

Abstract
Background
Reduced cell spreading is a prominent feature of aged dermal fibroblasts in human skin in vivo. Mitochondrial DNA (mtDNA) common deletion has been reported to play a role in the human aging process, however the relationship between age-related reduced cell spreading and mtDNA common deletion has not yet been reported.
Results
To examine mtDNA common deletion in the dermis of aged human skin, the epidermis was removed from full-thickness human skin samples using cryostat. mtDNA common deletion was significantly elevated in the dermis of both naturally aged and photoaged human skin in vivo. To examine the relationship between age-related reduced cell spreading and mtDNA common deletion, we modulated the shape of dermal fibroblasts by disrupting the actin cytoskeleton. Reduced cell spreading was associated with a higher level of mtDNA common deletion and was also accompanied by elevated levels of endogenous reactive oxygen species (ROS). Boosting cellular antioxidant capacity by using antioxidants was found to be protective against mtDNA common deletion associated with reduced cell spreading.
Conclusion
mtDNA common deletion is highly prevalent in the dermis of both naturally aged and photoaged human skin in vivo. mtDNA common deletion in response to reduced cell spreading is mediated, at least in part, by elevated oxidative stress in human dermal fibroblasts. These data extend current understanding of the mitochondrial theory of aging by identifying the connection between mtDNA common deletion and age-related reduction of cell spreading.
Keywords: Cell shape, Mitochondrial common deletion, Reactive oxygen species, Human skin connective tissue aging
Background
Human skin is the largest organ of the human body. Its functions include providing a protective barrier from environmental stressors including heat, solar ultraviolet (UV) irradiation, infection, injury, and water loss. Human skin, like all other organs, undergoes a natural aging process with time. Unlike other organs, human skin also continuously experiences harmful stress from environmental sources such as solar UV irradiation that leads to damage [1, 2]. Based on its causes, cutaneous aging is classified into two types: natural aging and photoaging [3, 4]. Natural aging refers to those changes observed in all individuals resulting from the passage of time, whereas photoaging refers to those changes attributable to habitual sun exposure. Both of these processes are cumulative, therefore photoaging is superimposed on intrinsic aging.
Histological and ultrastructural studies have revealed that the major alterations in aged skin are localized to the dermal connective tissue and are manifested as a thin and damaged dermis [5, 6]. These features are derived directly from deleterious alterations in collagen, the most abundant structural protein in skin. Alterations of the collagenous ECM microenvironment directly relate to the development of age-related skin pathologies by causing increased fragility, impaired vasculature support, poor wound healing, and a tissue microenvironment that promotes epithelial cancer [7–9].
In human skin, dermal fibroblasts are responsible for collagen homeostasis. Consequently, impaired dermal fibroblast function is a major contributing factor in human skin connective tissue aging [10–12]. We previously reported that a prominent characteristic of dermal fibroblasts in aged skin is reduced spreading and contact with collagen fibrils, causing cells to lose their typical elongated spindle-like morphology and become shorter with a rounded and collapsed morphology [6, 13, 14]. In young healthy skin, dermal fibroblasts attach to intact collagen fibrils and achieve normal cell spreading and shape. However, in aged dermis the collagen fibrils are fragmented, which impairs fibroblast-collagen interactions. These alterations impair fibroblast spreading and function. While cell shape is known to regulate many cellular functions [15–18], the molecular basis of their impact on dermal fibroblast function and skin connective tissue aging are not well understood.
Mitochondrial DNA (mtDNA) mutations and deletions have long been proposed to play a role in many human diseases [19–22] as well as the aging process [23–27]. mtDNA common deletion (4977 bp) is one of the best described mitochondrial deletions [22, 28]. mtDNA deletions including mtDNA common deletion have been reported to be prevalent in aged human skin [2, 23, 26, 29]. However, these existing data largely describe the levels of mtDNA deletions in epidermal keratinocytes, and the high turnover of keratinocytes would result in little time to accumulate mtDNA deletion. Although dermal fibroblasts are the major cell type responsible for the maintenance of dermal connective tissue homeostasis, little is known about the role of mtDNA common deletion in aging dermal fibroblasts. Dermal fibroblasts have a very low proliferative rate which would allow for an accumulation of mtDNA deletion. Additionally, the relationship between age-related reduced cell spreading, which is a prominent feature of aged dermal fibroblasts, and mtDNA common deletion has been virtually unexplored. Based on this information, we explored the possible connection between age-related reduced cell spreading and mtDNA common deletion in the dermis of human skin. We found that mtDNA common deletion is significantly increased in both naturally aged and photoaged human skin dermis in vivo, and that reduced fibroblast spreading induces the increase in mtDNA common deletion through increased endogenous reactive oxygen species (ROS). This mechanism provides new insight into the connection between age-related reduced cell spreading and the mtDNA common deletion/ROS axis, implicating a molecular basis for the pathophysiology of human skin connective tissue aging.
Methods
Procurement of human skin samples
Punch biopsies from sun-protected human buttock skin were obtained from clinically normal adult volunteers; 20–30 year-old males for the young group (N = 8, mean age 24 ± 3 years) and 80+ year-olds for the aged group (N = 8, mean age 82 ± 4 years). Samples of severely photoaged skin and subject-matched sun-protected skin were obtained from the extensor forearm and sun-protected underarm, respectively (N = 10, mean age 54 ± 3 years). The presence of severe photodamage was determined based on clinical criteria as previously described [30]. Skin samples were 4 mm diameter sections of full thickness skin. All procedures involving human subjects were approved by the University of Michigan Institutional Review Board, and all subjects provided written informed consent.
Cell culture
Primary adult human dermal fibroblasts were isolated from punch biopsies of sun-protected buttock skin from healthy volunteers (mean age 31 ± 4 years) [31]. Briefly, dermal fibroblasts were isolated by digesting skin with bacterial collagenase (Worthington Biochemical Corporation, Lakewood, NJ, USA). Early passage primary adult human dermal fibroblasts (less than nine passages) were cultured in Dulbecco’s Modified Eagle’s Media (DMEM, Invitrogen Life Technology, Carlsbad, CA, USA) with 10 % Fetal Calf Sera (FBS, Invitrogen Life Technology, Carlsbad, CA, USA) at 37 °C, 5 % CO2. The approximate population doubling time was 2 days. For latrunculin-A (Lat-A) treatment, cells were treated with Lat-A at a concentration of 30 nM for three days. For some studies, cultured dermal fibroblasts were treated with N-acetylcysteine (NAC, Sigma, St Louis, MO, USA) at a concentration of 10 mM immediately after Lat-A treatment for three days.
RNA quantitative real-time RT-PCR
RNA isolation and quantitative real-time RT-PCR were performed as previously described [31]. Briefly, total RNA was extracted by Trizol reagent and 100 ng total RNA was reverse transcribed using a Taqman Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real-time RT-PCR was performed by SYBR green real-time PCR using a 7300 Sequence Detector (Applied Biosystems, Foster City, CA, USA). Human keratin 14, type I procollagen, and 36B4 primers were described previously [32]. Target gene mRNA were normalized to the housekeeping gene 36B4 (a ribosomal protein used as an internal control for quantification).
Quantitative determination of mtDNA with 4977 bp deletion
Genomic DNA from human skin dermis and in vitro tissue culture cells was isolated using QIAamp DNA Minikit (QIAGEN, Inc., Hilden, Germany). To quantify mtDNA common deletion (4,977-bp deletion in mtDNA), we employed the quantitative “3 primer PCR” method [24, 33], which simultaneously detects both wild-type and common deletion, as illustrated in Fig. 1a. Oligonucleotide primers B and C corresponded to heavy strand positions 13720–13705 and 9028–9008, respectively; primer A corresponded to light strand positions 8273–8289. Primer B was designed in the region of mtDNA common deletion, whereas primers A and C flanked the region of mtDNA common deletion. By design, nested primers A and B only amplified wild-type mtDNA (755 bp), whereas nested primers A and C only amplified deleted mtDNA (470 bp) [33]. Primers A and C would not yield any PCR products if there was no deleted mtDNA, because the large flanking region (>5-kb) is too long to be amplified under PCR conditions of very short polymerase extension time, as described below. The PCR reaction mixture (25 μl total volume) contained 150 ng DNA template, 2 μM PCR primers, 10 μM dNTPs, and PCR master mix (Applied Biosystems, Foster City, CA, USA). The PCR conditions were pre-denaturation at 94 °C for 2 min, followed by 35 cycles at 94 °C for 25 s, 60 °C for 30s, and a final extension at 72 °C for 10 min. These PCR products were confirmed by electrophoresis on a 2 % agarose gel and sequencing (data not shown) [24, 33]. The levels of deleted and wild-type mtDNA were measured by real-time PCR using a 7300 Sequence Detector (Applied Biosystems, Foster City, CA, USA). The mtDNA common deletion levels (primers A/C) were normalized by simultaneously measuring wild-type mtDNA (primers A/B). PCR was performed and normalized against a serial dilution of a control DNA sample, and then each target PCR product in the samples was quantified based on the corresponding standard curve. All PCR experiments included a negative control with no template DNA (double-distilled water) and a positive control with PCR products from primers A and C (470 bp), which was cloned into pCDNA 3.1 (Invitrogen, Carlsbad, CA, USA).
Fig. 1.
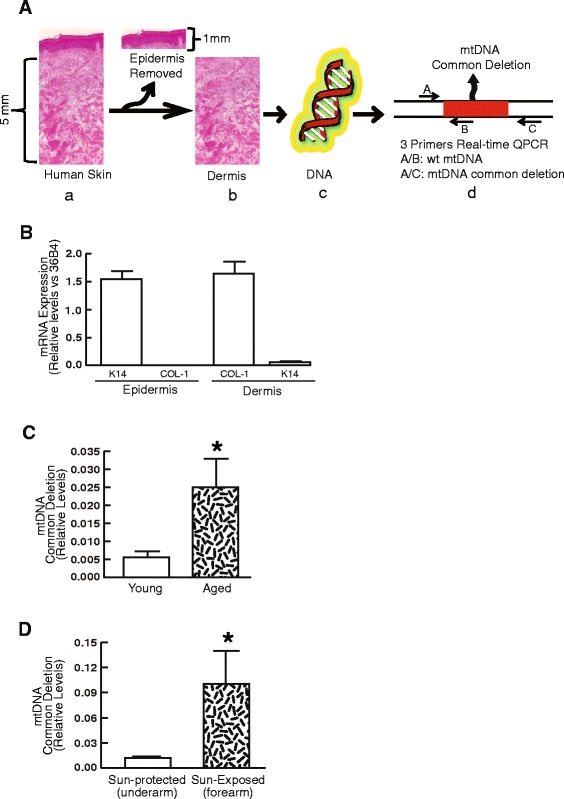
Accumulation of mtDNA common deletion in the dermis of naturally aged and photoaged human skin in vivo. a Schematic representation of the dissection of human skin dermis and detection of mtDNA common deletion in the dermis. a) Haematoxylin & Eosin staining of full-thickness human skin; b) Skin dermis was prepared by cutting off epidermis at a depth of 1 mm by cryostat; c) Purified total DNA from the dermis; d) Detection of mtDNA common deletion in the dermis. Real-time QPCR primers A, B, and C (indicated by arrows) were used to simultaneously PCR DNA fragments from both wild-type mtDNA (primers A/B) and deleted mtDNA (primers A/C). The PCR fragment from primers A/C was too large for wildtype mtDNA (>5 kb) to be amplified with the PCR conditions used for detection (see Methods for details). b Minimal contamination of keratinocyte in the dermis. Total RNA was prepared from epidermal and dermal sections, and keratin 14 (marker of epidermis) and type I procollagen (marker of dermal fibroblasts) mRNA levels were quantified by real-time RT-PCR. Keratin 14 and type I procollagen mRNA levels were normalized by the housekeeping gene (36B4, internal control). Mean ± SEM. N = 6, *p < 0.05. c mtDNA common deletion is increased in the dermis of naturally aged human skin in vivo. mtDNA common deletion was determined by real-time QPCR (see details for Methods). Young group (N = 8, mean age 24 ± 3 years) and aged group (N = 8, mean age 82 ± 4 years). Mean ± SEM. *p < 0.05. d mtDNA common deletion is increased in the dermis of photoaged human skin in vivo. mtDNA common deletion was determined by real-time QPCR (see Methods for details). Photoaged skin and subject matched sun-protected skin were obtained from the extensor forearm and sun-protected buttocks, respectively. Mean ± SEM. N = 10. *p < 0.05
Phalloidin staining, intracellular ROS measurements, and mitochondrial stain
Cell morphology was assessed by incubating cultures with Phalloidin. Cells were washed with PBS and fixed in 2 % paraformaldehyde for 30 min followed by Phalloidin stain (Sigma, St. Louis, MO, USA) for one hour. Relative cell surface areas were quantified by ImageJ (NIH, MD, USA). Intracellular ROS was measured by redox-sensitive fluorescent dye (Redox Sensor Red CC-1, Grand Island, NY, USA). Briefly, fibroblasts were incubated with Redox Sensor Red CC-1 (1 μM) at 37 °C for one hour in the dark. Cells were washed twice in PBS, then fixed with 2 % paraformaldehyde for 15 min. The cells were stained with DAPI to visualize nuclei, and cells were observed and photographed using Zeiss HBO100 fluorescence microscope. Intracellular ROS was quantified by ImageJ (NIH, MD, USA). Briefly, the intensity of the red area corresponding to oxidized RedoxSensor Red CC-1-positive staining was measured from 10 random fields per slide. Next, the software settings were programmed to quantify all nuclei (DAPI, blue staining) in the same fields, and the number of cells analyzed was considered. Finally, the intensity of the red area (indication of ROS levels) was normalized by the number of cells and the results expressed as a relative ROS level. To stain mitochondria, cells were incubated with MitoTracker® fluorescent dye (200 nM for 30 min, Molecular Probes, Inc.Eugene, OR, USA) according to the manufacture’s protocol. MitoTracker® passively diffuses across the plasma membrane and accumulates in active mitochondria of live cells.
Statistical analysis
Statistical significance between groups was determined with the Student’s t-test. All p values are two-tailed and considered significant when p < 0.05.
Results
mtDNA common deletion is accumulated in the dermis of naturally-aged and photoaged human skin in vivo
To assess mtDNA common deletion in the dermis, the epidermis was removed using cryostat to cut at a depth of 1 mm (Fig. 1a). To monitor separation of epidermis from dermis, the levels of keratin 14 mRNA (a marker of basal keratinocytes) and type I procollagen mRNA (a marker of dermal fibroblasts) were determined in each fraction. Keratin 14 mRNA levels in the dermal fraction were reduced 82-fold relative to the epidermis, indicating minimal contamination from keratinocytes in the dermal fraction, most likely from deeper appendages such as hair follicles and sweat glands (Fig. 1b). In contrast, type I procollagen mRNA levels were 152-fold greater in the dermis relative to epidermis, indicating collagen-rich dermis (Fig. 1b).
We next determined the levels of mtDNA common deletion in the dermis by purification of DNA from the dermis. As shown in Fig. 1c, the levels of mtDNA common deletion were significantly increased in aged human skin dermis compared to young skin dermis. The average level of mtDNA common deletion in aged dermis was 4.5-fold greater compared to young skin dermis. Similarly, the levels of mtDNA common deletion were much higher in sun-exposed photodamaged skin dermis (forearm) than in sun-protected skin dermis (underarm) (Fig. 1d). The average level of mtDNA common deletion in photodamaged skin dermis was 6.9-fold larger compared to sun-protected skin dermis. These results demonstrate the accumulation of mtDNA common deletion in the dermis of both naturally aged and photoaged human skin in vivo.
Reduced cell spreading induces mtDNA common deletion associated with increased reactive oxygen species (ROS) in human skin dermal fibroblasts
We next explored the possible connection between age-related reduced cell spreading, which is a prominent feature of aged dermal fibroblasts, and mtDNA common deletion in dermal fibroblasts from human skin. We modulated the shape of dermal fibroblasts by disrupting the actin cytoskeleton with latrunculin-A (Lat-A), which rapidly blocks actin polymerization [34]. As expected, disruption of the actin cytoskeleton impaired fibroblast spreading and resulted in a rounded shape (Fig. 2a, right panel). Staining of the actin cytoskeleton with phalloidin indicated a loss of actin cytoskeletal fibers and a reduced cell area (Fig. 2a, right panel). Quantification indicated that the surface areas of fibroblasts were reduced by 63 % in Lat-A treated cells compared to control cells (Fig. 2b). Interestingly, reduced cell spreading was associated with a significant elevation of mtDNA common deletion (Fig. 2c). The levels of mtDNA common deletion were increased 3.2-fold by reduced cell spreading (Fig. 2c). These data suggest that reduced cell spreading leads to elevated mtDNA common deletion in human dermal fibroblasts. As mitochondrial morphology is crucial for normal mitochondrial function, we assessed mitochondrial morphology by labeling mitochondria with MitoTracker fluorescent dye. These data indicated that the gross shape of mitochondria was similar between Lat-A treated cells and control cells (Fig. 2d). It has been reported that cellular damage from reactive oxygen species (ROS) likely plays an important role in mtDNA deletions as well as in the aging process [25, 35, 36]. We therefore examined the relative oxidant levels in fibroblasts using the redox-sensitive fluorescent dye RedoxSensor Red CC-1. Normal well-spreading fibroblasts displayed a very low level of oxidant-generated fluorescence (Fig. 2e, left panel). In contrast, reduced-spreading fibroblasts displayed intense oxidant-generated fluorescence (Fig. 2e, right panel). The level of oxidant-generated fluorescence was 4.2-fold greater in fibroblasts that had reduced spreading than in well-spreading fibroblasts (Fig. 2f). RedoxSensor Red is oxidized by a broad range of oxidizing species, therefore it reflects a general cellular redox state. To identify the source of endogenous ROS, the cells were double stained with mitochondrial markers Mitotracker and RedoxSensor. Figure 2g revealed a strong co-localization of Mitotracker and RedoxSensor, indicating that the mitochondria are the primary source of ROS induced by reduced cell spreading. Taken together, these data indicate that reduced cell spreading, due to disassembly of the actin cytoskeleton, leads to increased mtDNA common deletion accompanied by elevated mitochondrial ROS levels.
Fig. 2.
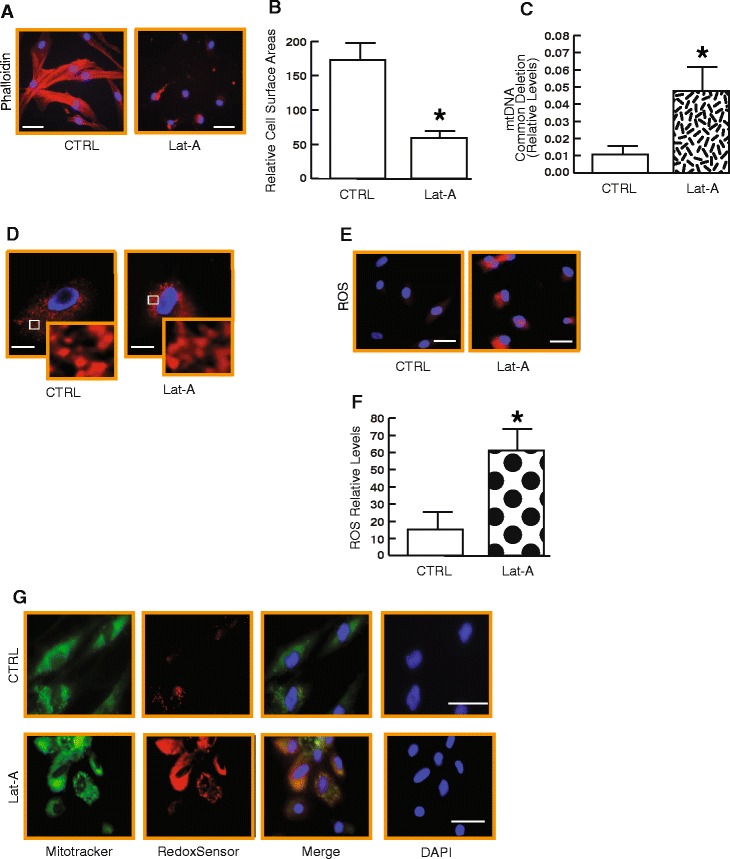
Reduced cell spreading induces mtDNA common deletion associated with increased reactive oxygen species (ROS) in human skin dermal fibroblasts. a Representative images of dermal fibroblasts treated with Lat-A (see Methods for details), which impairs actin polymerization and fibroblast spreading resulting in a rounded cell shape. Dermal fibroblasts were stained with phalloidin and were imaged by fluorescence microscopy. Red fluorescence delineates cell cytoplasm; blue fluorescence delineates nuclei. Bars = 50 μm. b The relative cell surface areas were quantified by ImageJ. Mean ± SEM, N = 4, *p < 0.05. c Reduced cell spreading induces mtDNA common deletion. mtDNA common deletion was determined by real-time QPCR (see details for Methods). Mean ± SEM. N = 4 *p < 0.05. d Cells were stained with MitoTracker fluorescent dye and were imaged by fluorescence microscopy. Red fluorescence delineates gross morphology of mitochondria; blue fluorescence delineates nuclei. Bars = 50 μm. e and f Reduced cell spreading induces ROS. Intracellular ROS levels were measured by Redox Sensor Red fluorescence and quantified by ImageJ (see Methods for details). Red fluorescence indicates ROS and blue fluorescence delineates nuclei. Mean ± SEM. N = 4, *p < 0.05. g Reduced cell spreading induces mitochondrial ROS. Cells were double stained with Mitotracker (green) and RedoxSensor (red); blue fluorescence delineates nuclei. Bars = 100 μm
Antioxidant treatment protects against mtDNA common deletion induced by reduced cell spreading in human dermal fibroblasts
We next investigated whether boosting cellular antioxidant capacity could protect against mtDNA common deletion associated with reduced cell spreading. We chose N-acetyl-cysteine (NAC), which is an antioxidant and metabolic precursor of glutathione [37]. Glutathione serves as a co-factor for the antioxidant enzyme glutathione peroxidase, which plays a critical role in protecting cells from oxidative damage by reducing lipid peroxides and converting hydrogen peroxide to water. Treatment of fibroblasts with NAC (10 mM) for three days immediately after Lat-A treatment markedly diminished the elevation of endogenous ROS levels (Fig. 3a). Treatment of fibroblasts with NAC effectively reduced ROS levels by 87 % in Lat-A treated cells (Fig. 3b). We next determined whether the addition of NAC could prevent the negative influence of oxidative stress on the levels of mtDNA common deletion associated with reduced cell spreading. NAC treatment partially but significantly prevented the elevated levels of mtDNA common deletion in a dose-dependent manner (Fig. 3c). Figure 3d further demonstrated that reduced cell spreading increased mtDNA common deletion in a time-dependent manner, and that the increase was significantly prevented by NAC treatment. These results indicate that the deleterious effects of endogenous oxidative exposure are responsible, at least in part, for reduced-cell-spreading-associated mtDNA common deletion.
Fig. 3.
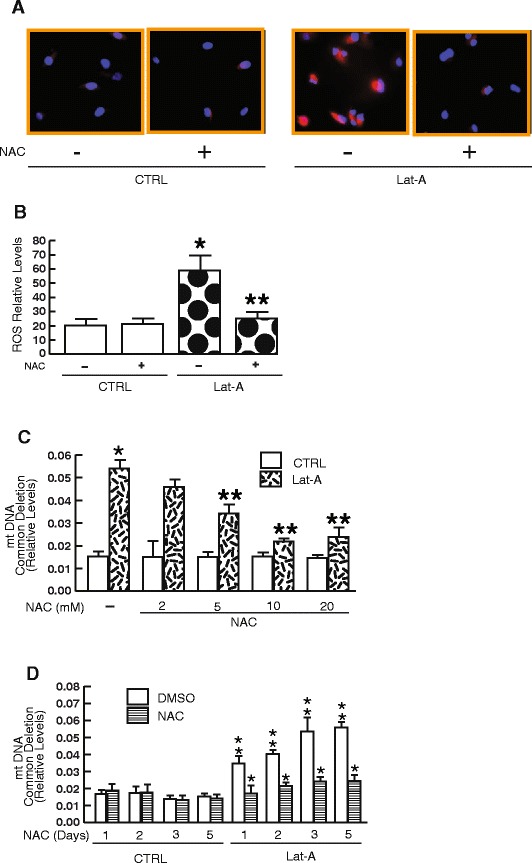
Antioxidant treatment protects against reduced-cell-spreading-associated mtDNA common deletion in human dermal fibroblasts. a and b Fibroblasts were treated with vehicle (CTRL, DMSO) or N-acetylcysteine (NAC, 10 mM) immediately after Lat-A treatment for three days. Intracellular ROS levels were measured by Redox Sensor Red fluorescence and quantified by ImageJ (see Methods for details). Red fluorescence indicates ROS and blue fluorescence delineates nuclei. Mean ± SEM. N = 4, *p < 0.05 vs CTRL. ** p < 0.05 vs Lat-A without NAC. c Anti-oxidant treatment protects against elevated mtDNA common deletion by oxidative exposure in a dose-dependent manner. Fibroblasts were treated with vehicle (CTRL, DMSO) or N-acetylcysteine (NAC, 2–20 mM) immediately after Lat-A treatment for three days. mtDNA common deletion was determined by real-time QPCR (see Methods for details). Mean ± SEM. *p < 0.05 vs CTRL. ** p < 0.05 vs Lat-A without NAC. d Time-course of anti-oxidant protection against mtDNA common deletion by oxidative exposure. Fibroblasts were treated with vehicle (CTRL, DMSO) or N-acetylcysteine (NAC, 10 mM) immediately after Lat-A treatment for indicated times. mtDNA common deletion was determined by real-time QPCR (see Methods for details). Mean ± SEM. *p < 0.05 vs Lat-A without NAC. ** p < 0.05 vs without Lat-A and NAC
Discussion
Human skin is largely composed of collagen-rich connective tissue, which provides structural and functional support. The collagen-rich connective tissue is produced, organized, and maintained by dermal fibroblasts. During aging, dermal collagen fibrils undergo progressive loss and fragmentation, leading to thin and structurally weakened skin. Age-related alterations of collagen fibrils impair skin structure and function, and create a tissue microenvironment that promotes age-related skin problems. Fibroblasts in young human dermis appear flattened and spread, and are in intimate contact with numerous intact collagen fibrils. In contrast, fibroblasts in aged human dermis have a collapsed appearance with little cytoplasm, and lack direct association with fragmented collagen fibrils [6, 13, 14]. Reduced fibroblast spreading has significant consequences in human skin connective tissue aging. Firstly, reduced fibroblast spreading significantly up-regulates matrix metalloproteinase-1 (MMP-1) expression through the activation of AP-1 transcription factor, a major driving force for MMP-1 expression [38]. Age-related elevation of MMP-1 causes fragmentation of collagen fibrils, which is considered an important process in human skin connective tissue aging [12, 38, 39]. Secondly, reduced fibroblast spreading significantly down-regulates collagen production through impairment of TGF-β signaling [10, 12, 40], which is a primary regulator of collagen and other ECM protein production [10, 41]. Reduced production of collagen and other ECM proteins contributes significantly to thinning of the skin, a prominent feature of aged human skin.
We recently demonstrated that increasing dermal fibroblast spreading through improved structural support of the dermal ECM microenvironment can activate fibroblasts to a more "youthful" state in aged human skin in vivo [40, 42]. Injection of dermal filler (cross-linked hyaluronic acid) into the skin of individuals over 70 years of age enhances cell spreading, leading to stimulated fibroblast collagen production and proliferation, expanded vasculature, and increased epidermal thickness. Therefore, fibroblasts in aged human skin retain their capacity for functional activation, which can be restored by enhancing cell spreading. These data also indicate that proliferation and function of other cell types, including endothelial cells and keratinocytes, can be enhanced in aged skin by enhancing cell spreading. These findings support the concept that cell spreading, along with tissue microenvironment, is critical for dermal fibroblast function.
In this report we show another aspect of the deleterious effect of age-related reduced dermal fibroblast spreading on skin connective tissue aging, through induction of mtDNA common deletion. Although the accumulation of mtDNA common deletion in aged human skin has been reported for more than two decades [23, 26, 29], our data provide two pieces of new information that give us a better understanding of the age-related accumulation of mtDNA common deletion in skin. First, we demonstrate that mtDNA common deletion is induced by age-related reduction of dermal fibroblast spreading. Our findings extend current understanding of the mitochondrial theory of aging by identifying reduction of cell spreading as a factor for age-related accumulation of mtDNA common deletion. Secondly, we demonstrate that mitochondria are the primary source of elevated ROS in response to age-related reduction of cell spreading. There are multiple sources of intracellular ROS in mammals, including mitochondria, NADPH oxidases (NOX), xanthine oxidase, monoamine oxidase, and nitric oxide synthase. Mitochondrial ROS are widely associated with several age-related chronic diseases and the health of many vital organ systems [43–45]. Although beyond the scope of this study, it is of interest to investigate whether mitochondrial ROS are associated with reduced cell spreading in age-related chronic diseases. It is also of interest to test whether mitochondrial-targeted antioxidants, such as MitoQ and SkQ1, effectively protect against mitochondrial damage induced by reduced cell spreading.
Accumulating evidence suggests that increased mtDNA mutations/deletions can lead to increased ROS production, while at the same time oxidative stress can also induce mtDNA mutations/deletions [36, 46–50]. Our data demonstrates that mtDNA common deletion in response to reduced cell spreading is mediated, at least in part, by elevated ROS, suggesting the possibility of a positive feedback loop between ROS and mtDNA common deletion in response to reduced cell spreading. The mechanism by which reduced cell spreading leads to increased endogenous ROS levels remains to be determined. Cell shape impacts a multitude of cellular processes including signal transduction, gene expression, and metabolism [15–18]. Recent evidence suggests that cytoskeletal tension plays a key role in translation of mechanical information into cell function [51–53]. In general, intracellular redox homeostasis is maintained by enzymatic antioxidant defenses such as superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT). SODs are responsible for the dismutation of superoxide radicals, generated by NAD(P)H oxidases, to hydrogen peroxide. CAT converts hydrogen peroxide into water and oxygen. Impairment or imbalance of the enzymatic antioxidant defenses could contribute to elevated ROS generation in response to reduced cell spreading. We are currently investigating the possibility of whether an imbalance of antioxidant enzyme levels may contribute to elevated ROS levels in dermal fibroblasts in response to reduced spreading. Currently, knowledge regarding the relationship among reduction of cell spreading, ROS, and mtDNA common deletion is in a nascent state. Further studies are needed to understand the mechanisms that couple reduced cell spreading to oxidative stress and mtDNA common deletion in human dermal fibroblasts, their role in skin connective tissue aging, and possible therapeutic implications. In addition, future work should focus on types of mtDNA deletions/mutations other than common deletion, which may be considerably more prevalent.
Accumulating evidence suggests that aging is associated with increased frequency of mitochondrial DNA mutations/deletions and increased production of ROS [36, 46–50]. However, such observations have been made primarily in tissues with high rates of oxidative metabolism such as brain and muscle. The extent to which mtDNA mutations/deletions contribute to age-related changes in skin dermis remains to be clarified. Although an increased frequency of mtDNA mutations has been reported in naturally and photoaged aged human skin [2, 23, 24, 26], the frequency of mtDNA common deletion in the dermal compartment in naturally and photoaged skin has not been well documented. Our results demonstrate the accumulation of mtDNA common deletion in the dermis of both naturally and photoaged human skin in vivo. These data are consistent with our previous report that oxidant levels are indeed increased in dermal fibroblasts in aged skin in vivo [38]. The magnitude of mtDNA common deletion seems to be 10-fold higher in photoaged skin (Fig. 1d) compared to naturally aged skin (Fig. 1c), which is consistent with the general concept that more severe dermal connective tissue damage occurs in photoaged skin than in naturally aged skin.
Antioxidant treatments to either retard the aging process or to treat age-related disorders are subjects of heightened interest. NAC is safe for human use, and we previously demonstrated that NAC penetrates human skin and effectively mitigates ROS-driven responses to acute UV irradiation in human skin in vivo [54]. Our data suggest that antioxidant NAC may be able to retard skin connective tissue aging through reduction of mtDNA common deletion. Slowing the accumulation of mtDNA common deletion would likely result in less functional decline in dermal fibroblasts over time. It is tempting to speculate that the combination of an antioxidant and an agent that promotes features of cell spreading would promote normal dermal fibroblast function and revitalize skin.
Conclusion
In conclusion, based on our findings in aged human skin dermis in vivo and in vitro, we postulate that age-related reduction of dermal fibroblast spreading brings about numerous alterations including increased ROS/oxidative stress, which promotes mtDNA common deletion (Fig. 4). Increased mtDNA common deletion could further induce ROS/oxidative stress through a positive feedback mechanism, forming a critical mechanism of human skin aging. This mechanism extends current understanding of the oxidative theory of aging by recognizing that age-related reduction of dermal fibroblast spreading induces mtDNA common deletion through ROS/oxidative stress.
Fig. 4.
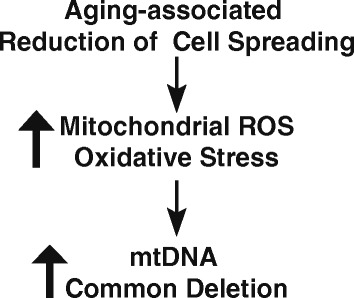
Proposed model for age-related reduction of dermal fibroblast spreading induces mtDNA common deletion through ROS/oxidative stress (see Discussion for details)
Acknowledgement
We thank Yushun Cui for graphics assistance. This work was supported by the National Institute of Health (ES014697 and ES014697 30S1 to TQ).
Abbreviations
- mtDNA
mitochondrial DNA
- Lat-A
Latrunculin-A
- ROS
Reactive oxygen species
- NAC
N-acetyl-cysteine
- UV
Ultraviolet
- COL-1
type I collagen
- K14
Keratin 14
- MMP-1
Matrix metalloproteinase 1
- ECM
Extracellular matrix
- PCR
Polymerase chain reaction
- AP-1
Activator protein-1
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CQ and MC performed the experiments, CQ wrote the manuscript. TQ designed the experiments and analyzed the data, and supervised the study. DP and TQ edited the manuscript. All authors commented on the manuscript. All authors read and approved the final manuscript.
Contributor Information
Chunji Quan, Email: chunjiquan@163.com.
Moon Kyun Cho, Email: mkcho2001@daum.net.
Daniel Perry, Email: danperry@med.umich.edu.
Taihao Quan, Phone: (734) 763-1469, Email: thquan@umich.edu.
References
- 1.Fisher GJ, Datta SC, Talwar HS, Wang ZQ, Varani J, Kang S, et al. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature. 1996;379(6563):335–9. doi: 10.1038/379335a0. [DOI] [PubMed] [Google Scholar]
- 2.Berneburg M, Plettenberg H, Krutmann J. Photoaging of human skin. Photodermatol Photoimmunol Photomed. 2000;16(6):239–44. doi: 10.1034/j.1600-0781.2000.160601.x. [DOI] [PubMed] [Google Scholar]
- 3.Yaar M, Eller MS, Gilchrest BA. Fifty years of skin aging. J Investig Dermatol Symp Proc. 2002;7(1):51–8. doi: 10.1046/j.1523-1747.2002.19636.x. [DOI] [PubMed] [Google Scholar]
- 4.Chung JH. Photoaging in Asians. Photodermatol Photoimmunol Photomed. 2003;19(3):109–21. doi: 10.1034/j.1600-0781.2003.00027.x. [DOI] [PubMed] [Google Scholar]
- 5.Fisher GJ, Wang ZQ, Datta SC, Varani J, Kang S, Voorhees JJ. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337(20):1419–28. [DOI] [PubMed]
- 6.Fisher GJ, Varani J, Voorhees JJ. Looking older: fibroblast collapse and therapeutic implications. Arch Dermatol. 2008;144(5):666–72. doi: 10.1001/archderm.144.5.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kudravi SA, Reed MJ. Aging, cancer, and wound healing. In Vivo. 2000;14(1):83–92. [PubMed] [Google Scholar]
- 8.Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–9. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bissell MJ, Kenny PA, Radisky DC. Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harb Symp Quant Biol. 2005;70:343–56. doi: 10.1101/sqb.2005.70.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quan T, Shao Y, He T, Voorhees JJ, Fisher GJ. Reduced expression of connective tissue growth factor (CTGF/CCN2) mediates collagen loss in chronologically aged human skin. J Invest Dermatol. 2010;130(2):415–24. [DOI] [PMC free article] [PubMed]
- 11.Quan T. Skin connective tissue aging and dermal fibroblasts. Dermal Fibroblasts: Histological Perspectives, Characterization and Role in Disease. 2013;31–55.
- 12.Quan T, Fisher GJ. Role of Age-Associated Alterations of the Dermal Extracellular Matrix Microenvironment in Human Skin Aging: A Mini-Review. Gerontology. 2015;61(5). PMID:25660807; doi:10.1159/000371708. [DOI] [PMC free article] [PubMed]
- 13.Varani J, Schuger L, Dame MK, Leonard C, Fligiel SE, Kang S, et al. Reduced fibroblast interaction with intact collagen as a mechanism for depressed collagen synthesis in photodamaged skin. J Invest Dermatol. 2004;122(6):1471–9. [DOI] [PubMed]
- 14.Qin Z, Voorhees JJ, Fisher GJ, Quan T. Age-associated reduction of cellular spreading/mechanical force up-regulates matrix metalloproteinase-1 expression and collagen fibril fragmentation via c-Jun/AP-1 in human dermal fibroblasts. Aging Cell. 2014;13(6):1028–37. [DOI] [PMC free article] [PubMed]
- 15.Janmey PA, Wells RG, Assoian RK, McCulloch CA. From tissue mechanics to transcription factors. Differentiation. 2013;86(3):112–20. [DOI] [PMC free article] [PubMed]
- 16.Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9(2):108–22. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iskratsch T, Wolfenson H, Sheetz MP. Appreciating force and shape-the rise of mechanotransduction in cell biology. Nat Rev Mol Cell Biol. 2014;15(12):825–33. doi: 10.1038/nrm3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mammoto A, Ingber DE. Cytoskeletal control of growth and cell fate switching. Curr Opin Cell Biol. 2009;21(6):864–70. doi: 10.1016/j.ceb.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Cha MY, Kim DK, Mook-Jung I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med. 2015;47 doi: 10.1038/emm.2014.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Enns GM. The contribution of mitochondria to common disorders. Mol Genet Metab. 2003;80(1–2):11–26. doi: 10.1016/j.ymgme.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125(7):1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Wallace DC. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science. 1992;256(5057):628–32. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 23.Berneburg M, Plettenberg H, Medve-Konig K, Pfahlberg A, Gers-Barlag H, Gefeller O, et al. Induction of the photoaging-associated mitochondrial common deletion in vivo in normal human skin. J Invest Dermatol. 2004;122(5):1277–83. [DOI] [PubMed]
- 24.Birch-Machin MA, Tindall M, Turner R, Haldane F, Rees JL. Mitochondrial DNA deletions in human skin reflect photo- rather than chronologic aging. J Invest Dermatol. 1998;110(2):149–52. [DOI] [PubMed]
- 25.Sastre J, Pallardo FV, Vina J. The role of mitochondrial oxidative stress in aging. Free Radic Biol Med. 2003;35(1):1–8. [DOI] [PubMed]
- 26.Yang JH, Lee HC, Lin KJ, Wei YH. A specific 4977-bp deletion of mitochondrial DNA in human ageing skin. Arch Dermatol Res. 1994;286(7):386–90. [DOI] [PubMed]
- 27.Lee HC, Wei YH. Mitochondria and aging. Adv Exp Med Biol. 2012;942:311–27. doi: 10.1007/978-94-007-2869-1_14. [DOI] [PubMed] [Google Scholar]
- 28.Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990;18(23):6927–33. doi: 10.1093/nar/18.23.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu CY, Lee HC, Fahn HJ, Wei YH. Oxidative damage elicited by imbalance of free radical scavenging enzymes is associated with large-scale mtDNA deletions in aging human skin. Mutat Res. 1999;423(1–2):11–21. [DOI] [PubMed]
- 30.Griffiths CE, Russman AN, Majmudar G, Singer RS, Hamilton TA, Voorhees JJ. Restoration of collagen formation in photodamaged human skin by tretinoin (retinoic acid). N Engl J Med. 1993;329(8):530–5. [DOI] [PubMed]
- 31.Quan T, He T, Kang S, Voorhees JJ, Fisher GJ. Connective tissue growth factor: Expression in human skin In vivo and inhibition by ultraviolet irradiation. J Invest Dermatol. 2002;118(3):402–8. [DOI] [PubMed]
- 32.Quan T, He T, Kang S, Voorhees JJ, Fisher GJ. Solar ultraviolet irradiation reduces collagen in photoaged human skin by blocking transforming growth factor-β type II receptor/Smad signaling. Am J Pathol. 2004;165(3):741–51. [DOI] [PMC free article] [PubMed]
- 33.Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3(1):13–9. [DOI] [PubMed]
- 34.Gieni RS, Hendzel MJ. Mechanotransduction from the ECM to the genome: are the pieces now in place? J Cell Biochem. 2008;104(6):1964–87. doi: 10.1002/jcb.21364. [DOI] [PubMed] [Google Scholar]
- 35.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–56. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 36.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320(5876):661–4. [DOI] [PubMed]
- 37.De Vries N, De Flora S. N-acetyl-l-cysteine. J Cell Biochem Suppl. 1993;17F:270–7. doi: 10.1002/jcb.240531040. [DOI] [PubMed] [Google Scholar]
- 38.Fisher GJ, Quan T, Purohit T, Shao Y, Moon KC, He T, et al. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am J Pathol. 2009;174(1):101–14. [DOI] [PMC free article] [PubMed]
- 39.Quan T, Little E, Quan H, Qin Z, Voorhees JJ, Fisher GJ. Elevated matrix metalloproteinases and collagen fragmentation in photodamaged human skin: impact of altered extracellular matrix microenvironment on dermal fibroblast function. J Invest Dermatol. 2013;133(5):1362–6. [DOI] [PMC free article] [PubMed]
- 40.Quan T, Wang F, Shao Y, Rittié L, Xia W, Orringer JS, et al. Enhancing structural support of the dermal microenvironment activates fibroblasts, endothelial cells, and keratinocytes in aged human skin in vivo. J Invest Dermatol. 2013;133(3):658–67. [DOI] [PMC free article] [PubMed]
- 41.Varga J, Rosenbloom J, Jimenez SA. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J. 1987;247(3):597–604. doi: 10.1042/bj2470597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang F, Garza LA, Kang S, Varani J, Orringer JS, Fisher GJ, et al. In vivo stimulation of de novo collagen production caused by cross-linked hyaluronic acid dermal filler injections in photodamaged human skin. Arch Dermatol. 2007;143(2):155–63. [DOI] [PubMed]
- 43.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43(3):332–47. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krause KH. Aging: a revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp Gerontol. 2007;42(4):256–62. doi: 10.1016/j.exger.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 45.Dai DF, Chiao YA, Marcinek DJ, Szeto HH, Rabinovitch PS. Mitochondrial oxidative stress in aging and healthspan. Longev Healthspan. 2014;3:6. [DOI] [PMC free article] [PubMed]
- 46.Cortopassi G, Shibata D, Soong N-W, Arnheim N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Nat Acad Sci USA. 1992;89:7370–4. [DOI] [PMC free article] [PubMed]
- 47.Lee H-C, Pang C-Y, Hsu H-S, Wei Y-H. Differential accumulations of 4,977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochima Biophysica Acta. 1994;1226:37–43. [DOI] [PubMed]
- 48.Linnane A, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;642–645. [DOI] [PubMed]
- 49.Terzioglu M, Larsson NG. Mitochondrial dysfunction in mammalian ageing. Novartis Found Symp. 2007;287:197–208. doi: 10.1002/9780470725207.ch14. [DOI] [PubMed] [Google Scholar]
- 50.Wei Y-H, Lee H-C. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med. 2002;227:671–682. doi: 10.1177/153537020222700901. [DOI] [PubMed] [Google Scholar]
- 51.Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. 2006;20(7):811–27. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- 52.Silver FH, Siperko LM, Seehra GP. Mechanobiology of force transduction in dermal tissue. Skin Res Technol. 2003;9(1):3–23. doi: 10.1034/j.1600-0846.2003.00358.x. [DOI] [PubMed] [Google Scholar]
- 53.Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260(5111):1124–7. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 54.Kang S, Chung JH, Lee JH, Fisher GJ, Wan YS, Duell EA, et al. Topical N-acetyl cysteine and genistein prevent ultraviolet-light-induced signaling that leads to photoaging in human skin in vivo. J Invest Dermatol. 2003;120(5):835–41. [DOI] [PubMed]