

Abstract
2-Arylethynyl derivatives of (N)-methanocarba adenosine 5′-uronamides are selective A3AR (adenosine receptor) agonists. Here we substitute a 1,2,3-triazol-1-yl linker in place of the rigid, linear ethynyl group to eliminate its potential metabolic liability. Docking of nucleosides containing possible short linker moieties at the adenine C2 position using a hybrid molecular model of the A3AR (based on the A2AAR agonist-bound structure) correctly predicted that a triazole would maintain the A3AR selectivity, due to its ability to fit a narrow cleft in the receptor. The analogues with various N6 and C2-aryltriazolyl substitution were synthesized and characterized in binding (Ki at hA3AR 0.3 – 12 nM) and in vivo to demonstrate efficacy in controlling chronic neuropathic pain (chronic constriction injury). Among N6-methyl derivatives, a terminal pyrimidin-2-yl group in 9 (MRS7116) increased duration of action (36% pain protection at 3 h) in vivo. N6-Ethyl 5-chlorothien-2-yl analogue 15 (MRS7126) preserved in vivo efficacy (85% protection at 1 h) with short duration. Larger N6 groups, e.g. 17 (MRS7138, >90% protection at 1 and 3 h), greatly enhanced in vivo activity. Thus, we have combined structure-based methods and phenotypic screening to identify nucleoside derivatives having translational potential.
Keywords: G protein-coupled receptor, nucleoside, click chemistry, neuropathic pain, molecular modeling
Introduction
Two nucleoside agonists of the A3 adenosine receptor (A3AR) are currently in clinical trials for the treatment of autoimmune inflammatory diseases (N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine, IB-MECA), including rheumatoid arthritis and plaque psoriasis, and hepatocellular carcinoma (2-chloro analogue, Cl-IB-MECA).1,2 An even broader range of conditions have been shown to be ameliorated by administration of A3AR agonists in animal models, including osteoarthritis, neutropenia, and other inflammatory and infective conditions and chronic neuropathic pain.3,4 Chronic neuropathic pain can result from injury or diseases such as cancer and diabetes or from pharmaceutical administration, and its treatment represents an unmet medical need. Thus, the design, synthesis and pharmacological characterization of novel drug-like A3AR agonists of high affinity, selectivity and in vivo efficacy are well justified.
The selective A3AR agonists that have advanced to clinical trials are adenine-9-riboside derivatives.1,2 As an extension of the structure activity relationship (SAR) of AR agonists, we have introduced nucleoside derivatives containing in place of ribose a bicyclo[3.1.0]hexane (methanocarba) ring system, which display increased affinity and/or selectivity for the A3AR compared to other AR subtypes.5 The rigid ribose ring substitute maintains a receptor-preferred conformation and thus decreases an entropic energy barrier for receptor binding. Prototypical nucleosides containing a methanocarba ring system in a North (N) envelope conformation are typically >100-fold more potent at the A3AR than the corresponding isomers in the South (S) conformation.6 A combination of A3AR-favoring modifications provided MRS5698 1 and MRS5980 2 (Chart 1A) as highly potent agonists with favorable protective properties in the sciatic nerve chronic constriction injury (CCI) model of neuropathic pain in mice.7-9 These rigidified nucleosides contain a C2-arylethynyl group,10 which is thought to be responsible for the high A3AR selectivity, based on the greater structural plasticity of the A3AR compared to the A2AAR, which is more constrained by disulfide bridges in the extracellular loops (ELs). In particular, we have proposed an outward movement of transmembrane helix 2 (TM2) in the A3AR in order to accommodate the C2-arylethynyl group and still to maintain conserved H-bonding interactions of the hydroxyls of the ribose-like moiety with the N6 and N7 of the adenine moiety.7,11
Chart 1.

A. Structures of recently reported highly selective A3AR agonists (1, 2) and the structural options considered in the present study (3). R is a small alkyl, cycloalkyl or substituted arylalkyl group; Ar refers to substituted phenyl or heterocyclic groups or ferrocene (See compound 4, Table 2). B. Hypothetical transient breakdown products 39 and 40 from acid treatment of the ferrocene derivatives 4 and 20, respectively. Only 39 was detected by mass upon acidification of 4; compound 20 was stable at pH 1.6.
However, the presence of an arylethynyl group might be an indication of potential liver toxicity due to its electrophilicity and possible reactivity with glutathione.12 Therefore, we explored alternative structures that would still maintain the proper geometry of the distal aryl group with respect to the adenosine core structure. A number of alternative C2 substituents were considered and compared by docking to an A3AR model: benzyl 3b, phenylethyl 3c, phenylcyclopropyl 3d, phenyl-trans-ethenyl 3e, and phenyl-triazolyl 3f (Chart 1A, Ar = C6H5). A 1,2,3-triazol-1-yl group was predicted to best maintain a similar geometry compared to the potent and selective C2-arylethynyl derivatives.
This work has multiple goals: 1) to identify a general class of compounds that are highly selective agonists of the A3AR but do not contain an arylethynyl group, based initially on a comparison of hypothetical structures using molecular modeling; 2) to determine the in vivo efficacy and duration of action in a mouse model of neuropathic pain in order to select preferred candidates for further development.
Results and discussion
We used molecular modeling to predict the effect of logical C2 modifications 3b-f (R = CH3, Ar = C6H5) on human (h) A3AR recognition. Because the rigid, elongated C2 substituent of the series of general formula 3a is key to the >3000-fold A3AR selectivity vs. the A2AAR, a substitution for the ethynyl group would need to maintain a similar extended conformation when bound to the receptor. The receptor model used for docking was our recently published hybrid homology model of the A3AR.7,11 The agonist-bound hA2AAR X-ray structure13 was used as a template for all of the TMs, except for the upper part of TM2.11 Figure 1 compares the effects of these substitutions when the nucleosides are docked in the hybrid model, and Table 1 summarizes the features of the different proposed linkers (substitutes for C≡C of 3a). Docking simulations showed that all the derivatives bearing the proposed alternative C2 substituents were able to fit the binding site of the A3AR hybrid model in an orientation analogous to the one observed for the original arylethynyl derivatives. Such a binding pose has been well validated in previous studies7,11 and shows all the interactions key for agonist binding at ARs.13 In particular, the pseudo-sugar moiety forms H-bonds with Thr94 (3.36), Ser271 (7.42) and His272 (7.43) (numbers in parenthesis follow the Ballesteros-Weinstein numbering system),14 the adenine core forms two H-bonds with Asn250 (6.55) and a π-π stacking with Phe168 (EL2), the N6 substituent interacts with hydrophobic residues on EL2 such as Val169 and the C2 group is directed towards TM2 (Figure 1A).
Figure 1.
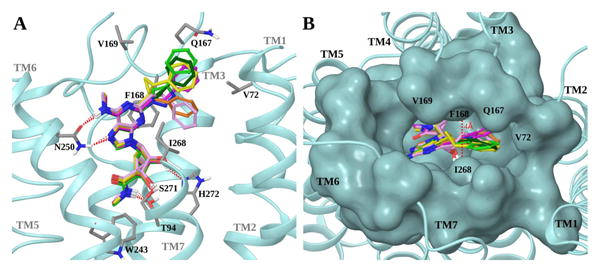
Superposition of the docking poses of trial compounds 3a – f (R = CH3, Ar = Ph) in the hA3AR hybrid homology model. A. Side view of the receptor (shown in cyan ribbon). Ligands are shown in sticks with different carbon colors (3a, dark green; 3b, orange; 3c, pink; 3d, yellow; 3e, magenta; 3f, light green). Side chains of residues important for ligand recognition are shown in sticks (grey carbons), and H-bonding interactions are indicated by red dashed lines. Non-polar hydrogen atoms are not displayed. The view of TM7 is partially omitted. B. Top view of the receptor showing the surface of the binding site residues in cyan. Ligands are shown in sticks with different carbon colors (color scheme as in panel A). The distance between Phe168 (EL2) and Ile268 (7.39) is highlighted.
Table 1.
Comparison of the features of C2 modifications 3a-f (Chart 1A, Ar = C6H5). A check mark indicates favorable (planarity and rigidity, with respect to the binding cleft in the C2-extended adenine region of the A3AR as in Figure 1) or unfavorable criteria (reactivity) as applied to each linker. All of the maximum lengths shown are within the range of the proposed outward displacement of TM2 in the hybrid A3AR model.
| Linker | Linker Features | ||||
|---|---|---|---|---|---|
| Planaritya | Rigidityb | Max Lengthc | Potential Reactivityd | ||
| a | C≡C | ✓ | ✓ | 8.00 | ✓ |
| b | CH2 | 6.37 | |||
| c | (CH2)2 | 5.84 | |||
| d | cyclopropyl | ✓ | 7.61 | ||
| e | CH=CH | ✓ | ✓ | 7.70 | ✓ |
| f | triazole | ✓ | ✓ | 8.80 | |
Ability to fit in a narrow space. This refers to the planarity of the linker only not of the overall C2 substituent.
Inability to bend.
Distance measured between the C2 of the adenine core and the most distal hydrogen of the phenyl ring.
Potential toxicity and/or reactivity with nucleophiles in vivo.
Thus, to select the best linker to be used in the new series of derivatives, we analyzed the various alternatives in a detailed manner. The rigid, elongated C2 substituent is a key feature for high A3AR selectivity, because of the greater structural plasticity of the A3AR compared to the A2AAR as previously proposed,7 therefore we predicted higher selectivity at the A3AR for those derivatives bearing rigid and elongated linkers, such as 3d-f. These substitutions might approximate the docked position of the terminal aryl group of known selective agonist series 3a (Figure 1A).
The terminal phenyl ring need not be coplanar with the adenine ring, but a substantial bending within the C2 substituent would not be favored. Compounds 3b and 3c, even though predicted to bind the A3AR, would probably interact also with the A2AAR because of flexibility in the position of the terminal phenyl ring, possibly resulting in reduced selectivity. Moreover, the C2 linker is accommodated in a narrow region between Phe168 (EL2) and Ile268 (7.39) (∼4 Å, Figure 1B). Therefore, we predicted a higher affinity at the A3AR for those derivatives bearing a planar linker, such as 3a, 3e and 3f, compared to those with wider and out-of-plane linkers, such as 3b-d. In light of these considerations, compounds 3e and 3f would be the best alternatives to preserve the high affinity and selectivity of the arylethynyl group. However, because of the potential chemical and photochemical reactivity of a styryl group, we decided not to synthesize derivatives of compound 3e but to focus on triazolyl derivatives. In fact, as highlighted in Table 1, triazole series 3f seemed to be the best alternative to alkynyl series 3a, because of its stability, its rigid, planar and elongated C2 group (Figure S1, Supporting information) and its synthetic feasibility. The terminal phenyl rings of 3a and 3f are largely overlapping when docked to the A3AR. However, 3f is slightly more elongated than 3a (by 0.8 Å), and it was shown previously that rigid C2 substituents more extended than phenylethynyl are tolerated at the A3AR.7
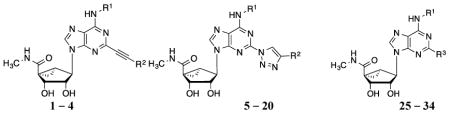
Nucleoside derivatives 5 - 20 (Table 2) were synthesized by the route shown in Scheme 1. The previous series of arylethynyl derivatives was prepared by a Sonogashira reaction15 of a substituted arylacetylene with a C2-iodo derivative of a (N)-methanocarba adenosine. Here we access the triazole derivatives via a click reaction, [3+2] cycloaddition of a congeneric set of 2-azidoadenosines (30 – 34) and arylacetylenes,16 which link adenine and C2-terminal aryl moieties (Scheme 1). The azide group is introduced on adenine via reaction of sodium azide with the corresponding 2-iodo intermediates (25 – 29), which were prepared as described from L-ribose.17 The product 2-(1,2,3-triazol-1-yl) nucleosides that were subsequently tested in binding assays were demonstrated using HPLC to be of high purity (>95%). Click chemistry of (N)-methanocarba adenosines was used previously to prepare various high affinity A3AR ligands.18
Table 2.
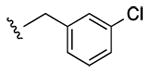
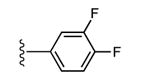
Structures and binding affinitiesa of AR agonists, including reference compounds 1-4, triazole derivatives 5-20 and their synthetic intermediates 25-34.
| |||||
|---|---|---|---|---|---|
| Compd. | R1 | R2 or R3 | A1AR% inhibition or Ki (nM)a | A2AAR% inhibitionc | A3AR% inhibition or Ki (nM)a |
| 1b,d |
|
|
6 ± 4% | 41 ± 10% | 3.49±1.84,3.08±0.23 (m) |
| 2b,d | CH3 |
|
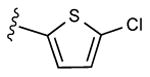
6 ± 1%, | 24 ± 13%, | 0.70 ± 0.11,36.1±4.7 (m) |
| 4b,d | CH3 |
|
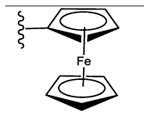
25 ± 2% | 47 ± 2% | 2.68 ± 0.44 |
| 5d | CH3 |
|
18 ± 4% | 35 ± 8% | 0.96 ± 0.07 |
| 6 | CH3 |
|
20 ± 5% | 31 ± 6% | 0.95 ± 0.50 |
| 7 | CH3 |
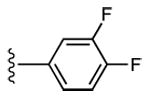
|
9 ± 4% | 39 ± 4% | 1.06 ± 0.10 |
| 8 | CH3 |
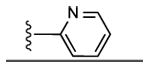
|
29 ± 7% | 26 ± 2% | 1.84 ± 0.38 |
| 9 | CH3 |
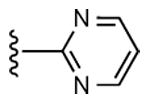
|
14 ± 7% | 27 ± 3% | 3.48 ± 0.97,334 ± 49 (m) |
| 10 | CH3 |
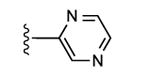
|
25 ± 10% | 8 ± 3% | 2.21 ± 0.34 |
| 11 | CH3 |
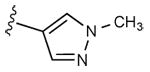
|
2 ± 2% | 1 ± 1% | 2.16 ± 0.32 |
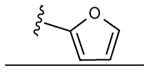
| 12 | CH3 |
|
43 ± 2% | 51 ± 1% | 0.96 ± 0.09 |
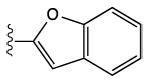
| 13 | CH3 |
|
12 ± 7% | 40 ± 6% | 1.25 ± 0.27 |
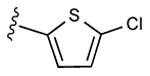
| 14 | CH3 |
|
27 ± 8% | 34 ± 5% | 0.73 ± 0.10 |
| 15d | C2H5 |
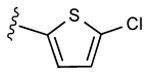
|
15 ± 11% | 32 ± 5% | 1.22 ± 0.26,9.64 ± 0.84 (m) |
| 16 |
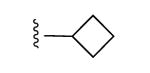
|
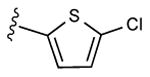
|
77% | ND | 7.05 ± 5.81,14.2 ± 0.2 (m) |
| 17 |
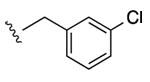
|
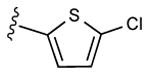
|
65% | ND | 9.02 ± 5.78,6.53 ± 0.58 (m) |
| 18 |
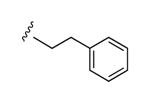
|
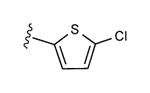
|
58% | ND | 6.66 ± 2.07 |
| 19 | CH3 |
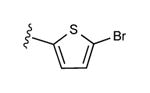
|
33 ± 7% | 7 ± 6% | 0.58 ± 0.17 |
| 20 | CH3 |
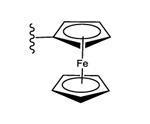
|
23 ± 3% | 54 ± 2% | 3.09 ± 0.21 |
| 25b | CH3 | I | ND | ND | 1.91 ± 0.85 |
| 26 | CH2CH3 | I | 1910 ± 300 | 11 ± 7% | 1.22 ± 0.21 |
| 27 |
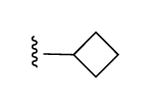
|
I | 93% | >10,000 | 1.53 ± 0.45 |
| 28b |
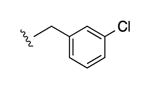
|
I | 2200e | >10,000 | 3.6 |
| 29 |
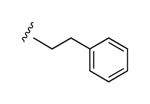
|
I | 68% | 42% | 0.91 ± 0.14 |
| 30 | CH3 | N3 | 77 ± 1% | 1 ± 1% | 0.54 ± 0.10 |
| 31 | CH2CH3 | N3 | 707 ± 152 | 11 ± 6% | 0.69 ± 0.08 |
| 32 |
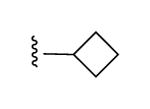
|
N3 | 100% | 46% | 0.85 ± 0.10 |
| 33 |
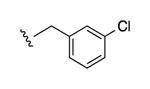
|
N3 | ND | 2770e | 1.08 ± 0.75 |
| 34 |
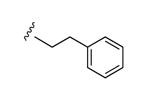
|
N3 | 93% | 1110e | 0.85 ± 0.10 |
Binding in membranes prepared from CHO or HEK293 (A2A only) cells stably expressing one of three hAR subtypes, unless noted. The binding affinity for hA1, A2A and A3ARs was expressed as Ki values (n = 3–4, unless noted), measured using agonist radioligands [3H]N6-R-phenylisopropyladenosine 35, [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamido-adenosine 36, or [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyl-uronamide 37, respectively. Additional values designated (m) are for mouse ARs. A percent in italics refers to inhibition of binding at 10 μM. Nonspecific binding was determined using 38 (10 μM). Values are expressed as the mean ± SEM. Ki values were calculated as reported.24
Percent of inhibition at 10 μM.
1, MRS5698; 2, MRS5980; 4, MRS5979; 5, MRS7110; 15, MRS7126; 17, MRS7138.
n = 1.
ND – not determined.
Scheme 1.

Synthesis of A3AR agonists containing triazolyl spacers between the C2 position of adenine and a terminal aryl group. For the structures of 5 - 20 see Table 2.
Radioligand binding assays were performed at three subtypes of hARs that are relevant for this chemical series and at the mouse (m) A3AR, which were stably expressed in mammalian cells, as previously reported (Table 2).7,8 An unusual ferrocene-ethynyl derivative 4 was included as a reference compound due to its prolonged in vivo activity and for comparison to its triazole analogue 20.8 Representative binding curves are shown in Supporting Information. As predicted using the 3D model of the hA3AR, the affinity and selectivity of the N6-methyl and N6-ethyl [1,2,3]triazolyl derivatives (5 – 15, 19, 20) were extremely high. All of the derivatives (5 – 34) displayed Ki values in the range of 0.3 – 12 nM at the A3AR, but larger N6 groups, i.e. 16 – 18, reduced selectivity by increasing A1AR affinity. The 2-iodo and 2-azido intermediates (25 – 34) were even less selective in comparison to the A1AR and A2AAR. Thus, the triazolyl spacer constitutes a suitable replacement for the ethynyl group with respect to the receptor binding profile. Also, a triazole group like an alkyne is uncharged at physiological pH. A set of A3AR-selective triazolyl derivatives in the 9-riboside series of adenosine agonists was reported previously,19 and this (N)-methanocarba series extends the finding of high A3AR affinity in the present set of analogues, and with comparable or greater selectivity. There is a correlation (r2 = 0.66) between the A3AR binding affinity of twelve N6-methyl C2-triazolyl derivatives reported here and the corresponding ethynyl derivatives (Figure 2). This is consistent with the hypothesis that the terminal aryl ring occupies a similar position in the two series when bound to the A3AR, as shown in Figure 2. Binding of selected compounds at the mA3AR indicated that a C2-triazolyl-5-chlorothien-2-yl group in compounds 15 – 17 maintained a relatively high affinity in the range of 7 – 14 nM with various N6 groups. Curiously, the C2-triazolyl-pyrimidin-2-yl derivative 9 was much less potent than other analogues at the mA3AR. The ligand efficiency20 with respect to A3AR affinity was favorable: 1, 0.23; 2, 0.33; 9, 0.27; 15, 0.31; 17, 0.24.
Figure 2.

Correlation of the binding affinities at the hA3AR of parallel series of triazolyl (Y-axis) and ethynyl (X-axis) derivatives.8 The values shown include those determined for triazolyl compounds 5 – 14, 19, and 20.
After confirming the A3AR-selectivity, the A3AR agonists were screened in vivo in the mouse CCI model.21 The protection by A3AR agonists against chronic neuropathic pain occurs at both central and peripheral sites of action.11 The methods used were as reported,7,8 and the agonist was given by oral gavage (3 μmol/kg) on day 7 after sciatic nerve ligation, i.e. the time when peak pain develops. The paw withdrawal threshold (PWT) was measured up to 5 h after drug administration. The maximal efficacy in vivo and the duration of action were indicated (Table 3). Each compound reached its peak protection against chronic neuropathic pain by 1 h after administration. In the earlier alkynyl series8 peak protection was achieved between 0.5 and 2 h after administration and maintained up to 3 h (as with 5-chlorothienyl derivative 2, which suppressed neuropathic pain to the degree of 83% protection at 3 h). By comparison, compound 1 at the same dose displayed only 24% protection after 3 h.8 Here, at 1 h after drug treatment the mechanoallodynia was a nearly completely reversed by some triazole derivatives, i.e. pyrimidin-2-yl 9, N-methylpyrazolyl 11, 5-chlorothien-2-yl 15 and 5-bromothien-2-yl 19 analogues and compounds 16 – 18. For N6-methyl analogues, the majority of the pain protection by the triazolyl derivatives waned by the 3 h time point (Figures 3A, B). In analogues containing the same terminal 5-chlorothiophene group found in very efficacious and long-acting agonist 2, an N6-ethyl group in 15 compared to the N6-methyl homologue 14 produced a higher maximal protection at 1 h, i.e. 85% vs. 67%, respectively. A 2-pyrimidyl group in 9 favored duration of action in vivo, with 36% of the protection remaining at 3 h, which would not have been predicted by its relatively weak mA3AR affinity. Larger N6 groups, e.g. 3-chlorobenzyl in 17 (>90% protection at 1 and 3 h), combined with a terminal chlorothienyl group greatly enhanced in vivo activity (Figure 3C). At a lower dose (1 μmol/kg), 17 produced the same duration of protection but the maximal effect reached a plateau at ∼70% (Figure S2, Supporting Information). N6-Cyclobutyl 16 and N6-(2-phenylethyl) 18 derivatives were also prolonged in activity.
Table 3.
Activity of orally administered A3AR agonists (3 μmol/kg) in CCI model of neuropathic pain (mechanoallodynia) in mice and physicochemical parameters.
| Compound | Max. effect Emax (%±SEM)a |
Effect at 3h (%±SEM) |
n | MW (D) |
cLogPb | tPSA (Å2) |
|---|---|---|---|---|---|---|
| 1 | 100 ± 0.0 | 23.7 ± 10.8 | 4 | 565 | 4.15 | 122 |
| 2 | 93.3 ± 6.7 | 82.8 ± 11.2 | 4 | 459 | 2.17 | 122 |
| 4 | 94.3 ± 5.7 | 78.1 ± 3.5 | 4 | 526 | 1.42 | 122 |
| 5 | 77.4±4.5 | 27.5±4.1 | 4 | 461 | 1.58 | 150 |
| 6 | 69.1±8.6 | 7.9±7.9 | 2 | 496 | 2.04 | 150 |
| 7 | 75.2±12.8 | 27.0±7.4 | 4 | 497 | 1.79 | 150 |
| 8 | 60.9±11.9 | 21.1±3.3 | 4 | 462 | 0.29 | 162 |
| 9 | 92.7±9.8 | 36.2±6.8 | 4 | 463 | -0.67 | 175 |
| 10 | 76.6±0.9 | 26.6±6.3 | 4 | 463 | -0.67 | 175 |
| 11 | 82.5±5.9 | 19.1±5.9 | 4 | 465 | -0.31 | 165 |
| 12 | 65.3±8.2 | 21.1±7.0 | 2 | 451 | 0.96 | 159 |
| 13 | 74.0±10.7 | 22.3±8.3 | 4 | 502 | 2.34 | 159 |
| 14 | 66.8±19.4 | 8.4±7.2 | 2 | 502 | 2.19 | 150 |
| 15 | 84.9±14.0 | 17.9±12.6 | 4 | 515 | 2.72 | 150 |
| 16 | 90.7±6.0 | 85.2±9.0 4 | 4 | 542 | 3.10 | 150 |
| 17 | 94.7±3.1 | 91.0±4.5 4 | 4 | 612 | 4.35 | 150 |
| 18 | 94.1±5.9 | 82.9±5.3 2 | 2 | 592 | 4.29 | 150 |
| 19 | 69.1±8.6 | 13.5±2.3 | 2 | 546 | 2.34 | 150 |
| 20 | 94.1±5.9 | 22.4±6.6 | 2 | 569 | 1.22 | 165 |
Percent represents reversal of reduction in PWT of the ipsilateral hind paw. The time of peak for compounds 5 – 20 is 1 h. Data for 1, 2 and 4 are from Tosh et al.8 No mechano-allodynia or A3AR agonist effect on PWT was observed on the contralateral side.
calculated using ChemBioDraw, v. 14.0.
Figure 3.

Protection against hind paw mechanoallodynia in mice resulting from CCI of the sciatic nerve. Triazolyl derivatives were administered at a single dose (p.o.) of 3 μmol/kg at the point of peak pain (7 days post surgery; representative curves of 2-4 determinations). A. Derivatives 5, 6 and 8 – 10, in which the terminal aryl group is a phenyl, substituted phenyl or six-membered heterocycle. B. Derivatives 7, 11, 13, 19 and 20, in which the terminal aryl group is a 3,4-difluorophenyl or five-membered heterocycle. C. Derivatives 16 – 18, with large N6 substitution and in which the terminal aryl group is 5-chlorothien-2-yl. Data are mean ± SEM of n=2 or 4 and analyzed by two-tailed, two-way ANOVA with Bonferroni comparisons to D0 or D7/BL. *P<0.05 vs. D0 and †P<0.05 vs. D7/BL.
There is some consistency of the in vivo results between triazole and ethynyl series. For example, a ferrocene group present in ethynyl derivative 4 and triazole 20 was associated with high efficacy in vivo, although in vitro stability tests indicated that 4 was subject to rapid degradation (t1/2 in human liver microsomes and pH 1.6 simulated stomach acid of 3.66 and 46.5 min, respectively).8 Therefore, we used mass spectrometry (Figures S3-S6, Supporting information) to identify the immediate, transient product of acidic decomposition of 4 upon exposure to aqueous HCl at pH 1.6. The result indicated a rapid loss of Fe-cyclopentadiene as detected within 5 min by mass, leaving a cyclopentadiene moiety remaining on the nucleoside (39, Chart 1B); however, 39 was highly unstable and unable to be isolated. The corresponding cyclopentadiene 40 in the triazole series was not detected upon incubation at pH 1.6 of 20, which was stable. Therefore, the substitution of the 2-alkyne spacer with triazole increased the chemical stability of the ferrocene derivative. This might result from greater delocalization of the ferrocene electron density in the direction of adenine moiety in the extended triazole-linked aromatic system, rendering the ferrocene moiety less readily protonated.
Off-target activities of several triazole derivatives (12, 14) were also determined in a broad receptor binding screen (Supporting Information).22 The only interactions at IC50 <10 μM were with H2 histamine (12, 50% inhibition at 10 μM) and sigma2 receptors (12, 50%; 14, 62%) and the peripheral benzodiazepine receptor (TSPO, 14, 54%). These few interactions reinforced the A3AR selectivity of these representative compounds.
Conclusions
Derivatives of selective A3AR agonists containing a triazole linker in place of an ethynyl group, a preferred substitution as predicted using molecular modeling, were prepared and shown to parallel the ethynyl series in receptor affinity and selectivity. Docking of possible linker moieties at the C2 position, using a molecular model of the A3AR, correctly predicted that the receptor affinity and selectivity would be maintained due to common structural features that favored selective binding at the A3AR. A planar C2-triazole linker fit well in a narrow cleft when docked in the receptor binding site. The effects of N6 substitution were also probed. All of these analogues contain a (N)-methanocarba ring system to optimize A3AR affinity, which improves the selectivity in comparison to the ribose series.5
The analogues were characterized in binding and in vivo assays to demonstrate efficacy in controlling chronic neuropathic pain in agreement with A3AR selectivity. An N6-ethyl group in 15 preserved in vivo activity compared to N6-methyl and binding selectivity, while larger groups reduced A3AR selectivity. In comparison to the activities of the corresponding 2-arylethynyl derivatives, the duration of action of many analogues tended to be shorter.8 Nevertheless, among N6-methyl derivatives a terminal pyrimidin-2-yl group in 9 extended the duration of action compared to other aryl groups. A highly prolonged and full efficacy in reversing mechanoallodynia was achieved in a few derivatives, i.e. 16 – 18 that contained larger N6 substitution. The most enhanced in vivo efficacy and duration was seen with N6-(3-chlorobenzyl derivative 17 (nearly full protection up to 4 h).
The use of X-ray structures of closely related GPCRs in homology modeling, docking and screening has facilitated understanding ligand recognition and drug discovery for the A3AR and for other receptors.23 Here, the effects of potential structural changes were probed computationally to select preferred structures to synthesize. The use of a phenotypic assay, i.e. the CCI model, provides much information on efficacy, duration of action and indirectly oral bioavailability. Thus, this study has progressed from the stage of structure-based design to a comparison of molecules having the desired pharmacological properties in in vitro and in vivo preclinical experiments. We have combined efficient methods to narrow the group of nucleoside derivatives of potential translational interest.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases, and NIH R01 HL077707). We thank Noel Whittaker (NIDDK) for mass spectral determinations. We thank Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health's Psychoactive Drug Screening Program (Contract # HHSN-271-2008-00025-C) for screening data.
Abbreviations
- AR
adenosine receptor
- cAMP
adenosine 3′,5′-cyclic monophosphate
- CCI
chronic constriction injury
- CHO
Chinese hamster ovary
- CNS
central nervous system
- DIC
N,N′-diisopropylcarbodiimide
- DMEM
Dulbecco's modified Eagle medium
- DMF
N,N-dimethylformamide
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- HEK
human embryonic kidney
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectroscopy
- MW
molecular weight
- NECA
5′-N-ethylcarboxamidoadenosine
- NMR
nuclear magnetic resonance
- PBS
phosphate buffered saline
- TSPO
translocator protein
- PWT
Paw Withdrawal Threshold
- RMS
root-mean-square
- SAR
structure-activity relationship
- TBAP
tetrabutylammonium dihydrogenphosphate
- TEA
triethylamine
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- TM
transmembrane helix
- tPSA
total polar surface area
Footnotes
Supporting information: Supplementary data (chemical synthesis, characterization data, calculations and results of in vivo experiments, modeling procedures and off-target screening) associated with this article can be found in the online version, at doi:.
Notes and references
- 1.Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. J Rheumatol. 2008;35:41. [PubMed] [Google Scholar]
- 2.Bar-Yehuda S, Stemmer SM, Madi L, Castel D, Ochaion A, Cohen S, Barer F, Zabutti A, Perez-Liz G, Del Valle L, Fishman P. Int J Oncol. 2008;33:287. [PubMed] [Google Scholar]
- 3.Borea PA, Varani K, Vincenzi F, Baraldi PG, Tabrizi MA, Merighi S, Gessi S. Pharmacol Rev. 2015;67:74. doi: 10.1124/pr.113.008540. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z, Janes K, Chen C, Doyle T, Tosh DK, Jacobson KA, Salvemini D. FASEB J. 2012;26:1855. doi: 10.1096/fj.11-201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tosh DK, Jacobson KA. Med Chem Comm. 2013;4:619. doi: 10.1039/C2MD20348K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobson KA, Ji XD, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. J Med Chem. 2000;43:2196. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. J Med Chem. 2012;55:4847. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tosh DK, Finley A, Paoletta S, Moss SM, Gao ZG, Gizewski E, Auchampach J, Salvemini D, Jacobson KA. J Med Chem. 2014;57:9901. doi: 10.1021/jm501021n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Little JW, Ford A, Symons-Liguori AM, Chen Z, Janes K, Doyle T, Xie J, Luongo L, Tosh DK, Maione S, Bannister K, Dickenson A, Vanderah TW, Porreca F, Jacobson KA, Salvemini D. Brain. 2015;138:28. doi: 10.1093/brain/awu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dal Ben D, Buccioni M, Lambertucci C, Marucci G, Volpini R, Cristalli G. Curr Med Chem. 2011;18:1444. doi: 10.2174/092986711795328391. [DOI] [PubMed] [Google Scholar]
- 11.Paoletta S, Tosh DK, Finley A, Gizewski E, Moss SM, Gao ZG, Auchampach JA, Salvemini D, Jacobson KA. J Med Chem. 2013;56:5949. doi: 10.1021/jm4007966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards PJ, Sturino C. Curr Med Chem. 2011;18:3116. doi: 10.2174/092986711796391714. [DOI] [PubMed] [Google Scholar]
- 13.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens R. Science. 2011;332:322. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366. [Google Scholar]
- 15.Chinchilla R, Nájera C. Chem Rev. 2007;107:87. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 16.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 17.Joshi BV, Melman A, Mackman RL, Jacobson KA. Nucleos Nucleot Nucleic Acids. 2008;27:279. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tosh DK, Chinn M, Yoo LS, Kang DW, Luecke H, Gao ZG, Jacobson KA. Bioorg Med Chem. 2010;18:508. doi: 10.1016/j.bmc.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cosyn L, Palaniappan KK, Kim SK, Duong HT, Gao ZG, Jacobson KA, Van Calenbergh S. J Med Chem. 2006;49:7373. doi: 10.1021/jm0608208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hopkins AL, Groom CR, Alex A. Drug Discov Today. 2004;9:430. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 21.Bennett GJ, Xie YK. Pain. 1988;33:87. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 22.Paoletta S, Tosh DK, Salvemini D, Jacobson KA. PLoS ONE. 2014;9:e97858. doi: 10.1371/journal.pone.0097858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mason JS, Bortolato A, Weiss DR, Deflorian F, Tehan B, Marshall FH. In Silico Pharmacology. 2013;1:23. doi: 10.1186/2193-9616-1-23. [DOI] [Google Scholar]
- 24.Cheng YC, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 25.Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Bioorg Med Chem Lett. 2008;18:2813. doi: 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.