

Abstract
Actinic keratosis (AKs) is one of the most common skin lesions leading to an increased risk of developing squamous cell carcinoma and other skin malignancies. The lesions principally arise as a result of excessive ultraviolet (UV) exposure. AKs may regress spontaneously, remain stable or evolve to invasive squamous cell carcinoma. The risk of squamous cell carcinoma is significantly increased patients with more than 5 AKs. The main mechanisms involved in the formation of AK are inflammation, mutagenesis, oxidative stress, impaired apoptosis, immunosuppression, disregulation of cell growth and proliferation, and tissue remodeling. Human papilloma virus has also been correlated with the formation of some AKs. As an individual ages, his skin is exposed to increasing cumulative amounts of UV light and other environmental insults. This is especially true for the head, neck and forearms. These insults do not target only the skin where individual lesions develop, but also the surrounding area. In this area undetectable preclinical AK lesions or dysplastic cells may be present. The whole affected area is known as the 'field'. Therefore, management is divided into lesion-directed and field-directed therapies. Currently, the therapies in use are lesion-directed cryotherapy and/or excision, and field-directed topical agents: 5-fluorouracil, diclofenac, photodynamic therapy, imiquimod, and ingenol mebutate. Combining lesion- and field-directed therapies showed good results and several novel therapies are under investigation. Treatment is variable and personalized, what makes a gold standard management algorithm difficult to design. This review aims to describe the rationale behind the available treatment options for AKs based on current understanding of pathophysiology and epidemiology.
Keywords: 5-fluorouracil, actinic keratoses, actinic keratosis, diclofenac, field cancerization, imiquimod, ingenol mebutate, treatment, photodynamic therapy
Introduction
Actinic keratoses (AKs) are the most common lesions within the continuum of SCC. AK may follow 3 different pathways: regression, persistence or progression to in situ or invasive SCC, which rarely metastasizes. Even though today the risk of an AK’s progression to invasive SCC is unclear, the rate varies from the lowest 0,1% to the highest as 20%. For a patient affected by multiple AK lesions, the risk to develop an invasive cutaneous SCC is between 0.15% and 80%. The relative risk of SCC increases for patients affected by more than five AK lesions. It has been shown by histologic evidence that most of SCC arises from AK lesions whereas it is not possible to predict which AK lesions will advance to SCC; for these reasons they should be treated as early as possible.[1] AKs are common among fair-skinned individuals, and they typically appear on sun-exposed areas. In Australia, for example, 40-50% of the Caucasian population over the age of 40 years develops AKs.[2-3]
In the United States, the prevalence ranges from 11 to 26%, whereas in Europe 15% of men and 6% of women have been reported to be affected[4] (in Italy the prevalence is 1,4%, however it is believed that this estimate is largely biased by underreport). People who have the tendency to sunburn and have difficulty to tan (Fitzpatrick II) are exposed to high levels of risk of adverse effects of UV radiation. Older age, male gender, cumulative UV exposure, proximity to the equator and immunosuppression are other factors of risk to develop AKs.[5] Individuals with AKs tend to evolve into 6-8 lesions on average and they appear commonly on the head [Fig. 1], bald scalp, face, dorsal forearms and hands.
Figure 1.
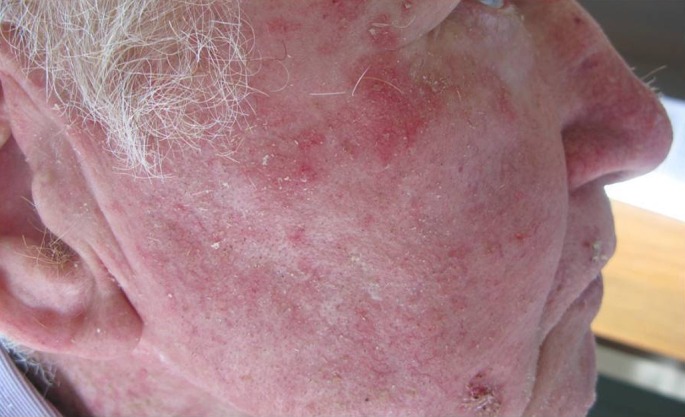
Actinic keratosis of the face.
Immunocompromised patients (undergone, for example, organ transplantation and, therefore, immunosuppressive therapy) are up to 250 times more likely to develop AK. Typically they occur as 1-3 mm erythematous scaly papules with a hyperkeratotic texture, therefore, they are best identified through a touch inspection rather than visual inspection alone. Single AKs lesions are also present, and both single and multiple lesions grow slowly, from small (less than 1 cm in diameter), dry, flesh-coloured, or erythematous papules they advance to large plaques, with telangiectasias, often covered by yellow or brown adherent scales, and, except in their hypertrophic form, they show little infiltration.[6-7] The surrounding areas may also show evidence of chronic sun damage with telangiectasias and yellow discoloration. AKs on the back of the hands or on the forearms are often thick and hyperkeratotic wherease on the head and neck are usually thinner. Occasionally, lesions develop marked hyperkeratosis and may occur with cutaneous horns. When the horn-like material is removed, a reddened and partly fissured substrate appears and 15.7% of these cutaneous horns 15.7% are squamous cell carcinoma.[8] According to the clinical presentation, we distinguish three different grade of AK: grade I visible and slightly palpable; grade II visible and palpable; grade III frankly visible and hyperkeratotic. The normal AK’s development is for hard horny proliferations ("verrucous keratosis").[9-10] Other AK’s clinical variants include lichen planus-like keratosis and actinic cheilitis. The clinical features usually suggest the diagnosis of AK; however, in order to exclude deeper involvement, particularly when there are large, bleeding, ulcerated, or indurated lesions, the biopsy is used.
Genetics and histology
The adverse effects of UV radiation on keratinocyte DNA results into actinic keratosis. reducing the skin immunity.[11-12] Mutations involved in the development of AK and increased risk of AK’s progression into SCC are p16 (INK4a) (on chromosome 9p21)[13], p14 (ARF), p15 (INK4b) and p53[14]. UV-A (320-400 nm), the most abundant source of solar radiation, penetrates the skin more deeply than UV-B, leading to the nucleic acids’ oxidative damage, membrane lipids and cell proteins damage, producing reactive oxygen species (ROS).[15] These ROSs affect the cellular transduction pathways and cell-cell signaling, causing altered proliferation.[16] Thymine (T) — guanine (G) is the signature mutation of UV-A, due to the formation of 8-hydroxyguanine adducts. UV-B (290-320 nm) radiation directly leads to the formation of cyclobutane pyrimidine dimers and 6-4 photoproducts, which in turn give rise to the characteristic cytosine (C) - T and CC -TT mutations.[17] Further, p53 (a tumor suppressor protein) inactivation by UV-B light is a crucial step in the AKs development, creating genetically unstable keratinocytes. Mutations in the TP53 gene have been reported in 90% of human cutaneous SCCs.[18] It has been shown that the main function of p53 in mice is to protect the skin cancer induction by UV light. In fact, the absence of functional repair genes, such as p53, other DNA mutations go on to promote the carcinogenesis. Moreover, absorbed UV light increases the arachidonic acid production and its metabolites, and other pro-inflammatory cytokines. Furthermore, reactive oxygen species induce lipid peroxidation and cellular destruction.[19-20] Therefore, the main mechanisms in the development of AK are inflammation, oxidative stress, immunosuppression, impaired apoptosis, mutagenesis, cell growth dis-regulation and proliferation, and tissue remodeling.[21] This knowledge is the basis of AK medical management. The AK’s ability to regress or progress to SCC, it has been considered as premalignant, however many publications suggested as more appropriate its reclassification as "cancerous".[22-23] Cockerell proposed to use the "keratinocyte intraepidermal neoplasia" (KIN) nomenclature, conforming to the classification used for cervical intraepithelial neoplasia.[24] Recently, cutaneous human papilloma virus (HPV) infection has been associated with AK development.[25] The exact mechanism has not been completely understood, but the E6 protein of cutaneous HPV seemed to contribute to the reduction of Bak protein levels. It has been reported that E6 protein shows pro-apoptotic effects and it is activated as a protective mechanism in keratinocytes exposed to UV light.[26] It is a challenge to distinguish between AK and early SCC because they share several cellular and histopathological features. A common definition is based on the relative depth of dysplastic cells within the skin. AKs can be anatomically defined as a basal layer epidermal lesion that may extend involving the both granular and cornfield layers.[27] Since the epidermis basal layer is the site of dividing cells the disease begins there. Bowen’s disease (squamous cell carcinoma in situ) lies at the more extensive end of the epidermal involvement spectrum, but it remains largely indistinguishable from AK at cellular level. Invasiveness of the SCC is defined by its extension beyond the epidermis, through the basement membrane and into the dermis. However, at the diagnosis the Bowen’s disease must be considered a malignant tumor, contained within the epidermis with potential for lateral spread and of becoming invasive. Distinguishing histologically between Bowen’s disease and SCC, especially when the SCC is very well differentiated and dermal protrusions are smooth edged, it is still a challenge. Padilla et al,[28] by a gene expression profiling, demonstrated that AK is a precursor lesion of the SCC, and that they are closely related genetically. This study shows that AK lies on a spectrum between normal skin and SCC. It has also been discovered that reduced p53 immuno-staining analysis of AK is associated with a greater probability that those lesions might develope into SCC.[29] This may help in the future to predict which biopsied lesion is more likely to require early or surgical intervention.
Therapy and field cancerization
Removal of the lesion, which is the most common therapy for individual AKs, should always be considered for isolated lesions or early presentations of AKs. Other therapies include the destructive kinds such as liquid nitrogen cryotherapy, curettage with or without electrodessication, and shave excision.[30] These are procedurally simple, quick and provide adequate clearance of abnormal tissue although they fail to address field cancerization.
Cryotherapy
Cryotherapy to the affected area works by destroying atypical AK cells by lowering the skin temperature. This technique is ideal if lesions are scattered or limited in number, or for patients who are non-compliant with topical regimes. Reported cure rates range from 39 to 83%. Cryotherapy with liquid nitrogen is the most frequently used technique. Cryotherapy is generally well-tolerated and does not require local anesthetic but it can be painful and frequently result in permanent hypopigmentation.[31] Blisters, scarring, textural skin changes, infection, and hyperpigmentation are some of the potential side-effects.
Curettage
Curettage involves using a curette to mechanically remove atypical cells. A shave excision using a surgical blade can also be applied to excise the abnormal cells. Currettage and shave excision may also be followed by electrocautery to destroy additional atypical cell layers as well as provide hemostasis. They are most appropriate for treating individual AKs, cases where a biopsy is required to rule out frank carcinoma, or for hypertrophic AKs that are refractory to other treatments. Studies that document cure rates using these treatment modalities are lacking. Potential side-effects may include scarring, infection, anesthetic related side-effects, and dyspigmentation.
Topical Field Therapy
Generally, physicians are faced with field cancerization where the patients are covered in actinic damage. This describes both clinical and subclinical lesions within a given anatomical region, for which patients need a different therapeutic approach, known as field therapy.
The term originally coined by Slaughter et al. in 1953 referred to histologically abnormal epithelium adjacent to tumor tissue within the aero digestive region to explain the occurrence of multiple primary tumors local recurrences.
Field therapy involves eradication of both the clinically visible and subclinical AKs within the treatment area.[32]
Topical 5-fluorouracil
The antimetabolite 5-fluorouracil (5-FU) was the first approved topical field therapy serendipitously discovered when AKs were observed to become inflamed and subsequently resolved in cancer patients receiving systemic 5-FU as a chemotherapeutic agent. It was eventually designed into an effective topical formulation which acts as a thymidylate synthase inhibitor by blocking a methylation reaction which then in turn disrupts DNA and RNA synthesis and effectively stops the growth of the rapidly proliferating atypical cells over normal cutaneous tissue.[33] The average cure rate is 62.5% however, for optimal results, full patient adherence is necessary. Also, concurrent treatment with topical tretinoin has been shown to enhance 5-FU’s effectiveness.
Patients undergoing successful treatment generally experience erythema, inflammation, and erosions. Other common side-effects include pain, pruritus, photosensitivity, and burning at the site of application. Topical 5-FU should not be used in cases where patients also suffer from other cutaneous conditions, such as melasma or acne rosacea.[34-35]
Diclofenac
The nonsteroidal anti-inflammatory drug, Diclofenac (3% gel) is believed to exert its effects through the inhibition of cyclooxygenase (COX), especially COX-2. Without COX, prostaglandin production that suppresses the immune system allowing tumor formation is reduced and the cascade is disrupted. The treatment regimen is vigorous (twice-daily for 90 days) but only mild to moderate local skin reactions have been observed. Rare, drug-induced hepatotoxicity has been reported. Transaminases should therefore be measured periodically in patients receiving long-term therapy.[33]
Imiquimod
Topical 5% imiquimod cream (Aldara®) originally indicated as a treatment for genital and perianal warts[36] and used off label to treat Bowen’s disease, invasive SCC, lentigo maligna, molluscum contagiosum, keloid scars, and other diseases, was later approved for treating AKs and superficial basal cell carcinomas. Imiquimod disrupts tumor proliferation by acting as a toll-like receptor-7 agonist that modifies immune response and stimulates apoptosis. Stockfleth et al.[4] showed that 84% of treated AKs showed clinical clearance with one 12-week cycle of 5% imiquimod therapy but local irritant reactions are common. That was associated with long duration of application (twice-weekly for 16 weeks), makes treatment adherence challenging. Both visible and subclinical AKs can be targeted by administration to the lesion and surrounding tissue targets. Systemic effects, such as fatigue, flu-like symptoms, headaches, myalgias, and angioedema are rare. Recently, regulatory approval was granted by Health Canada in December 2009 and by the US FDA in March 2010 to imiquimod 3.75% (Zyclara™) for the treatment of AKs on the face or balding scalp. Two identical placebo-controlled trials have evaluated the safety and efficacy of imiquimod 3.75%. In the trial carried out by Swanson et al., creams were applied daily to the entire face or balding scalp for two 2-week treatment cycles separated by a 2-week interval without treatment. Patients applying imiquimod 3.75% achieved a median lesion reduction of 82%, while just over one-third demonstrated complete clearance. These efficacy data rival those achieved using imiquimod 5% twice-weekly for 16 weeks, with the advantage of significantly improved patient tolerance exhibited by the lower dosage. The therapy was found to be safe and did not result in any serious adverse events. Erythema was observed in most patients, with about 25% developing severe erythema. However, no patients withdrew from the study because of this; compliance rates were noted to be greater than 90%. Overall, the newly approved imiquimod 3.75% is a reasonable alternative to imiquimod 5%, as it demonstrates comparable efficacy, allows for a much simplified, shorter dosing regimen, and seemingly yields less severe adverse effects. Furthermore, imiquimod 3.75%[37] is approved for the treatment of a larger surface area of up to 200 cm2, when compared with 25 cm2 for the 5% formulation and is thus able to target more AKs.
Ingenol mebutate
In January 2012, the FDA approved ingenol mebutate gel (Picato, Leo Pharma, Inc.) for the topical treatment of AK. Ingenol mebutate, an active compoundfound in the sap of the Euphorbia peplus plan is known forits dermatological uses, including the treatment of cancerous lesions. This medication treats AK lesions by rapidly inducing cell death.
The P-glycoprotein absorptive drug transport enables topical ingenol mebutate to pass the stratum corneum barrier and exerts its action in the dermis and hypodermis directly and indirectly through local production of inflammatory cytokines. This causes initial tumor ablation, which is characterized by rapid disruption of plasma membrane and subsequent mitochondrial swelling followed by cell death via primary necrosis.[38] The second phase is marked by local acute inflammatory response due to neutrophil infiltration. During the third and last phase, tumor-reactive antibodies are induced and relapses are avoided through antibody-dependent neutrophil cytotoxicity eliminating residual cancer cells. Ingenol mebutate is a protein kinase C (PKC) pathway activator. Consequently, it targets and damages the sub-epidermal intrinsic tumor vasculature. PKC delta activation slows cell proliferation, induces cell cycle arrest, and enhances differentiation in various undifferentiated cell lines. Additionally, it promotes apoptosis of caspases, increases stability of p53, and promotes phosphorylation of signaling molecules.
The application of the gel results in rapid destruction of AK lesions, so that treatment is needed for only 2-3 days.
Systemic absorption following topical application of ingenol mebutate is negligible. Blood levels of the drug and its two metabolites are below the lower limits, defined as less than 0.1 ng/mL, and its metabolites have been shown to have no effect on the cytochrome P450 (CYP) enzyme system. No drug interactions have been attributed to ingenol mebutate. Ingenol mebutate is available in two strengths, 0.015% and 0.05%.[39] The 0.015% strength is indicated for AKs of the face and scalp [Fig. 2]. The gel is applied to the affected area once a day for 3 consecutive days ona surface area of 25 cm2. AK of the trunk and extremities (i.e. body, arms, hands, and legs) should be treated with the 0.05% gel and should be applied to the affected area once a day for 2 consecutive days on a surface area of 25 cm2. Regardless of the strength, patients should be advised to use a new tube for each day of treatment. All tubes should be stored in the refrigerator.
Figure 2.
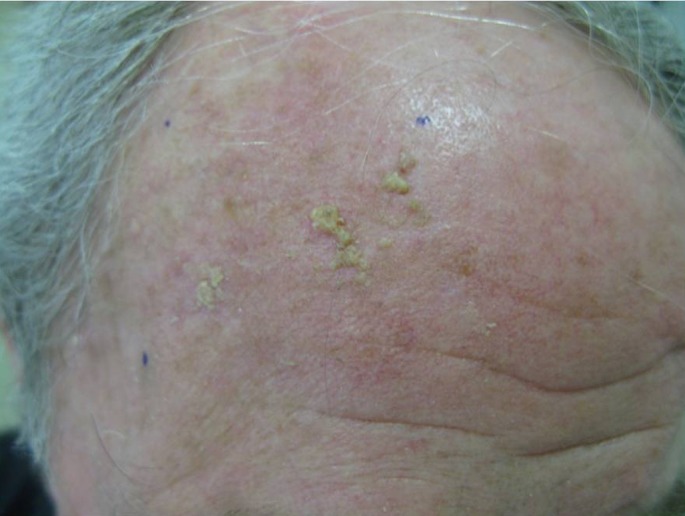
Actinic keratosis of the scalp.
Application-site reactions such as pain, itching, irritation, infection, periorbital edema, nasopharyngitis and headache are some of the adverse reactions commonly observed in clinical trials.[40-41] The most common local skin responses were dose-related erythema, flaking/scaling/dryness, and scabbing/crusting that resolved within one month. Given the dual mechanism of action involving primary necrosis and concurrent inflammation; these adverse effects are not entirely unexpected. Moreover, important safety end points such as treatment-related scarring and pigmentary changes were not evident with the topical therapy.
Clinical findings ranging from mild epithelial keratoconjunctivitis to severe keratitis have made periocular area unsuitable for application. Safety in pregnant females and children less than 18 years of age has not been established.
The main advantage of the ingenol mebutate therapy is that similar degrees of efficacy can be achieved with only two or three daily applications. Two immediate benefits evidently derive from the short exposure to ingenol mebutate. The first benefit is the relatively rapid resolution of local reactions. On the face or scalp, where irritation is most noticeable, the peak local-skin-response score was recorded on day 4: the score declined rapidly thereafter and local reactions were almost resolved by the day 15 visit [Fig. 3]. The second benefit regards the short duration of treatment, which may result in very high (>98%) adherence to the therapy, thus contributing to the effectiveness of ingenol mebutate.[42] Several patients find it difficult to adhere to the currently available regimens of topical treatment that last for periods of 1 to 4 months — this may result in "real-world" effectiveness lower than that that achieved in supervised and patient- compensated clinical trials. The lack of response to treatments with imiquimod and fluorouracil may be due to failure in completing the regimens.
Figure 3.
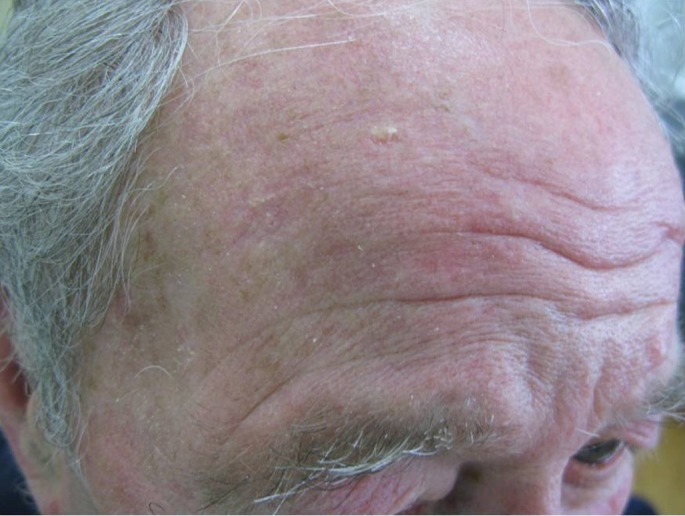
Successful teatment of actinic keratosis with ingenol mebutate after 15 days.
Ingenol mebutate, a new topical field therapy to address this common, chronic skin condition, can offer several advantages over other AK treatments currently on the market.
Procedural Field Therapy
Procedural field therapies may be an appropriate option for patients who: require minimal down time, are unlikely to adhere to a topical approach, have AKs resistant to topical therapy, or favor an optimal cosmetic result. Treatment options for procedural field therapy include: photodynamic therapy, manual dermabrasion, laser resurfacing, cryopeeling, and chemical peels. Each of these techniques treats AKs by destroying the superficial layers of the skin through physical or chemical means.
Photodynamic Therapy
Photodynamic therapy (PDT) is a procedural field therapy that uses topical 5-aminolevulinic acid (ALA) or methyl aminolevulinate (Metvix®/Metvixia®) to target AKs. These molecules preferentially find their way into the hyperproliferating cells, which lack normal cell to cell adhesion junctions, and are converted intracellularly to protoporphyrin IX (PpIX). This photosensitizer is then exposed to blue or red light, which corresponds to the peaks in the absorption spectrum of PpIX and results in a phototoxic reaction destroying the abnormal cell. 20 PDT is effective for the treatment of multiple and diffuse AKs, and its cosmetic results are generally excellent.[43] However, it is not ideal for treating thicker or deeper AKs and is normally reserved for patients who exhibit an inadequate response to topical field therapy or cryosurgery. Patients may experience erythema, edema, and a burning sensation during the light therapy.[44]
Conclusion
There is no widely accepted algorithm for the treatment of AKs. In many patients various different treatment regiments must be employed in order to successfully manage AKs, especially in widespread or resistant cases. As usual, the best way to manage AKs is prevention, i.e. avoiding exposure to significant or unnecessary UV radiation. Encouraging patients to wear broad-based sunscreens, widebrimmed hats, sunglasses, and to avoid exposure during peak hours may help prevent recurrence or limit the progression of AKs.
References
- Zalaudek I, Giacomel J, Schmid K, Bondino S, Rosendahl C, Cavicchini S, Tourlaki A, Gasparini S, Bourne P, Keir J, Kittler H, Eibenschutz L, Catricalà C, Argenziano G. Dermatoscopy of facial actinic keratosis, intraepidermal carcinoma, and invasive squamous cell carcinoma: a progression model. J Am Acad Dermatol. 2012;66:589–597. doi: 10.1016/j.jaad.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Marks R, Jolley D, Dorevitch AP, Selwood TS. The incidence of non-melanocytic skin cancers in an Australian population: results of a five-year prospective study. Med J Aust. 1989;150:475–478. doi: 10.5694/j.1326-5377.1989.tb136588.x. [DOI] [PubMed] [Google Scholar]
- Frost CA, Green AC. Epidemiology of solar keratoses. Br J Dermatol. 1994;131:455–464. doi: 10.1111/j.1365-2133.1994.tb08544.x. [DOI] [PubMed] [Google Scholar]
- Stockfleth E, Sibbring GC, Alarcon I. New Topical Treatment Options for Actinic Keratosis: a Systematic Review. Acta Derm Venereol. 2015. doi: 10.2340/00015555-2167. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Harvey I, Frankel S, Marks R, Shalom D, Nolan-Farrell M. Non-melanoma skin cancer and solar keratoses II analytical results of the South Wales Skin Cancer Study. Br J Cancer. 1996;74:1308–1312. doi: 10.1038/bjc.1996.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Foley P, Freeman M, Chong AH. Solar keratosis: epidemiology, pathogenesis, presentation and treatment. Australas J Dermatol. 2007; 48: 67– 74; quiz 75-6. Review. [DOI] [PubMed] [Google Scholar]
- Frost C, Williams G, Green A. High incidence and regression rates of solar keratoses in a Queensland community. J Invest Dermatol. 2000;115:273–237. doi: 10.1046/j.1523-1747.2000.00048.x. [DOI] [PubMed] [Google Scholar]
- Yu RC, Pryce DW, Macfarlane AW, Stewart TW. A histopathological study of 643 cutaneous horns. Br J Dermatol. 1991;124:449–452. doi: 10.1111/j.1365-2133.1991.tb00624.x. [DOI] [PubMed] [Google Scholar]
- Butani AK, Arbesfeld DM, Schwartz RA. Premalignant and early squamous cell carcinoma. Clin Plast Surg. 2005; 32: 223– 235. Review. Erratum in: Clin Plast Surg. 2006; 33: 497 [DOI] [PubMed] [Google Scholar]
- Rossi R, Mori M, Lotti T. Actinic teratosi. Int J Dermatol. 2007;46:895–904. doi: 10.1111/j.1365-4632.2007.03166.x. [DOI] [PubMed] [Google Scholar]
- Simon JC, Tigelaar RE, Bergstresser PR, Edelbaum D, Cruz PD Jr. Ultraviolet B radiation converts Langerhans cells from immunogenic to tolerogenic antigen-presenting cells. Induction of specific clonal anergy in CD4+ T helper 1 cells. J Immunol. 1991;146:485–491. [PubMed] [Google Scholar]
- Yoshikawa T, Rae V, Bruins-Slot W, Van den Berg JW, Taylor JR, Streilein JW. Susceptibility to effects of UVB radiation on induction of contact hypersensitivity as a risk factor for skin cancer in humans. J Invest Dermatol. 1990;95:530–536. doi: 10.1111/1523-1747.ep12504877. [DOI] [PubMed] [Google Scholar]
- Mortier L, Marchetti P, Delaporte E, Martin de Lassalle E, Thomas P, Piette F, Formstecher P, Polakowska R, Danzé PM. Progression of actinic keratosis to squamous cell carcinoma of the skin correlates with deletion of the 9p21 region encoding the p16(INK4a) tumor suppressor. Cancer Lett. 2002;176:205–214. doi: 10.1016/s0304-3835(01)00757-1. [DOI] [PubMed] [Google Scholar]
- Kanellou P, Zaravinos A, Zioga M, Stratigos A, Baritaki S, Soufla G, Zoras O, Spandidos DA. Genomic instability, mutations and expression analysis of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in actinic keratosis. Cancer Lett. 2008;264:145–161. doi: 10.1016/j.canlet.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Timares L, Katiyar SK, Elmets CA. DNA damage, apoptosis and langerhans cells--Activators of UV-induced immune tolerance. Photochem Photobiol. 2008;84:422–436. doi: 10.1111/j.1751-1097.2007.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman B, Cockerell CJ. Pathobiology of actinic keratosis: ultraviolet-dependent keratinocyte proliferation. J Am Acad Dermatol. 2013; 68 (1 Supp 1): PA1– A4, S10–S19 [DOI] [PubMed] [Google Scholar]
- Brash DE, Ziegler A, Jonason AS, Simon JA, Kunala S, Leffell DJ. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Investig Dermatol Symp Proc. 1996; 1: 136– 142. Review. [PubMed] [Google Scholar]
- Schmults CD, Karia PS, Carter JB, Han J, Qureshi AA. Factors predictive of recurrence and death from cutaneous squamous cell carcinoma: a 10-year, single institution cohort study. JAMA Dermatol. 2013;149:541–547. doi: 10.1001/jamadermatol.2013.2139. [DOI] [PubMed] [Google Scholar]
- Wang L, Eng W, Cockerell CJ. Effects of ultraviolet irradiation on inflammation in the skin. Adv Dermatol. 2002;18:247–286. [PubMed] [Google Scholar]
- Hruza LL, Pentland AP. Mechanisms of UV-induced inflammation. J Invest Dermatol. 1993;100:35S–41S. doi: 10.1111/1523-1747.ep12355240. [DOI] [PubMed] [Google Scholar]
- Berman B, Cockerell CJ. Pathobiology of actinic keratosis: ultraviolet-dependent keratinocyte proliferation. J Am Acad Dermatol. 2013;68 (1 Suppl 1):S10–S19. doi: 10.1016/j.jaad.2012.09.053. [DOI] [PubMed] [Google Scholar]
- Lober BA, Lober CW, Accola J. Actinic keratosis is squamous cell carcinoma. J Am Acad Dermatol. 2000;43 (5 Pt 1):881–882. doi: 10.1067/mjd.2000.108373. [DOI] [PubMed] [Google Scholar]
- Heaphy MR Jr, Ackerman AB. The nature of solar keratosis: a critical review in historical perspective. J Am Acad Dermatol. 2000;43 (1 Pt 1):138–150. doi: 10.1067/mjd.2000.107497. [DOI] [PubMed] [Google Scholar]
- Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”) J Am Acad Dermatol. 2000;42 (1 pt 2):11–17. doi: 10.1067/mjd.2000.103344. [DOI] [PubMed] [Google Scholar]
- Lebwohl MG, Rosen T, Stockfleth E. The role of human papillomavirus in common skin conditions: current viewpoints and therapeutic options. Cutis. 2010; 86: Suppl 1-11; quiz suppl 12. [PubMed] [Google Scholar]
- Jackson S, Harwood C, Thomas M, Banks L, Storey A. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 2000;14:3065–3073. doi: 10.1101/gad.182100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma ("actinic keratosis"). J Am Acad Dermatol. 2000; 42 (1 Pt 2): 11– 17. Review. [DOI] [PubMed] [Google Scholar]
- Padilla RS, Sebastian S, Jiang Z, Nindl I, Larson R. Gene expression patterns of normal human skin, actinic keratosis, and squamous cell carcinoma: a spectrum of disease progression. Arch Dermatol. 2010;146:288–293. doi: 10.1001/archdermatol.2009.378. [DOI] [PubMed] [Google Scholar]
- Neto PD, Alchorne M, Michalany N, Abreu M, Borra R. Reduced P53 Staining in Actinic Keratosis is Associated with Squamous Cell Carcinoma: A Preliminary Study. Indian J Dermatol. 2013;58:325. doi: 10.4103/0019-5154.113935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds A, Chia A, Shumack S. Actinic keratosis: Rationale and Managment. Dermatol Ther (Heidelb. 2014;4:11–31. doi: 10.1007/s13555-014-0049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai KE, Fergin P, Freeman M, Vinciullo C, Francis D, Spelman L, Murrell D, Anderson C, Weightman W, Reid C, Watson A, Foley P. A prospective study of the use of cryosurgery for the treatment of actinic keratosis. Int J Dermatol. 2004;43:687–692. doi: 10.1111/j.1365-4632.2004.02056.x. [DOI] [PubMed] [Google Scholar]
- Kaufman R. The concept of field cancerization. Melanoma Res. 2010;20:e13–e14. [Google Scholar]
- de Berker D, McGregor JM, Hughes BR, British Association of Dermatologists Therapy Guidelines and Audit Subcommittee. Guidelines for the management of actinic keratosis. Br J Dermatol. 2007; 156: 222– 230 (erratum in Br J Dermatol. 2008; 158: 873). [DOI] [PubMed] [Google Scholar]
- McIntyre WJ, Downs MR, Bedwell SA. Treatment options for actinic keratosis. Am Fam Physician. 2007;76:667–671. [PubMed] [Google Scholar]
- Rahvar M, Lamel SA, Maibach HI. Randomized, vehicle-controlled trials of topical 5-fluorouracil therapy for actinic keratosis treatment: an overview. Immunotherapy. 2012; 4: 939– 945. Review. Erratum in: Immunotherapy. 2012; 4: 1898. Rhavar, Maral [corrected to Rahvar, Maral]. [DOI] [PubMed] [Google Scholar]
- Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007; 157 Suppl 2: 8– 13. Review. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Cooper EA, Abramovits W. Zyclara (imiquimod) cream, 3.75% Skinmed. 2010;8:227–229. [PubMed] [Google Scholar]
- Rosen RH, Gupta AK, Tyring SK. Dual mechanism of action of ingenol mebutate gel for topical treatment of actinic keratosis: rapid lesion necrosis followed by lesion-specific immune response. J Am Acad Dermatol. 2012;66:486–493. doi: 10.1016/j.jaad.2010.12.038. [DOI] [PubMed] [Google Scholar]
- Siller G, Gebauer K, Welburn P, Katsamas J, Ogbourne SM. PEP005 (ingenol mebutate) gel, a novel agent for the treatment of actinic keratosis: results of a randomized, double-blind, vehicle-controlled, multicentre, phase IIa study. Australas J Dermatol. 2009;50:16–22. doi: 10.1111/j.1440-0960.2008.00497.x. [DOI] [PubMed] [Google Scholar]
- Anderson L, Schmieder GJ, Werschler WP, Tschen EH, Ling MR, Stough DB, Katsamas J. Randomized, double-blind, double-dummy, vehicle-controlled study of ingenol mebutate gel 0.025% and 0.05% for actinic keratosis. J Am Acad Dermatol. 2009;60:934–943. doi: 10.1016/j.jaad.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Lebwohl M, Swanson N, Anderson LL, Melgaard A, Xu Z, Berman B. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010–1019. doi: 10.1056/NEJMoa1111170. [DOI] [PubMed] [Google Scholar]
- Lebwohl M, Shumack S, Stein Gold L, Melgaard A, Larsson T, Tyring SK. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratosis. JAMA Dermatol. 2013;149:666–670. doi: 10.1001/jamadermatol.2013.2766. [DOI] [PubMed] [Google Scholar]
- Serra-Guillén C, Nagore E, Hueso L, Traves V, Messeguer F, Sanmartín O, Llombart B, Requena C, Botella-Estrada R, Guillén C. A randomized pilot comparative study of topical methyl aminolevulinate photodynamic therapy versus imiquimod 5% versus sequential application of both therapies in immunocompetent patients with actinic keratosis: clinical and histologic outcomes. J Am Acad Dermatol. 2012;66:e131–e137. doi: 10.1016/j.jaad.2011.11.933. [DOI] [PubMed] [Google Scholar]
- Van der Geer S, Krekels GA. Treatment of actinic keratosis on the dorsum of the hands: ALA-PDT versus diclofenac 3% gel followed by ALA-PDT. A placebo-controlled, double-blind, pilot study. J Dermatolog Treat. 2009;20:259–265. doi: 10.1080/09546630902882048. [DOI] [PubMed] [Google Scholar]