

Abstract
The vascular adventitia is emerging as an important modulator of vessel remodeling. Adventitial myofibroblasts migrate to the neointima after balloon angioplasty, contributing to restenosis. We postulated that angiotensin II (Ang II) enhances adventitial myofibroblast migration in vitro via reduced nicotinamide-adenine dinucleotide phosphate oxidase–derived H2O2 and that Nox4-based oxidase promotes migration. Ang II increased myofibroblast migration in a concentration-dependent manner, with a peak increase of 1023±83%. Rat adventitial myofibroblasts were cotransfected with human Nox4 and human p22-phox plasmids or an empty vector. PCR showed an 8-fold increase in human Nox4 and human p22-phox plasmid expression. Using RT-PCR with primers specifically designed for rat reduced nicotinamide-adenine dinucleotide phosphate oxidases, endogenous Nox levels were determined. Ang II decreased endogenous Nox4 and Nox1 mRNA to 41% and 27% of control, respectively, but had no effect on Nox2. Cotransfection with human Nox4 and human p22-phox plasmids combined with Ang II reduced endogenous Nox4 mRNA levels (37±5% of control; P<0.05), whereas it had no significant effect on Nox1 or Nox2. In empty vector–transfected cells, Ang II increased myofibroblast migration by 192±32% versus vehicle (P<0.01) while increasing H2O2 (473±22% versus control; P<0.001). Cotransfection with human Nox4 and human p22-phox plasmids decreased Ang II-induced migration (46±6%; P<0.001) in parallel with attenuation of H2O2 production (23±8% versus empty vector; P<0.05). Our data suggest that Nox4 promotes Ang II-induced myofibroblast migration via an H2O2-dependent pathway. The data also suggest that Nox4 causes feedback inhibition of its own expression in adventitial myofibroblasts.
Keywords: adventitia, fibroblasts, myofibroblasts, migration, neointima, restenosis, NADPH oxidase
Neointimal hyperplasia is a major response to vascular injury.1 Proliferation and migration of vascular smooth muscle cells and, more recently, myofibroblasts are implicated in narrowing of the arterial lumen in response to injury, mimicking one of the hallmark characteristics of atherosclerosis.2–5 This process of vascular cell migration and proliferation in response to injury and atherosclerosis is reportedly promoted by proto-oncogenes6 and various growth factors, including angiotensin II (Ang II).7 Most recent studies have implicated reactive oxygen species (ROS) derived from reduced nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase in vascular cell proliferation and neointimal growth8,9 and in a number of growth- and migration-related signaling pathways.10
NADPH oxidase is a well-characterized ROS-generating system that generally catalyzes the 1-electron reduction of oxygen to O2-, precursor of a variety of other ROS. The classical phagocyte NADPH oxidase is a multicomponent enzyme complex that includes the 2 plasma membrane–spanning polypeptide subunits, p22-phox and the anchoring component Nox2 (previously known as gp91-phox), along with 4 essential cytoplasmic subunits, p40-phox, p47-phox, p67-phox, and p21rac.11–13 The vascular Nox2-based oxidase is sometimes constitutive but is activated by hormone-induced interaction of cytosolic and membrane-associated components. A novel homologue of Nox2-based oxidase, Nox4, interacts with p22-phox; the formed cytochrome is reportedly self-sufficient and constitutive in its production of ROS.14 Ang II is the prototype hormonal stimulant of this class of enzymes in the vasculature15–17 and is a major vascular growth factor released by tissue and cellular stretch, eg, in balloon angioplasty-induced injury.18
Recently, the NADPH oxidase isoforms Nox1 and Nox2 have been implicated in the proliferation and migration of cells from the vascular wall to the neointima.4,5,19,20 Nox1 upregulation has been postulated to promote vascular cell proliferation and migration during restenosis.20 Schröder et al21 reported recently that migration of vascular smooth muscle cells induced by fibroblast growth factor is mediated by Nox1. We reported that the vascular adventitia expresses the Nox2-based oxidase system, which is involved in the production of O2- by adventitial fibroblasts22,23 and neointimal hyperplasia,5,24 with the latter process being partially attributed to the migration of adventitial myofibroblasts.5,19,24–27
The unique location of Nox4 in focal adhesions suggests a distinct role of this isoform in the migration of vascular smooth muscle cells28; however, its role in adventitial myofibroblasts is not known.29 Cucoranu et al30 showed recently that Nox4 is involved in differentiation of cardiac fibroblasts to their migratory phenotype myofibroblasts. Thus, we postulated that Nox4 would have a promigratory effect on vascular adventitial myofibroblasts. In this study, we focused on examining the ability of Ang II to stimulate vascular myofibroblast migration and testing the hypothesis that over-expression of Nox4 and p22-phox in adventitial myofibroblasts promotes Ang II-induced myofibroblast migration.
Materials and Methods
Culture of Adventitial Myofibroblasts From the Rat Aorta
Adventitial fibroblasts were harvested from thoracic aortas of male Sprague-Dawley rats (Charles River), as described previously.22 They were grown in DMEM containing 1000 mg/L of D-glucose and L-glutamine and 110 mg/L of sodium pyruvate (Gibco) supplemented with 1% penicillin/streptomycin (Invitrogen) and 10% FBS (heat-inactivated; Mediatech). Once the fibroblasts reached 80% to 90% confluence, they were passaged by trypsinization; passages 5 to 9 were used, because Western blot showed that they broadly expressed smooth muscle α-actin, indicating the myofibroblast phenotype (data not shown).
Migration of Adventitial Myofibroblasts
An in vitro Transwell system (Corning Life Sciences) was used to determine whether Ang II stimulates the migration of adventitial myofibroblasts and whether transfection of myofibroblasts with human Nox4 (h-Nox4) and human p22-phox (h-p22) plasmid (h-nox4/p22) could alter myofibroblast migration. Transfected or nontransfected cells were harvested with 0.05% trypsin and sedimented at 148g. The cell pellet was resuspended in serum-free DMEM and placed in the upper chamber of an 8-µm pore-size Transwell system. Ang II (Sigma-Al-drich) or its vehicle (0.01 N acetic acid in saline) was added to both chambers of the well. A concentration range of 1 to 300 nmol/L of Ang II was tested based on preliminary studies and other published works determining 100 nmol/L of Ang II to be the optimal dose for ROS production. After 6 hours, cells that did not migrate were aspirated and wiped from the upper well with a cotton-tipped applicator. After preliminary experiments, we chose a 6-hour incubation period because it demonstrated the largest differences in migration between Ang II- and vehicle-treated cells without their replication. Migratory cells adherent to the bottom surface of the membrane were fixed with buffered formalin (Sigma-Aldrich, 4.3%) for 1 hour and stained with hematoxylin (Sigma-Aldrich) for 10 minutes, washing away excess hematoxylin with tap water for 2 minutes. Membranes were allowed to dry overnight, cut from the Transwell with a scalpel and affixed to a glass slide using Crystal Mount (Biomeda). Five random X200 fields were counted per membrane.
Transfection of Myofibroblasts With NADPH Oxidase Components Nox4 and p22-phox
At 80% to 90% confluence, the medium was changed to DMEM containing 10% FBS without antibiotics, and cells were allowed to stabilize overnight. Then, they were either transfected with an empty vector control (2 µg of pcDNA3, Invitrogen) or cotransfected with pcDNA plasmids expressing the open-reading frames for human Nox4 and human p22-phox (1 µg of h-Nox4 and 1 µg of p22, kindly provided by Dr J.D. Lambeth, Emory University). This was done using lipofectamine 2000 (Invitrogen) at a ratio of 5 µL of lipofectamine to 2 µg of DNA. For a 12-well plate, 2 µg of total plasmid and 5 µL of lipofectamine were each diluted into separate aliquots of 50 µL of OptiMEM (Gibco) per well. Both were allowed to equilibrate for 5 minutes, mixed at a 1:1 ratio, and again equilibrated for 20 minutes. A total of 100 µL of the lipofectamine-plasmid mix was then added to each well. In some experiments, vehicle control or 3000 U/mL of catalase was added to the cells. This was done by mixing catalase with Bioporter (Genlantis), a lipid-based protein delivery system, as per the manufacturer’s directions, and adding it to the transfection reactions. Transfection was allowed to proceed for 24 hours, and cells were harvested for assay. Transfection had no effect on cell viability as determined by Trypan blue exclusion.
RNA Purification and cDNA Preparation
Total RNA was purified from rat adventitial myofibroblasts using TRIzol reagent (Invitrogen) and treated with DNAse as recommended by the manufacturer. First-strand cDNA synthesis was performed using total RNA, oligo dT primers, and Superscript II reverse transcriptase (Invitrogen). cDNA samples were also treated with RNase H.
Conventional PCR
Both h-Nox4 and h-p22 were quantified by conventional PCR of the cDNA using a PTC-100 programmable thermal controller (MJ Research, Inc). Primers for h-Nox4 were designed using Primer3 software (h-Nox4, h4 forward: AGCAATAAGCCAGTCACCATCATTT and h4 reverse: GAGTAAATCTGCAAACCAACGGAAG), and primers for h-p22 were as described by Dr David Lambeth (Emory University; h-p22, p22 forward: AACGAGCAGGCGCTGGCGTCCG and p22 reverse: GCTTGGGCTCGATGGGCGTCCACT). Band intensity on agarose gels was analyzed using National Institutes of Health ImageJ software.
RT-Polymerase Chain Reaction
Endogenous rat Nox1, Nox2, Nox4 and 18S rRNA were quantified by amplification of the same cDNA using a LightCycler (Roche) real-time thermocycler and a QuantiTect Probe PCR kit from Qiagen with primers and probes designed by TIBmol. Sequences are as follows: for rat Nox1, 1F1: ACCAATGCCCAGGATCG, 1R1: CGGTTTGCCTAATTCGTCC, 1FL: CCACTTCGTACTGGAAGACATCCTCAC-FL, and 1LCR: LC Red640-GACTGTGCCAAAGGGACCATCCA-PH; for rat Nox2, 2F2: CTGTGATAAGCAGGAGTTCCAA, 2R2: CCTGCACAGCCAGTAGAAGT, 2FL: CATCACCACCTCATAGCTGAACACA-FL, and 2LCR: LC Red640-CTTCACTGGCTGTACCAAAGGGC-PH; for rat Nox4, 4F1: GCTTGTTGAAGTATCAAACCAAT, 4R2: TCCAGAAATCCAAATCCAGGT, 4FL: AACACTGGTGAAGATTTGCCTGGA-FL, and 4LCR: LC Red640-GAACCCAAGTTCCAAGCTCATTTCC-PH; and for rat 18sRNA, 18sF1: ACGAACCAGAGCGAAAGCAT, 18sR3: TGAGGTTTCCCGTGTTGAGT, 18sFL: TCGGAACTACGACGGTATCTGATCTGATCGTC, and 18sLCR: LC Red640-CGAACCTCCGACTTTCGTTCTTGAT. Amplification conditions were as follows: 500 nmol/L of forward and reverse primers, 100 nmol/L of fluorescein- and LCR640-labeled probes, and 4 mmol/L of MgCl2. Annealing temperatures was 59°C for Nox1, Nox2, and 18sRNA and 56°C for Nox4. Copy numbers were calculated by the instrument software from standard curves generated for genuine rat Nox1, Nox4, Nox2, p22-phox, and 18S templates.
Measurement of Myofibroblast H2O2 Production and the Effect of h-Nox4/p22 Expression
Cells were transfected with h-Nox4/p22 or an empty vector control and made quiescent overnight in DMEM containing 0.67% FBS. Cells were harvested with trypsin (0.05%), rinsed with DMEM, and pelleted at 148 g and then were washed once and resuspended in Hank’s balanced salt solution containing calcium and magnesium (Gibco). We added 2×105 cells to each well of a 96-well Microlite 1+ luminometry plate (Thermo-Scientific) containing 200 µL of Hank’s balanced salt solution and 0.2 mmol/L of luminol (Acros Organics), 0.31 U/mL of horseradish peroxidase (Pierce), and 100 nM of Ang II (Sigma-Aldrich) or its vehicle. Catalase (3000 U/mL) was used to quench the reaction. Readings were made using Fluoroskan FL (Thermo-Scientific). Measurements are reported as the maximal value per well minus the lowest value after catalase was added.
Statistical Analysis
Data are expressed as means±SEMs. Paired values were compared using a 2-tailed t test. Multiple comparisons were performed by 1-way ANOVA followed by posthoc pairwise comparisons using Bonferroni corrections or Hochberg’s step-up method to adjust for multiple comparisons so that familywise α levels of 0.05 were maintained.
Results
Effect of Ang II on Myofibroblast Migration
We examined the effect of 1 to 300 nmol/L of Ang II on adventitial myofibroblast migration across 8 µm Transwell membranes. As seen in Figure 1, Ang II increased adventitial myofibroblast migration in a concentration-dependent manner. One hundred nanomoles per liter Ang II caused an increase from 2.3±0.4 to 25.8±1.9 cells per field (10.2±0.8-fold peak stimulation compared with vehicle; P<0.001; n=5), significantly greater than the increase caused by 1 and 10 nmol/L Ang II (6.5±1.3 and 11.4±2.0 cells per field or 1.8±0.6-fold and 4.0±0.9-fold increase versus vehicle, respectively; P<0.001; n=5). At 300 nmol/L Ang II, we observed a migration of 24.6±1.2 cells per field (a 9.7±0.5-fold increase over vehicle; P<0.001; n=5); however, it was not significantly different from the response obtained with 100 nmol/L of Ang II.
Figure 1.
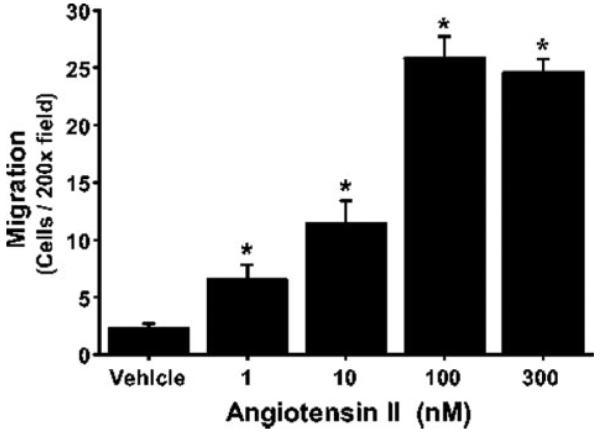
Effect of Ang II on adventitial myofibroblast migration. Adventitial myofibroblasts were plated in 8-µm pore-size Transwells, and either vehicle or Ang II (1 to 300 nmol/L) was added. After 6 hours, cells that migrated through the Transwell membrane were fixed, stained, and counted (5 random X200 fields). Data are expressed as the number of cells per X200 field±SEM and were considered significantly different when P<0.05.
*P<0.001; n=4 to 5.
Effects of Ang II and Transfection With h-Nox4/p22 on Levels of NADPH Oxidase Isoforms in Adventitial Myofibroblasts
Conventional and real-time PCR were used to determine the effectiveness of transfection and the effects of overexpressing Nox4 on endogenous Nox levels, respectively. Species-specific primers were used to differentiate between endogenous and exogenous levels of Nox isoforms.
Exogenous h-Nox4/p22 Expression
Cotransfection with h-Nox4/p22-phox–expressing plasmids resulted in an 8-fold increase in the mRNA for both h-Nox4 and h-p22 compared with the empty vector control, as assessed by conventional PCR (Figure 2). We observed a 7.80±1.26-fold increase in h-Nox4 mRNA compared with control and an increase of 7.34±1.08-fold in h-p22 mRNA expression. Treatment with Ang II did not significantly affect the levels of either transcript.
Figure 2.

Effect of Nox4/p22-phox cotransfection on cellular mRNA levels. Adventitial myofibroblasts were transfected with either an empty-vector control plasmid or cotransfected with plasmids expressing human Nox4 and p22-phox. The cells were stimulated with either Ang II (100 nmol/L for 3 hours) or vehicle and mRNA levels assessed by conventional PCR. A, Agarose gels of PCR products using human Nox4 and p22 primers and rat 18sRNA primers. B, Quantification of band intensity using ImageJ software. Nox4 and p22-phox mRNA levels are shown by the shaded and filled bars, respectively. Data are expressed as fold change±SEM from their corresponding controls (empty vector plus vehicle; n=8 to 9). *Statistical significance where P<0.05.
Endogenous Nox Expression
Using rat-specific primers and probes for Nox1, 2, and 4, we determined the effect of expressing h-Nox4/p22 on endogenous Nox mRNA levels by real-time PCR. In empty vector–transfected cells treated with vehicle, we detected 1.4±0.4 copies of Nox4 mRNA per 10 000 18S copies (Table). Ang II treatment decreased these levels to 0.7±0.2 copies of Nox4 mRNA per 10 000 18S copies (Figure 3; 41±8% of empty vector control; P<0.05). Ang II decreased Nox1 mRNA from 0.5±0.1 to 0.2±0.04 (vehicle versus Ang II; P<0.05) but had no effect on Nox2 mRNA.
Table.
Copy No. of Nox Homologues Expressed in Myofibroblasts Transfected With Empty Vector or h-Nox4/p22 Plasmid and Treated With Ang II or Its Vehicle
| Nox Homologues |
Vector+ Vehicle |
Vector+ Ang II |
h-Nox4/p22+ Vehicle |
h-Nox4/p22+ Ang II |
|---|---|---|---|---|
| Nox4 | 1.392±0.366 | 0.687±0.232 | 0.876±0.227 | 0.567±0.162 |
| Nox1 | 0.496±0.115 | 0.159±0.036 | 0.540±0.154 | 0.365±0.114 |
| Nox2 | 0.004±0.002 | 0.003±0.001 | 0.003±0.001 | 0.007±0.004 |
Copy No. of mRNA for Nox4, Nox1, and Nox2 were obtained by real-time PCR of cDNA from myofibroblasts treated as indicated. Data are expressed as copies per 10 000 copies of 18sRNA. Values are means±SEMs (n=6).
Figure 3.
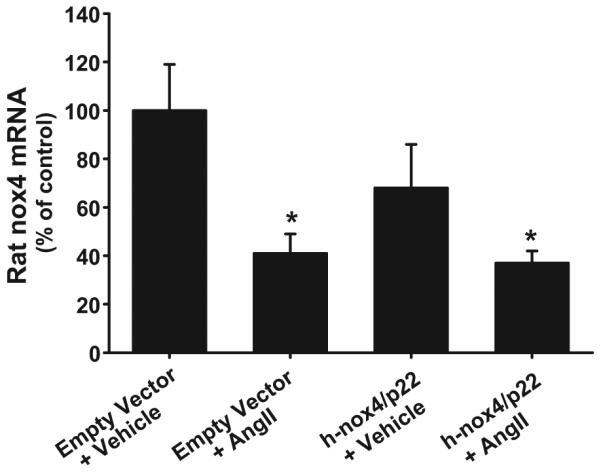
Effects of Nox4/p22-phox expression on endogenous levels of Nox4 mRNA. Adventitial myofibroblasts were transfected with either plasmids expressing human Nox4 and p22- phox or an empty vector control. Then they were stimulated with either Ang II (100 nmol/L) or vehicle, and mRNA levels for endogenous Nox4 and 18sRNA were determined using real-time PCR with rat-specific primers and probes. Data are expressed as the ratio of Nox4 mRNA vs 18sRNA and were plotted as a percentage of empty vector plus vehicle control on the day of assay±SEM. * P<0.05; n=6.
Cotransfection with h-Nox4/p22 had no significant effect on Nox4 mRNA, reducing levels to 0.9±0.2 copies per 10 000 18S copies (68±18% of empty vector control). Ang II treatment of cotransfected cells reduced these mRNAs further to 0.6±0.2 copies per 10 000 18S copies (Figure 3; 37±5% of empty vector control; P<0.05). These treatments had no effect on Nox1 or Nox2 mRNA (Table).
Effect of Ang II and h-Nox4/p22 Overexpression on H2O2 Generation in Adventitial Myofibroblasts
Ang II substantially increased H2O2 in empty vector–transfected cells by 473±22% compared with vehicle (0.0160± 0.00062 versus 0.0028±0.00007 arbitrary units; P<0.001; n=36; Figure 4). Similarly, Ang II increased H2O2 production in myofibroblasts transfected with h-Nox4/p22 by 371±38% compared with vehicle (0.0123±0.00099 versus 0.0026±0.00003 arbitrary units; P<0.001; n=36; Figure 4). However, induction by Ang II was significantly lower in h-Nox4/p22–transfected cells compared with empty vector controls (decreased by 23±8%; P<0.05).
Figure 4.
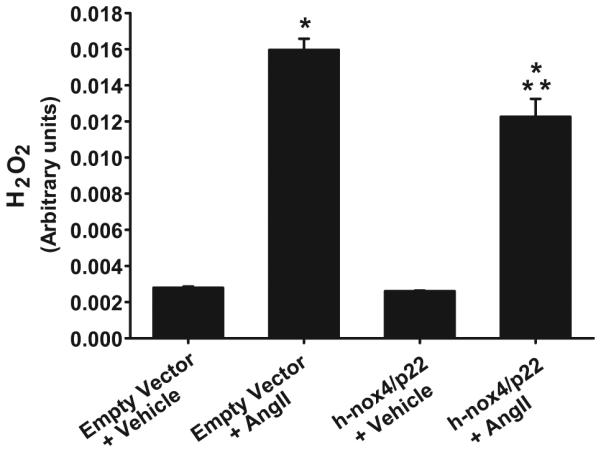
Effects of Nox4/p22-phox overexpression on Ang II-dependent H2O2 production. Adventitial myofibroblasts were transfected with either plasmids expressing Nox4 and p22-phox or an empty vector control. They were then stimulated with either Ang II (100 nmol/L) or vehicle and peak production of H2O2 measured using luminol. Data are expressed as arbitrary units of luminescence±SEM. *P<0.001 vs empty vector plus vehicle; **P<0.001 vs empty vector plus Ang II.
Effect of Ang II and h-Nox4/p22 Overexpression on Adventitial Myofibroblast Migration
We next examined the effect of h-Nox4/p22 expression on Ang II-stimulated myofibroblast migration. In empty vector–transfected myofibroblasts, Ang II stimulation caused a 2-fold increase in migration compared with vehicle (192±32%; P<0.001; n=12; Figure 5). This induction was suppressed by 46±6% when the myofibroblasts were transfected with h-Nox4/p22 (104±11% versus empty vector vehicle control; P<0.001; n=12). Ang II-induced migration of adventitial myofibroblasts was significantly attenuated by catalase (3000 U/mL) in both empty vector- and h-nox4/p22–transfected adventitial myofibroblasts (P<0.01; n=15).
Figure 5.
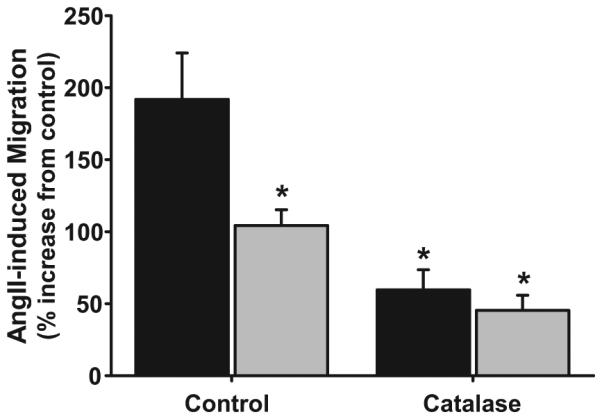
Effects of h-Nox4/p22 overexpression on Ang-II–induced migration of adventitial myofibroblasts. Adventitial myofibroblasts transfected with an empty vector control (black bars) or plasmids expressing Nox4 and p22-phox (gray bars) were treated with vehicle or Ang II in the presence or absence of catalase, and migration was measured after 6 hours. Data are expressed as the percentage increase in migration induced by Ang II (100 nmol/L) vs vehicle control. *P<0.01 vs empty vector control; n=15.
Discussion
Previous studies report a role for vascular adventitial myofibroblast migration in balloon angioplasty–induced restenosis and suggest that ROS derived from NADPH oxidases in myofibroblasts promote this process. Ang II is well established as a prototype activator of NADPH oxidase in vascular cells, including fibroblasts and myofibroblasts. We, therefore, proposed that Ang II-induced H2O2 production in myofibroblasts promotes migration. In this study we focused on the ability of Nox4 NADPH oxidase (Nox4 and p22-phox) to promote Ang II-stimulated myofibroblast migration, postulating that Ang II stimulates vascular adventitial myofibroblast migration via Nox4 oxidase and H2O2. To our knowledge, we report here for the first time that (1) Ang II induces the migration of vascular adventitial myofibroblasts in a concentration-dependent manner, and (2) this migration is mediated via production of H2O2. Moreover, we report for the first time the relative Nox mRNA levels in adventitial myofibroblasts (Nox4>Nox1>Nox2). We also show that overexpression of exogenous Nox4 and p22-phox leads to attenuation of endogenous Nox4 levels concomitant with reduced myofibroblast H2O2 production and migration. Our data indicate that cellular overexpression of Nox4 inhibits endogenous Nox4, which appears to lead to a reduced adventitial myofibroblast migration.
Effect of Ang II on Myofibroblast Migration
We showed that Ang II induces adventitial myofibroblast migration potently and in a concentration-dependent manner, with maximal stimulation occurring at 100 nmol/L. One hundred nanomoles per liter Ang II produced a 10-fold increase in the migration of serum-deprived myofibroblasts versus vehicle control (Figure 1). Under similar conditions, Li et al31 observed a 3-fold increase in migration in response to Ang II at this concentration. Differences in the magnitude of stimulation may be attributed to the varying genetic background of both rat populations (Sprague-Dawley versus WKY rats, respectively). By comparison, Dubey et al32 showed that a 10-fold higher Ang II concentration (1 µmol/L) caused a 5.8-fold increase in rat aortic smooth muscle cell migration in a similar Boyden chamber assay. In another study, 100 nmol/L Ang II caused a 65% increase in migration of wounded rat aortic smooth muscle cell monolayers.33 Thus, although it is difficult to make comparisons among cell preparations from different laboratories, the data may suggest that myofibroblasts are more responsive to Ang II with regard to migration. Consistent with this notion is the previous finding that ROS levels are markedly higher in adventitial fibroblasts versus smooth muscle cells.34
Endogenous Nox1, 2, and 4 mRNA levels in vascular adventitial myofibroblasts were strikingly different. In empty vector controls, Nox4 mRNA levels were 3-fold higher than Nox1 mRNA levels and 350-fold higher than Nox2 mRNA. Relatively high levels of adventitial myofibroblast Nox4 mRNA are consistent with previous reports of the important role of Nox4 in cardiac fibroblast differentiation into myofibroblasts.30 The data are also consistent with the relative order of abundance reported in proliferating aortic smooth muscle cells.35 Although it is not known whether the relative abundance that we observed in adventitial myofibroblasts is similar to that in fibroblasts, it is important to point out that mRNA levels for the various Nox isoforms do not necessarily indicate similar differences in protein levels or activity. Thus, caution is necessary when interpreting the significance of differences in mRNA, especially when correlations are being drawn. On initial consideration, the data may suggest that Nox1 and Nox2 play less important roles than Nox4 in myofibroblast migration; we can infer from previous reports that this is probably incorrect. For example, Szöcs et al20 reported that, after balloon angioplasty of the rat aorta (which involves marked myofibroblast migration), aortic Nox2 and Nox1 mRNA rose 15-fold and 2.7-fold versus sham controls, respectively, higher than the 2-fold increase observed for Nox4. Although those measurements were taken from whole aortas, they are consistent with a significant role for Nox2 in this process. In fact, our previous findings also support an important role for the Nox2-based oxidase in the process of myofibroblast migration, because both our inhibitor targeting Nox2 function and a dominant negative targeting the cytosolic activating factor p67-phox of Nox2-based oxidase both effectively inhibited this process in vivo.5,24 However, it is not clear at this point whether Nox2 plays a role in promoting myofibroblast migration in vivo, per se, or whether it is another aspect of this process that is mediated by Nox2, ie, earlier fibroblast proliferation.
Effects of Ang II on Levels of NADPH Oxidase Isoforms and H2O2 in Adventitial Myofibroblasts
Ang II treatment increased H2O2 levels =6-fold compared with vehicle in empty vector-treated myofibroblasts. We showed that this induction of H2O2 accounted for Ang II-induced migration, because catalase markedly diminished this response (Figure 5). The magnitude of this induction is in agreement with previous results by our group and others establishing Ang II as a potent stimulant of Nox expression and NADPH oxidase–derived ROS in vascular fibroblasts and in endothelial and smooth muscle cells.16,17,22,35–37 Clearly, Ang II stimulates the migration of vascular endothelial and smooth muscle cells in vitro.31,38 However, a link between Ang II-induced NADPH oxidase–derived ROS and vascular cell and, in particular, myofibroblast migration has not been established to our knowledge.
Although we demonstrated that H2O2 induces adventitial myofibroblast migration, we were unable to show a correlation of Ang II-induced migration with increased Nox levels. In contrast, our data show that Ang II decreased Nox1 and Nox4 but not Nox2 mRNA in control cells. The likely explanation for this effect is that prolonged exposure to Ang II-elevated H2O2 (6 hours) in culture led to downregulation of these 2 isoforms at the mRNA level. That is, it is very likely that these components increased at earlier time points after the administration of Ang II and then were downregulated. However, it is unclear why Ang II did not affect Nox2 mRNA levels. One possible explanation could be that quantification of Nox2 mRNA was at the limits of detection, and, therefore, we were unable to discern differences at such a low level. Future experiments will be necessary to confirm such an effect.
Effects of Transfection With h-Nox4/p22 on Levels of NADPH Oxidase Isoforms and H2O2 in Adventitial Myofibroblasts
h-Nox4/p22 transfection alone failed to elevate basal myofibroblast H2O2 production. This may suggest that, in adventitial myofibroblasts, overexpressed Nox4 was not constitutively active but required hormonal stimulation. In fact, Ang II treatment increased H2O2 by =4.7-fold levels compared with vehicle in empty vector–treated myofibroblasts. On the other hand, Ang II increased H2O2 3.7-fold in h-Nox4/p22, a 23% lower Ang II stimulation. We expected that h-Nox4/p22 overexpression would increase Ang II stimulation of H2O2 levels in these cells. Two possible explanations for the lack of such an increase could be that (1) the human isoforms of Nox4 and p22-phox are not as active as their endogenous counterparts, and/or (2) the decrease in H2O2 is a result of a net decrease in overall Nox4 levels brought about by a greater decrease in endogenous Nox4. Nevertheless, the fact that catalase suppressed migration in this group of cells confirmed the role of H2O2 (Figure 5).
We report novel findings that h-Nox4/p22 overexpression attenuated Nox4 mRNA. Even more interesting was the fact that h-Nox4/p22 transfection suppressed endogenous Nox4 mRNA levels in the absence of significant changes in Nox1 or Nox2 mRNA. Endogenous Nox4 mRNA showed a tendency toward a decrease in untreated h-Nox4/p22-treated cells versus vector controls, although these data did not reach statistical significance. A statistical power analysis showed that additional studies would likely have revealed a significant decrease. After treatment with Ang II, h-Nox4/p22-treated cells did illustrate a statistically significant decrease in Nox4 mRNA to 37% of control. Nevertheless, we believe that our report is the first to show that overexpression of a particular Nox isoform leads to specific downregulation of its endogenous analogue. Two possible explanations for this include the idea that (1) the specific ROS produced by Nox4 (H2O2 versus superoxide anion for Nox1 and Nox239) is capable of uniquely downregulating Nox4, and (2) elevated levels of Nox4 message or protein led to feedback inhibition of the endogenous Nox4 mRNA. In fact, Lassègue et al35 indicated that Ang II reduces Nox4 mRNA in rat vascular smooth muscle cells, although the mechanism controlling this effect is not known. Moreover, the specificity of this feedback inhibition may be explained by the process of autoregulated mRNA decay, in which the stability of a given mRNA is downregulated by its interaction with the encoded protein.40 In addition, recent data show that Nox1, Nox2, and Nox4 are located in distinct subcellular compartments.28 Such compartmentalization and juxtaposition to unique signaling pathways may explain the specific regulation of one Nox isoform over its counterparts. Experiments discerning such regulation will be necessary but are currently outside the scope of this study. Most importantly, the fact that reductions in Nox4 mRNA levels coincided with a significant reduction in Ang II-induced H2O2 and myofibroblast migration supports the role of Nox4 oxidase-derived H2O2 in myofibroblast motility. However, an important limitation of this study is that a direct involvement of endogenous Nox4 in migration was not tested. The latter is the focus of ongoing studies comparing the direct role of various vascular Nox isoforms in myofibroblast migration.
From a technical standpoint, it is important to point out that transfection with h-Nox4/p22 plasmids resulted in an ≈8-fold increase in exogenous Nox4 and p22-phox. The magnitude of this increase raised initial concerns as to its biological relevance or a possible toxic effect because of a large increase in ROS. In support of our experimental model, however, the literature reveals a 15-fold increase in Nox4 in response to transforming growth factor-β, which is involved in the physiological differentiation of cardiac fibroblasts.30 We also ruled out a toxic effect of this treatment, because cell viability was only reduced by 15% as measured by trypan blue exclusion (data not shown).
In conclusion, we have shown that Ang II potently stimulates adventitial myofibroblast migration via an H2O2-dependent mechanism. Adventitial myofibroblast migration appears to be highly sensitive to Ang II stimulation in comparison with that observed in vascular smooth muscle cells. We also showed that in the differentiated adventitial myofibroblast Nox4 mRNA was considerably more abundant than that for Nox1 and Nox2. These data alone suggest an important role for Nox4 in myofibroblast migration. Moreover, endogenous nox4 expression appeared to be uniquely inhibited by overexpression of exogenous Nox4. Taken together with the concomitant reduction in H2O2 production, the data suggest that endogenous Nox4 may play a significant role in adventitial myofibroblast migration. However, it must be noted that in vivo experiments are likely to involve considerably more complex mechanism(s) and may be confounded by the paracrine effect(s) of various cell types. Nonetheless, the current findings are novel in that, to our knowledge, they are the first to suggest the importance of Nox4 in adventitial myofibroblast migration and that overexpression of Nox4-based oxidase can uniquely inhibit endogenous Nox4-mediated cell signaling.
Perspectives
The role of individual NADPH oxidase–based systems in vascular wall biology is both timely and highly significant. ROS produced by these systems have been widely shown to promote a broad range of signaling pathways and alter both vascular physiology and function. Emerging evidence supports specific roles of the various Nox isoforms in different vascular cell types. Attenuation of Nox4 and its downstream signaling associate with reduced myofibroblast migration and, thus, may be expected to decrease neointimal hyperplasia and restenosis after balloon angioplasty. An enhanced understanding of the means to manipulate these NADPH oxidase isoforms could lead to the development of new drugs or transgene therapies aimed at halting the progression of restenosis, promoting long-term patency of blood vessels.
Acknowledgments
We thank Drs Kathy Griendling and J. David Lambeth for their generous contribution of the Nox4 and p22-phox plasmids, respectively. We also thank Dr Pablo Ortiz for his critical review of the article.
Sources of Funding
This work was supported by National Institutes of Health grants HL55425, HL079207, and HL28982 and the Fund for Henry Ford Hospital.
Footnotes
Disclosures
P.J.P. is an Established Investigator of the American Heart Association. The remaining authors report no disclosures.
Contributor Information
Mounir J. Haurani, Department of General Surgery, Henry Ford Health System, Detroit, Mich
M. Eugenia Cifuentes, Hypertension and Vascular Research Division, Henry Ford Health System, Detroit, Mich.
Alexander D. Shepard, Department of General Surgery, Henry Ford Health System, Detroit, Mich
Patrick J. Pagano, Hypertension and Vascular Research Division, Henry Ford Health System, Detroit, Mich
References
- 1.Schwartz SM, deBlois D, O’Brien ERM. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- 2.Clowes AW, Reidy MA, Clowes MM. Kinetics of cellular proliferation after arterial injury. I. Smooth muscle growth in the absence of endothelium. Lab Invest. 1983;49:327–333. [PubMed] [Google Scholar]
- 3.Schwartz SM, Campbell GR, Campbell JH. Replication of smooth muscle cells in vascular disease. Circ Res. 1986;58:427–444. doi: 10.1161/01.res.58.4.427. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 5.Dourron HM, Jacobson GM, Park JL, Liu J, Reddy DJ, Scheel ML, Pagano PJ. Perivascular gene transfer of NADPH oxidase inhibitor suppresses angioplasty-induced neointimal proliferation of rat carotid artery. Am J Physiol Heart Circ Physiol. 2005;288:H946–H953. doi: 10.1152/ajpheart.00413.2004. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz SM, Murry CE. Proliferation and the monoclonal origins of atherosclerotic lesions. Annu Rev Med. 1998;49:437–460. doi: 10.1146/annurev.med.49.1.437. [DOI] [PubMed] [Google Scholar]
- 7.Farhy RD, Carretero OA, Ho K-L, Scicli AG. Role of kinins and nitric oxide in the effects of angiotensin converting enzyme inhibitors on neointima formation. Circ Res. 1993;72:1202–1210. doi: 10.1161/01.res.72.6.1202. [DOI] [PubMed] [Google Scholar]
- 8.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 9.Souza HP, Souza LC, Anastacio VM, Pereira AC, Junqueira MdL, Krieger JE, da Luz PL, Augusto O, Laurindo FRM. Vascular oxidant stress early after balloon injury: evidence for increased NAD(P)H oxidoreductase activity. Free Radic Biol Med. 2000;28:1232–1242. doi: 10.1016/s0891-5849(00)00240-9. [DOI] [PubMed] [Google Scholar]
- 10.Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 11.DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 12.Zhan S, Vazquez N, Zhan Sh, Wientjes FB, Budarf ML, Schrock E, Ried T, Green ED, Chanock SJ. Genomic structure, chromosomal localization, start of transcription, and tissue expression of the human p40-phox, a new component of the nicotinamide adenine dinucleotide phosphate-oxidase complex. Blood. 1996;88:2714–2721. [PubMed] [Google Scholar]
- 13.Knaus UG, Morris S, Dong H-J, Chernoff J, Bokoch GM. Regulation of human leukocyte p21-activated kinases through G protein-coupled receptors. Science. 1995;269:221–223. doi: 10.1126/science.7618083. [DOI] [PubMed] [Google Scholar]
- 14.Geiszt M, Kopp JB, Várnai P, Leto TL. Identification of Renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pagano PJ, Chanock SJ, Siwik DA, Colucci WS, Clark JK. Angiotensin II induces p67phox mRNA expression and NADPH oxidase superoxide generation in rabbit aortic adventitial fibroblasts. Hypertension. 1998;32:331–337. doi: 10.1161/01.hyp.32.2.331. [DOI] [PubMed] [Google Scholar]
- 16.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 17.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase. Role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 18.Leri A, Claudio PP, Li Q, Wang X, Reiss K, Wang S, Malhotra A, Kajstura J, Anversa P. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest. 1998;101:1326–1342. doi: 10.1172/JCI316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang F, Dusting GJ. Natural phenolic compounds as cardiovascular therapeutics: potential role of their antiinflammatory effects. Curr Vasc Pharmacol. 2003;1:135–156. doi: 10.2174/1570161033476736. [DOI] [PubMed] [Google Scholar]
- 20.Szöcs K, Lassègue B, Sorescu D, Hilenski LL, Valppu L, Couse TL, Wilcox JN, Quinn MT, Lambeth JD, Griendling KK. Upregulation of nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol. 2002;22:21–27. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- 21.Schröder K, Helmcke I, Palfi K, Krause KH, Busse R, Brandes RP. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27:1736–1743. doi: 10.1161/ATVBAHA.107.142117. [DOI] [PubMed] [Google Scholar]
- 22.Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci U S A. 1997;94:14483–14488. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang HD, Hope S, Du Y, Quinn MT, Cayatte A, Pagano PJ, Cohen RA. Paracrine role of adventitial superoxide anion in mediating spontaneous tone of the isolated rat aorta in angiotensin II-induced hypertension. Hypertension. 1999;33:1225–1232. doi: 10.1161/01.hyp.33.5.1225. [DOI] [PubMed] [Google Scholar]
- 24.Weaver M, Liu J, Pimentel D, Reddy DJ, Harding P, Peterson EL, Pagano PJ. Adventitial delivery of dominant-negative p67phox attenuates neointimal hyperplasia of the rat carotid artery. Am J Physiol Heart Circ Physiol. 2006;290:H1933–H1941. doi: 10.1152/ajpheart.00690.2005. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, O’Brien JE, Jr, Fard A, Mannion JD, Wang D, Zalewski A. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation. 1996;94:1655–1664. doi: 10.1161/01.cir.94.7.1655. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y, Niculescu R, Wang D, Patel S, Davenpeck KL, Zalewski A. Increased NAD(P)H oxidase and reactive oxygen species in coronary arteries after balloon injury. Arterioscler Thromb Vasc Biol. 2001;21:739–745. doi: 10.1161/01.atv.21.5.739. [DOI] [PubMed] [Google Scholar]
- 27.Li G, Chen S-J, Oparil S, Chen Y-F, Thompson JA. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation. 2000;101:1362–1365. doi: 10.1161/01.cir.101.12.1362. [DOI] [PubMed] [Google Scholar]
- 28.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 29.Sorescu D, Weiss D, Lassègue B, Clempus RE, Szöcs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 30.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 31.Li L, Zhu DL, Shen WL, Gao PJ. Increased migration of vascular adventitial fibroblasts from spontaneously hypertensive rats. Hypertens Res. 2006;29:95–103. doi: 10.1291/hypres.29.95. [DOI] [PubMed] [Google Scholar]
- 32.Dubey RK, Jackson EK, Luscher TF. Nitric oxide inhibits angiotensin II-induced migration of rat aortic smooth muscle cell. Role of cyclic-nu-cleotides and angiotensin1 receptors. J Clin Invest. 1995;96:141–149. doi: 10.1172/JCI118014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang M, Foster E, Kahn AM. Insulin-stimulated NAD(P)H oxidase activity increases migration of cultured vascular smooth muscle cells. Am J Hypertens. 2005;18:1329–1334. doi: 10.1016/j.amjhyper.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 34.Chamseddine AH, Miller FJ., Jr. gp91phox contributes to NADPH oxidase activity in aortic fibroblasts but not smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285:H2284–H2289. doi: 10.1152/ajpheart.00459.2003. [DOI] [PubMed] [Google Scholar]
- 35.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91phox homologues in vascular smooth muscle cells. Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 36.Cifuentes ME, Rey FE, Carretero OA, Pagano PJ. Upregulation of p67phox and gp91phox in aortas from angiotensin II-infused mice. Am J Physiol Heart Circ Physiol. 2000;L279:H2234–H2240. doi: 10.1152/ajpheart.2000.279.5.H2234. [DOI] [PubMed] [Google Scholar]
- 37.Zhang H, Schmeisser A, Garlichs CD, Plotze K, Damme U, Mugge A, Daniel WG. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells: role of membrane-bound NADH-/ NADPH-oxidases. Cardiovasc Res. 1999;44:215–222. doi: 10.1016/s0008-6363(99)00183-2. [DOI] [PubMed] [Google Scholar]
- 38.Rossi F, Bertone C, Petricca S, Santiemma V. Ghrelin inhibits angiotensin II-induced migration of human aortic endothelial cells. Atherosclerosis. 2006;192:291–297. doi: 10.1016/j.atherosclerosis.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 39.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kyrpides NC, Ouzounis CA. Mechanisms of Specificity in mRNA Degradation: Autoregulation and Cognate Interactions. J Theor Biol. 1993;163:373–392. doi: 10.1006/jtbi.1993.1126. [DOI] [PubMed] [Google Scholar]