal neurogenesis and learning. Neurobiology of Aging. 2005;26:939–946. doi: 10.1016/j.neurobiolaging.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Morris RGM, Garrud P, Rawlins JNP, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Moscovitch M, Rosenbaum RS, Gilboa A, Addis DR, Westmacott R, Grady C, McAndrews MP, Levine B, Black S, Wincour G, Nadel L. Functional neuroanatomy of remote episodic, semantic and spatial memory: a unified account based on multiple trace theory. Journal of Anatomy. 2005;207:35–66. doi: 10.1111/j.1469-7580.2005.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 2002;110:429–441. doi: 10.1016/s0092-8674(02)00862-0. [DOI] [PubMed] [Google Scholar]
- Nguyen L, Rigo JM, Rocher V, Belachew S, Malgrange B, Rogister B, Leprince P, Moonen G. Neurotransmitters as early signals for central nervous system development. Cell Tissue Research. 2001;305:187–202. doi: 10.1007/s004410000343. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Goto T, Waeber C, Bhide PG. Dopamine modulates cell cycle in the lateral ganglionic eminence. Journal of Neuroscience. 2003;23:2840–2850. doi: 10.1523/JNEUROSCI.23-07-02840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrozek TA, Sari Y, Singh RP, Zhou FC. Neurotransmitters and substances of abuse: effects on adult neurogenesis. Current Neurovascular Research. 2004;1:251–260. doi: 10.2174/1567202043362225. [DOI] [PubMed] [Google Scholar]
- Roberge MC, Hotte-Bernard J, Messier C, Plamondon H. Food restriction attenuates ischemia-induced spatial learning and memory deficits despite extensive CA1 ischemic injury. Behavioral Brain Research. 2008;187:123–132. doi: 10.1016/j.bbr.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Rola R, Raber J, Rizk A, Otsuka S, Van den Berg SR, Morhardt DR, Fike JR. Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Experimental Neurology. 2004;188:316–330. doi: 10.1016/j.expneurol.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Rossi MA, Mash DC, deToledo-Morrell L. Spatial memory in aged rats is related to PKCgamma-dependent G-protein coupling of the M1 receptor. Neurobiology of Aging. 2005;26:53–68. doi: 10.1016/j.neurobiolaging.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Rössler M, Zarski R, Bohl J, Ohm TG. Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer’s disease. Acta Neuropathologica. 2002;103:363–369. doi: 10.1007/s00401-001-0475-7. [DOI] [PubMed] [Google Scholar]
- Rossner S. Cholinergic immunolesions by 192IgG-saporin – useful tool to stimulate pathogenic aspects of Alzheimer’s disease. International Journal of Developmental Neuroscience. 1997;15:835–850. doi: 10.1016/s0736-5748(97)00035-x. [DOI] [PubMed] [Google Scholar]
- Rozengurt E. Early signals in the mitogenic response. Science. 1986;234:161–166. doi: 10.1126/science.3018928. [DOI] [PubMed] [Google Scholar]
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. Journal of Neurology, Neurosurgery and Psychiatry. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JM, Barnes CA, Wenk GL, McNaughton BL. Differential effects of selective immunotoxic lesions of medial septal cholinergic cells on spatial working and reference memory. Behavioral Neuroscience. 1996;110:1181–1186. doi: 10.1037//0735-7044.110.5.1181. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Miesagaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult rat is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Squire LR. The neuropsychology of human memory. Annual Review of Neuroscience. 1982;5:241–273. doi: 10.1146/annurev.ne.05.030182.001325. [DOI] [PubMed] [Google Scholar]
- Stepanichev MY, Zdobnova IM, Zarubenko II, Moiseeva YV, Lazareva NA, Onufriev MV, Gulyaeva NV. Amyloid-β(25–35)-induced memory impairments correlate with cell loss in rat hippocampus. Physiology & Behavior. 2004;80:647–655. doi: 10.1016/j.physbeh.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Touzani K, Marighetto A, Jaffard R. Fos imaging reveals ageing-related changes in hippocampal response to radial arm maze discrimination testing in mice. European Journal of Neuroscience. 2003;17:628–640. doi: 10.1046/j.1460-9568.2003.02464.x. [DOI] [PubMed] [Google Scholar]
- Van der Borght K, Mulder J, Keijser JN, Eggen BJL, Luiten PGM, Van der Zee EA. Input from the medial septum regulates adult hippocampal neurogenesis. Brain Research Bulletin. 2005;6:117–125. doi: 10.1016/j.brainresbull.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Van Kampen JM, Eckman CB. Dopamine D3 receptor agonist delivery to a model of Parkinson’s disease restores the nigrostriatal pathway and improves locomotor behaviour. Journal of Neuroscience. 2006;26:7272–7280. doi: 10.1523/JNEUROSCI.0837-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kampen JM, Hagg T, Robertson HA. Induction of neurogenesis in the adult rat subventricular zone and neostriatum following dopamine D3 receptor stimulation. European Journal of Neuroscience. 2004;19:2377–2387. doi: 10.1111/j.0953-816X.2004.03342.x. [DOI] [PubMed] [Google Scholar]
- Van Kampen JM, Robertson HA. A possible role for dopamine D3 receptor stimulation in the induction of neurogenesis in the adult rat substantia nigra. Neuroscience. 2005;136:381–386. doi: 10.1016/j.neuroscience.2005.07.054. [DOI] [PubMed] [Google Scholar]
- Vann SD, Brown MW, Aggleton JP. Fos expression in the rostral thalamic nuclei and associated cortical regions in response to different spatial memory tests. Neuroscience. 2000;101:983–991. doi: 10.1016/s0306-4522(00)00288-8. [DOI] [PubMed] [Google Scholar]
- von Gunten A, Kövari E, Bussière T, Rivara CB, Gold G, Bouras C, Hof PR, Giannakopoulos P. Cognitive impact of neuronal pathology in the entorhinal cortex and CA1 field in Alzheimer’s disease. Neurobiology of Aging. 2006;27:270–277. doi: 10.1016/j.neurobiolaging.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Walsh TJ, Herzog CD, Gandhi C, Stackman RW, Wiley RG. Injection of IgG 192-saporin into the medial septum produces cholinergic hypofunction and dose-dependent working memory deficits. Brain Research. 1996;726:69–79. [PubMed] [Google Scholar]
- Wang SZ, Zhu SZ, Mash DC, el-Fakahany EE. Comparison of the concentration of messenger RNA encoding four muscarinic receptor subtypes in control and Alzheimer brains. Brain Research Molecular Brain Research. 1992;16:64–70. doi: 10.1016/0169-328x(92)90194-g. [DOI] [PubMed] [Google Scholar]
- Wang B, Hu Q, Hearn MG, Shimizu K, Ware CB, Liggitt DH, Jin LW, Cool BH, Storm DR, Martin GM. Isoform-specific knockout of FE65 leads to impaired learning and memory. Journal of Neuroscience Research. 2004;75:12–24. doi: 10.1002/jnr.10834. [DOI] [PubMed] [Google Scholar]
- Wang HY, Freidman E. Receptor-mediated activation of G proteins is reduced in postmortem brains from Alzheimer’s disease patients. Neuroscience Letters. 1994;173:37–39. doi: 10.1016/0304-3940(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Wiley RG. Neural lesioning with ribosome-inactivating proteins: suicide transport and immunolesioning. Trends in Neuroscience. 1992;15:285–290. doi: 10.1016/0166-2236(92)90078-m. [DOI] [PubMed] [Google Scholar]
- Wiley RG, Berbos TG, Deckwerth TL, Johnson EMJ, Lappi DA. Destruction of the cholinergic basal forebrain using immunotoxin to rat NGF receptor: modeling the cholinergic degeneration of Alzheimer’s disease. Journal of Neurological Science. 1995;128:157–166. doi: 10.1016/0022-510x(94)00226-e. [DOI] [PubMed] [Google Scholar]
- Wrenn CC, Wiley RG. The behavioral functions of the cholinergic basal forebrain: lessons from 192IgG-saporin. International Journal of Developmental Neuroscience. 1998;16:595–602. doi: 10.1016/s0736-5748(98)00071-9. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Alkon DL, Ma C. C-src protein tyrosine kinase activity is required for muscarinic receptor-mediated DNA synthesis and neurogenesis via ERK1/2 and CREB signaling in neural precursor cells. Journal of Neuroscience Research. 2003;72:334–342. doi: 10.1002/jnr.10591. [DOI] [PubMed] [Google Scholar]
- Ziabreva I, Perry E, Perry R, Minger SL, Ekonomou A, Przyborski S, Ballard C. Altered neurogenesis in Alzheimer’s disease. Journal of Psychosomatic Research. 2006;61:311–316. doi: 10.1016/j.jpsychores.2006.07.017. [DOI] [PubMed] [Google Scholar]