

Abstract
MicroRNAs (miRNAs) are small RNAs (~22 nt long) that post-transcriptionally regulate the expression of thousands of genes in a broad range of organisms, in both normal physiologic and disease contexts. MiRNA expression profiling is gaining popularity because miRNAs, as key regulators in gene expression networks, can influence many biological processes and have also shown promise as biomarkers for disease. Technological advances have enabled the development of various platforms for miRNA profiling, and an understanding of the strengths and pitfalls of different approaches can aid in the effective use of miRNA profiling for diverse applications. We review here the major considerations for carrying out and interpreting results of miRNA profiling studies, as well as current and emerging applications of miRNA profiling.
Keywords: miRNA, microRNA, methods, profiling
Introduction
Since their discovery1–3, microRNAs (miRNAs) have come to be recognized as critical regulators of gene expression in plants and animals. MiRNAs are short (~22 nucleotide) non-coding RNAs that regulate gene expression by binding to specific messenger RNA (mRNA) targets and promoting their degradation and/or translational inhibition. In most organisms, there are a relatively limited number of miRNAs compared to the number of mRNAs and proteins (for example, the human genome is believed to encode ~1000 miRNAs, whereas the number of mRNAs is typically estimated at >30,000). However, one miRNA may regulate hundreds of mRNAs and as a result have substantial effects on gene expression networks. Consequently, miRNA expression patterns can be especially rich in biological information, since variation in expression of hundreds of messenger RNAs may, to an extent, be reflected in the expression patterns of one or a few miRNAs that regulate them.
MicroRNA expression profiling has proven to be useful for identifying miRNAs that are important in regulation of a range of processes, including organismal development and establishment and maintenance of tissue differentiation. As a result, miRNA expression biomarkers are finding application in diagnosis of tissue differentiation state in cancers of unknown origin, and miRNAs are being investigated as reagents for re-programming of cell fate in stem cell applications. For investigators studying mechanisms of gene regulation, measuring miRNA expression can inform systems-level models of gene regulation, especially when miRNA information is combined with mRNA profiling and other genome-scale data. Finally, miRNAs have been shown to be much more stable than mRNAs in a range of specimen types – including blood plasma/serum, urine and formalin-fixed issue blocks – and are also measurable with much greater sensitivity than proteins (due to the fact that they can be amplified by RT-PCR). This has led to considerable interest in development of miRNAs as biomarkers for diverse molecular diagnostic applications, including in cancer, cardiovascular and autoimmune diseases, as well as to development of miRNA-based methods for forensic analysis where highly sensitive measurement of these molecules may provide information such as the cellular composition of forensic samples, for example.
Accordingly, miRNA profiling has become of interest to investigators working in diverse research areas of biology and medicine. Here we define miRNA profiling as measurement of the relative abundance of a cohort of miRNAs, ranging from a group of several miRNAs of specific biological interest, to comprehensive profiling of all miRNAs in a given species (typically numbering in the several hundreds). Profiling of such small RNA molecules poses some inherent challenges, but substantial technological advances in recent years have overcome many of these barriers and a wide range of approaches and platforms are now available for miRNA profiling.
In this article we aim to explain fundamental details of miRNAs relevant to miRNA profiling experiments, describe established and emerging methods for measuring miRNA expression profiles in a variety of biological samples (e.g., cells, tissues, and body fluids), and highlight strengths and limitations of different profiling approaches for specific biological applications. Our goal is to provide the reader an informed perspective for embarking on miRNA profiling studies or interpreting miRNA profiling results from the literature, as well as to highlight some of the biological and clinical applications that miRNA profiling is being applied to.
MicroRNA Characteristics
The considerations for planning and interpreting miRNA profiling experiments are related to the characteristics of these small RNAs and their mode of biogenesis and function. The basic pathway of miRNA biogenesis is described in Figure 1 and is well-summarized in recent reviews.4,5 Key points to bear in mind are: although mature miRNAs are typically ~22 nt in length, they originate from much longer primary transcripts that may be hundreds to thousands of nucleotides in length; biogenesis of mature miRNAs occurs through a multi-step process which begins with cleavage of the primary transcript (known as the pri-miRNA) to yield an approximately 70–100 nucleotide hairpin microRNA precursor (known as the pre-miRNA), and further cleavage to produce the mature miRNA, meaning that pre- and pri- miRNAs need to be distinguished from mature miRNAs in profiling analyses; and mature miRNAs are loaded into the RNA-induced silencing complex (RISC) where they are bound by a member of the Argonaute (AGO) family of RNA-binding proteins.
Figure 1. miRNA Biogenesis.
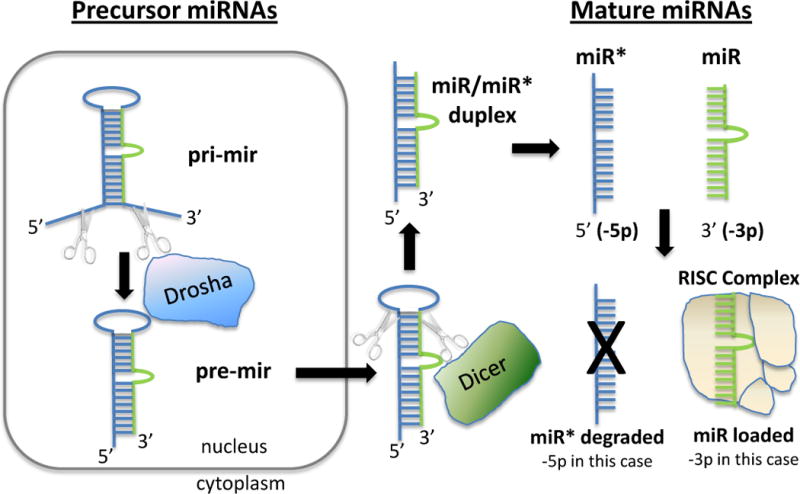
Primary microRNAs (pri-mir) are processed by Drosha to pre-miRNA (pre-mir) in the nucleus. Pre-miRNA are exported to the cytoplasm and further processed by Dicer to an miRNA:miRNA* duplex comprised single stranded miRNA derived from the 5′ (5p) and 3′ (3p) regions of the precursor. The duplex is unwound, and one of the strands is loaded into the RISC complex (3p in the figure), while the other passenger strand (miR*) is usually degraded (5p in the figure).
Although microRNA represents only ~0.01% of total RNA by weight, the average copy number of an individual miRNA species has been estimated at 500/cell, which may be higher than the average expression of mRNA species.6 Individual miRNA species in cells vary in concentration over a dynamic range of at least four orders of magnitude, with some cell type-restricted miRNAs present at greater than 10,000 copies per cell.7 The reason miRNAs are so numerous while representing only a tiny fraction of total RNA quantity is that miRNAs have a much lower molecular weight compared to mRNA, and are also much less diverse. To date, the total number of mature miRNAs in humans is estimated at ~1,000, and numbers in other well-studied organisms such as D. melanogaster, C. elegans, and A. thaliana range between ~200–300 (mirBase v18, accessed January 22, 2012).
It is important to consider that different mature miRNA species can be produced from a single pre-miRNA molecule (Figure 1). Distinct miRNAs are generated from the 3′ and 5′ arms of the pre-mRNA duplex.12 In addition, each mature miRNA often comprises a distribution of sizes centered around 22 nt, rather than a discrete single length. As an example, RNA sequencing data deposited on miRBase for human miR-16 shows that 77.2% of sequencing reads are 22 nt, 19.4% are 23 nt, and the remaining ~4% of miR-16 species are less than 22 nt.13 In many cases, this variation in mature miRNA length is due to 3′ post-transcriptional modifications (including additions and/or deletion of nucleotides), which have in some cases been shown to affect miRNA stability or function.14,15 Understanding the current and historical nomenclature used to describe related miRNAs (e.g., miR* vs. miR-3p/-5p nomenclature) is important and a summary of the key elements of miRNA nomenclature is given in Box 1.
Box 1. MicroRNA nomenclature.
|
| ||
| Naming Convention | Meaning | Example(s) |
|
| ||
| 3 letter prefix | Species identification. This prefix is sometimes dropped in some journals, with the species under consideration mentioned in the adjoining text. | hsa (human) dme (fruit fly) |
|
| ||
| pri-mir or pre-mir (not lower case “r”) | Used to indicate primary transcript of miRNA (pri-mir) or precursor form after Drosha processing (pri-mir) | pri-mir-16, pre-mir-16 |
|
| ||
| miR | Mature miRNA | hsa-miR-16 |
|
| ||
| −3p or −5p | Mature miRNA originating from the 3′ or 5′ end of the pre-miRNA, respectively | hsa-miR-142-3p hsa-miR-142-5p |
|
| ||
| a or b | Closely related miRNAs that are related in sequence and evolutionary origin | hsa-miR-20a hsa-miR-20b |
|
| ||
| −1, or −2 | Identical mature miRNA sequences that originate from different genomic loci | hsa-miR-16-1 hsa-miR-16-2 |
|
| ||
| miR* (star) | ‘Passenger strand’ found at lower concentration, usually degraded (retired in miRBase 17) | hsa-miR-9* |
|
| ||
Naming conventions for miRNAs have been evolving and can be confusing to newcomers in the field. Most miRNAs are named with a species prefix and a number that designates the specific miRNA. For example, hsa-miR-21 indicates Homo sapiens microRNA number 21, as hsa is the prefix for Homo sapiens. Many journals, including this one, do not include species prefixes in miRNA names, so we drop species prefixes in this review. Prefixes may also be added to the name to convey information about mature vs. miRNA primary transcript (e.g., pri-mir-21 and pre-mir-21). In addition, suffixes are sometimes added to designate whether the mature miRNA arose from the 3′ or 5′ arm of the pre-miRNA. For example, miR-486-5p designates a mature form based on the capitalization of the ‘R’ in ‘miR’ that arose from the 5 prime (i.e.,-5p) arm of the pre-miRNA hairpin.88 MiRNAs that comprise families that are related in sequence may have lower case letters following the name (e.g. miR-20a and miR-20b). In some cases, multiple transcriptional units at different loci in the genome encode miRNAs that are identical in sequence in their mature form; in this case a numerical suffix is appended by a dash to designate the different genomic origins (e.g. miR-16-1 and miR-16-2). The ‘*’ (star) that is appended to some miRNA names (e.g. miR-9*), especially in datasets generated using older miRNA profiling platforms. The designation of ‘*’ is meant to indicate the “minor species” of the two mature miRNAs that are produced from the 3′ and 5′ arms of the pre-miRNA duplex (i.e., forming the miRNA:miRNA* duplex). It is now recognized that both the dominant (non-star) and ‘*’ forms can be functional89 and may be present at comparable concentrations in the cell, or the miRNA* form might even be at higher concentration depending on the precursor gene used and the cell, tissue, or species90 being examined. For these reasons, it has been recommended the miRNA/miRNA* nomenclature be dropped in favor of using ‘−3p’ or ‘−5p’ suffix in every case.88
MiRbase (http://www.mirbase.org)8,9 has emerged as a definitive repository of miRNA sequences as well as an authoritative source of miRNA nomenclature that is a valuable resource for miRNA profiling studies. Originally called the microRNA Registry10, the current version of miRBase has a total of 18,226 entries of hairpin pre-cursor miRNAs expressing 21,643 mature miRNA products (miRBase version 18, accessed December 9, 2011) encompassing a range of plant and animal species. In addition to providing information about predicted precursor hairpin sequences and experimentally identified mature miRNA sequences, miRBase provides an interface to quantitative RNA sequencing data deposited in the NCBI gene expression omnibus (GEO) as well as links to miRNA target prediction databases.11
Sample Considerations
Sample processing and RNA extraction methods can have a significant impact on the results of miRNA profiling, particularly for samples prone to miRNA degradation.16–19 In this section we highlight specimen considerations (Table 1) and the major methodologies for miRNA isolation and quality control issues as they relate to different sample types such as tissues and body fluids (Figure 2). Although the study of plant miRNAs is a dynamic and highly productive area of ongoing research, we will focus here on profiling of miRNAs in animals.
Table 1.
Sample Considerations
| Sample type | Applications | miRNA Yield* | Considerations |
|---|---|---|---|
| Cell lines | Gene Regulation | +++ | High quality miRNA usually |
| Fluorescence activated cell sorted cells | Translational, Basic Science | ++ | Lower yield, less heterogeneity |
| Fresh tissue (e.g., tumor) | Translational Research | +++ | Cell heterogeneity |
| Formalin-fixed, paraffin-embedded tissue | Clinical, Translational Research | ++ | More reliable than mRNA as an analyte in FFPE |
| Laser-capture microdissected tissue | Translational Research | + | Less heterogeneity, but lower yeild, never completely pure |
| Plasma, Serum | Biomarkers | + | RNAses, low yield, typically cannot evaluate quality or quantity |
| Urine | Biomarkers | + | Can evaluate cell pellet vs. supernatant |
+ <1ng, ++ 1–10ng, +++ >10ng
Figure 2. miRNA Profiling Workflow.
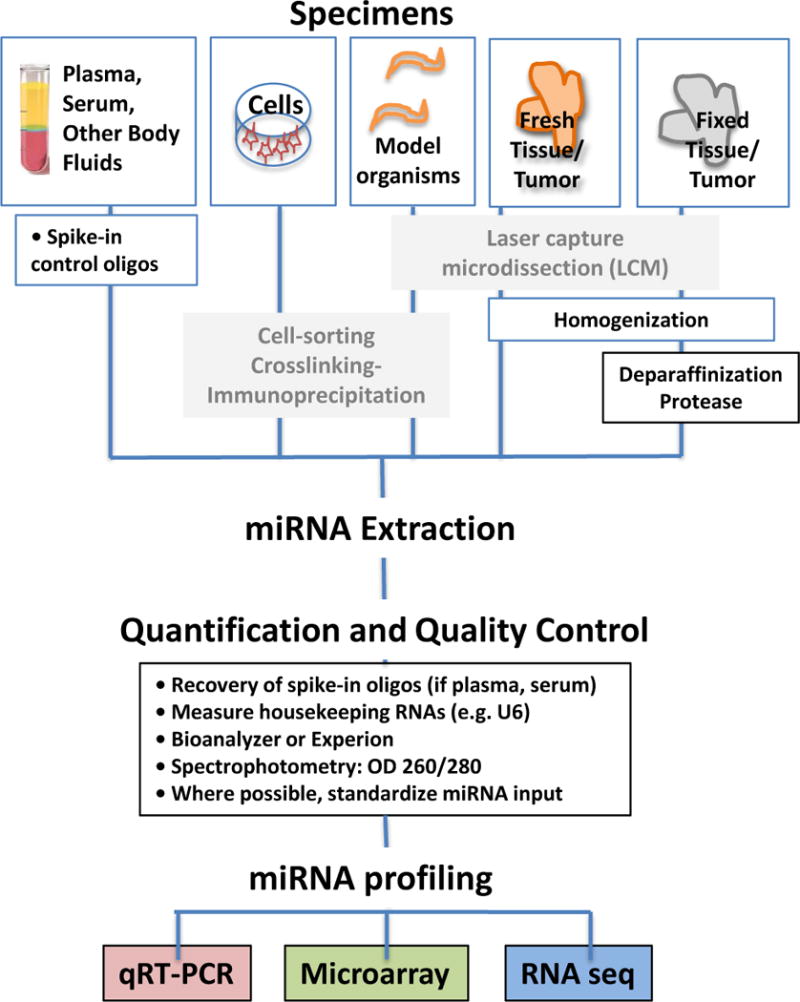
A) MicroRNAs can be extracted from a variety of specimen types, highlighted here are plasma and serum, cells in culture, fresh tissue/tumor, or fixed tissue/tumors. B) Methods for purifying miRNA populations of interest include size purification by gel electrophoresis, and AGO2 immunoprecipitation (AGO2-IP), with or without UV crosslinking (CLIP). Laser capture microdissection (LCM) can be used to purify material from fresh-frozen or formalin-fixed paraffin embedded (FFPE) tissue sections. C) MicroRNA isolation methods are similar to total RNA isolation, and commercially available extraction kits are listed in the light orange boxes. These typically use a chemical extraction combined with a purification step involving binding and eluting from a silica column. Formalin fixed (FF), or formalin fixed, paraffin embedded (FFPE) tissue/tumor require additional deparaffinization and protease treatment, and commercially available kits designed for these sample types are also listed in the light orange boxes. We recommend adding spike-in oligos prior to RNA extraction to enable a quality control check, particularly when using plasma or serum samples. D) A variety of methods can be used to assess miRNA quality after extraction. For most samples, these methods include spectrophotometry, automated capillary electrophoresis with the Bioanalyzer or Experion, and/or determining expression of housekeeping miRNAs. For serum and plasma, which usually have total RNA yields too low to accurately quantify, determining the recovery of spiked-in oligos can be a useful surrogate.
Sample Types and miRNA Extraction
It is possible to extract high-quality microRNA from a wide range of cell and tissue sources, including cell lines, fresh tissues, formalin-fixed paraffin embedded tissues, plasma, serum, urine, and other body fluids (Table 1).20,21 The principles for isolating miRNA are in general the same as for isolation of total RNA, except that miRNA isolation protocols are slightly modified to retain (and sometimes enrich) the small RNA fraction.21 Widely used commercially-available products such as miRNeasy (Qiagen), mirVana™ (Ambion), and PureLink™ (Invitrogen) miRNA isolation kits are based on chemical extraction using concentrated chaotropic salts such as guanidine thiocyanate (e.g. Trizol and QIAzol® reagents) followed by a solid-phase extraction procedure on silica columns.
Tissues and cell lines generally yield high-quality miRNA that is suitable for profiling studies. This is true even for formalin-fixed paraffin embedded tissue (FFPE), in which profiles of extracted miRNA have been shown to correlate well with those obtained from matched frozen reference samples.22 Unlike mRNA, which is highly fragmented and generally less reliable in FFPE compared to fresh tissue, miRNA is surprisingly stable and intact in FFPE, which appears to be largely independent of formalin fixation time and duration of tissue block storage.22,23 This stability offers a distinct advantage of miRNA over mRNA as a tissue analyte in the clinical setting, where FFPE may be the only sample type available. Commercial kits such as the High Pure miRNA isolation kit (Roche), RecoverAll™ total nucleic acid kit (Ambion), and miRNeasy FFPE kit (Qiagen) recover miRNA from FFPE samples following deparaffinization and protease treatment.22,23 At least two groups have compared different miRNA FFPE kits and described quantitative and qualitative differences in miRNA profiling results that could be attributed to the extraction method.22,24
Cell heterogeneity is an important consideration in miRNA profiling studies involving tissues because shifts in the proportion of different cell types (for example, variation in the number of infiltrating inflammatory cells in tissue samples) may influence the expression profiles obtained. This is especially pertinent to miRNA profiling because many miRNAs are expressed in a tissue-specific manner. Methods such as laser-capture microdissection or fluorescent-activated cell sorting can be used to enrich cells of interest when cell-specific miRNA signatures are sought.25
For some specimen types the preparation and miRNA extraction methods may need to be specially optimized, based on an understanding of the sample type being investigated and knowledge of variables that influence extraction efficiency or cause qualitative shifts in miRNA profiles. For example, human blood plasma is a challenging specimen type due to high levels of endogenous RNase activity and additional sources of variation. Although endogenous plasma miRNAs are protected from RNase in their native state, they are degraded within seconds if extracted and spiked back into plasma, suggesting that RNA extraction methods that fail to completely and rapidly inactivate RNase are not suitable for plasma or serum specimens.26,27 In addition, for these specimen types, other “pre-analytic” variables such as centrifugation conditions, white blood cell counts, and red blood cell hemolysis may also dramatically influence the nature and quantity of miRNA that is extracted from plasma, indicating that these need to be considered in the design of a plasma profiling study.28–30 Furthermore, miRNAs exist in at least two distinct physical states in blood plasma – within vesicles/exosomes, or associated with small AGO2-containing protein complexes – suggesting that specimen processing conditions that alter vesicle content will influence miRNA profiles.31 Although RNA extraction from tissues is well established now as a scalable procedure amenable to high-throughput methods, for highly proteinaceous liquid samples (e.g., plasma and serum) a method for highly efficient miRNA extraction that can be easily scaled up for large volumes and/or number of samples remains elusive.
For most profiling applications it is not necessary to enrich the miRNA component of the cellular RNA because many profiling platforms are designed to distinguish between miRNAs and the many other, more abundant RNA species present in total cellular RNA. If enrichment is required, low molecular weight RNA (e.g., in the 18–24 nt range to broadly capture miRNAs) can be recovered by size fractionating and gel-purifying the desired size range fraction on a polyacrylamide gel27; for more crude enrichment, column-based protocols also exist.
Quality and Quantity Assessment
Assessment of the quality and quantity of extracted RNA is important for reproducibility and accuracy in miRNA profiling studies. Because many profiling methods can be performed using total RNA, assessment of the miRNA population specifically is not always necessary and it is routine to assess yield and degree of overall RNA integrity using spectrophotometry and automated capillary electrophoresis instruments such as the Bioanalyzer 2100 (Agilent) and Experion (Bio-Rad). A small RNA chip is available for the Bioanalyzer 2100 for estimating miRNA abundance, expressed as the quantity of RNA in the 15–40 nt window relative to total RNA abundance. However, the estimation of miRNA abundance by this method may only be accurate when overall RNA integrity is very high.32
Where assessment of RNA extraction efficiency is important (e.g., in acellular liquid specimens like plasma or serum where extraction efficiency can vary from sample to sample), a known amount of synthetic miRNAs that are not expressed in the biological sample can be “spiked-in” at an early step in RNA isolation (e.g. a mixture of three non-conserved C. elegans miRNAs has been used for plasma samples33). Measurements of these spiked-in miRNAs in the recovered RNA can serve as a control for RNA extraction efficiency.
MicroRNA Profiling Methods
In this section, we review general concepts and special challenges relevant to miRNA profiling. We will briefly discuss methodology, highlight strengths and limitations and cite examples of successful use. Table 2 contains a summary of important features and lists commercial vendors for many of the platforms that are in common use.
Table 2.
MicroRNA Profiling Technologies
| Advantages | Disadvantages | Assay/Platform | Vendor | RNA* required | material costs per sample** | reference | |
|---|---|---|---|---|---|---|---|
| qRT-PCR | Established method, sensitive and specific. Easily adapted to existing real-time PCR workflow. Can be used to determine absolute quantification. Customizable | Cannot identify novel miRNA (including in less frequently studied species) | TaqMan® individual Assays | ABI | + – ++ | $ – $$ | Chen, et al 2005, PMID 21642990 |
| miRcury LNA™ qPCR Primers | Exiqon | ||||||
| TaqMan® OpenArray | ABI | ||||||
| TaqMan® TLDA Microfluidics Card | ABI | Junker, et al 2009 | |||||
| Biomark™ HD System | Fluidigm | Petriv, et al 2010 | |||||
| SmartChip Human MicroRNA | Wafergen | Keller, et al 2011 | |||||
| miScript miRNA PCR Array | SABiosciences/Qiagen | ||||||
| MicroRNA Microarray | Established method. Easily adapted to existing microarray workflow. | Lower specificity than RNAseq. Cannot be used to determine absolute quantification. | Geniom Biochip miRNA | CBC (febit) | ++ | $ | Keller, et al 2011 |
| miRCURY LNA™ microRNA Array | Exiqon | ||||||
| μParaFlo™ Biochip Array | LC Biosciences | ||||||
| MicroRNA Microarray | Agilent | ||||||
| GeneChip® miRNA Array | Affymetrix | ||||||
| OneArray® | Phalanx Biotech | ||||||
| Sentrix® Array Matrix and BeadChips | Illumina | ||||||
| GenoExplorer™ | Genosensor | ||||||
| RNA Sequencing | High accuracy and sensitvity. Can detect novel miRNAs | Significant computational support needed for data analysis. Cannot be used to determine absolute quantification. | High Throughput Next Generation Sequencing Platforms | +++ | $$$ | ||
| HiSeq™ 2000 (Genome Analyzer IIX) | Illumina | Kato, et al 2011 | |||||
| SOLiD™ | ABI | Ramsingh, et al 2010, Schulte, et al 2010 | |||||
| GS FLX+ (454) | Roche | Wyman, et al 2009; Rajagopalan et al 2006, Soares et al 2012 | |||||
| Smaller Scale Next Generation Sequencing Platforms | |||||||
| Ion Torrent™ | Invitrogen | ||||||
| MiSeq™ | Illumina | ||||||
| GS Junior (454) | Roche | ||||||
| Amplification not required. Potential to determine absolute quantification | Expensive, currently less sensitive | Single Molecule Sequencing Technologies | $$$$ | ||||
| tSMS™ | Helicos | Kapranov, et al 2010 | |||||
| SMRT™ | Pacific Biosciences | ||||||
+ <ng, ++ ng-μg, +++ >μg
excludes cost of instrument and RNA extraction
General Concepts and Special Challenges in miRNA Profiling
Many properties unique to miRNAs pose challenges to their accurate detection and quantification.34 For instance, the ~22nt length of mature miRNAs is insufficient for annealing to traditional primers designed for reverse transcription and PCR. In addition, unlike mRNAs, miRNAs lack a common sequence such as a poly(A) tail that can be used for selective enrichment or as a universal primer binding site for reverse transcription. This is significant because miRNAs represent a small fraction (~0.01%) of the total RNA mass and must therefore be selectively detected in a background of other, diverse RNA species35, including pri- and pre-miRNA precursors that also contain the RNA sequence of the mature miRNA species. In addition, miRNAs within a family (e.g. let-7 family) can differ by as little as a single nucleotide, making the ability to discriminate between forms with single nucleotide differences important. Finally, many miRNAs have variants (so-called “isomiRs”) resulting from post-transcriptional nucleotide additions to 3′ ends of mature miRNAs and measurement of different forms may be needed in some cases.14,36 Another challenge for profiling hundreds of miRNAs in parallel is that owing to their short length, variance in miRNA GC content leads to wide variance in melting temperatures (Tm) for annealing reactions, creating significant miRNA-specific biases. Despite these challenges, three major approaches are currently well-established: qRT-PCR, hybridisation-based methods (e.g., DNA microarrays), and high-throughput sequencing (i.e., RNAseq). We discuss these in detail below, and provide in Figure 4 a decision-tree to aid in determining which approach is best-suited for a given application.
Figure 4.
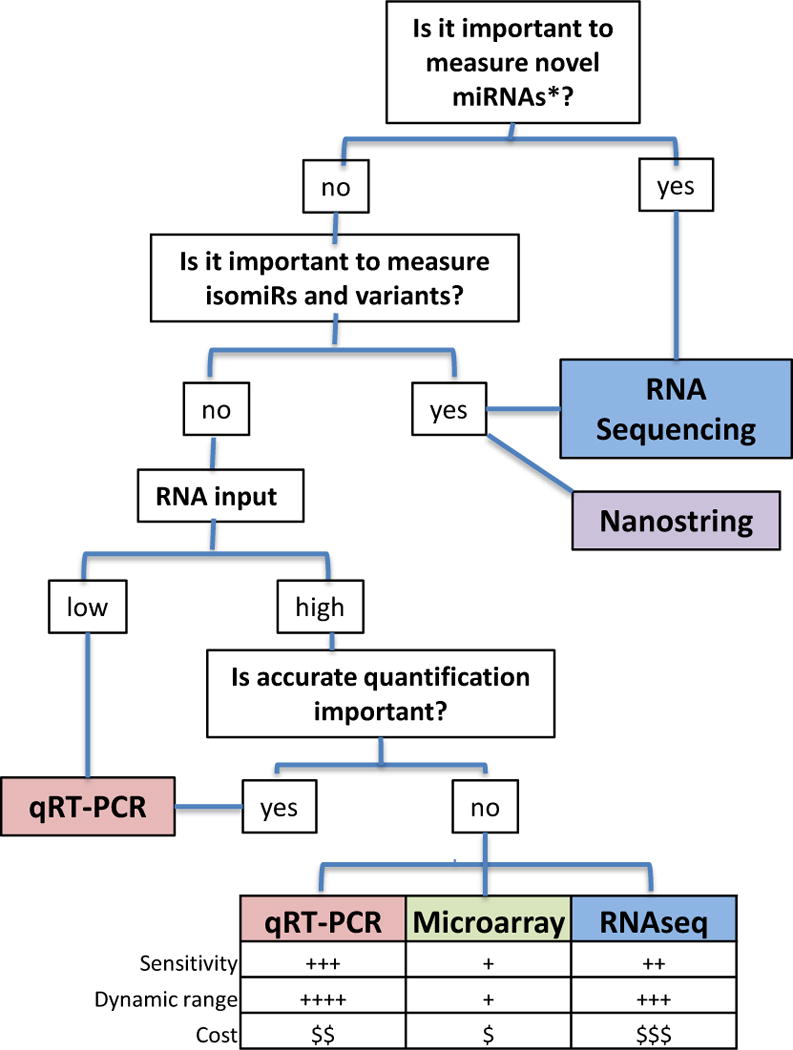
Choosing an miRNA Profiling Platform.
Quantitative Reverse Transcription-PCR-based Methods
One major approach relies on reverse transcription of miRNA to cDNA, followed by quantitative polymerase chain reaction (qRT-PCR), with real-time monitoring of reaction product accumulation (i.e., “real-time PCR”). An appealing aspect is ease of incorporation into workflow for laboratories familiar with real-time PCR. In order to scale this approach for miRNA profiling, reactions are carried out in a highly parallel, high-throughput form (i.e., hundreds of qRT-PCR reactions measuring different miRNAs using the same reaction conditions). Commercially available customizable plates and cards can be designed to examine a small set of miRNAs regulating a pathway of interest, for example, or more comprehensive coverage (see Table 2). Two main strategies are used for priming the reverse transcription reaction to generate cDNA: addition of a poly(A) tail using E. coli poly(A) polymerase and generation of a reverse transcription primer binding site using a stem-loop primer (Figure 3A).
Figure 3. Approaches to miRNA Profiling.
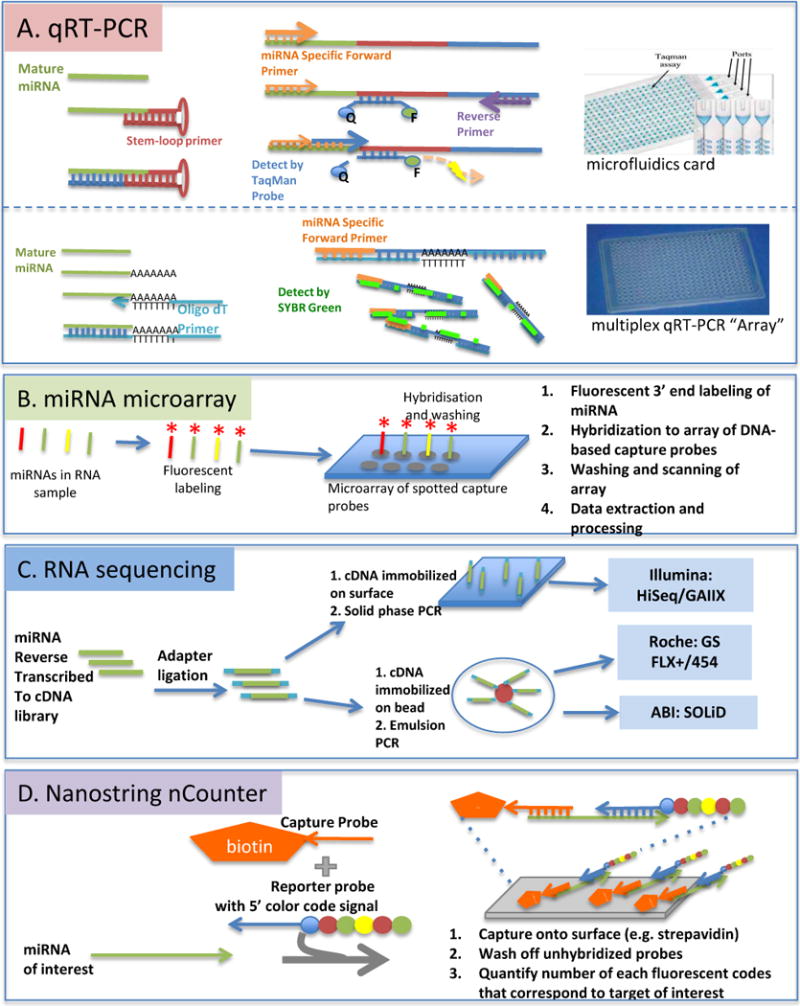
(A) qRT-PCR. In TaqMan qRT-PCR the reverse transcription (RT) reactions use stem loop primers specific to the 3′ end of the miRNA for specificity (A, top left). Amplicons are generated using an miRNA-specific forward primer, where DNA polymerase proceeds along template, the TaqMan probe is hydrolyzed so the quencher is freed from fluorescent dye, resulting in light emission (A, top middle). In SYBR green-based qRT-PCR miRNA is polyadenylated at the 3′ end and Oligo d(T) used as an RT-primer (A, bottom left). An miRNA-specific forward primer and oligo d(T) enable PCR amplification with dsDNA-intercalating SYBR green dye as the detector (A, bottom middle). Both TaqMan and SYBR green-based qRT-PCR are available in medium format “array” (A, right). (B) miRNA Microarray. DNA-based capture probes (which may or may not incorporate LNA-modified bases) are used to capture fluorescently-tagged miRNAs, followed by scanning of slides and quantification of fluorescence. Several variations on this approach exist. (C) RNA Sequencing. Current established RNA sequencing platforms begin with reverse transcription of miRNA to a cDNA library. Adaptor ligation then allows the library to either be affixed to a solid phase as in the Illumina platform or to beads for emulsion PCR as in the Roche and ABI platforms. For details of RNA sequencing chemistry see Metzker 2010.40 (D) Nanostring NCounter: Two target-specific probes are designed for each miRNA of interest: a 3′ capture probe containing biotin to allow adsorbance to solid phase via strepavidin, and a second 5′ reporter probe with an individual color-coded sequence. No amplification or labeling is required with this method.
A hurdle in performing highly parallel qRT-PCR is that optimal reaction conditions may vary substantially between miRNAs due to sequence-specific differences in primer annealing. Although different vendors have sought to solve this problem using a variety of approaches, one effective strategy has been the incorporation of locked nucleic acids (LNAs) into primers to standardize optimal miRNA primer hybridisation conditions for the hundreds of PCR assays to be run simultaneously.
For large-scale miRNA profiling (i.e., hundreds of miRNAs) using qRT-PCR, several medium-throughput platforms are available that use pre-plated PCR primers distributed typically across multiwell dishes, or alternatively microfluidic cards containing nanoliter-scale wells (Figure 3A). Because qPCR array plates can accommodate a larger RNA input volume than microfluidics-based approaches where reaction volume is exceptionally small, they may be better-suited for profiling applications in samples with very little input or low concentration miRNA such as plasma, serum, and other body fluids.30,31
Hybridisation-Based Methods
Microarrays were among the first methods used for parallel analysis of large numbers of microRNAs. After RNA purification, miRNAs are tagged with fluorophore-labeled nucleotides at their 3′ end, often using T4 RNA ligase (Figure 3A). Detection of individual miRNAs occurs by hybridization of the labeled miRNA to discretely arranged capture probes (complementary synthetic DNA oligonucleotides), which are “arrayed” into spots on a slide or on beads. Locked nucleic acids (LNAs) can be incorporated into capture probes to effectively normalize otherwise highly variable Tm.38 Commercially available microarrays differ in miRBase content, species coverage, and incorporation of LNA-modified capture probes (Table 2).
MicroRNA microarrays have the advantage of generally being less expensive than the other profiling methods discussed and yet allow a larger number of parallel measurements. Most research institutions have access to experienced microarray facilities, and adapting existing workflow to miRNA profiling is relatively straightforward. MicroRNA microarrays are best used for comparing relative abundance of specific microRNAs between two states (e.g., treatment vs. control or disease vs. healthy), and, importantly, cannot be used to determine absolute quantification. Because of limited specificity, initial observations are typically validated by a second method, such as qRT-PCR or northern blot.
A recent innovation in miRNA profiling based on hybridization is the Nanostring nCounter® in which a multiplexed probe library is created using two sequence-specific capture probes tailored to an miRNA of interest (Figure 3B).39 Advantages of this method are the direct measurement of RNA without need for amplification or cloning, multiplexed detection of multiple miRNAs in a single reaction, and the ability to discriminate between similar variants with high accuracy. For example, the technology has been used to discriminate 3′ end nucleotide variants of mature miRNAs.14 Current limitations may include limited accessibility of the instrument and the need for increased open source software tools for data analysis.
RNASeq
The advent of “next-generation sequencing” platforms has enabled a third major approach for miRNA expression profiling, “RNA-seq”. The general approach begins with the preparation of a small RNA cDNA library from the RNA sample of interest, followed by the “massively parallel” sequencing of millions of individual cDNA molecules from the library on a single run (Figure 3C). Bioinformatic analysis of the sequence reads identifies both known and novel microRNAs in the datasets, and provides relative quantification using a digital approach (i.e., the number of sequence reads for a given miRNA relative to the total reads in the sample is an estimate of relative abundance of the miRNA). The principal next-generation technologies have been well-described in the literature.40 A summary of the important distinguishing features of these platforms with respect to miRNA profiling is in Table 2.
The major advantages of next-generation sequencing for miRNA profiling are: detection of known and novel miRNAs; and precise identification of miRNA sequences (e.g., can readily distinguish between variants differing by a single nucleotide, as well as isomiRs of varying length). It should be noted, however, that RNAseq-based miRNA profiling studies typically identify a plethora of small RNAs of novel sequence (i.e., putative miRNAs), yet not all of these may not be bona fide miRNAs. Criteria have been established for the annotation of small RNA sequences as miRNAs, and these include sequence length of approximately 22nt, genomic origin that predicts a precursor RNA sequence that can fold into a duplex structure, identification in the data of both a −3p and −5p version, and conservation across species.41,42
Drawbacks to next-generation sequencing include high cost, the computational infrastructure required for data analysis and interpretation, as well as the platform-independent biases introduced by library-preparation methods which preferentially capture different miRNAs.43–45 However, smaller, faster and more affordable next-generation sequencing instruments are currently being introduced (Table 2), which should make sequencing increasingly available.
Single molecule real-time sequencing (SMRT) or single molecule sequencing (SMS) methods promise faster and less biased output than methods currently in use (Table 2). However, in their current versions, they are hampered by both higher error rates and cost, and are not yet widely available. While they have been used to study short RNA species, to our knowledge, they have yet to be applied to profiling of miRNAs.46
Data Analysis and Interpretation
In this section we briefly discuss the general approach for analysis of microRNA profiling data. For a discussion of experimental and computational approaches to miRNA target prediction and associated software tools, we point readers to recent reviews.47,48
The processes comprising basic miRNA profiling data analysis can be summarized as: data processing, data quality assessment, data normalization, and calculation of differential expression. The optimal approach to data analysis depends on the platform selected for miRNA profiling and the nature of the experiment.
For processing of the raw data, the software being used should be carefully investigated before embarking on miRNA profiling studies. For example, it is useful to know if there is any pre-processing of the data by instrument software (e.g., automatic ROX dye normalization used in TaqMan qRT-PCR). For microarray image capture, repeated scanning using different settings is recommended to assess the variance that can be attributed to this step. Similarly, for RNA-seq applications, testing different software for sequencing read alignment can help to validate an optimal approach.
Once raw data has been output in a useable form, quality assessment can be performed, typically by assessing the performance of internal controls and analyzing replicates to detect biases.49 For example, microarrays have well-known geographic biases (i.e., some areas of the array perform differently than others) and this can also be a problem for qRT-PCR plates. Such biases can be assessed, however, by analyzing replicates of internal controls distributed throughout the array or PCR plate. Batch effects related to run-to-run variation should can also be detected by running a reference standard sample across different batches; although batch effects often cannot be eliminated, their effects can be minimized by making sure that each batch of samples included a mix of samples from different groups that are to be compared. Finally, building in “check functions” to ensure a data handling error has not occurred50 is useful, as is systematic recording of all data manipulation steps to permit traceability.
Normalization, the next step after quality assessment, refers to adjusting for variation in the data that are due to known factors (usually technical factors, for example variations in amount of RNA introduced into the profiling workflow due to limits of precision of pipetting) and not related to the biological differences that are being investigated. The optimal data normalization method is dependent on the experimental question, specimen type, profiling platform, and the number of miRNAs being simultaneously profiled.
Normalization may be to a single RNA or set of RNAs or use the entire set of profiling data. Normalization to a reference housekeeping small nuclear or nucleolar RNA such as U6, U24, or U26, or invariant miRNAs51 is effective in many cases, but we emphasize that this approach requires validation that the reference RNA is not influenced by the condition being studied. Spike-in controls at the RT step to normalize for RNA extraction efficiency33, and plate calibrator controls at the PCR step are common in medium throughput PCR array applications such as those offered by Exiqon and Applied Biosystems. We recently showed that a mixture of 15 synthetic miRNA spike-in controls could be used to effectively normalize miRNA signatures on a solid-phase microarray platform.49 Global mean or median normalization is another commonly applied approach that relies on the assumption that the majority of miRNA do not change between conditions. One study that examined 430 miRNAs and 18 small RNA controls by RT-PCR reported that mean normalization was superior to using the reference RNA controls.52 Global normalization is often applied in a similar fashion to normalize microarray and RNA-seq data, although we caution users that this approach can be significantly influenced by outliers due to the relatively limited number of miRNA measurements.
MiRNA profiling experiments typically involve making comparisons between two or more groups, and therefore the next stage of analysis is usually the calculation of differential miRNA expression between groups. It is important consider the dynamic range and accuracy of quantitation of the platform when performing these calculations. qRT-PCR generally has the widest dynamic range, highest accuracy, and is the only method that has the potential for absolute miRNA quantification (e.g., by generating standard curves from dilutions of synthetic miRNA oligos of known concentration). MicroRNA microarrays are inexpensive but have the lowest sensitivity and dynamic range, and are therefore best used as discovery tools, where promising observations are validated by an alternate method.
For RNA-seq, miRNA quantification is expressed as a value relative to the total number of sequence reads for a given sample; thus comparisons between samples with high variance in miRNA distribution of expression are currently unreliable. However, as newer technologies such as single molecule sequencing that do not require PCR-based amplification become more established, absolute quantification by sequencing may become possible.
Applications: Mechanisms of Gene Regulation
In this section and the next we shift our focus to potential applications for miRNA profiling and accompanying considerations.
Developmental Biology
Studying developmental processes through miRNA profiling has been a particularly fruitful area. Studies are typically designed to compare miRNA profiles between different stages of development, with the aim of identifying miRNAs causally involved in developmental transitions or cell type specification. A recent study of the expression profiles of 288 miRNAs in 27 phenotypically distinct hematopoietic blood cells sorted by fluorescence activated cell sorting (FACS), including single rare progenitor and stem cells, was performed using a microfluidics-based multiplexed qRT-PCR assay.53 In this study design, the extremely limited number of rare cells demanded high sensitivity. qRT-PCR is the most sensitive of the platforms, and the use of microfluidics allowed evaluation of hundreds of miRNAs from single cells. One of the themes emerging from miRNA profiling studies and subsequent functional analyses in developmental biology is that miRNA expression patterns can be tissue-specific and that specific miRNAs function to buffer developmental transitions and/or to maintain differentiation states. Thus, there is particular interest in miRNA profiling for understanding stem cell biology and cellular reprogramming.
Novel miRNA Discovery
Many investigators study gene regulation in organisms which are not well-characterized at genomic or transcriptomic levels. In such cases, miRNAs may not be known and therefore qRT-PCR or microarray products may be unavailable. In such scenarios, RNA-seq is a powerful approach for both discovering and profiling miRNAs. For example, a recent study of miRNA and isomiRs in Atlantic halibut development used a combination of SOLid deep sequencing for discovery followed by Sanger sequencing for validation.59 Next generation sequencing has also been particularly valuable in the study of miRNA gene and host evolution within different plant species, as the majority of miRNAs are family- or species-specific.60
Studying miRNA:mRNA and miRNA:Protein Interactions
For identifying targets of miRNAs, an increasingly popular approach is to analyze miRNAs associated with mRNA targets and/or to an RNA binding protein of interest. For example, immunoprecipitation using antibodies to the RISC complex can be performed prior to RNA isolation, with subsequent analysis of both miRNAs and mRNAs.31 A method called crosslinking immunoprecipitation (CLIP)55,56 uses ultraviolet irradiation to create covalent bonds between RNA binding proteins and RNA.57 Using crosslinking with immunoprecipitation has identified miRNAs and mRNAs bound to Ago by RNA-sequencing and bioinformatics. This elegant combined approach is referred to as “HITS-CLIP” for high-throughput sequencing crosslinking immunoprecipitation.58
Integrative analyses of miRNAs in the context of gene regulatory networks
A major area of interest in miRNA profiling analysis is integration of miRNA profiles with other large-scale genomic datasets and with gene regulatory network models. In silico tools are increasingly becoming available for functional analysis of altered miRNA subsets and interrogation of biological networks. MAGIA83 (http://gencomp.bio.unipd.it/magia) and mirConnX84 (http://www.benoslab.pitt.edu/mirconnx) are examples of online tools that can integrate miRNA expression profiles with matched mRNA expression data to provide integrated analyses of biological networks. These programs both interface with Cytoscape85, a popular network visualization tool, as well as with online databases of miRNA expression patterns in human disease such as miR2disease86 (http://www.mir2disease.org/). miConnX also interfaces with the miR-Ontology database87 (http://ferrolab.dmi.unict.it/miro), a compendium miRNA-phenotype associations in humans.
Post-Transcriptional Modifications of miRNA
RNA sequencing has the ability to identify new variants of known miRNA, and can thereby serve as a tool for examining regulation of miRNAs themselves. For example, 3′ non-templated nucleotide additions were identified as a potential regulatory enzymatic modification to miRNAs by using a combination of RNA sequencing and Nanostring nCounter technology.14
Applications: Disease Biomarkers
Many features of miRNA profiles, including rich information content, high stability and accessibility in specimen types such as formalin-fixed tissue and plasma/serum, and potential for highly sensitive measurement, have made the study of miRNAs as disease biomarkers a highly popular and productive area.
Tissue-Based miRNA Biomarkers
A common study design is to define differences in miRNA profiles between normal and affected tissues to look for biomarkers of disease, which can further be correlated with prognosis, or therapeutic response. Each of the three main miRNA profiling platforms has been used for biomarker discovery efforts, in diseases as diverse as Huntingtons62, and microarrays for multiple sclerosis63, qRT-PCR for schizophrenia64, kidney disease65, diabetes66,67, rheumatoid arthritis68, and preeclampsia69, colon cancer70, hepatocellular carcinoma (HCC)71. Because tissue yields ample miRNA, all three platforms are viable options. One of the greatest successes which as led to clinical translation is the identification of miRNA profiles that can classify the organ of origin of cancers that are histologically undifferentiated and difficult to otherwise classify77,78 A clinical diagnostic assay is now available based on a 48-miRNA classifier generated from microarray studies of known tumor types.78 Predictive accuracy of the test was estimated at 89–100%.79,80 Additional diagnostic assays are also on the market that reportedly distinguish between different histological types of pulmonary cancer.
Circulating Biomarkers
MicroRNAs, including specific cancer-derived miRNAs are detectable in the cell-free circulation (i.e., in serum and plasma)27 and thus circulating microRNAs have been eagerly sought as noninvasive biomarkers in diseases such as HCC72, esophageal cancer73, and non-small cell lung cancer.74 Similarly, miRNAs entering the circulation as a result of tissue injury are prompting the development of new clinical tests for conditions such as stroke and myocardial infarction. Although this is a growing clinical application of miRNA profiling, it is worth noting that there is increasing concern around the need for rigorous control of pre-analytic and analytic variables when considering potential circulating miRNA markers.28,30,75,76
Forensics
Recently, microRNA profiles from different body fluids have been characterized and found to be robust distinguishers of different body fluids in the forensic setting.81,82 These studies were performed using microarrays for discovery of candidate markers, and qRT-PCR for further validation of candidate miRNAs.
Conclusions and Perspective
In this review we have focused exclusively on methods for measuring mature miRNA species. However, just as integrating miRNA profiling data with messenger RNA data is yielding better understanding of gene regulation and improving systems-level modeling, the measurement of pri-mir and pre-mir profiles is also likely to be useful in the future, especially given reports that regulation of miRNA biogenesis is important in cancer and potentially other diseases as well. In addition, most miRNA profiling is currently done using total RNA extracted from the whole cell. It is becoming increasingly appreciated that miRNA is sub-compartmentalized within cells (e.g., nuclear vs. cytoplasmic, miRNAs present in different protein complexes) and the higher resolution view afforded by miRNA profiling of specific sub-cellular compartments may provide new insights. Profiling of miRNAs bound to their targets is likely to improve with improved experimental methods and improvements in sequencing technology. This could pay large dividends for basic science investigations of biological processes where miRNAs play a significant regulatory role. We should also mention that other small non-coding RNAs, such as Piwi-interacting RNAs (piRNA), as well as long non-coding RNA (lncRNA) are increasingly recognized to play important roles in cellular physiology, and it is likely that new classes of non-coding RNA remain to be discovered. In the future, profiling methods such as RNA-seq that have the potential to detect all classes of RNA are likely to shed light on the entirety of the transcriptome.
One of the most exciting applications of miRNA profiling is in the development of clinically useful molecular diagnostic tests. There are significant hurdles with respect to understanding sample-to-sample biological variability that is not related to the phenotype of interest, but if these barriers can be overcome the richness of information associated with miRNA profiles and the high stability of miRNAs in clinical samples suggest strong potential for eventual clinical translation. In the more distant future, inexpensive, point-of-care miRNA diagnostic tests may become available, given that miRNA stability in samples such as dried blood spotted on filter paper and isothermal amplification methods for miRNAs have been demonstrated. Nanopore-based methods for miRNA measurement are also being developed that, if perfected, could enable simple, rapid and highly sensitive miRNA assays that could pave the way for broadly available clinical tests based on miRNA profiling.
Footnotes
Conflict of Interest Statement: Dr. Tewari is an inventor on patent applications related to circulating microRNA. He has served on the Scientific Advisory Boards of Wafergen, Inc. and Combimatrix, Inc. within the last three years and has had a past research collaboration with scientists at Nanostring, Inc.
Movies: None
- miRBase: http://www.mirbase.org/
- Discussion of miRNA naming conventions: http://www.mirbase.org/blog/2011/04/whats-in-a-name/
- Bionetworks analysis: MAGIA83 (http://gencomp.bio.unipd.it/magia), mirConnX84 (http://www.benoslab.pitt.edu/mirconnx), miR2disease86 (http://www.mir2disease.org/), miR-Ontology database87 (http://ferrolab.dmi.unict.it/miro)
- Target prediction: miRanda (http://www.microRNA.org), Targetscan (http://www.targetscan.org), PicTar (http://pictar.mdc-berlin.de)
References
- 1.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 2.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–62. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 3.Reinhart BJ, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–6. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 4.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nature reviews. Molecular cell biology. 2009;10:126–39. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 5.Davis-Dusenbery BN, Hata A. Mechanisms of control of microRNA biogenesis. Journal of biochemistry. 2010;148:381–92. doi: 10.1093/jb/mvq096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ragan C, Zuker M, Ragan MA. Quantitative prediction of miRNA-mRNA interaction based on equilibrium concentrations. PLoS computational biology. 2011;7:e1001090. doi: 10.1371/journal.pcbi.1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC Genomics. 2007;8:166. doi: 10.1186/1471-2164-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids research. 2006;34:D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic acids research. 2008;36:D154–8. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffiths-Jones S. The microRNA Registry. Nucleic acids research. 2004;32:D109–11. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bar M, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem cells. 2008;26:2496–505. doi: 10.1634/stemcells.2008-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nature reviews. Genetics. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic acids research. 2011;39:D152–7. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wyman SK, et al. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome research. 2011;21:1450–61. doi: 10.1101/gr.118059.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cloonan N, et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome biology. 2011;12:R126. doi: 10.1186/gb-2011-12-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibberson D, Benes V, Muckenthaler MU, Castoldi M. RNA degradation compromises the reliability of microRNA expression profiling. BMC biotechnology. 2009;9:102. doi: 10.1186/1472-6750-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Podolska A, Kaczkowski B, Litman T, Fredholm M, Cirera S. How the RNA isolation method can affect microRNA microarray results. Acta biochimica Polonica. 2011 [PubMed] [Google Scholar]
- 18.Wang WX, et al. Focus on RNA isolation: obtaining RNA for microRNA (miRNA) expression profiling analyses of neural tissue. Biochimica et biophysica acta. 2008;1779:749–57. doi: 10.1016/j.bbagrm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammerle-Fickinger A, et al. Validation of extraction methods for total RNA and miRNA from bovine blood prior to quantitative gene expression analyses. Biotechnology letters. 2010;32:35–44. doi: 10.1007/s10529-009-0130-2. [DOI] [PubMed] [Google Scholar]
- 20.Weber JA, et al. The microRNA spectrum in 12 body fluids. Clinical chemistry. 2010;56:1733–41. doi: 10.1373/clinchem.2010.147405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Accerbi M, et al. Methods for isolation of total RNA to recover miRNAs and other small RNAs from diverse species. Methods in molecular biology. 2010;592:31–50. doi: 10.1007/978-1-60327-005-2_3. [DOI] [PubMed] [Google Scholar]
- 22.Doleshal M, et al. Evaluation and validation of total RNA extraction methods for microRNA expression analyses in formalin-fixed, paraffin-embedded tissues. The Journal of molecular diagnostics: JMD. 2008;10:203–11. doi: 10.2353/jmoldx.2008.070153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xi Y, et al. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA. 2007;13:1668–74. doi: 10.1261/rna.642907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dahlgaard J, et al. Analytical variables influencing the performance of a miRNA based laboratory assay for prediction of relapse in stage I non-small cell lung cancer (NSCLC) BMC research notes. 2011;4:424. doi: 10.1186/1756-0500-4-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoefig KP, Heissmeyer V. Measuring microRNA expression in size-limited FACS-sorted and microdissected samples. Methods in molecular biology. 2010;667:47–63. doi: 10.1007/978-1-60761-811-9_4. [DOI] [PubMed] [Google Scholar]
- 26.Chim SS, et al. Detection and characterization of placental microRNAs in maternal plasma. Clin Chem. 2008;54:482–90. doi: 10.1373/clinchem.2007.097972. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell PS, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–8. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald JS, Milosevic D, Reddi HV, Grebe SK, Algeciras-Schimnich A. Analysis of Circulating MicroRNA: Preanalytical and Analytical Challenges. Clinical chemistry. 2011;57:833–40. doi: 10.1373/clinchem.2010.157198. [DOI] [PubMed] [Google Scholar]
- 29.Duttagupta R, Jiang R, Gollub J, Getts RC, Jones KW. Impact of Cellular miRNAs on Circulating miRNA Biomarker Signatures. PLoS One. 2011;6:e20769. doi: 10.1371/journal.pone.0020769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pritchard CC, et al. Blood cell origin of circulating microRNAs: a cautionary note for cancer biomarker studies. Cancer prevention research. 2011 doi: 10.1158/1940-6207.CAPR-11-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arroyo JD, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A. 2011;108:5003–8. doi: 10.1073/pnas.1019055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becker C, Hammerle-Fickinger A, Riedmaier I, Pfaffl MW. mRNA and microRNA quality control for RT-qPCR analysis. Methods. 2010;50:237–43. doi: 10.1016/j.ymeth.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Kroh EM, Parkin RK, Mitchell PS, Tewari M. Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR) Methods. 2010;50:298–301. doi: 10.1016/j.ymeth.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wark AW, Lee HJ, Corn RM. Multiplexed detection methods for profiling microRNA expression in biological samples. Angewandte Chemie. 2008;47:644–52. doi: 10.1002/anie.200702450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 36.Newman MA, Mani V, Hammond SM. Deep sequencing of microRNA precursors reveals extensive 3′ end modification. RNA. 2011;17:1795–803. doi: 10.1261/rna.2713611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benes V, Castoldi M. Expression profiling of microRNA using real-time quantitative PCR, how to use it and what is available. Methods. 2010;50:244–9. doi: 10.1016/j.ymeth.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 38.Castoldi M, et al. A sensitive array for microRNA expression profiling (miChip) based on locked nucleic acids (LNA) RNA. 2006;12:913–20. doi: 10.1261/rna.2332406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geiss GK, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nature biotechnology. 2008;26:317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 40.Metzker ML. Sequencing technologies – the next generation. Nature reviews. Genetics. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 41.Gunaratne PH, Coarfa C, Soibam B, Tandon A. miRNA Data Analysis: Next-Gen Sequencing. Methods in molecular biology. 2012;822:273–88. doi: 10.1007/978-1-61779-427-8_19. [DOI] [PubMed] [Google Scholar]
- 42.Yang JH, Qu LH. deepBase: Annotation and Discovery of MicroRNAs and Other Noncoding RNAs from Deep-Sequencing Data. Methods in molecular biology. 2012;822:233–48. doi: 10.1007/978-1-61779-427-8_16. [DOI] [PubMed] [Google Scholar]
- 43.Linsen SE, et al. Limitations and possibilities of small RNA digital gene expression profiling. Nature methods. 2009;6:474–6. doi: 10.1038/nmeth0709-474. [DOI] [PubMed] [Google Scholar]
- 44.Tian G, et al. Sequencing bias: comparison of different protocols of microRNA library construction. BMC biotechnology. 2010;10:64. doi: 10.1186/1472-6750-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hafner M, et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011;17:1697–712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kapranov P, et al. New class of gene-termini-associated human RNAs suggests a novel RNA copying mechanism. Nature. 2010;466:642–6. doi: 10.1038/nature09190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas M, Lieberman J, Lal A. Desperately seeking microRNA targets. Nature structural & molecular biology. 2010;17:1169–74. doi: 10.1038/nsmb.1921. [DOI] [PubMed] [Google Scholar]
- 48.van Rooij E. The art of microRNA research. Circulation research. 2011;108:219–34. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- 49.Sarkar D, et al. Quality assessment and data analysis for microRNA expression arrays. Nucleic acids research. 2009;37:e17. doi: 10.1093/nar/gkn932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hutson S. Data handling errors spur debate over clinical trial. Nature medicine. 2010;16:618. doi: 10.1038/nm0610-618a. [DOI] [PubMed] [Google Scholar]
- 51.Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA. 2008;14:844–52. doi: 10.1261/rna.939908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mestdagh P, et al. A novel and universal method for microRNA RT-qPCR data normalization. Genome biology. 2009;10:R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petriv OI, et al. Comprehensive microRNA expression profiling of the hematopoietic hierarchy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15443–8. doi: 10.1073/pnas.1009320107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kato M, Chen X, Inukai S, Zhao H, Slack FJ. Age-associated changes in expression of small, noncoding RNAs, including microRNAs, in C. elegans. RNA. 2011;17:1804–20. doi: 10.1261/rna.2714411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ule J, et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–5. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 56.Ule J, Jensen K, Mele A, Darnell RB. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005;37:376–86. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 57.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–86. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Licatalosi DD, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–9. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bizuayehu TT, et al. Differential expression patterns of conserved miRNAs and isomiRs during Atlantic halibut development. BMC Genomics. 2012;13:11. doi: 10.1186/1471-2164-13-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cuperus JT, Fahlgren N, Carrington JC. Evolution and functional diversification of MIRNA genes. The Plant cell. 2011;23:431–42. doi: 10.1105/tpc.110.082784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marti E, et al. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic acids research. 2010;38:7219–35. doi: 10.1093/nar/gkq575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guerau-de-Arellano M, Alder H, Ozer HG, Lovett-Racke A, Racke MK. miRNA profiling for biomarker discovery in multiple sclerosis: From microarray to deep sequencing. Journal of neuroimmunology. 2011 doi: 10.1016/j.jneuroim.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lai CY, et al. MicroRNA expression aberration as potential peripheral blood biomarkers for schizophrenia. PLoS One. 2011;6:e21635. doi: 10.1371/journal.pone.0021635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saal S, Harvey SJ. MicroRNAs and the kidney: coming of age. Current opinion in nephrology and hypertension. 2009;18:317–23. doi: 10.1097/MNH.0b013e32832c9da2. [DOI] [PubMed] [Google Scholar]
- 66.Zampetaki A, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circulation research. 2010;107:810–7. doi: 10.1161/CIRCRESAHA.110.226357. [DOI] [PubMed] [Google Scholar]
- 67.Zhu H, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tili E, Michaille JJ, Costinean S, Croce CM. MicroRNAs, the immune system and rheumatic disease. Nature clinical practice. Rheumatology. 2008;4:534–41. doi: 10.1038/ncprheum0885. [DOI] [PubMed] [Google Scholar]
- 69.Gunel T, et al. Serum microRNA expression in pregnancies with preeclampsia. Genetics and molecular research: GMR. 2011;10 doi: 10.4238/2011.November.8.5. [DOI] [PubMed] [Google Scholar]
- 70.Buffa FM, et al. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer research. 2011;71:5635–45. doi: 10.1158/0008-5472.CAN-11-0489. [DOI] [PubMed] [Google Scholar]
- 71.Ji J, et al. MicroRNA expression, survival, and response to interferon in liver cancer. The New England journal of medicine. 2009;361:1437–47. doi: 10.1056/NEJMoa0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou J, et al. Plasma MicroRNA Panel to Diagnose Hepatitis B Virus-Related Hepatocellular Carcinoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011 doi: 10.1200/JCO.2011.38.2697. [DOI] [PubMed] [Google Scholar]
- 73.Komatsu S, et al. Circulating microRNAs in plasma of patients with oesophageal squamous cell carcinoma. British journal of cancer. 2011;105:104–11. doi: 10.1038/bjc.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boeri M, et al. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc Natl Acad Sci U S A. 2011;108:3713–8. doi: 10.1073/pnas.1100048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watson AK, Witwer KW. Do Platform-Specific Factors Explain MicroRNA Profiling Disparities? Clinical chemistry. 2011 doi: 10.1373/clinchem.2011.175281. [DOI] [PubMed] [Google Scholar]
- 76.Kim DJ, et al. Plasma Components Affect Accuracy of Circulating Cancer-Related MicroRNA Quantitation. The Journal of molecular diagnostics: JMD. 2012;14:71–80. doi: 10.1016/j.jmoldx.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 78.Rosenfeld N, et al. MicroRNAs accurately identify cancer tissue origin. Nature biotechnology. 2008;26:462–9. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 79.Ferracin M, et al. MicroRNA profiling for the identification of cancers with unknown primary tissue-of-origin. The Journal of pathology. 2011;225:43–53. doi: 10.1002/path.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Varadhachary GR, et al. Prospective gene signature study using microRNA to identify the tissue of origin in patients with carcinoma of unknown primary. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:4063–70. doi: 10.1158/1078-0432.CCR-10-2599. [DOI] [PubMed] [Google Scholar]
- 81.Courts C, Madea B. Specific micro-RNA signatures for the detection of saliva and blood in forensic body-fluid identification. Journal of forensic sciences. 2011;56:1464–70. doi: 10.1111/j.1556-4029.2011.01894.x. [DOI] [PubMed] [Google Scholar]
- 82.Zubakov D, et al. MicroRNA markers for forensic body fluid identification obtained from microarray screening and quantitative RT-PCR confirmation. International journal of legal medicine. 2010;124:217–26. doi: 10.1007/s00414-009-0402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sales G, et al. MAGIA, a web-based tool for miRNA and Genes Integrated Analysis. Nucleic acids research. 2010;38:W352–9. doi: 10.1093/nar/gkq423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang GT, Athanassiou C, Benos PV. mirConnX: condition-specific mRNA-microRNA network integrator. Nucleic acids research. 2011;39:W416–23. doi: 10.1093/nar/gkr276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lopes CT, et al. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010;26:2347–8. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jiang Q, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic acids research. 2009;37:D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lagana A, et al. miRo: a miRNA knowledge base. Database: the journal of biological databases and curation. 2009 doi: 10.1093/database/bap008. bap008 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Griffiths-Jones S. What’s in a Name? 2011 http://www.mirbase.org/blog/2011/04/whats-in-a-name/
- 89.Yang JS, et al. Widespread regulatory activity of vertebrate microRNA* species. RNA. 2011;17:312–26. doi: 10.1261/rna.2537911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Griffiths-Jones S, Hui JH, Marco A, Ronshaugen M. MicroRNA evolution by arm switching. EMBO reports. 2011;12:172–7. doi: 10.1038/embor.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]