

Abstract
G protein-coupled estrogen receptor (GPER) has been shown to be important in several disease states such as estrogen sensitive cancers. While several selective ligands have been identified for the receptor, little is known about how they interact with GPER and how their structures influence their activity. Specifically, within one series of ligands, whose structure varied only at one position, the replacement of a hydrogen atom with an acetyl group changed a potent antagonist into a potent agonist. In this study, two GPER homology models were constructed based on the x-ray crystal structures of both the active and inactive β2-adrenergic receptors (β2AR) in an effort to characterize the differences of binding modes between agonists and antagonists to the receptor, and to understand their activity in relation to their structures. The knowledge attained in this study is expected to provide valuable information on GPER ligands structure activity relationship to benefit future rational design of potent agonists and antagonists of the receptor for potential therapeutic applications.
Keywords: GPER, homology modeling, agonism, antagonism
Introduction
Recent advances in G protein-coupled receptor (GPCR) structural biology have provided a wealth of knowledge stemming from a dramatic increase in the number of diverse GPCR crystal structures, especially agonist bound and activated ones. [1–19] The latter is of special significance because there is still much uncertainty about the exact activation mechanism of GPCRs. Traditionally, GPCRs were thought of being in one of two states, either an inactive or an active state. However, recent biophysical evidence seems to support the hypothesis that GPCRs may have multiple active conformational substates.[20] For example, several available crystal structures of agonist-bound GPCRs have shown significant conformational variety.[21]
A number of key observations about the active state of GPCRs have been derived from the available agonist-bound crystal structures. When comparing the inactive and active state crystal structures of the β2-adrenergic receptor (β2AR, PDB codes: 2RH1 and 3SN6 respectively), upon activation there were several movements in the transmembrane helices and changes in residue interactions.[7,22] Notably, there was a conformation rearrangement between helix 5 and 7, and intracellularly, an outward movement of helix 6.[2] Concurrently, the ionic lock between D/E6.30 and R3.50 in the conserved DRY sequence was interrupted along with movement of W6.48 (“toggle switch”) from TM7 toward TM5 (amino acids represented in the Ballesteros-Weinstein nomenclature).[1,2,21–24] However, such observations were not commonly seen for all activated GPCR crystal structures due to a variety of factors such as varied crystallization techniques.
The recent advances in GPCR crystal structures allow for a better understanding of inactive and active state differences, which can also be beneficial to GPCR homology modeling studies. By constructing the conformation of both the inactive and active state of a receptor, a direct comparison between states can be conducted, and in turn help illustrate critical ligand-receptor interaction to facilitate ligand design. In this report, such an analysis was pursued with the G protein-coupled estrogen receptor (GPER, formally GPR30) in order to elucidate how it interacts with its selective ligands, particularly its agonists.
GPER was first discovered due to its overexpression in several breast cancer cell lines and the rapid non-genomic signaling that estrogens produced but could not be explained through their interaction with either estrogen receptor α or β (ERα or ERβ).[25–27] While its exact functions are still being elucidated, it has been shown to be involved in several body systems and have clinical relevance in breast cancer diagnosis.[27–42] Recently, its endogenous ligand 17β-estradiol (Figure 1) was used as a starting point for virtual screening in which three selective and high affinity ligands based on the same “G-scaffold” were identified (Figure 1).[43–45] The first one, G-1, was found to be an agonist with a Ki of 11 nM for GPER and no significant binding to ERα or ERβ.[43] Later, two antagonists, G-15 and G-36, carrying similar affinity and selectivity were reported.[44,45] All three ligands share the same scaffold and only differ in their substitutions on the 6-position of the tetrahydroquinoline ring. Another endogenous ERα agonist, estriol (Figure 1), has been shown to also bind to GPER as an antagonist instead.[42,46,47] However, it only binds to GPER with a much lower binding affinity compared to 17β-estradiol.[42] Homology modeling study (using bovine rhodopsin as the template) of GPER and docking efforts have shown that estriol may bind to GPER in a similar manner as 17β-estradiol does.[46,48,49]
Figure 1.

Chemical structures of known GPER ligands and their binding affinity to the receptor.
Since no crystal structure of GPER is available, homology modeling has to be relied on for molecular modeling studies, and thus far, the homology models reported are all based on the bovine rhodopsin crystal structure.[46,48,49] A putative binding pocket consisting of four polar residues and ten hydrophobic residues was proposed and correlated well with experimental data.[46] While these models have been useful for preliminary study of ligand-GPER interactions they represent mainly the inactive state of the receptor and therefore may not be able to fully explain agonist interactions with GPER.
In order to explore the structure activity relationship of GPER ligands, including agonists and antagonists, both inactive and active homology models of GPER were constructed based on the inactive (antagonist-bound) and active (agonist-bound) crystal structures of β2AR (2RH1 and 3SN6 respectively). β2AR was preliminary chosen because it shares higher homology with GPER compared to bovine rhodopsin and has been crystallized in both its active and inactive forms. Once the homology models were optimized, ligands were docked into both models and analyzed with the final objective of clarifying how the seemingly minor difference in ligand structure between G-1, G-15, and G-36 may induce their distinct functional activity.
Computational Methods
Sequence alignment and model building
All molecular modeling was collected using the SYBYL-X 1.3 molecular modeling package (Tripos LP, St. Louis, MO) on dual-core AMD Opteron(tm) 2.4 GHz processors. The amino acid sequence of G protein estrogen receptor 1 (GPER) was obtained from UniProtKB/Swiss-Prot (Q99527). The amino-acid sequences of all the currently available GPCR crystal structures were collected and compared to the sequence of GPER using ClustalX 2.0.[50] Within ClustalX a multiple alignment was performed with a gap opening penalty of 15 using the BLOSUM protein weight matrix series. Additional manual adjustment of the multiple sequence alignment was done to eliminate any gaps in transmembrane helices and to align disulfide-bond forming cysteine residues in extracellular loop 2 (ECL2) and transmembrane helix 3 (TM3). Both overall homology levels and homology between individual regions were used to select the most appropriate template (Table 1). β2AR was chosen as the template structure of choice and an inactive and active crystal structures were chosen (PDB codes: 2RH1 and 3SN6) respectively. Sequence alignment between GPER and β2AR was further optimized based on the most conserved residues among most GPCRs and used for model construction for both the inactive and active models. The comparative modeling software, MODELLER 9v8, was used to generate 100 homology models for each state using the default parameters.[51]
Table 1.
Regional homology between GPER and all available GPCR crystal structures.
| GPCR templatesa | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OPSD | ADRB2 | DRD3 | CXCR4 | AA2AR | HRH1 | ADRB1 | ACM2 | S1PR1 | OPRK | OPRM | OPRX | OPRD | |
| N-term | 23 | 28 | 29 | 24 | 17 | 0 | 24 | 14 | 13 | 18 | 17 | 28 | 27 |
| TM1 | 50 | 51 | 39 | 65 | 45 | 30 | 41 | 47 | 40 | 56 | 53 | 45 | 50 |
| IL1 | 29 | 33 | 25 | 40 | 20 | 33 | 33 | 33 | 33 | 60 | 50 | 29 | 43 |
| TM2 | 57 | 57 | 47 | 57 | 48 | 53 | 48 | 47 | 38 | 59 | 50 | 46 | 48 |
| EL1 | 0 | 0 | 10 | 25 | 33 | 20 | 33 | 20 | 10 | 33 | 17 | 29 | 20 |
| TM3 | 49 | 54 | 63 | 77 | 57 | 41 | 56 | 44 | 48 | 64 | 59 | 56 | 60 |
| IL2 | 38 | 40 | 30 | 33 | 38 | 57 | 31 | 44 | 20 | 44 | 50 | 36 | 42 |
| TM4 | 54 | 68 | 39 | 33 | 46 | 24 | 38 | 17 | 29 | 38 | 38 | 26 | 36 |
| El2 | 20 | 44 | 7 | 46 | 40 | 0 | 15 | 6 | 11 | 13 | 15 | 14 | 15 |
| TM5 | 44 | 42 | 40 | 53 | 48 | 43 | 31 | 34 | 25 | 41 | 42 | 42 | 38 |
| IL3 | 41 | 13 | 43 | 20 | 67 | 41 | 9 | 9 | 10 | 25 | 19 | 27 | 22 |
| TM6 | 56 | 62 | 83 | 77 | 49 | 56 | 57 | 9 | 52 | 61 | 61 | 47 | 62 |
| EL3 | 17 | 29 | 32 | 0 | 29 | 17 | 20 | 50 | 17 | 30 | 14 | 0 | 0 |
| TM7 | 52 | 52 | 44 | 47 | 56 | 52 | 17 | 19 | 19 | 16 | 19 | 14 | 15 |
| C-term | 29 | 31 | 15 | 22 | 26 | 29 | 7 | 0 | 14 | 19 | 18 | 0 | 19 |
| average of TMs | 52 | 55 | 51 | 58 | 50 | 43 | 31 | 26 | 25 | 38 | 35 | 29 | 33 |
Shown are homology of each individual helixes (TM1-7), intra (IL1-3) and extracellular (EL1-3) loops. Percentages shown are derived from ratios of residues that have identity+similarity/total number of residues in that region. Bolded numbers represent the largest percentage in the specified region.
Small molecule construction
All ligands used in the docking studies were built with standard bond lengths and angles using the molecular modeling package SYBYL-X 1.3. The small molecules were assigned Gasteiger-Huckel charges and energy minimized with the Tripos Force Field (TFF).
Model selection and quality assessments
Model screening was performed by using the genetic-algorithm docking program GOLD 5.1 (Cambridge Crystallographic Data Centre, Cambridge, UK) to dock G-1, G-15, G-26, 17β-estradiol, 17α-estradiol, and estriol into both the 100 active and 100 inactive state GPER homology models using GOLD score as the fitness function.[52] From each group of the 100 active and inactive state models, one receptor model was chosen based upon the discrete optimized protein energy (DOPE) scores, fitness function values, and the electronic and steric interactions between the ligands and receptor. For the active model, the homology model chosen bound G-1 with the highest GOLD score; while for the inactive model, the homology model chosen bound both G-15 and G-26 with the highest GOLD scores. For both models chosen, model refinement was done by using molecular mechanics based energy minimization in Sybyl-X 1.3. Briefly, both models were minimized using a Tripos Force Field with Gasteiger-Hückel charges, a non-bonded interaction cutoff of 8 Å with a distance-dependent dielectric constant of ε = 4 being terminated at 0.05 kcal/(mol Å). Both minimized models were then analyzed using PROCHECK and ProTable within SYBYL-X 1.3 to ensure the overall quality of the models (i.e. acceptable torsion angles, steric clashes, bond lengths, etc).
Molecule docking
After energy minimization of the active and inactive receptor models, both of them were then subjected to another round of docking of the agonists and antagonists (G-1, G-15, G-26, 17β-estradiol, 17α-estradiol, and estriol) to assure that they still bound in the same manner. Using GOLD 5.1 the ligands were all docked into both the active and inactive GPER models. The putative binding area was restricted to a 15 Å radius around N310 and each ligand was docked into the receptors a total of 20 iterations using the generic GOLD docking parameters. Ligand docking poses having the highest GOLD scores were then merged into either the active or inactive GPER model and the subsequent receptor-ligand complex was energy minimized using SYBYL-X 1.3 with the previously described parameters.
Results and Discussion
GPER active and inactive homology model construction
Template structure choice is the foundation of any homology modeling study since it directly impacts the reliability and quality of the prospective model.[23,53–56] With the advent of a large number of available GPCR crystals structures, that choice can be difficult or even controversial. In order to define the possibly most suitable templates for GPER, a multi sequence alignment was done with all the currently available GPCR crystal structures.[1–19] Figure 2 shows some of the representative structures’ sequence alignment with GPER. Further analysis, as seen in Table 1, was done by comparing the homology of the individual domains of the template sequences to GPER for all of the available GPCR structures. The results suggest that both CXCR4 and β2AR may be reasonable choices based upon their overall percentage of homology to GPER (58% and 55% in the TM domains respectively). Both templates have the GPCR highly conserved residues aligned with GPER and are well aligned in their transmembrane helices with no gaps in the alignment. However, only the inactive state structure for CXCR4 is available, whereas there are several active and inactive crystal structures for β2AR. For the purposes of this study, having both the active and inactive templates from the same receptor would be advantageous since it might lessen differences between two models to only the intrinsic differences between the two receptor states. Therefore, β2AR was chosen as the template of choice to model both the active and inactive GPER states.
Figure 2.

Multisequence alignment of GPER with representative sequences. RHOD, bovine rhodopsin; ADB2, human β2-adrenergic receptor; ADB1, human β1-adrenergic receptor; AA2A, human A2a- adenosine receptor; CXCR4, human chemokine receptor CXCR4; DAD3, human D3-dopamine receptor. The most conserved residues among GPCRs superfamily were marked in dark grey while the potential activation involved residues (N6.55 and N6.52) on TM6 were marked in light grey.
Currently, there are several crystal structures of both active and inactive β2AR. In all, six inactive crystal structures (PDB codes: 2RH1, 2R4R, 3D4S, 3KJ6, 3NY8, 3NYA) and three active crystal structures (PDB codes: 3P0G, 3PDS, 3SN6) are available.[6,7,22,57–61] Of the inactive structures, 2RH1 was chosen since it has the highest resolution of 2.4 Å and the co-crystalized ligand, carazolol, is an inverse agonist.[7] Being co-crystalized with an inverse agonist may ensure that the receptor might be stabilized in its inactive state. All three active structures of β2AR represent unique conformations that can be representative of one of the active substates. Since any given crystal structure may be only a snapshot of one single conformation in a dynamic process, it is impossible to judge how accurate one crystal structure is compared to others.[23] Therefore, even though the active crystal structures of β2AR differ from each other, they still might represent one of the activated substates. The Gαs-coupled crystal structure (PDB code: 3SN6) was chosen since it is the only structure coupled to a G protein and thus might represent a conformation of the active state during the signal transduction process, which is a key event in agonist binding. Since GPER also can couple to the same type of G protein, 3SN6 might simulate a similar conformation of GPER when bound to the G protein.[62] Consequently, adopting 3SN6 as a template structure may produce the conformation of the receptor that the agonist may induce in order to initiate the activation process.
Models for both the inactive and active states were generated from the same alignment which required minimal adjustment from the multi sequence alignment as seen in Figure 2. Generated models were screened based upon their conformational stability (DOPE score) and their ability to bind to antagonists and agonists (GOLD score). Specifically, the top ten models that had the highest GOLD scores for the ligands were scrutinized by comparing their DOPE scores and the favorable interactions that were formed between the receptor and ligand(s). The inactive and active models having the highest GOLD scores (which indicating reasonable receptor-ligand interactions), and a low DOPE score relative to the other models were chosen for further refinement. Extracellular loop 2 (EL2) was remodeled due to its low homology with that of β2AR.[23] Figure 3 shows both the active (3A) and inactive (3B) GPER homology models overlapped with their corresponding templates with RMSDs of 3.74 Å and 6.15 Å respectively. As expected, the models closely mimicked their template structures and showed very similar structural differences between the active and inactive states. The overall RMSD values observed between the active and inactive states were 2.96 Å for the β2AR structures and 4.29 Å for the GPER models. Major structure characteristics such as the outward movement of TM6 and lack of an ionic lock between D/E6.30 and R3.50 are still present in the active state homology model of GPER (Figure 3C, D).
Figure 3.

A) The active GPER homology model (light) overlapped with the template structure (3SN6, dark) with a RMSD of 3.74 Å; B) The inactive GPER homology model (light) overlapped with the template structure (2RH1, dark) with a RMSD of 6.15 Å; C) The crystal structures for active (3SN6, light) and inactive (2RH1, dark) β2-adrenergic receptors overlapped with a RMSD of 2.96 Å; D) The active (dark) and inactive (light) GPER homology models based on 3SN6 and 2RH1 respectively with a RMSD of 4.29 Å.
G-1 binding studies in GPER activated model
The docking studies of G-1 in the active GPER model revealed a key relationship between the receptor and its selective agonist G-1. As shown in figure 4 (A, B), G-1 may adopt two distinct binding modes with GOLD scores of 60.8 and 60.3 for individual binding modes I and II (Table 2). Binding mode I represents G-1 with its acetyl group pointing upward, reaching toward the extracellular domain of the binding pocket, and binding mode II has the acetyl group pointing downward, toward TM5 and 6, as shown in Figures 4C and 4D respectively.
Figure 4.

A) Consensus of the binding modes of G-1 in the activated GPER model after 20 docking runs; B) Conformation overlapping of two distinguished binding modes of G-1; Zoom-in of the active GPER homology model with G-1, figures are oriented with TM7 in top middle and TM3 spanning the bottom: C) Binding mode I of G-1 and D) Binding mode II of G-1.
Table 2.
GOLD docking scores of GPER ligands bound into both the active and inactive GPER homology models.
| GPER Model | Ligand | Binding Mode | Fitness | S(hb_ext) | S(vdw_ext) | S(hb_int) | S(int) |
|---|---|---|---|---|---|---|---|
| Active | 17β-estradiol | Mode I | 42.3 | 4.84 | 33.16 | 0 | -8.14 |
| Mode II | 45.0 | 7.03 | 34.14 | 0 | -9.01 | ||
|
| |||||||
| G-1 | Mode I | 67.6 | 0.03 | 54.22 | 0 | -5.17 | |
| Mode II | 71.63 | 2 | 54.34 | 0 | -5.09 | ||
|
| |||||||
| Inactive | G-15 | Mode I | 57.9 | 0.12 | 42.19 | 0 | -0.24 |
|
| |||||||
| G-36 | Mode I | 64.9 | 0.25 | 47.72 | 0 | -0.92 | |
Within binding mode I (Figure 4C) a hydrophobic pocket is formed by L1192.63 in TM2, M1333.28, L1373.32, and M1413.36 in TM3, F2786.55 and I2796.56 in TM6, P3037.33 and H3077.37 in TM7. The two aromatic groups in G-1 may form π–π stacking interactions with F206 and F208 on EL2. The secondary amine in the tetrahydroquinoline moiety may form a hydrogen bond with the 2.6 Å away carbonyl oxygen atom of the amide moiety of N3107.40 on TM7. Additional hydrogen bonding opportunities may also exist: the amide moiety of N1182.62 with the carbonyl group of G-1 that is 3.3 Å away, and E2756.52 with the oxygen atoms in the 1,3-benzodioxole ring of G-1.
In binding mode II a similar hydrophobic pocket was formed by the same residues in TM2, TM3, TM6, and TM7 seen in binding mode I (Figure 4D). Additionally, the two aromatic groups in G-1 may still form π stacking interactions with F206 and F208 on EL2. However, due to the different binding mode the hydrogen bonding interactions between G-1 and the receptor completely changed. The secondary amine shifted upward and was 4.6 Å away from N3107.40 and therefore, may no longer form a hydrogen bond with it, but that interaction is replaced by a carbonyl-halogen bond between the carbonyl oxygen atom of N3107.40 and the bromine atom of G-1 which was 3.1 Å away. An oxygen atom in G-1’s benzodioxole ring can form a hydrogen bond with the amide nitrogen of N1182.62 that was 2.5 Å away. The acetyl group oxygen atom of G-1 may act as a hydrogen bond acceptor for the amide nitrogen of N2766.53 on TM6 at a distance of 3.1 Å which was not seen in binding mode I.
Altogether, binding mode I and II of G-1 shared several hydrophobic and aromatic interactions with GPER, but differed in their hydrogen bonding networks. Within binding mode I, the amine, carbonyl oxygen, and one of the benzodioxole oxygens in G-1 showed favorable interactions with N3107.40, N1182.62, and E2756.52 respectively. In binding mode II, G-1’s bromine and benzodioxole oxygen, shared interactions with N3107.40 and N1182.62, respectively. Additionally, the carbonyl oxygen of G-1 may interact with N2766.53 in binding mode II. The conservation of most of the residues that G-1 interacted with between the two modes was reflected by their almost identical GOLD scores of 60.8 and 60.3 for binding modes I and II. To further test the favorability of one binding mode over the other, G-1 was docked 1000 times into the active GPER model. While binding mode I and II were still the only two modes observed, mode II was favored at a ratio of almost 5:1 compared to mode I.
Dual binding modes of G-1 in the activated model
Superimposing the two binding modes of G-1 revealed the apparent pseudosymmetry of the molecule’s electronic and structural state (Figure 5A). The overall conformation of G-1 is maintained along the ligand’s backbone. In both modes, while the tetrahydroquinoline amino group stayed in the relatively same position and the electronic properties of the carbonyl oxygen atom was comparable to the ones in the dioxolane ring, there were hydrogen-bonding oxygens at both ends of G-1 which adds to its symmetry. Since the acetyl group can freely rotate on the phenyl ring it would match with either of the two oxygen atoms on the dioxolane ring on the other end of the molecule, leading to an even greater degree of symmetry. It is important to note that the dual binding modes of G-1 may be unique to the active GPER model; when G-1 was docked into the inactive GPER model, only binding mode I was observed (see Supplemental information).
Figure 5.
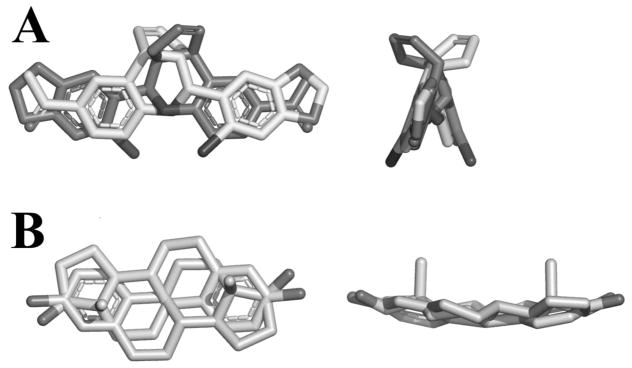
A) Overlap of binding mode I and II of G-1 observed in the active GPER model; B) Overlap of 17β-estradiol showing the pseudosymmetry of the ligand.
In order to see if other GPER agonists had a similar pseudosymmetry, 17β-estradiol was rotated 180° and superimposed on itself (Figure 5B). At the first glance, 17β-estradiol’s structure seemed to share similar type of pseudosymmetry as G-1 due to the two polar hydroxyl groups at either end of the molecule while the overall conformation of the molecule was maintained. However, unlike G-1, the existence of only one aromatic group in 17β-estradiol decreased its overall symmetry. The aromatic group did overlay well the aliphatic portion the molecule, but their electronic properties differed. Overall, these results suggest the importance of GPER agonists to have a rigid scaffold with polar moieties at both ends in a symmetrical manner. Further evidence of this key feature is the fact that among the “G-scaffold” series of compounds, G-1 is the only molecule carrying this type of pseudosymmetry.
17β-estradiol binding studies in GPER activated model
From the G-1 binding studies, active GPER conformations were obtained from binding modes I and II. Subsequently, 17β-estradiol was docked into both models in order to validate the importance of structural symmetry in GPER ligand agonism (Figure 6). For both the mode I and mode II GPER models, 17β-estradiol mimicked the corresponding G-1 binding poses. In binding mode I the 17-hydroxyl group pointed upwards, whereas for binding mode II the 17-hydroxyl group pointed downward toward TM5 and 6.
Figure 6.

Docking studies of the energy minimized, active GPER homology models from G-1 binding modes I and II with 17β-estradiol. Figures are oriented with TM7 in top middle and TM3 spanning the bottom. (A) Binding mode I of 17β-estradiol. (B) Binding mode II of 17β-estradiol.
Analysis of binding mode I, Figure 6A, of 17β-estradiol indicated that it shared the same hydrophobic pocket as G-1, but lacked the π stacking with F206 and F208 on EL2 as seen in G-1 binding. Additionally, two hydrogen bonds may be formed with GPER: the 17 position hydroxyl group may act as a hydrogen bond acceptor for the amide of N1182.62 (2.5 Å), and the phenoxyl group may act as a hydrogen bond donor with E2756.52 on TM6 (2.6 Å). In all, this showed that 17β-estradiol can form several similar interactions with GPER as G-1 in binding mode I. The same held true for 17β-estradiol when docked into the active GPER binding mode II model, Figure 6B. As before, the hydrophobic pocket seen for G-1 was maintained when docking 17β-estradiol. However, the aromatic portion of the molecule may now form a π–π stacking interaction with F206 on EL2. Hydrogen bonding is also maintained to N1182.62 and E2756.52. Subsequently, 17β-estradiol’s phenoxyl group acted as a hydrogen bond acceptor with the amide of N1182.62 (2.6 Å) and the hydroxyl group at the 17 position acted as a hydrogen bond donor to E2756.52 (3.0 Å). The same hydrogen bonding network was maintained in both binding modes of 17β-estradiol and they compared well with the ones seen for G-1 binding modes I and II.
Comparison of G-1 and 17β-estradiol’s hydrogen bonding networks for binding mode I and II revealed several similarities. Within all four binding poses (G-1 binding mode I and II, and 17β-estradiol binding mode I and II) there was a hydrogen bonding interaction with N1182.62 on TM2 and a polar or hydrogen bonding interaction with either E2756.52 or N2766.53 on TM6. When 17α-estradiol, which does not bind to GPER, was docked into the active GPER model the change in stereochemistry of the 17 position hydroxyl group leads to a loss of interaction with one of those residues in both GPER binding modes (Supplemental Information). Overall, these results suggest the importance of interaction with those residues for GPER ligand agonism.
Inactive GPER model binding studies
To fully characterize GPER’s interactions with its ligands, the two GPER selective antagonists G-15 and G-36 were also docked into the inactive model of GPER along with estriol (see Supplemental Information). As seen, G-15, and G-36 only bound in a manner similar (Figure 7) to the binding mode I observed for G-1 in the active model. For both ligands, a hydrophobic pocket was formed by V1162.60 and L1192.63 on TM2, M1333.28 and L1373.32 in TM3, F206 on EL2, together with I2796.56 and F2786.55 on TM6. In addition, F208 (EL2) and H3077.37 may form π–π stacking with the benzodioxole ring and the tetrahydroquinoline rings for both ligands respectively. Hydrogen bonding between the carbonyl oxygen of N3107.40 and the ligands’ secondary amines, and between E2756.52 and the oxygens in the benzodioxole rings were conserved in both ligands. The only differences in binding between the two occur at their substitution site on the tetrahydroquinoline ring. Since G-15 lacks a substitution at the 6-position, it showed no additional interactions with that side of the binding pocket (Figure 7A); whereas the isopropyl group of G-36 fit into a hydrophobic pocket consisting of L1192.63 and several other residues (Figure 7B). The loss of the extra hydrophobic interactions for G-15 is reflected in its GOLD score being 57.9 compared to the higher score of 64.9 for G-36 (Table 2). Overall, there was a slight shift downward in the binding pocket for these two ligands when compared to the active GPER model.
Figure 7.
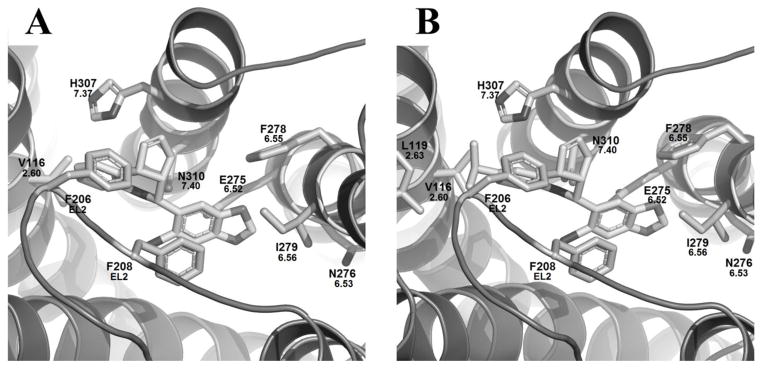
Docking studies of the inactive GPER homology model with G-15, and G-36. Figures are oriented with TM7 in top middle and TM3 spanning the bottom. (A) G-15 bound. (B) G-36 bound.
Comparison of active and inactive GPER binding studies
When comparing the binding of G-1 to the inactive and active GPER models, the main distinction is the presence of two binding modes for the active model and their hydrogen bonding networks. While binding mode I is shared by both the active and inactive docking studies for both agonists and antagonists alike, binding mode II is unique to the active model. Furthermore, when the antagonists G-15 and G-36 were docked into the active model (see supplemental information) only binding mode I was observed, making binding mode II exclusive to the agonist G-1.
The key difference between hydrogen bonding networks of GPER agonists and antagonists were interactions with TM2 and TM6. While all of the “G-scaffold” ligands may interact with E2756.52 on TM6 through a polar interaction between the carboxylic acid of E2756.52 and one of the oxygen atoms of benzodioxole ring, the antagonists lack interactions with N1182.62 and N2766.53. Both G-1 and 17β-estradiol can form hydrogen bonds with N1182.62 whereas the antagonists lack any polar interactions with it. A hydrogen bonding interaction with N2766.53 was only seen for G-1 binding mode II which, in combination with the fact that binding mode II was only seen for agonists, suggests its importance in receptor activation.
GPER activation
The exact mechanism of GPCR activation is still not yet fully known, but there is a consensus that several movements tend to occur in the transmembrane helices as well as changes in intermolecular interactions (i.e. hydrogen bonding networks). The two main interactions are: a change in the conformation state of W6.48 (“toggle switch”) and disruption of an “ionic lock” formed between R3.50 and D/E6.30.[2,23] Currently, the available crystal structures of activated GPCRs show an outward movement of the intracellular end of TM6 with a rearrangement of TM5 and TM7.[2,22] These observations agree with biophysical data that shows the macroscopic movement of activated receptors.[2,23] However, changes in the specific intermolecular interactions are more elusive in the available active crystal structures. Only subtle changes in the position of W6.48 are seen in the adrenergic structures and the broken “ionic lock” is only seen in a handful of structures and even varies between receptors in the same crystal.[1,21] Therefore, it is hard to tell which crystal structures are more accurate representations of an active state due to all of the substates that have been observed.
Only two crystal structures are thought to represent fully active states, a Nb80 stabilized (nanobody 80) β2AR bound to agonist BI-167107 and a heterotrimeric G protein complexed with β2AR bound to agonist BI-167107 (PDB codes: 3PDS and 3SN6 respectively).[22,61] Since the active GPER model was modeled after one of those structures (3SN6) it too showed the same activation features. While the docking studies performed on the active GPER model are not definitive, they do offer plausible explanations why an acetyl group substituent produces agonism in the “G-scaffold” ligands. In all, the key residues for ligand induced GPER activation can be inferred based upon the structure-activity relationships observed through the docking studies.
Comparison of the binding pocket interactions between GPER agonists and antagonists revealed key differences in their hydrogen bonding capabilities to the receptor. Unlike the antagonists, G-1 could form hydrogen bonds with either N1182.62 in TM2 or N2766.53 in TM6 while in binding mode I or II respectively. While both helices have been implicated within the activation mechanism of GPCRs,[63–65] the interaction of the G-1 with TM6 might better explain GPER activation mechanism. During activation and subsequent G protein binding, an agonist may help stabilize the large displacement in TM6 that occurs along with other movements in the receptor.[1,21,63–65] The stabilization, which Schwartz et al. has proposed to be a part of a global toggle switch, has been shown to occur through interaction of an agonist with TM6 using engineered GPCRs.[64] Furthermore, the crystallized active GPCRs, adenosine A2A, β1-adrenergic, and β2-adrenergic receptors have all shown that their agonists interact with a asparagine residue (N6.55) that is crucial for their activation.[8,61,66] Molecular modeling studies have also shown the importance of N6.55 to stabilize the activated β1-adrenergic and β2-adrenergic receptors.[67,68] Comparing the sequences of those receptors to that of GPER, N2766.53 in GPER is only one helical turn directly below N6.55 in the other three receptors (Figure 2). Therefore, the interactions observed for N6.55 in agonist bound adenosine A2A, β1-adrenergic, and β2-adrenergic receptors may serve as an explanation for GPER agonist binding mode involving N6.53. Within GPER agonist binding pocket the hydrogen bond between the carbonyl oxygen atom of G-1 in binding mode II and the amide hydrogen atom of N2766.53 may serve the purpose to stabilize the global TM movements in the receptor activation process similar to the aforementioned receptors. Further site-directed mutagenesis study should help to fully elucidate this observation.
Conclusion
The current study was aimed at elucidating plausible structure-activity relationship of GPER ligands by applying homology models for both the inactive and active states of the receptor. From the docking studies of the active state model, two binding modes for the agonist G-1 were observed. These binding modes overlapped with each other and showed an apparent symmetry of the ligand that the antagonists, G-15 and G-36, lacked. Unlike the active state model, only binding mode I was observed while docking G-15 and G-36 into the inactive GPER model. Comparison of the agonist and antagonist binding pocket interactions revealed only G-1 can interact with N1182.62 and N2766.53 (binding mode II) which might aid in stabilizing the active state of the receptor. Because the antagonists of GPER binding mode did not involve such interaction with N2766.53 while similar conserved interactions have been observed for several crystallized agonist bound GPCRs, we may conclude that these residues may be important in GPER activation process. The direct comparison between the active and inactive binding states of the receptor not only provides a plausible explanation for the structure activity relationship for the GPER selective “G-scaffold” ligands, but also may be helpful in future rational ligand design for drug development goals.
Supplementary Material
References
- 1.Congreve M, Langmead CJ, Mason JS, Marshall FH. J Med Chem. 2011;54:4283–4311. doi: 10.1021/jm200371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kontoyianni M, Liu Z. Curr Med Chem. 2012;19:544–556. doi: 10.2174/092986712798918824. [DOI] [PubMed] [Google Scholar]
- 3.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 4.Patny A, Desai PV, Avery MA. Curr Med Chem. 2006;13:1667–1691. doi: 10.2174/092986706777442002. [DOI] [PubMed] [Google Scholar]
- 5.Bizzantz C, Bernard P, Hibert M, Rognan D. Proteins: Struct, Funct, Bioinf. 2003;50:5–25. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen SGF, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, et al. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 7.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 10.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, et al. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, et al. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desal H, Clemons B, Cahalan SM, Schuerer SC, et al. Science. 2012;335:852–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XH, Carroll FI, et al. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, et al. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park PSH. Curr Med Chem. 2012;19:1146–1154. doi: 10.2174/092986712799320619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fanelli F, Benedetti PGD. Chem Rev. 2011;111:PR438–PR535. doi: 10.1021/cr100437t. [DOI] [PubMed] [Google Scholar]
- 22.Rasmussen SGF, DeVree YZBT, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, et al. Nature. 2011;477:549–557. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park PSH. Curr Med Chem. 2012;19:1146–1154. doi: 10.2174/092986712799320619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ballestesteros JAWH. Methods Neuosci. 1995;25:366–428. [Google Scholar]
- 25.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 26.Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- 27.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 28.Prossnitz ER, Barton M. Prostaglandins other Lipid Mediators. 2009;89:89–97. doi: 10.1016/j.prostaglandins.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prossnitz ER, Sklar LA, Oprea TI, Arterburn JB. Trends Pharmacol Sci. 2008;29:116–123. doi: 10.1016/j.tips.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Prossnitz ER, Barton M. Nat Rev Endocrinol. 2011;7:715–726. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, Steinhoff MM, Sabo E. Clin Cancer Res. 2008;12:6359–6366. doi: 10.1158/1078-0432.CCR-06-0860. [DOI] [PubMed] [Google Scholar]
- 32.Wang D, Hu L, Zhang G, Zhang L, Chen C. Endocrinology. 2010;38:29–37. [Google Scholar]
- 33.Arias-Pulido H, Royce M, Gong Y, Joste N, Lomo L, Lee SJ, Chaher N, Verschraegen C, Lara J, Prossnitz ER, et al. Breast Cacner Res Treat. 2010;123:51–58. doi: 10.1007/s10549-009-0631-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo HJ, Luo P, Yang GL, Peng QL, Liu MR, Tu G. J Breast Cancer. 2011;14:185–190. doi: 10.4048/jbc.2011.14.3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filardo EJ, Quinn JA, Bland KI, Frackelton ARJ. Mol Endocrinol. 2000:14. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 36.Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pessi V, Montanaro D, Musti AM, Picard D, Ando S. J Biol Chem. 2004;279:27008–27016. doi: 10.1074/jbc.M403588200. [DOI] [PubMed] [Google Scholar]
- 37.Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Ando S, Maggiolini M. Mol Endocrinol. 2006;20:631–646. doi: 10.1210/me.2005-0280. [DOI] [PubMed] [Google Scholar]
- 38.Vivacqua A, Lappano R, De Marco P, Sisci D, Aquila S, De Amicis F, Ando S, Maggiolini M. Mol Endocrinol. 2009;23:1815–1826. doi: 10.1210/me.2009-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vivacqua A, Bonofiglio D, Albanito L, Madeo A, Rago V, Carpino A, Musti AM, Picard D, Ando S, Maggiolini M. Mol Endocrinol. 2006;70:1414–1423. doi: 10.1124/mol.106.026344. [DOI] [PubMed] [Google Scholar]
- 40.Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Ando S, et al. Cancer Res. 2007;67:1859–1866. doi: 10.1158/0008-5472.CAN-06-2909. [DOI] [PubMed] [Google Scholar]
- 41.Barton M. Steroids. 2012;77:935–942. doi: 10.1016/j.steroids.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 42.Thomas P, Pang Y, Filardo EJ, Dong J. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 43.Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, et al. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 44.Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, et al. Nat Chem Biol. 2009;5:421–427. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaquchi Y, Hayashi SI, Sklar LA, et al. J Steroid Biochem Mol Biol. 2011;127:358–366. doi: 10.1016/j.jsbmb.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lappano R, Rosano C, De Marco P, De Francecso EM, Pezzi V, Maggiolini M. Mol Cell Endocrinol. 2010;320:162–170. doi: 10.1016/j.mce.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Pang Y, Dong J, Thomas P. Endocrinology. 2008;149:3410–3426. doi: 10.1210/en.2007-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lappano R, Rosano C, Santolla MF, Pupo M, De Francesco EM, De Marco P, Ponassi M, Spallarossa A, Ranis A, Maggiolini M. Curr Cancer Drug Targets. 2012;12:531–542. doi: 10.2174/156800912800673284. [DOI] [PubMed] [Google Scholar]
- 49.Lappano R, Santolla MF, Pupo M, Sinicropi MS, Caruso A, Rosano C, Maggiolini M. Breast Cancer Res. 2012;14:R12–R24. doi: 10.1186/bcr3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 51.Sali A, Blundell TL. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 52.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 53.Sela I, Golan G, Strajbl M, Rivenzon-Segal D, Bar-Haim S, Bloch I, Inbal B, Shitrit A, Ben-Zeev E, Fichman M, et al. Curr Top Med Chem. 2010;10:638–656. doi: 10.2174/156802610791111498. [DOI] [PubMed] [Google Scholar]
- 54.Mobarec JC, Sanchez R, Filizola M. J Med Chem. 2009;52:5207–5216. doi: 10.1021/jm9005252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Michino M, Abola E, Brooks CLI, Dixon JS, Moult J, Stevens RC GD. 2008 Participants. Nat Rev Drug Disc. 2009;8:455–463. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R G. D. 2010 Participants. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EYT, Velasquez J, Kuhn P, Stevens RC. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bokoch MP, Zou Y, Ramussen SGF, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi HJ, Thian FS, Kobilka TS, et al. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi HJ, DeVee BT, Sunahara RK, et al. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rasmussen SGF, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, DeVree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dorsam R, Gutkind S. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 63.Kolinski M, Plazinska A, Jozwiak K. Curr Med Chem. 2012;19:1155–1163. doi: 10.2174/092986712799320547. [DOI] [PubMed] [Google Scholar]
- 64.Schwartz TW, Fimurer TM, Holst B, Rosenkilde MM, Elling CE. Ann Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- 65.Deupi X, Standfuss J, Schertler G. Biochem Soc Trans. 2012;40:383–388. doi: 10.1042/BST20120001. [DOI] [PubMed] [Google Scholar]
- 66.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, Tate CJ. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vanni S, Rothlisberger U. Curr Med Chem. 2012;19:1135–1145. doi: 10.2174/092986712799320493. [DOI] [PubMed] [Google Scholar]
- 68.Katritch V, Abagyan R. Trends Pharmacol Sci. 2011;32:637–643. doi: 10.1016/j.tips.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.