

Abstract
Methylibium sp. strain T29 was isolated from a gasoline-contaminated aquifer and proved to have excellent capabilities in degrading some common fuel oxygenates like methyl tert-butyl ether, tert-amyl methyl ether and tert-butyl alcohol along with other organic compounds. Here, we report the draft genome sequence of M. sp. strain T29 together with the description of the genome properties and its annotation. The draft genome consists of 608 contigs with a total size of 4,449,424 bp and an average coverage of 150×. The genome exhibits an average G + C content of 68.7 %, and contains 4754 protein coding and 52 RNA genes, including 48 tRNA genes. 71 % of the protein coding genes could be assigned to COG (Clusters of Orthologous Groups) categories. A formerly unknown circular plasmid designated as pT29A was isolated and sequenced separately and found to be 86,856 bp long.
Keywords: Methylibium, Betaproteobacteria, Draft genome, Fuel oxygenates, Bioremediation
Introduction
Fuel oxygenates like MTBE, ETBE and TAME have been blended into gasoline for decades to boost octane ratings and to improve the efficiency of fuel combustion in engines. But being the most water-soluble components of gasoline they have simultaneously become some of the most frequently detected pollutants in groundwater posing a serious threat to drinking water supplies [1]. Moreover, recent studies have reported that they can be carcinogenic in humans [2], so remediation of the sites polluted with these compounds became an important issue. Several microbial consortia and individual bacterial strains were isolated so far being capable of their degradation to various extents [3, 4]. However, only a few of them were studied in detail and there are even fewer cases where the genetic and enzymatic background of the degradation is elucidated at least in some aspects.
Methylibium petroleiphilum PM1 was one of the first isolated individual MTBE-degrading strains originated from a compost-filled biofilter in Los Angeles, California, USA [5]. To date it is the only representative of the genus identified at the species level [6, 7]. During laboratory experiments it proved to have outstanding MTBE-degrading ability and it was tested in a bioaugmentation field study, too [8]. Afterwards, a number of bacteria closely related to M. petroleiphilum PM1 were detected based on 16S rDNA sequences at MTBE-contaminated sites at different geographic locations suggesting that the genus might have an important role in MTBE biodegradation [8, 9]. Later its complete genome sequence was published which revealed that besides the 4 Mb circular chromosome, M. petroleiphilum PM1 possesses a ~600 kb megaplasmid carrying the genes involved in MTBE degradation [10]. At present, no genome sequence information is available for other members of the Methylibium genus. As part of a French-Hungarian project aiming to characterize novel fuel oxygenate-degrading bacteria at the genomic level, we have isolated a novel Methylibium strain. The MTBE-degrading capacity of the strain was as high as the M. petroleiphilum PM1’s but some of its genetic and metabolic characteristics were found to be significantly different. Here we present the classification and features of Methylibium sp. T29 together with the description of the draft genome sequence and annotation compared to the reference strain M. petroleiphilum PM1.
Organism information
Classification and features
A novel potent MTBE-degrading bacterial strain designated as T29 was isolated from a mixed bacterial culture enriched from gasoline-contaminated groundwater samples collected from the area of Tiszaújváros, Hungary. The enrichment culture was supplemented with tert-butyl alcohol (TBA), one of the known key intermediates of MTBE biodegradation, as the sole carbon source. The strain was found to be able to utilize the following compounds provided as the sole carbon and energy sources: MTBE, TAME, TBA, 2-HIBA, benzene, methanol, ethanol, 1-propanol, 1-butanol, formate, piruvate and acetate, but cannot grow on ETBE, DIPE, n-alkanes, toluene, ethylbenzene, o-, m- and p-xylene, 2-propanol, acetone, formaldehyde, lactate, citrate and glucose. Strain T29 was routinely maintained in mineral salts medium (124 mg/l (NH4)2SO4, 50 mg/l MgSO4 · 7H2O, 12.5 mg/l CaCl2 · 2H2O, 350 mg/l KH2PO4, 425 mg/l K2HPO4, 1 mg/l FeSO4 · 7H2O, 1 mg/l CoCl2 · 6H2O, 1 mg/l MnSO4 · H2O, 1 mg/l ZnSO4 · 7H2O, 1 mg/l Na2MoO4 · 2H2O, 1 mg/l Na2WO4 · 2H2O, 0.25 mg/l NiCl2 · 6H2O, 0.1 mg/l H3BO3, 0.1 mg/l CuSO4 · 5H2O and 1.5 % agar if necessary) containing 200 mg/l MTBE or in ½ × TSB medium (8.5 g/l pancreatic digest of casein, 1.5 g/l papaic digest of soybean meal, 2.5 g/l NaCl, 1.25 g/l K2HPO4, 1.25 g/l glucose and 1.5 % agar if necessary) at 28 °C. Cells of strain T29 form pale yellow, shiny colonies on minimal agar plates and cream colored ones on ½ × TSA plates while secreting a brownish pigment molecule (Fig. 1, panel c) reminiscent of pyomelanin produced by certain Pseudomonas spp. and other strains belonging mainly to Gammaproteobacteria [11, 12]. Strain T29 stained Gram-negative and according to transmission electron micrographs (Fig. 1, panel a and b) the cell shape is coccobacillus. A smaller fraction of the cell population possesses a single polar flagellum (Fig. 1, panel b). Possible intracellular poly-β-hydroxyalkanoate granules (white spots) and possible protein inclusion bodies (dark spots) can also be observed.
Fig. 1.
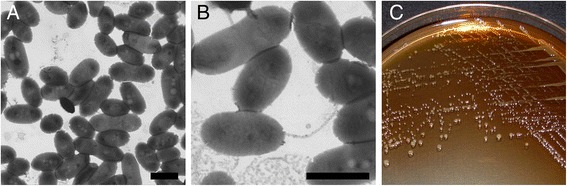
Transmission electron micrographs (a and b) and extracellular pigment production (c) of Methylibium sp. T29. For TEM examination the cells were suspended in 18 MΩ ultra-pure water, and 10 μl of the cell suspension was placed on carbon- and Formvar-coated 300 Mesh copper grids. Single 10 μl drops of 1 % (w/v) aqueous uranyl acetate were added to the grid for 15 s. The images were taken on a Hitachi S-4800 type (FEG) scanning electron microscope in transmission mode using 25 kV acceleration voltage. Scale bars represent 1 μm. The morphology of the cells is similar to M. petroleiphilum PM1’s [6]. While grown on ½ × TSA plates M. sp. T29 secreted a brownish pigment resembling pyomelanin produced by certain Pseudomonas spp
Initial taxonomic assignment of the strain was established by comparing its 16S ribosomal RNA gene sequence to the nonredundant Silva SSU Ref database [13, 14]. Phylogenetic analysis was conducted using MEGA 6 [15]. According to the phylogenetic analysis, strain T29 belongs to the genus Methylibium (Table 1). The closest relative of strain T29 is M. petroleiphilum PM1 (Fig. 2).
Table 1.
Classification and general features of Methylibium sp. strain T29 according to the MIGS recommendation [37]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [38] | |
| Phylum Proteobacteria | TAS [39] | ||
| Class Betaproteobacteria | TAS [40, 41] | ||
| Order Burkholderiales | TAS [41, 42] | ||
| Family Comamonadaceae | TAS [43, 44] | ||
| Genus Methylibium | TAS [6, 7] | ||
| Species Methylibium sp. | IDA | ||
| Strain T29 | IDA | ||
| Gram stain | Negative | IDA | |
| Cell shape | Coccobacillus | IDA | |
| Motility | Motile | IDA | |
| Sporulation | Not reported | NAS | |
| Temperature range | Mesophilic | IDA | |
| Optimum temperature | 28 °C | IDA | |
| pH range; Optimum | Not determined; routinely grown at pH 6.5 | IDA | |
| Carbon source | MTBE; TAME; TBA; methanol; ethanol | IDA | |
| MIGS-6 | Habitat | Soil; Groundwater | IDA |
| MIGS-6.3 | Salinity | Not reported | NAS |
| MIGS-22 | Oxygen requirement | Aerobic | IDA |
| MIGS-15 | Biotic relationship | Free living | NAS |
| MIGS-14 | Pathogenicity | Non-pathogenic | NAS |
| MIGS-4 | Geographic location | Tiszaújváros, Hungary | IDA |
| MIGS-5 | Sample collection | Nov-2010 | IDA |
| MIGS-4.1 | Latitude | 47.9179167 | IDA |
| MIGS-4.2 | Longitude | 21.0285667 | IDA |
| MIGS-4.4 | Altitude | 94 m | IDA |
aEvidence codes – IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [45]
Fig. 2.
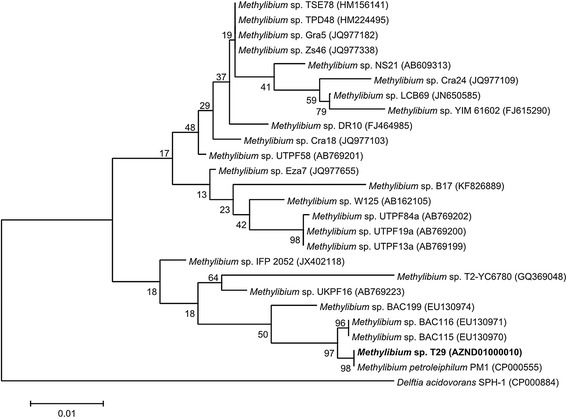
Dendrogram indicating the phylogenetic relationships of Methylibium sp. T29 relative to other Methylibium isolates. The maximum likelihood tree was inferred from 1329 aligned positions of the 16S rRNA gene sequences and derived based on the Tamura-Nei model using MEGA 6 [15]. Delftia acidovorans SPH-1 was used as an outlier. Bootstrap values (expressed as percentages of 1000 replicates) are shown at branch points. Bar: 0.01 substitutions per nucleotide position. The corresponding GenBank accession numbers are displayed in parentheses
Despite its close relatedness based on 16S rDNA sequences, the new strain differs from the type strain M. petroleiphilum PM1 in several aspects. For example, unlike M. petroleiphilum PM1, strain T29 is resistant to tetracycline, ampicillin [16] and mercury, and cannot grow on n-alkanes [10]. Moreover, PCR primers designed for mdpA and other known genes involved in MTBE degradation in M. petroleiphilum PM1 [17] failed to detect any related sequences in strain T29 suggesting that the genetic makeup of MTBE metabolism in this strain differs significantly from the one in M. petroleiphilum PM1. Pulsed field gel electrophoresis of restriction enzyme digested genomic DNA of strain T29 and M. petroleiphilum PM1 revealed major differences in the genomic sequences of the two strains (data not shown). Based on the evidences above, the new strain was named as Methylibium sp. T29.
Genome sequencing information
Genome project history
The genome of M. sp. T29 was sequenced by using Ion Torrent technology in our facility. The draft genome was assembled de novo using the overlap layout consensus methodology by the freely available software GS De Novo Assembler 2.9 (Roche). This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession number AZND00000000. The version described in this paper is AZND01000000. The plasmid pT29A was isolated and sequenced separately by the same technology. The assembly was performed by a different approach using SPAdes 3.0 [18]. The sequence was circularized and finished by manual editing. The full sequence of the plasmid pT29A is also available in GenBank under the accession number NC_024957.1.
Growth conditions and genomic DNA preparation
M. sp. T29 was isolated from a mixed bacterial culture enriched from gasoline-contaminated groundwater samples collected from the area of Tiszaújváros, Hungary, in November 2010. The strain was deposited into the National Collection of Agricultural and Industrial Microorganisms (NCAIM) [19] under the accession number NCAIM B.02561.
For genomic DNA preparation, bacteria were grown under aerobic conditions in a tightly sealed bottle at 28 °C for 14 days in mineral salts medium supplemented with 200 mg/l MTBE. Genomic DNA was isolated using UltraClean Microbial DNA Isolation Kit (MO BIO) according to the protocol provided by the manufacturer.
Genome sequencing and assembly
The genomic library was prepared using IonXpress Plus Fragment Library Kit (Life Technologies) and was sequenced using Ion PGM 200 Sequencing Kit v2 with an Ion Torrent PGM Sequencer. The raw data were processed using Torrent Suite 4.0.1. The number of usable reads was 3,100,682 with a total base number of 690,903,502. The mean read length was 222.82 ± 41.88 bp, the mode length was 243 bp. Contigs were built de novo using GS De Novo Assembler 2.9 (Roche). The assembly resulted in 608 contigs, the largest contig size was 98,303 bp, the minimum contig size was 505 bp. The half of the genome consists of contigs larger than 15,441 bp (N50). The average coverage was 150 × (Table 2).
Table 2.
Genome sequencing project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Draft |
| MIGS-28 | Libraries used | One 200 bp Ion Torrent library |
| MIGS-29 | Sequencing platforms | Ion Torrent PGM |
| MIGS-31.2 | Fold coverage | 150× |
| MIGS-30 | Assemblers | GS De Novo Assembler 2.9 |
| MIGS-32 | Gene calling method | Prodigal 2.6, Barrnap 0.3, Aragorn 1.2 (as part of Prokka 1.8) |
| Locus Tag | X551 | |
| Genbank ID | AZND00000000 | |
| Genbank Date of Release | 2014/02/20 | |
| GOLD ID | Gp0074688 | |
| BIOPROJECT | PRJNA229978 | |
| MIGS-13 | Source Material Identifier | SAMN02422539 |
| Project relevance | Environmental, biotechnology |
The pT29A plasmid was purified using a modified plasmid miniprep method [20] and treated with Plasmid-Safe™ ATP-dependent DNase (Epicentre) before sequencing with Ion Torrent technology using the kits mentioned above. 40,770 reads were obtained with a total base number of 8,500,697. The mean read length was 208.50 ± 51.50 bp, the mode length was 234 bp. The reads were assembled into an 86,856 bp circular sequence with SPAdes 3.0 [18] and manual editing.
Genome annotation
The assembled draft genome and the pT29A sequences were annotated using Prokka 1.8 [21]. For the prediction of signal peptides and transmembrane domains SignalP 4.1 Server [22, 23] and TMHMM Server v. 2.0 [24] were used, respectively. Assignment of genes to the COG database [25, 26] and Pfam domains [27] was performed with WebMGA server [28].
Genome properties
The total size of the draft genome of M. sp. T29 is 4,449,424 bp and has a G + C content of 68.7 % which is similar to the genome of the type strain M. petroleiphilum PM1 (4,643,669 bp, G + C content of 67.6 %). For M. sp. T29 a total of 4806 genes, whilst for M. petroleiphilum PM1 4477 genes were predicted. 3 rRNA, 48 tRNA and 1 tmRNA genes were detected in the genome of M. sp. T29. We could make functional prediction for 72.8 % of the protein coding genes, while the rest were named as hypothetical proteins. Of the coding genes, 71 % could be assigned to COG categories and 71.4 % has Pfam domains (for detailed statistics see Tables 3 and 4). The map of the draft genome of M. sp. T29 aligned to the full genome of the closest relative M. petroleiphilum PM1 is illustrated in Fig. 3 and Fig. 4. The plasmid pT29A carries 90 protein coding genes, of which 72.2 % has functional prediction and 70 % could be assigned to COG categories (Table 5). The most abundant functional category was the coenzyme transport and metabolism (Table 6). The map of the plasmid is shown in Fig. 5.
Table 3.
Genome statistics
| Attribute | Value | %age of total |
|---|---|---|
| Genome size (bp) | 4,449,424 | 100 |
| DNA coding (bp) | 3,743,112 | 84.1 |
| DNA G + C (bp) | 3,057,506 | 68.7 |
| DNA scaffolds | 608 | n/a |
| Total genes | 4806 | n/a |
| Protein coding genes | 4754 | 98.9 |
| RNA genes | 52 | 1.1 |
| Pseudo genes | 196 | 4.1 |
| Genes in internal clusters | N.D. | N.D. |
| Genes with function prediction | 3498 | 72.8 |
| Genes assigned to COGs | 3376 | 71.0 |
| Genes with Pfam domains | 3395 | 71.4 |
| Genes with signal peptides | 381 | 8.0 |
| Genes with transmembrane helices | 1014 | 21.3 |
| CRISPR repeats | 0 | 0 |
Table 4.
Number of genes associated with general COG functional categories in the whole genome
| Code | Value | %age | Description |
|---|---|---|---|
| J | 169 | 3.5 | Translation, ribosomal structure and biogenesis |
| A | 2 | 0.0 | RNA processing and modification |
| K | 276 | 5.8 | Transcription |
| L | 190 | 4.0 | Replication, recombination and repair |
| B | 4 | 0.1 | Chromatin structure and dynamics |
| D | 32 | 0.7 | Cell cycle control, Cell division, chromosome partitioning |
| V | 59 | 1.2 | Defense mechanisms |
| T | 284 | 6.0 | Signal transduction mechanisms |
| M | 218 | 4.6 | Cell wall/membrane biogenesis |
| N | 100 | 2.1 | Cell motility |
| U | 122 | 2.6 | Intracellular trafficking and secretion |
| O | 170 | 3.6 | Posttranslational modification, protein turnover, chaperones |
| C | 292 | 6.1 | Energy production and conversion |
| G | 126 | 2.6 | Carbohydrate transport and metabolism |
| E | 295 | 6.2 | Amino acid transport and metabolism |
| F | 72 | 1.5 | Nucleotide transport and metabolism |
| H | 196 | 4.1 | Coenzyme transport and metabolism |
| I | 177 | 3.7 | Lipid transport and metabolism |
| P | 236 | 5.0 | Inorganic ion transport and metabolism |
| Q | 118 | 2.5 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 456 | 9.6 | General function prediction only |
| S | 337 | 7.1 | Function unknown |
| - | 823 | 17.3 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Fig. 3.
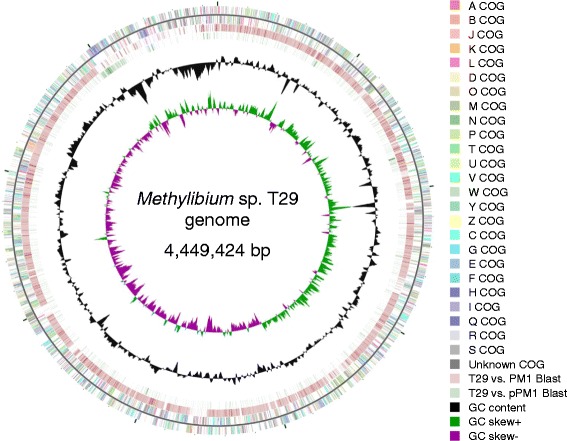
Circular representation of the draft genome of Methylibium sp. T29 displaying relevant genome features. The contigs of M. sp. T29 were reordered by Mauve [35] using the genome sequence of M. petroleiphilum PM1 as the reference. The COG categories were assigned to genes by WebMGA [28]. The circular map was visualized by CGView [36]. The features are the following from outside to center: (A) genes on forward strand; genes on reverse strand (colored by COG categories); blast alignment of the M. petroleiphilum PM1 chromosome and megaplasmid to the draft genome of M. sp. T29; GC content; GC skew
Fig. 4.
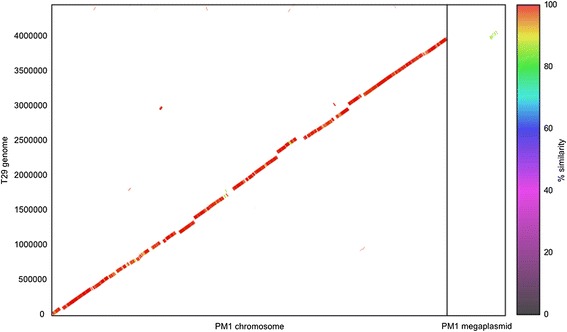
Genome sequence similarity plot of Methylibium sp. T29 and Methylibium petroleiphilum PM1. Contigs from the draft genome assembly of M. sp. T29 were reordered with Mauve 2.3.1 [35] using the complete genome of M. petroleiphilum PM1 as the reference. The alignment and plotting were performed with MUMmer 3.0 [29]
Table 5.
Statistics for the pT29A plasmid
| Attribute | Value | %age of total |
|---|---|---|
| Genome size (bp) | 86,856 | n.a. |
| DNA coding (bp) | 75,837 | 87.3 |
| DNA G + C (bp) | 58,265 | 67.1 |
| DNA scaffolds | 1 | 100.0 |
| Total genes | 90 | 100.0 |
| Protein coding genes | 90 | 100.0 |
| RNA genes | 0 | 0.0 |
| Pseudo genes | 1 | 1.1 |
| Genes in internal clusters | N.D. | N.D. |
| Genes with function prediction | 65 | 72.2 |
| Genes assigned to COGs | 63 | 70.0 |
| Genes with Pfam domains | 67 | 74.4 |
| Genes with signal peptides | 12 | 13.3 |
| Genes with transmembrane helices | 17 | 18.9 |
| CRISPR repeats | 0 | 0.0 |
Table 6.
Number of genes associated with general COG functional categories in the pT29A plasmid genome
| Code | Value | %age | Description |
|---|---|---|---|
| J | 0 | 0.0 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.0 | RNA processing and modification |
| K | 8 | 8.9 | Transcription |
| L | 10 | 11.1 | Replication, recombination and repair |
| B | 4 | 0.1 | Chromatin structure and dynamics |
| D | 1 | 1.1 | Cell cycle control, Cell division, chromosome partitioning |
| V | 0 | 0.0 | Defense mechanisms |
| T | 7 | 7.8 | Signal transduction mechanisms |
| M | 0 | 0.0 | Cell wall/membrane biogenesis |
| N | 0 | 0.0 | Cell motility |
| U | 0 | 0.0 | Intracellular trafficking and secretion |
| O | 0 | 0.0 | Posttranslational modification, protein turnover, chaperones |
| C | 3 | 3.3 | Energy production and conversion |
| G | 0 | 0.0 | Carbohydrate transport and metabolism |
| E | 1 | 1.1 | Amino acid transport and metabolism |
| F | 0 | 0.0 | Nucleotide transport and metabolism |
| H | 19 | 21.1 | Coenzyme transport and metabolism |
| I | 0 | 0.0 | Lipid transport and metabolism |
| P | 5 | 5.6 | Inorganic ion transport and metabolism |
| Q | 0 | 0.0 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 4 | 4.4 | General function prediction only |
| S | 10 | 11.1 | Function unknown |
| - | 22 | 24.4 | Not in COGs |
The total is based on the total number of protein coding genes in the plasmid genome
Fig. 5.
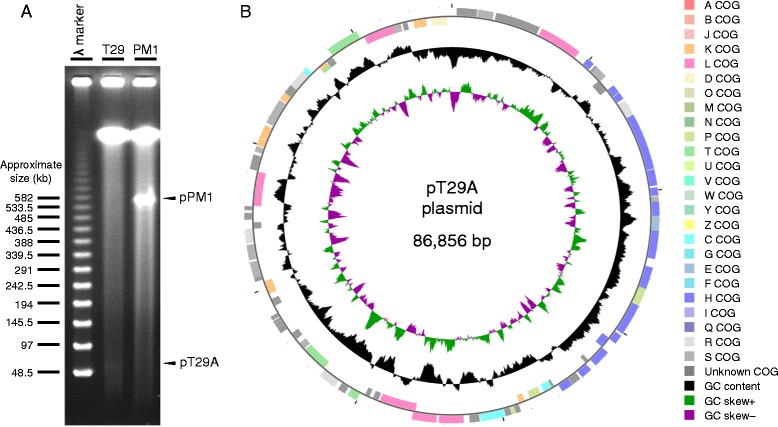
Detection and features of the pT29A plasmid. a Separation of megaplasmids of M. petroleiphilum PM1 and M. sp. T29 by pulsed field gel electrophoresis. The experiment was conducted according to Barton et al. [30]. The arrows show the ~600 kb partially linearized megaplasmid of M. petroleiphilum PM1 described in [10], and the ~87 kb partially linearized pT29A plasmid described in this paper. b Circular representation of the pT29A plasmid of M. sp. T29 displaying relevant features. The circular map was visualized by CGView [36]. The features are the following from outside to center: genes on forward strand, genes on reverse strand (colored by COG categories), GC content and GC skew
Conclusions
On average, the draft genome of M. sp. T29 shows 97 % identity to the M. petroleiphilum PM1 chromosome and 85 % identity to a small part of the M. petroleiphilum PM1 megaplasmid at the nucleotide level as measured by NUCmer [29] (Fig. 4) but significant differences were also found. Notably, most parts of the 600 kb megaplasmid are missing from M. sp. T29. A pulsed field gel electrophoretic analysis to detect megaplasmids [30] revealed that unlike M. petroleiphilum PM1 our isolate does not harbor the megaplasmid which carries the genes for MTBE-degradation [10]. Instead, a ~87 kb plasmid is present (Fig. 5) that we named pT29A.
The fact that in M. petroleiphilum PM1 the genes for MTBE-metabolism are located on the pPM1 megaplasmid suggested that in M. sp. T29 these genes are also carried by the pT29A plasmid. Surprisingly, no known genes associated with MTBE-degradation were found among the plasmid coded genes besides a cobalamin-synthesis operon which differs from the one in M. petroleiphilum PM1. Cobalt ions or cobalamin are required for complete MTBE-degradation in some strains for the utilization of 2-HIBA which is a key intermediate in the metabolic pathway [31, 32]. However, we were able to identify the putative components of the MTBE-degradation pathway in the whole genome of the M. sp. T29 including orthologous genes coding for the MTBE monooxygenase [16] and the TBA monooxygenase [33] showing only 84 and 81 % identity at the amino acid level to their M. petroleiphilum PM1 counterparts, respectively (Table 7). As opposed to the considerably high similarity of the majority of the two genomes, the significantly lower sequence conservation of the MTBE-degradation pathway components and the fact that these genes are not linked to the pT29A plasmid indicate that the gene cluster for MTBE-metabolism is probably located on a transposon which resides on the megaplasmid and the chromosome in M. petroleiphilum PM1 and M. sp. T29, respectively. There are unique sequences in the M. sp. T29 genome missing from M. petroleiphilum PM1 conferring different functions, i.e. resistances to different antibiotics (ampicillin, meticillin, tetracycline, sulfonamide), heavy metals (mercury, copper, cobalt, nickel, zinc, cadmium, tellurium) and other toxic compounds (i.e. arsenic). Other unique sequences code for various metabolic enzymes, transcriptional regulators, sensor proteins, components of restriction modification systems, phage- and transposon-related proteins and hypothetical proteins. The MTBE monooxygenase function for the candidate gene mdpA and the resistances to ampicillin, tetracycline and mercury were verified experimentally. According to the gene annotations, M. sp. T29 can utilize other environmentally polluting compounds as well (i.e. chlorinated aromatic hydrocarbons, haloacids and certain polycyclic aromatic hydrocarbons) but these functions have not been tested yet. The organism was predicted as non-human pathogen (probability of being a human pathogen is 0.083) by PathogenFinder 1.1 [34], therefore it can be safely applied during in situ bioremediation experiments. Based on the genome sequence described here we designed PCR primers specific to the M. sp. T29-type mdpA to track our strain in the field at MTBE-contaminated sites in Hungary. The nucleotide sequences of other genes in the MTBE-degradation pathway can also be used to construct better oligonucleotide chips to detect the potentially active genes in environmental samples.
Table 7.
Genes involved in the degradation of MTBE in Methylibium petroleiphilum PM1 and Methylibium sp. T29
| Gene function | Gene ID in M. petroleiphilum PM1 | Gene ID in M. sp. T29 | %age identity at the nucleic acid level | %age identity at the amino acid level |
|---|---|---|---|---|
| MTBE monooxygenase | Mpe_B0606 | X551_03232 | 79 | 84 |
| Rubredoxin | Mpe_B0602 | X551_03234 | no significant similarity | 43 |
| Rubredoxin reductase | Mpe_B0597 | X551_01331 | no significant similarity | 29 |
| ATP-dependent transcriptional regulator | Mpe_B0601 | X551_04638 | 74 | 85 |
| Hydroxymethyl tert-butyl ether dehydrogenase | Mpe_B0558 | X551_02800 | 86 | 91 |
| tert-butyl formate carboxylesterase | Mpe_A2443 | X551_01122 | 99 | 99 |
| tert-butyl alcohol hydroxylase | Mpe_B0555 | X551_02402 | 79 | 81 |
| Iron-sulfur oxidoreductase | Mpe_B0554 | X551_02401 | 82 | 82 |
| 2-methyl-2-hydroxy-1-propanol dehydrogenase | Mpe_B0561 | X551_02804 | 83 | 85 |
| Hydroxyisobutyraldehyde dehydrogenase | Mpe_A0361 | X551_03863 | Partial homology | 36 |
| 2-hydroxy-isobutyryl-CoA ligase | Mpe_B0539 | X551_02557 | 85 | 94 |
| 2-hydroxy-isobutyryl-CoA mutase | Mpe_B0541 | X551_02559 | 89 | 92 |
| 2-hydroxy-isobutyryl-CoA mutase C-terminal domain | Mpe_B0538 | X551_02556 | 86 | 91 |
| 3-hydroxybutyryl-CoA dehydrogenase | Mpe_B0547 | X551_02564 | 79 | 84 |
| Acetyl-CoA acetyltransferase | Mpe_A3367 | X551_00431 | Partial homology | 45 |
Acknowledgements
This work has been funded by the Hungarian National Development Agency and was conducted as part of the MiOxyFun project: “Biodegradability of fuel oxygenates (ETBE and MTBE): Microorganisms - Monooxygenases - Functionality (TÉT_10-1-2011-0376)”.
Abbreviations
- MTBE
Methyl tert-butyl ether
- ETBE
Ethyl tert-butyl ether
- TAME
Tert-amyl methyl ether
- TBA
Tert-butyl alcohol
- 2-HIBA
2-hydroxyisobutyric acid
- DIPE
Diisopropyl ether
- TSA
Tryptic soy agar
- TSB
Tryptic soy broth
Footnotes
Zsolt Szabó and Péter Gyula contributed equally to this work
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ZS isolated the strain, performed the metabolic characterization and all the microbiological work and significantly contributed to the writing of the manuscript. PG carried out the molecular characterization and all the bioinformatic analysis including phylogenetic analysis, the genome assembly, annotation, functional genome analysis and finding the components of the MTBE-degradation pathway. He is also a major contributor to writing of the manuscript. HR and EB carried out the sample preparation, the genome sequencing and quality control of the data. BG participated in the genome comparison analysis. P Pach coordinated and supervised the bioinformatic analysis. P Pekker performed the electron microscopy experiments. IP and ZB were the supervisors of the project and were responsible for finishing the manuscript. All authors read and approved the final version of the manuscript.
References
- 1.Johnson R, Pankow J, Bender D, Price C, Zogorski J. MTBE - To what extent will past releases contaminate community water supply wells? Environ Sci Technol. 2000;34:210A–7. doi: 10.1021/es003268z. [DOI] [PubMed] [Google Scholar]
- 2.Burns KM, Melnick RL. MTBE: recent carcinogenicity studies. Int J Occup Env Heal. 2012;18:66–9. doi: 10.1179/107735212X13293200778947. [DOI] [PubMed] [Google Scholar]
- 3.Fayolle F, Vandecasteele JP, Monot F. Microbial degradation and fate in the environment of methyl tert-butyl ether and related fuel oxygenates. Appl Microbiol Biotechnol. 2001;56:339–49. doi: 10.1007/s002530100647. [DOI] [PubMed] [Google Scholar]
- 4.Hyman M. Biodegradation of gasoline ether oxygenates. Curr Opin Biotechnol. 2013;24:443–50. doi: 10.1016/j.copbio.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Hanson JR, Ackerman CE, Scow KM. Biodegradation of methyl tert-butyl ether by a bacterial pure culture. Appl Environ Microbiol. 1999;65:4788–92. doi: 10.1128/aem.65.11.4788-4792.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakatsu CH, Hristova K, Hanada S, Meng XY, Hanson JR, Scow KM, Kamagata Y. Methylibium petroleiphilum gen. nov., sp. nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. Int J Syst Evol Microbiol. 2006;56:983–9. doi: 10.1099/ijs.0.63524-0. [DOI] [PubMed] [Google Scholar]
- 7.Stackebrandt E, Verbarg S, Frühling A, Busse HJ, Tindall BJ. Dissection of the genus Methylibium: reclassification of Methylibium fulvum as Rhizobacter fulvus comb. nov., Methylibium aquaticum as Piscinibacter aquaticus gen. nov., comb. nov. and Methylibium subsaxonicum as Rivibacter subsaxonicus gen. nov., comb. nov. and emended descriptions of the genera Rhizobacter and Methylibium. Int J Syst Evol Microbiol. 2009;59:2552–60. doi: 10.1099/ijs.0.008383-0. [DOI] [PubMed] [Google Scholar]
- 8.Smith AE, Hristova K, Wood I, Mackay DM, Lory E, Lorenzana D, Scow KM. Comparison of biostimulation versus bioaugmentation with bacterial strain PM1 for treatment of groundwater contaminated with methyl tertiary butyl ether (MTBE) Environ Health Perspect. 2005;113:317–22. doi: 10.1289/ehp.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hristova K, Gebreyesus B, Mackay D, Scow KM. Naturally occurring bacteria similar to the methyl tert-butyl ether (MTBE)-degrading strain PM1 are present in MTBE-contaminated groundwater. Appl Environ Microbiol. 2003;69:2616–23. doi: 10.1128/AEM.69.5.2616-2623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kane SR, Chakicherla AY, Chain PSG, Schmidt R, Shin MW, Legler TC, Scow KM, Larimer FW, Lucas SM, Richardson PM, Hristova KR. Whole-genome analysis of the methyl tert-butyl ether-degrading beta-proteobacterium Methylibium petroleiphilum PM1. J Bacteriol. 2007;189:1931–45. doi: 10.1128/JB.01259-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yabuuchi E, Ohyama A. Characterization of “pyomelanin”-producing strains of Pseudomonas aeruginosa. Int J Syst Bacteriol. 1972;22:53–64. doi: 10.1099/00207713-22-2-53. [DOI] [Google Scholar]
- 12.Turick CE, Knox AS, Becnel JM, Ekechukwu AA, Milliken CE. Properties and function of pyomelanin. In: Elnashar MM, editor. Biopolymers. Rijeka: Sciyo; 2010. pp. 449–72. [Google Scholar]
- 13.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.SILVA SSU Ref NR (Non-Redundant) Database [http://www.arb-silva.de/projects/ssu-ref-nr/]
- 15.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt R, Battaglia V, Scow K, Kane S, Hristova KR. Involvement of a novel enzyme, MdpA, in methyl tert-butyl ether degradation in Methylibium petroleiphilum PM1. Appl Environ Microbiol. 2008;74:6631–8. doi: 10.1128/AEM.01192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopes Ferreira N, Malandain C, Fayolle-Guichard F. Enzymes and genes involved in the aerobic biodegradation of methyl tert-butyl ether (MTBE) Appl Microbiol Biotechnol. 2006;72:252–62. doi: 10.1007/s00253-006-0494-3. [DOI] [PubMed] [Google Scholar]
- 18.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.National Collection of Agricultural and Industrial Microorganisms (NCAIM) [http://ncaim.uni-corvinus.hu]
- 20.Heringa SD, Monroe JD, Herrick JB. A simple, rapid method for extracting large plasmid DNA from bacteria. Nature Precedings 2007 doi:10.1038/npre.2007.1249.1.
- 21.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 22.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 23.SignalP 4.1 Server [http://www.cbs.dtu.dk/services/SignalP/]
- 24.TMHMM Server v. 2.0: Prediction of transmembrane helices in proteins [http://www.cbs.dtu.dk/services/TMHMM/]
- 25.COGs - Clusters of Orthologous Groups [http://www.ncbi.nlm.nih.gov/COG/]
- 26.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer ELL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–22. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu S, Zhu Z, Fu L, Niu B, Li W. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics. 2011;12:444. doi: 10.1186/1471-2164-12-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barton BM, Harding GP, Zuccarelli AJ. A general method for detecting and sizing large plasmids. Anal Biochem. 1995;226:235–40. doi: 10.1006/abio.1995.1220. [DOI] [PubMed] [Google Scholar]
- 31.François A, Mathis H, Godefroy D, Piveteau P, Fayolle F, Monot F. Biodegradation of methyl tert-butyl ether and other fuel oxygenates by a new strain, Mycobacterium austroafricanum IFP 2012. Appl Environ Microbiol. 2002;68:2754–62. doi: 10.1128/AEM.68.6.2754-2762.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohwerder T, Breuer U, Benndorf D, Lechner U, Müller RH. The alkyl tert-butyl ether intermediate 2-hydroxyisobutyrate is degraded via a novel cobalamin-dependent mutase pathway. Appl Environ Microbiol. 2006;72:4128–35. doi: 10.1128/AEM.00080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuster J, Schäfer F, Hübler N, Brandt A, Rosell M, Härtig C, Harms H, Müller RH, Rohwerder T. Bacterial degradation of tert-amyl alcohol proceeds via hemiterpene 2-methyl-3-buten-2-ol by employing the tertiary alcohol desaturase function of the Rieske nonheme mononuclear iron oxygenase MdpJ. J Bacteriol. 2012;194:972–81. doi: 10.1128/JB.06384-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cosentino S, Larsen MV, Aarestrup FM, Lund O. PathogenFinder - Distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0077302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36:W181–4. doi: 10.1093/nar/gkn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, Ashburner M, Axelrod N, Baldauf S, Ballard S, Boore J, Cochrane G, Cole J, Dawyndt P, De Vos P, dePamphilis C, Edwards R, Faruque N, Feldman R, Gilbert J, Gilna P, Glöckner FO, Goldstein P, Guralnick R, Haft D, Hancock D, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2. Part B. 2. New York: Springer; 2005. p. 1. [Google Scholar]
- 40.Garrity GM, Bell JA, Lilburn T. Class II. Betaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2. Part C. 2. New York: Springer; 2005. p. 575. [Google Scholar]
- 41.Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006;56:1–6. doi:10.1099/ijs.0.64188-0. [DOI] [PubMed]
- 42.Garrity GM, Bell JA, Lilburn T. Bergey's Manual of Systematic Bacteriology. Volume 2. Part C. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2. Part C. 2. New York: Springer; 2005. p. 575. [Google Scholar]
- 43.Willems A, De Ley J, Gillis M, Kersters K. Comamonadaceae, a new family encompassing the acidovorans ribosomal RNA complex, including Variovorax paradoxus gen. nov., comb. nov., for Alcaligenes paradoxus (Davis 1969) Int J Syst Bacteriol. 1991;41:445–50. doi: 10.1099/00207713-41-3-445. [DOI] [Google Scholar]
- 44.Willems A, Gillis M. Family IV. Comamonadaceae Willems, De Ley, Gillis and Kersters 1991a, 447VP. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2. Part C. 2. New York: Springer; 2005. pp. 686–8. [Google Scholar]
- 45.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]