

Fecal, oral, and skin biomes of isolated Amerindians show higher human bacterial diversity including antibiotic resistance genes.
Keywords: Antibiotic, Amerindian, Microbiome, Resistome, Westernization
Abstract
Most studies of the human microbiome have focused on westernized people with life-style practices that decrease microbial survival and transmission, or on traditional societies that are currently in transition to westernization. We characterize the fecal, oral, and skin bacterial microbiome and resistome of members of an isolated Yanomami Amerindian village with no documented previous contact with Western people. These Yanomami harbor a microbiome with the highest diversity of bacteria and genetic functions ever reported in a human group. Despite their isolation, presumably for >11,000 years since their ancestors arrived in South America, and no known exposure to antibiotics, they harbor bacteria that carry functional antibiotic resistance (AR) genes, including those that confer resistance to synthetic antibiotics and are syntenic with mobilization elements. These results suggest that westernization significantly affects human microbiome diversity and that functional AR genes appear to be a feature of the human microbiome even in the absence of exposure to commercial antibiotics. AR genes are likely poised for mobilization and enrichment upon exposure to pharmacological levels of antibiotics. Our findings emphasize the need for extensive characterization of the function of the microbiome and resistome in remote nonwesternized populations before globalization of modern practices affects potentially beneficial bacteria harbored in the human body.
INTRODUCTION
Host-microbial interactions are important determinants of host physiology, including immune responses, metabolic homeostasis, and behavior (1–4). Microbiota transplantation can transfer phenotypes such as nutritional status from donor to recipient (5, 6), indicating that altered microbial communities can therefore cause, as well as result from, altered physiological states. Despite increasing evidence that the microbiome has important roles in human health, we do not yet know the extent to which the human microbiome has changed during the adoption of life-styles associated with westernization. Here, we described the microbiome from Yanomami subjects in the Amazon with no previous report of contact with non-Yanomami. The Yanomami were originally mountain people who were first contacted in the mid-1960s, and who continue to live seminomadic hunter-gatherer life-styles in the Amazon jungle. In Venezuela, they inhabit a region protected from development, and many still inhabit uncharted villages in the vast mountainous Yanomami territory (7). These remote populations of hunter-gatherers have life-styles similar to those of our human ancestors, and are unexposed to modern practices known to exert antimicrobial effects.
For instance, pharmacologic-dose antibiotic exposure is nearly ubiquitous worldwide, and all human microbiota studied to date, remote or industrialized, harbor diverse antibiotic resistance (AR) genes (8, 9). Although AR genes have been computationally predicted in ancient oral microbiota (10), functional resistance from the preantibiotic era remains largely uncharacterized. These subjects from an uncontacted community therefore represent a unique proxy for the preantibiotic era human resistome. Here, we characterized the microbiome and resistome of these subjects, and compared them to those of other non-isolated populations. Samples of the oral cavity (n = 28), forearm skin (n = 28), and feces (n = 12) were obtained from 34 of the 54 villagers (table S1) at the time of the first medical expedition to an isolated, previously uncharted village in the High Orinoco state of Venezuela. The age of the subjects was between 4 and 50 years, as estimated by Yanomami health workers.
RESULTS
Unprecedented high bacterial and functional diversity in the microbiome of uncontacted Amerindians
We sequenced the V4 region of the 16S rRNA gene from fecal, oral, and skin samples and compared these data with those from previous studies (11, 12). The microbiome of the uncontacted Amerindians exhibited the highest diversity ever reported in any human group. Diversity in feces and skin, but not in the mouth, was significantly higher in Yanomami than in U.S. subjects (Fig. 1, A, E, and I). Noticeably, fecal diversity was even higher than in the semitransculturated Guahibo Amerindians and Malawians [analysis of variance (ANOVA) and Tukey’s honest significant difference (HSD) test, P < 0.001; Fig. 1A]. Principal coordinate analysis (PCoA) of UniFrac distances reveals that bacterial communities from each body site segregate westernized people from agrarian and hunter-gatherer groups (Fig. 1, B, F, and J, and fig. S1). Accumulation curves of fecal operational taxonomic units (OTUs) and phylogenetic species richness (γ diversity) confirmed that the Yanomami harbor significantly higher diversity than other populations (unpaired t test with Bonferroni correction, P < 0.0001; fig. S2A) and, at the same time, have the lowest microbiome variability (fig. S2B). Within-population distances in the Yanomami were lower than those in the semi-accultured Guahibo Amerindians (fig. S2C). Prevalence/abundance curves further confirm that the Yanomami share a higher proportion of their fecal microbiota despite their higher diversity (Fig. 1D). These results were consistent with those obtained sequencing the V2 region of the 16S rRNA gene of the same set of samples extracted, amplified, and sequenced together with samples from U.S. subjects (fig. S3).
Fig. 1. Microbiota diversity in fecal, oral, and skin samples from uncontacted Yanomami in relation to other human groups.
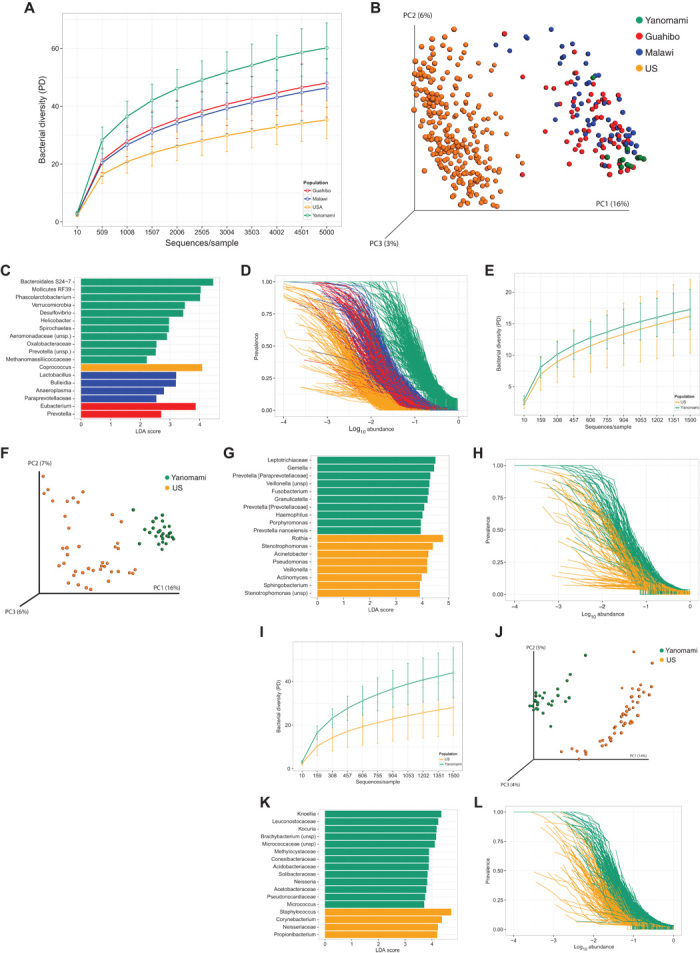
(A) Faith’s phylogenetic diversity (PD) (average ± SD) of fecal samples from Yanomami and Guahibo Amerindians, Malawians, and U.S. subjects. OTU tables rarefied at 5000 sequences per sample. Interpopulation differences were significant (P < 0.001, ANOVA with Tukey’s HSD) for all but Guahibo-Malawi comparison (P = 0.73). (B) PCoA plot based on UniFrac distances calculated on the OTU table of fecal samples rarefied at 5000 sequences per sample. (C) Top discriminative bacteria among populations in fecal samples as determined by linear discriminant analysis (LDA) effect size (LEfSe) analysis. (D) Normalized prevalence/abundance curves for all OTUs found at 1% abundance or more in fecal samples. (E) Faith’s phylogenetic diversity (average ± SD) of oral samples from Yanomami and U.S. subjects. OTU tables rarefied at 1500 sequences per sample. Interpopulation differences were not significant (P = 0.296, ANOVA with Tukey’s HSD). (F) PCoA plot based on UniFrac distances calculated on OTU tables of oral samples rarefied at 1500 sequences per sample. (G) Top discriminative bacteria among populations in oral samples as determined by LEfSe analysis. (H) Normalized prevalence/abundance curves for all OTUs found at 1% abundance or more in oral samples. (I) Faith’s phylogenetic diversity (average ± SD) of skin samples from Yanomami and U.S. subjects. OTU tables rarefied at 1500 sequences per sample. Interpopulation differences were significant (P < 0.001, ANOVA with Tukey’s HSD). (J) PCoA plot based on UniFrac distances calculated on OTU tables of skin samples rarefied at 1500 sequences per sample. (K) Top discriminative bacteria among populations in skin samples as determined by LEfSe analysis. (L) Normalized prevalence/abundance curves for all OTUs found at 1% abundance or more in skin samples.
On the basis of taxa detectable at the obtained sequencing depth, the Yanomami fecal microbiota was characterized by high Prevotella and low Bacteroides, contrary to what was observed in U.S. subjects and similar to that in Guahibo Amerindians, Malawians (fig. S4), and African hunter-gatherers (13). Analyses of discriminative bacteria in relation to U.S., Malawian, and Guahibo populations (14) indicated that Yanomami had higher intestinal Bacteroidales S24-7 (a poorly characterized family close to the Parabacteroides), Mollicutes and Verrucomicrobia, members of the families Aeromonadaceae, Oxalobacteraceae, and Methanomassiliicoccaceae, and genera Phascolarctobacterium, Desulfovibrio, Helicobacter, Spirochaeta, and Prevotella (Fig. 1C). Although diet plays a significant role in shaping the microbiome, factors other than nutrition also influence microbiota composition. For example, Helicobacter was absent in the feces of U.S. subjects, and intestinal Oxalobacter and Desulfovibrio were markedly increased in the Yanomami in relation to U.S. subjects (fig. S5).
In contrast to the fecal microbiome, the Yanomami had similar levels of oral bacterial diversity to those found in U.S. subjects (Fig. 1E) despite a distinct composition (Fig. 1F), and higher prevalence of oral taxa (Fig. 1H). Although both populations were dominated by Streptococcus, the Yanomami had higher proportions of several oral Prevotella, Fusobacterium, and Gemella, and lower Rothia, Stenotrophomonas, Acinetobacter, or Pseudomonas (Fig. 1G and fig. S4). The Yanomami cutaneous microbiota had significantly higher diversity (Fig. 1I) and clustered apart from that of U.S. subjects (Fig. 1J). No single taxon dominated the Yanomami skin microbiota, in contrast to the low skin bacterial diversity in U.S. subjects and high relative proportions of Staphylococcus, Corynebacterium, Neisseriaceae, and Propionibacterium (Fig. 1K and fig. S4). Some bacterial taxa previously reported as environmental, such as Knoellia or Solibacteraceae (15–17), appeared to be enriched in the skin microbiota of the Yanomami. As with fecal and oral microbiota, skin bacterial prevalence was also higher in the Yanomami (Fig. 1L).
Metagenomic predictions using PICRUSt (18) showed distinct microbiome functional profiles for different human populations (Fig. 2, B, F, and J), with the Yanomami having higher fecal and skin (Fig. 2, A and I), but not oral (Fig. 2E), functional diversity, higher gene prevalence (Fig. 2, D, H, and L), and lower intragroup variation (fig. S2D). Pathways involving amino acid metabolism, glycosyltransferases, and biosynthesis of lipopolysaccharides, terpenoid-quinones, and vitamins [including riboflavin, previously reported to be increased in non-Western populations (11)] were found to be overrepresented in the Yanomami (Fig. 2C). Their oral mucosa also had significant overrepresentation of pathways of fatty acid biosynthesis, glycosyltransferases, and methane metabolism (mostly derived from members of the Methylocystaceae) (Fig. 2G). Their skin was enriched in organic acids, amino acids, vitamins, and methane (originating from Methylobacter, Methylosinus, and Methylocystaceae) pathways (Fig. 2K). Validation of the predicted bacterial functions by comparing against the sequenced metagenome of the same samples and using a previously reported metagenome from Guahibo Amerindians (11) confirmed the robustness of our results (see Materials and Methods).
Fig. 2. Metagenomic diversity of fecal and oral samples from uncontacted Yanomami in relation to other human groups.
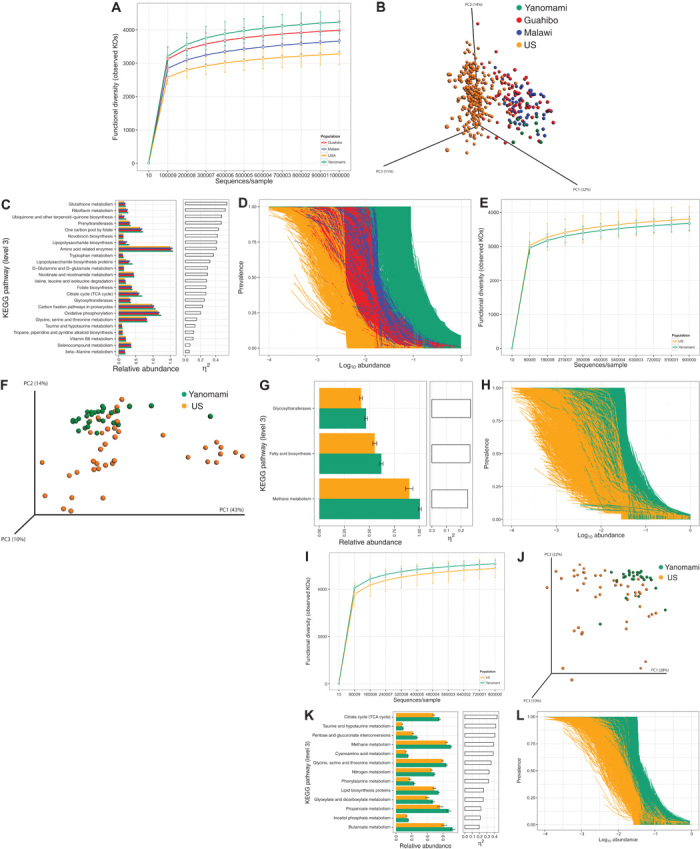
(A) Functional diversity (average ± SD) measured by the observed number of KEGG orthologs in fecal samples of Yanomami and Guahibo Amerindians, Malawians, and U.S. subjects. Metagenomic tables rarefied at 1 million sequences per sample. Interpopulation differences were significant for all comparisons (P < 0.001, ANOVA and Tukey’s HSD). (B) PCoA plot based on Bray-Curtis distances calculated on the metagenomic table of fecal samples rarefied at 1 million sequences per sample. (C) Top discriminative metabolic pathways in Yanomami fecal samples as determined by STAMP; pathways ranked by effect size (η2). (D) Normalized prevalence/abundance curves for all KOs found at 1% abundance or more in fecal samples. (E) Functional diversity (average ± SD) measured by the observed number of KEGG orthologs in oral samples of Yanomami and Guahibo Amerindians, Malawians, and U.S. subjects. Metagenomic tables rarefied at 900,000 sequences per sample. Interpopulation differences were not significant (P = 0.07, t test). (F) PCoA plot based on Bray-Curtis distances calculated on the metagenomic table of oral samples rarefied at 900,000 sequences per sample. (G) Top discriminative metabolic pathways in Yanomami oral samples as determined by STAMP; pathways ranked by effect size (η2). (H) Normalized prevalence/abundance curves for all KOs found at 1% abundance or more in oral samples. (I) Functional diversity (average ± SD) measured by the observed number of KEGG orthologs in skin samples of Yanomami and U.S. subjects. Metagenomic tables rarefied at 1 million sequences per sample. Interpopulation differences were significant for all comparisons (P < 0.001, ANOVA and Tukey’s HSD). (J) PCoA plot based on Bray-Curtis distances calculated on the metagenomic table of skin samples rarefied at 1 million sequences per sample. (K) Top discriminative metabolic pathways in Yanomami skin samples as determined by STAMP; pathways ranked by effect size (η2). (L) Normalized prevalence/abundance curves for all KOs found at 1% abundance or more in skin samples.
To provide an archive to study human microbial evolution, we isolated 1097 bacterial strains from Yanomami fecal or oral samples. Characterization of 24 Yanomami Escherichia coli strains using multilocus sequence typing (MLST) based on seven housekeeping genes (19) showed that they differ substantially from strains circulating in Western societies, and that none of the isolates belonged to phylogenetic group B2 common in Western populations (fig. S6).
Presence of AR genes in the microbiome of uncontacted Amerindians
Phenotypic resistance was tested in 131 Amerindian E. coli strains isolated from 11 fecal specimens. We found that all E. coli isolates were sensitive to all 23 antibiotics tested. However, functional libraries created from the genomic DNA of 38 of these E. coli isolates, including the 24 typed strains (table S12), yielded AR genes targeted against eight antibiotics, including four antibiotics from within the phenotypic screen (table S3). Overexpression of chromosomally encoded E. coli genes was likely responsible for most of the resistance (89% have homologs in E. coli K-12 MG1655; table S4).
For a culture-independent interrogation of the commensal Amerindian resistome, we created shotgun metagenomic libraries from fecal and oral microbiota of four subjects (table S5) (8, 20) after multiple displacement amplification (MDA) and screened them against 15 antibiotics (table S6). We identified 28 functional AR genes in the microbiota of these people naïve to anthropogenic antibiotics (table S4), including genes resistant to semisynthetic and synthetic antibiotics, to which the villagers and their surroundings are highly unlikely to have ever been exposed. These genes include an extended-spectrum cblA β-lactamase that confers resistance to five later-generation antibiotics, including a fourth-generation cephalosporin and a synthetic monobactam, and penicillin-binding proteins (PBPs) resistant to the third-generation cephalosporin ceftazidime (Fig. 3A). The six PBPs have 81 to 100% amino acid identity to PBPs from Neisseria and Kingella, oral commensals in both Western and Amerindian hosts (21).
Fig. 3. Representative AR genes captured through functional metagenomic selection of Amerindian oral and fecal microbiota.
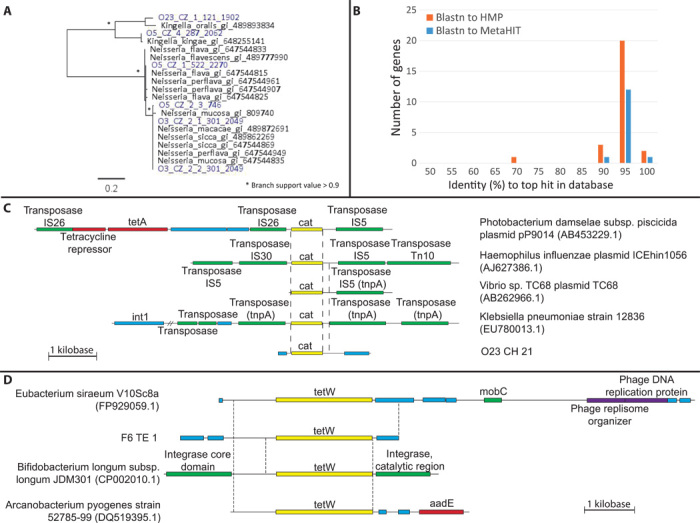
(A) Maximum likelihood (ML) tree of ceftazidime-resistant PBPs from Amerindian oral microbiota (blue) and their top blastx hits in NCBI nr (black). (B) Identity (%) of the 28 AR genes to their top hits in HMP and MetaHIT (blastn). Fourteen aligned to MetaHIT, all isolated from fecal microbiota. (C) Chloramphenicol acetyltransferase (cat) from O23_CH_21 and homologs in NCBI nt (nucleotide database). Dashed lines indicate 99% identical sequences. Dashes indicate omitted sequence (8487..25716). (D) tetW from F6_TE_1 and homologs in NCBI nt. Dashed lines indicate 98 to 99% identical sequences. Sequences are to scale. Yellow, AR gene; red, other resistance genes; green, mobile genetic elements; purple, bacteriophage genes; blue, other.
Most of the AR genes had close homologs in human microbiota from industrialized societies. Seventy-nine percent of the AR genes aligned to the Human Microbiome Project (HMP) assemblies with ≥95% nucleotide identity, and all genes that aligned to HMP (87% of fecal genes) also aligned to MetaHIT assemblies (Fig. 3B). Only one gene (F3_TE_1), a ribosomal protection protein (RPP) encoding resistance to tetracycline, did not align to HMP, MetaHIT, or any protein in National Center for Biotechnology Information (NCBI) nr (at >70% amino acid identity) and may be exclusive to this population. Thus, despite different antibiotic exposures, the microbiota from antibiotic-naïve and industrialized people share a common resistome.
Whole metagenome shotgun sequencing of fecal and oral specimens from the Amerindian village was performed to quantify the abundance of the functionally selected AR genes from the Amerindian microbiota and from age- and gender-matched individuals from an industrialized setting (Puerto Rico). Genes from all resistance classes except RPPs were restricted to their body site of origin, either the fecal or oral microbiota (Fig. 4), perhaps due to tight linkage with a microbial host specific to that habitat. Antibiotic-resistant PBPs and transporters functionally selected from Puerto Rican metagenomes were no less abundant in the Amerindian metagenomes (Wilcoxon rank sum test, Bonferroni correction, P > 0.05), and 72% share ≥95% amino acid identity with commensal Neisseria and Kingella species. However, 14% of the Amerindian AR genes are syntenic with mobilization elements, including plasmids and transposases, highlighting the risk for horizontal gene transfer in response to antibiotic selection (see the Supplementary Materials) (22). This is likely an underestimation of the mobilization risk, because many of the AR genes not found to be syntenic with mobilization elements in the Amerindian context are associated with such elements in industrialized settings and are widespread in commensals and pathogens (Fig. 3, C and D).
Fig. 4. Abundance of functionally selected AR genes in Amerindian fecal and oral metagenomes.

The heatmap displays the abundances in RPKM (reads per 1-kb gene sequence per million reads mapped) of functionally selected AR genes in the fecal and oral metagenomes of Amerindians. The AR genes were functionally selected from the oral and fecal microbiota of the Yanomami individuals and matched Puerto Rican controls. The shotgun-sequenced metagenomes are from the Yanomami villagers, including individuals not investigated with functional metagenomic selections. Shotgun-sequenced metagenomes were clustered by UPGMA (unweighted pair group method with arithmetic mean) on the AR gene abundance profiles. Fecal and oral microbiota cluster apart.
DISCUSSION
The characterization of an isolated group of Yanomami hunter-gatherers here reported represents a unique opportunity to understand a human microbiome and resistome minimally exposed to modern practices, which can exert significant antimicrobial effects with lasting consequences in the host (23, 24). The Yanomami here described exhibit unprecedented levels of bacterial diversity. Despite the small population of this village and the limited sample size of our study, they had significantly higher levels of fecal and skin diversity, both bacterial and functional, than any other human population reported so far. In contrast with the large infrequent meals characteristic of Western diets, frequent meals and food seasonality in the Yanomami would, according to disturbance theory (25, 26), lead to oscillating microbial populations depending on resource availability. The microbiome diversity is more evenly shared among the Yanomami across all body sites than in other populations, a phenomenon that could partially be due to cohabitation (27). However, Guahibo Amerindians living in two different Amazonian villages had less shared fecal taxa and higher within-village distances (fig. S2C). Together, these results suggest that the isolation and lack of transculturation of the Yanomami can also account for their higher bacterial diversity and prevalence.
The similarity in oral diversity levels between Yanomami and U.S. individuals, despite their clear differences in bacterial composition, is intriguing. Oral hygiene in the Yanomami is limited to occasional washing of the mouth with water. This factor combined with the ancestral habit of chewing tobacco constantly and from early ages (28) might explain why the oral microbiota is equally diverse between these populations.
In the highly diverse skin microbiota of the Yanomami, the presence of environmental bacterial taxa is consistent with their higher body environmental exposure in relation to westernized people who wear clothes and spend much time indoors. The extent to which this environmental exposure contributes to the observed increased skin diversity is currently not known.
Because the Yanomami have remained relatively isolated for more than 11,000 years since the arrival of their ancestors in the Americas (29), genomic divergence from Western strains is to be expected. Assuming a doubling rate of once per hour, an estimated 100 million bacterial generations would have occurred with little gene migration in the Yanomami microbiome, in relation to the much higher rates in the westernized world. The bacterial archive gathered from these unique specimens will be of great value to study evolution of human-associated microbial strains.
Even with no known exposure to pharmacologic-dose antibiotics, AR genes were present in the microbiome of these uncontacted Amerindians. AR genes against natural antibiotics could have entered the human microbiota through exchange with antibiotic-producing soil microbes and neighboring microbes (30, 31) or evolved in a soil-dwelling ancestor of a human commensal. Resistance to later-generation antibiotics may represent cross-resistance or a housekeeping gene that provides resistance only in a novel regulatory context (32). Alternatively, AR genes could have reached the Yanomami via traded objects or a chain of human contact that reached antibiotic-exposed populations. These routes would require survival of commensal or cosmopolitan microbes in the environment during transfer and/or horizontal gene transfer into the human microbiota multiple times in the recent past (33). Regardless of their origin, the AR genes have been maintained in the absence of apparent antibiotic selection pressure in this very diverse Amerindian microbiome. The carriage of a pool of mobilizable resistance genes to next-generation antibiotics in antibiotic-naïve microbiota may help explain the rapidity with which clinical resistance has arisen to each new antibiotic class after its introduction (30, 34). Because antibiotics were introduced to the village after the first medical visit, future studies in this unique setting would help elucidate their effect on the human microbiome and resistome.
The high shared gene identity with commensal taxa of the source body site suggests that many of the AR genes are encoded by commensals. Continued carriage of microbes through industrialization may partially explain a common resistome in uncontacted and industrialized people. Our discovery of functional AR genes encoded by antibiotic-sensitive E. coli isolates confirms that culture-dependent screening severely underestimates the potential community resistome (9, 35, 36). Nevertheless, most of the identified AR genes were part of the E. coli core genome, in contrast with previous studies of traditional communities in the Peruvian Amazonas and in Bolivia (9, 37). In these populations, which have access to antibiotics and to urban areas, high levels of acquired resistance to antibiotics and a remarkable diversity of AR genes and plasmids have been detected in commensal E. coli isolates. Although AR genes are presumably silenced in Yanomami E. coli isolates of the current study, selection for mutant regulatory regions could readily occur with the introduction of antibiotic selective pressure, as has been observed clinically (34).
Our work emphasizes the value of deep characterization of microbiomes of people living ancestral life-styles, particularly if practices in industrialized societies might eradicate potentially beneficial microbes and their encoded functions. Characterization of the commensal resistome of diverse people will also inform the design and prudent deployment of antibiotics to minimize enrichment for preexisting resistance. Causation and functional consequences of microbiome changes need to be understood before functional restoration of the microbiome is possible to attempt to reverse the current global trends in metabolic and inflammatory diseases.
MATERIALS AND METHODS
Subject and community characterization
The High Orinoco area of Amazonas state in Venezuela is a vast area (~80,000 km2) of jungle, which is the Venezuelan homeland for about 15,000 Yanomami people living in the country. Most of the villages are near rivers, but isolated villages still exist in mountainous areas, and they are described for the first time in the Annual Reports of the Venezuelan Ministry of Health. In 2008, an unmapped village was sighted by an army helicopter, and a medical mission landed there in 2009. Ó.N.-A. was the only author at the site. To protect the identity of the villagers, the name and coordinates of the village are not provided. The Yanomami from the uncontacted village were hunters and gatherers, with no agriculture or domestication of livestock. Trade was evidenced by the presence of machetes, cans, or clothing that are commonly exchanged for arrows with other Yanomami.
The age of the 34 subjects was between 4 and 50 years, as estimated by Yanomami health workers on the medical team (table S1). The villagers were observed to eat wild bananas and seasonal fruits, plantain, palm hearts, and cassava. Hunting consists mostly of birds and small mammals, as well as small crabs, frogs, and small fish from nearby streams, and occasionally peccary, monkey, and tapir. Water is collected from a stream about 5-min walking distance from the village. They also knew the word Medicina from other Yanomami, but the only mission with a medical dispensary (Mahecoto) is estimated to be at least 2 weeks by foot through mountainous terrain.
For comparative purposes, we use data from our published studies of the Guahibo Amerindians and Malawians (11), from villages already in transition toward urban life-styles, with domestication of animals, agricultural practices, some degree of market economy, medical outposts, and access to towns.
Human specimens
Volar forearm skin, oral mucosa, and fecal samples were collected before the health team vaccinated children and administered antibiotics. Samples were immediately frozen in liquid nitrogen and remained frozen until DNA extraction and bacterial culturing were performed.
Samples were cultured on diverse media under aerobic and anaerobic conditions (see below). DNA was also extracted from samples and bacterial cultures from the Yanomami and from five Puerto Rican individuals (to examine their resistome), using the PowerSoil DNA Isolation Kit (MO BIO) following the manufacturer’s recommendations, with the addition of 30-min incubation after the addition of C6 buffer.
16S rRNA analysis
The V4 region of the 16S rRNA gene was amplified with barcoded primers and sequenced as previously described (38). α Diversity was estimated using Faith’s phylogenetic diversity (39) on rarefied tables at 5000 (fecal), 1500 (oral), and 1500 (skin) sequences per sample. β Diversity was measured using unweighted UniFrac (40) on the rarefied tables. γ Diversity (total species diversity within an environment) was estimated using diversity accumulation curves for both mean bacterial genera and phylogenetic species richness as a function of the number of subjects in each population. LEfSe (14) was used with default parameters on species-level OTU tables to determine taxa that best characterize each population; only features with LDA score >2.0 were kept. For presentation purposes in Fig. 1, features with deeper taxonomic annotation (up to the genus level) are presented. A complete list of all significant features is presented in tables S7 to S9. Data from the same set of Yanomami samples generated in combination with a different group of U.S. subjects (41) using the Illumina Genome Analyzer IIx and the V2 region (27 F-338R) of the 16S rRNA gene were analyzed using the same parameters as described above, producing consistent results with those obtained with the V4 region (fig. S3).
Although the main driver of differences in microbiome composition among fecal samples reflected Western versus non-Western life-styles, PCoA of the non-Western subjects alone also revealed differences between these populations, with the Yanomami appearing more similar to the Guahibo than to the Malawians (fig. S7). Because the major differences between Western and non-Western populations were due to changes within the Bacteroidetes phylum (in particular changes in the relative abundances of the Bacteroides and Prevotella), we analyzed non-Bacteroidetes OTUs alone. PCoA analysis of unweighted UniFrac distances and estimates of bacterial diversity (Faith’s phylogenetic diversity) confirm that taxa outside of the Bacteroidetes phylum are also responsible for the differences between the Western and non-Western fecal microbiota (fig. S8).
Normalized prevalence/abundance plots were used to determine whether the proportion of subjects carrying an OTU is higher in a population regardless of its abundance: For each OTU, a curve was constructed measuring the percentage of a population that carries the OTU at at least a given abundance (normalized over the total abundance of the OTU).
Age correlation with bacterial diversity (Faith’s phylogenetic diversity) was examined in all populations and body sites, and only diversity in Malawian fecal samples had a significant but weak positive correlation with age (fig. S9). This was expected in our village because sampled subjects were all older than 4 years, when the fecal microbiota already exhibits an adult-like structure (11).
We expanded the scope of cultures and degree of westernization by performing a meta-analysis combining our study with data from subjects living in urban (United States, Italy), agrarian (Guahibo, Malawi), and hunter-gatherer (Hazda, Tanzania) cultures (11, 42) (fig. S1). Data for Italian and Tanzania subjects were generated using the V4 region and a 454 GS-FLX Titanium sequencer, which resulted in longer reads than those obtained with Illumina technologies. Because there is only a short overlap [~40 base pairs (bp)] in the sequenced region from these two technologies, we did not trim sequence length for either data set, and note that this meta-analysis has some vulnerability to study effects because of a lack of fully overlapping sequence data. OTU tables were rarefied at 1000 sequences per sample, and supervised classification was performed using random forests with an out of bag error model.
Metagenomic prediction and validation
We used PICRUSt 1.0.0 (18) to predict metagenomic content from the 16S rRNA data and STAMP to perform pathway analysis STAMP (43). Because PICRUSt predictions rely on existing sequenced genomes that are mainly derived from Western populations, we first compared the sequenced fecal metagenomes of a Western (n = 18) and a non-Western (n = 10) population (11) against their PICRUSt-predicted metagenomes. Metagenomic reads were mapped against KEGG Orthology (KO)–annotated amino acid sequences as previously described (44), and PICRUSt-predicted metagenomes were estimated using default parameters. To determine the extent to which PICRUSt accuracy is affected by biases toward Western reference genomes, we also calculated the weighted nearest sequenced taxon index (NSTI), which measures the average branch length between OTUs in a sample and a reference genome (higher scores indicating longer distances and thus a less characterized microbial environment). The average Spearman correlation was 0.75 (±0.02) in the Guahibo Amerindians (fig. S2E), close to 0.78 (±0.01) in the U.S. subjects, and to the 0.80 correlation reported in the original PICRUSt paper (18). Mean NSTI values are not significantly different (P = 0.158, t test), indicating that the lower correlation between the predicted and sequenced Guahibo metagenomes is not necessarily due to biases in reference genomes toward those found in Western populations.
In addition, we performed shotgun metagenomic sequencing on 8 fecal and 14 oral samples. Reads passing quality filtering were mapped as previously described (44). An average of 12.9 ± 8.7% of the reads were assigned, similar to or higher than values reported elsewhere [for example, 6.1% in (45)]. Coverage per sample was highly uneven, and we filtered out low-quality samples as follows. We constructed four subsets of the samples for which both 16S and metagenomic data were available: all samples (“all”), samples with ≥10,000 mapped metagenomic sequences (“10 K”), ≥20,000 mapped sequences (“20 K”), and ≥100,000 mapped sequences (“100 K”). For each of these subsets, we performed 100 rarefactions at increasing sequencing depth levels (all: 100/500/1000/5000; 10 K, 20 K, 100 K: 1000/5000/10,000) and calculated Bray-Curtis similarity and PCoA. For each sample subset and each of the 200 (100 for 16S and 100 for metagenomic) PCoA plots thus obtained, we performed Procrustes analysis (46). We found that the 10 K subset achieved the best fit at any rarefaction level (fig. S10), as shown by the lower average M2 (goodness of fit between 16S and metagenomic data, with lower values indicating better fit), and samples with less than 10,000 reads mapped (F23, F36, F49, F50, and O18) were excluded from further analyses.
Pearson correlation between the sequenced and PICRUSt metagenome was high except for oral samples O4 and O13 (fig. S11A). In addition, we performed Procrustes analysis using Bray-Curtis distances on 100 rarefied tables for sequenced and PICRUSt-predicted metagenomes. One hundred Monte Carlo simulations were additionally performed on each of the PICRUST versus shotgun comparisons to estimate a P value. All permutations were significant (P < 0.01) and M2 values were generally low (0.27 ± 0.01), further indicating that there is a good fit between shotgun and PICRUSt data (fig. S11, B and C).
We investigated whether, despite the low and uneven coverage obtained, bacterial contigs could be assembled from the Yanomami data. We used velvet (47) to assemble the metagenomic reads into contigs (k = 31 to 91). The obtained assembly was fragmentary and composed mostly of short contigs (fig. S12A). Bacterial taxonomy was assigned to each contig by aligning them to IMG genomes using nucmer (48) (minimum identity >90% over at least 80% of the query length). Contigs were clustered using CONCOCT (49), and cluster quality was assessed on the basis of the taxonomic assignments for contigs within a cluster using scripts provided in CONCOCT. Although most clusters could not be unequivocally assigned to a single species, we could confirm some of the taxa identified in the Yanomami samples through 16S sequencing (table S10). Cluster quality was measured for all k-mer lengths and at three minimum contig lengths (100, 200, and 500 bp) using NMI (normalized mutual information), F1 score, Rand index, and adjusted Rand index (fig. S12B). A subset of k-mer values were selected for each contig length upon inspection of the quality metrics, and a PCoA plot was generated for clusters obtained with those k-mer sizes and contig lengths (fig. S12C). Overall, these results indicate that we might lack coverage to faithfully reconstruct bacterial genomes using current data. The functional uniqueness of the microbiota in these uncontacted Amerindians may be a result of their isolation for more than 11,000 years since the arrival of their ancestors in the Americas, and genomic divergence from bacterial strains found in Western populations is therefore to be expected. Assuming a doubling rate of once per hour to once per day, an estimated 4 to 100 million bacterial generations would have occurred with little gene migration in the Yanomami microbiome, in contrast to the higher rates in the westernized world.
Cultures and E. coli analyses
We isolated 1097 bacterial strains from frozen fecal or oral samples from the uncontacted Amerindians to provide an archive to study human microbial evolution. Specimens were cultured in seven different media [blood, chocolate, McConkey, Columbia, trypticase soy agar (TSA), CDC anaerobic blood agar (CDC), and Brucella Laked Blood Agar with Kanamycin and Vancomycin (LKV)] under aerobic and anaerobic (jars, bags, and anaerobic chamber) conditions at 37°C.
After 16S rRNA sequencing of 329 isolates (table S11), we characterized 24 E. coli isolates from fecal samples by MLST of seven housekeeping genes (19). MLST analysis was performed using the scheme at the University College of Cork (UCC) E. coli MLST Web site (http://mlst.ucc.ie), which is based on the housekeeping genes adk, fumC, gyrB, icd, mdh, purA, and recA (19). Polymerase chain reaction (PCR) was performed according to protocol (http://mlst.ucc.ie/mlst/dbs/Ecoli/documents/primersColi_html). All PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and sequenced in both directions. Nucleotide sequences were quality-controlled, edited, aligned, and trimmed with the SeqMan (DNASTAR Inc.). All nucleotides within the consensus sequence template were supported by at least two sequence chromatograms. A unique allele number was assigned to each distinct sequence within a locus, and a distinct sequence type (ST) number was assigned to each unique combination of alleles. Allele and ST assignments were made and deposited at the publicly accessible E. coli UCC MLST database (http://mlst.ucc.ie/mlst/dbs/Ecoli) (19). Genetic relationships of Amerindian E. coli isolates were assessed by comparing allelic profiles of Amerindian isolates with existing profiles present in the MLST database. The BURST clustering algorithm was used to define clonal complexes (CCs) of related isolates derived from a common ancestor, the patterns of descent linking them together, and the ancestral genotype (50, 51). Isolates were grouped into CCs if they differed at no more than one locus from at least one other member of the group (51). Founder genotypes of CCs were defined as the ST of the CC with the highest number of neighboring single-locus ST variants. All samples were confirmed to be E. coli by sequencing the 16S rRNA gene. Isolates belonged to 19 STs (table S12), of which 12 were novel, indicating that uncontacted people harbor unique strains. eBURST comparison of 19 STs found in Amerindians with that of the 3544 STs that represent the full global diversity of this species revealed that our isolates represented a broad diversity. This analysis also revealed that most of our isolates were distributed in six CCs, with three STs appearing as singletons (ST-3316, ST-164, and ST-3415) (table S12). These singletons were assigned to CC-10 (differing in less than one locus from at least one other member of the group) when less stringent conditions were used (that is, STs sharing five or more alleles). Two CCs in particular had more than five Amerindian isolates (CC-10 and CC-58, respectively). These data suggest ongoing strain or genetic transfer among the E. coli populations of the individuals in our study, probably facilitated by the fact that all lived in the same village with limited external contact.
The ECOR collection of 72 reference strains (52), which represent the breadth of genetic diversity within the species E. coli, was included for phylogenetic analysis. In-frame–concatenated sequences were aligned using MUSCLE (53), and the ML tree was reconstructed with the HKY85 model of DNA evolution (100 bootstrap replicates) using PhyML (54). In PhyML, the transition/transversion ratio, proportion of invariant sites, and the γ shape parameter were estimated, and subtree-pruning and regrafting and nearest-neighbor interchange tree topology search methods were used. Escherichia fergusonii was used as an outgroup to root the E. coli phylogeny. Gene sequences from all seven MLST loci were also analyzed using ClonalFrame version 1.2 (55), a Bayesian method of constructing evolutionary histories that takes both mutation and recombination into account. Five independent runs were performed, each consisting of 100,000 iterations, including a 50,000 burn-in. Analyses from five independent runs were concatenated to construct a 50% majority-rule consensus tree. MEGA5 (56) was used to draw the consensus phylogenetic tree.
AR phenotypes in E. coli from Amerindians
Of the 329 fecal bacterial isolates (table S11), 131 were E. coli. Of these, all were tested for susceptibility to 23 antibiotics.
MDA of metagenomic DNA
We performed MDA on fecal and oral metagenomic DNA from Amerindian subjects 3, 5, 6, and 23 and the five Puerto Rican subjects with the illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare Life Sciences). The recommended protocol was followed with a 2-hour incubation. We pooled three to six reactions per sample to reduce MDA template amplification bias.
Creation of metagenomic libraries
Small-insert metagenomic libraries were constructed from the MDA-amplified metagenomic DNA in the vector pZE21 as previously described (table S5) (20, 31, 57). The DNA fragments cloned into pZE21 contain several bacterial genes and are situated downstream of the constitutively active pLTetO-1 promoter, enabling expression and phenotypic selection of the shotgun-cloned fragments.
Creation of E. coli isolate genomic libraries
The E. coli isolate genomic libraries were created from 38 isolates from the 11 individuals who provided fecal samples. The number of isolates per individual ranged from 1 to 7. Genomic DNA (500 ng each) from 38 E. coli isolates was combined into two pools, each containing DNA from 19 isolates. Genomic DNA was not amplified with MDA. Small-insert libraries (libraries A and B) were created from the pooled genomic DNA using the procedure above (table S2).
Functional selection of libraries for AR
Metagenomic and genomic libraries were plated on Mueller-Hinton agar supplemented with kanamycin (50 μg/ml) (the selection marker in the pZE21 vector) and another antibiotic (table S6). Puerto Rican and E. coli isolate libraries were not selected against nitrofurantoin. Antibiotic concentrations were clinically relevant concentrations recommended by the Clinical and Laboratory Standards Institute or the minimum inhibitory concentration of the library host MegaX transformed with self-ligated pZE21. In all cases, antibiotic concentrations inhibited MegaX transformed with self-ligated pZE21. Surviving colonies were pooled. In some cases, resistant colonies were confirmed by replating on solid media (F6 cefepime and O23 tetracycline selections) or by inoculating into liquid media (F6 cefepime, 238 penicillin, F3, F6, F23, T1001 tetracycline, O3 chloramphenicol, 238, 337, T1001 piperacillin-tazobactam, O23, T1001, library B ceftazidime, and library A and B aztreonam selections) containing the selective antibiotic at the same concentration. Reselected clones were used as the template for sequencing.
Sequencing and assembly of resistance-conferring inserts
Resistance-conferring inserts were isolated from pooled surviving colonies via PCR with vector-specific primers, barcoded, and sequenced in parallel with the Illumina HiSeq 2000 (2 × 101-bp reads). Reads were assembled into contigs, and open reading frames were identified with PARFuMS and then annotated with Resfams (31, 58). The sequences of the F6 cefepime, O23 tetracycline, and E. coli library A ceftazidime-resistant inserts were determined by paired-end Sanger sequencing performed at the Protein and Nucleic Acid Chemistry Laboratory (PNACL) at Washington University School of Medicine. For F6_AZ_9, F6_CT_1, O3_CZ_2_1, O3_CZ_2_2, O3_TE_1, O23_TE_1, F23_TE_1, F6_CH_4, O23_CH_19, and F23_CH_11, Sanger sequencing was performed at PNACL or at Genewiz with specific primers to confirm portions of the sequence, and the Sanger sequence was used for all analyses.
Computational filtering of assembled contigs from metagenomic libraries
Nonspecific amplification is frequently observed for MDA with commercial MDA reagents, particularly with small amounts of starting template (59). A total of 171 ng of DNA was amplified from three no-template control (NTC) reactions performed in parallel with the Amerindian sample amplifications, ~15× less output DNA per reaction than for the fecal and oral samples (57 ± 25 ng versus 854 ± 67 ng). There was no detectable amplification in the NTCs for the Puerto Rico sample amplifications (<0.25 ng each). We shotgun-sequenced 84 ng of the Amerindian NTC product without antibiotic selection and assembled the reads with the same procedure as the sample reads. To ensure the authenticity of the genes identified in the metagenomic functional selections, we excluded any sample contig from analysis that aligned to an NTC contig with at least 99% nucleotide identity over at least 200 bp (113 Amerindian and 4 Puerto Rican contigs). Additionally, we excluded any sample contig whose best hit in NCBI nt (16 August 2013) was a vector or mouse/human DNA, provided that the alignment was at least 200 bp long and had 99% sequence identity (four and two Amerindian contigs, respectively, and no Puerto Rican contigs). E. coli isolate libraries were not filtered because they did not undergo MDA. We did not consider any contigs under 500 bp.
AR gene identification
From the contigs that passed computational filtering, AR genes were identified on the basis of Resfams annotations specific to that antibiotic class, similarity to known resistance genes (for transporters), or subcloning and functional confirmation. The genes F23_TE_1:69-1403, O23_TE_1:1-1845, O23_CH_19:550-804, and F23_CH_11:1298-1636 were PCR-amplified from their respective antibiotic selections with primers encompassing the entire gene. They were ligated into pZE21 at an optimal distance from the RBS (ribosome binding site) and transformed into E. coli DH10B. The resistance phenotype was confirmed by plating on the selective antibiotic at the same concentration used in the initial selection and amplification of the fragment from surviving colonies with specific primers.
Alignment to NCBI
AR genes were compared to NCBI nr (non-redundant protein database) (downloaded 15 September 2014; including uncultured/environmental sample sequences) via blastx. The top hit was the hit with the lowest e value; any hits with the same e value and score were also considered top hits. Alignments 200 bp or shorter were excluded from analysis. Global %ID (table S4) was calculated from estwise (Wise2 package) sequence alignments. To compare the context of AR genes in contigs isolated from Amerindian metagenomes and in industrialized settings, genes and the surrounding contigs were compared to NCBI nt (15 August 2013).
Tree building for Amerindian PBPs
For each gene, the top blastx hit in NCBI nr and any hit with amino acid identity ≥99% and coverage ≥90% of the gene was downloaded from NCBI. Proteins were clustered with cd-hit at 99.5% identity (60), and the representative sequence for each cluster was used for tree building. Functionally isolated AR genes were translated with EMBOSS Transeq (61, 62). The phylogenetic tree was created with the Phylogeny.fr pipeline (63). Proteins were aligned with MUSCLE (Full Mode) (53), and PhyML3.0 (54, 64) was used to create the phylogenetic tree (default parameters, default substitution model, SH-like approximate likelihood ratio test for branch support), which was visualized with Geneious version 7.0 (Biomatters).
Alignment of functionally selected AR genes to the HMP and MetaHIT
AR genes were compared to the HMP (65) and MetaHIT (66) shotgun assemblies (HMP: 749 assemblies, http://www.hmpdacc.org/HMASM/; MetaHIT: 124 assemblies, NCBI/European Bioinformatics Institute accession ERA000116, http://www.bork.embl.de/~arumugam/Qin_et_al_2010/) via blastn. The reported sequence identity for each gene is that of the top alignment (lowest e value) in that database. Alignments 200 bp or shorter were excluded from analysis.
Alignment to E. coli MG1655
All AR genes isolated from the E. coli genomic libraries were compared to E. coli str. K-12 substr. MG1655 (RefSeq: NC_000913.3) on 19 September 2014 to determine the gene identity. All genes with MG1655 alignments (39 of 44 genes) aligned with ≥96% nucleotide identity.
Filtering of human sequences from shotgun reads
Human sequences were filtered from the forward-only trimmed shotgun reads by mapping to the human reference genome (GRCh38) with DeconSeq (67). Filtered reads were uploaded to the NCBI Sequence Read Archive (SRA) (SRP049631) and mapped to resistance genes.
Comparison of shotgun reads to functionally selected resistance genes
All functionally selected AR genes from the Amerindian and Puerto Rican oral and fecal metagenomic libraries and the Amerindian E. coli isolate genomic DNA libraries were clustered with cd-hit-est at 100% nucleotide identity over the length of the smaller gene (60). Forward-only trimmed and filtered shotgun reads from Amerindian fecal and oral microbiota were mapped to the clustered AR genes with bowtie2 (end-to-end alignment, one alignment per read) (68). Alignments were filtered to those with an alignment score ≥−0.3278 × the length of the read (~95% ID). Gene abundance in the metagenome was calculated as the number of reads mapping to the gene by the length of the gene in kilobases by millions of reads mapped. A Bray-Curtis dissimilarity matrix was calculated in R (package vegan, function vegdist) (69) from the gene × sample abundance table. Samples were clustered on the dissimilarity matrix with UPGMA (function hclust). Heatmaps of gene abundance were generated with R packages NMF and RColorBrewer (70, 71). Statistical differences in abundance between categories were calculated with Wilcoxon rank sum tests and corrected for multiple hypotheses with a Bonferroni correction.
Supplementary Material
Acknowledgments
We thank the Venezuelan “Fuerza Armada Nacional Bolivariana” and the members of the Health expedition, including medical technologists C. Ordaz, whose support made possible the collecting of these unique specimens. We are also grateful to the people of the newly contacted village for their trust and our shared desire that the unavoidable contact with our culture will bring health benefits and protection to their people. We thank M. Charbonneau, H. J. Lee, and J. Gu for optimization of the MDA metagenome amplification protocols. D. Wendel, J. Stombaugh, and G. Ackermann assisted with data processing. D. Berg-Lyons, G. Humphrey, and C. Lauber assisted with sample processing. Funding: This work was supported in part by the C&D Fund (M.G.D.-B.), the Emch Fund (M.G.D.-B.), NIH SCORE-PES RISE (M.G.D.-B.), SUCCESS (J.C.C.), Helmsley Charitable Trust HSP-007 (J.C.C.), NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) DK062429 (J.C.C.), NAKFI Synthetic Biology—SB2 (G.D. and R.K.), NIH Director’s New Innovator Award NIDDK DP2-DK098089 (G.D.), NIH National Institute of General Medical Sciences R01-GM099538 (G.D.), Washington University I-CARES award (G.D.), UH2AR057506 and the Diane Belfer Program for Human Microbial Ecology (M.J.B.), National Defense Science and Engineering Graduate fellowship (E.C.P.), and Howard Hughes Medical Institute Early Career Scientist (R.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. Author contributions: M.M. and M.G.D.-B. planned the study; O.N.-A., M.M., G.H., M. Contreras, O.L., and M.G.D.-B. implemented the sample collection and preservation; Z.G., K.S., J. McDonald, M. Cox, S.R., J.F.R., J.W., P.L.O., and M.G.D.-B. characterized bacterial isolates; E.C.P., B.W., and G.D. characterized the resistome; R.K., Z.G., K.S., J. McDonald, M.G.D.-B., J.W., P.L.O., S.R., B.W., E.C.P., and G.D. generated data; J.C.C., R.K., M.J.B., E.C.P., G.D., N.S., S.J.S., J. Metcalf, and M.G.D.-B. performed analyses and interpreted results; J.C.C., E.C.P., G.D., M.J.B., R.K., and M.G.D.-B. wrote the paper. Competing interests: The authors declare that they have no competing financial interests. Data and materials availability: 16S rRNA sequence data has been deposited to EBI with IDs ERA387449 and ERP008799. Assembled functional metagenomic contigs have been deposited to GenBank with IDs KJ910904–KJ910951, KJ910953–KJ910954, KJ910958–KJ910959, KJ910961–KJ910973, KJ910975–KJ910989, KJ910991–KJ911003, KJ911005–KJ911006, KJ911008–KJ911009, KJ911011, and KP063058, and raw shotgun metagenomic reads to SRA with ID SRP049631.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/3/e1500183/DC1
Text
Table S1. Samples obtained from the 34 Amerindian subjects included in this study: skin from right (BD) or left (BI) forearm (n = 28), oral (n = 28), and fecal (n = 12) specimens.
Table S2. Composition of Amerindian E. coli isolate genomic libraries.
Table S3. Functional capture of AR genes from Amerindian E. coli isolate genomic libraries.
Table S4. AR genes identified from functional selections of Amerindian E. coli isolate genomic libraries and Amerindian and Puerto Rican metagenomic libraries.
Table S5. Composition of metagenomic libraries from Amerindian and Puerto Rican fecal and oral microbiota.
Table S6. Antibiotics used in genomic and metagenomic library selections for AR genes.
Table S7. List of all taxa significantly associated with each population in fecal samples as determined by LEfSe analysis.
Table S8. List of all taxa significantly associated with each population in oral samples as determined by LEfSe analysis.
Table S9. List of all taxa significantly associated with each population in skin samples as determined by LEfSe analysis.
Table S10. List of clusters at different k-mer and minimum contig length and their associated taxonomy.
Table S11. Taxa of 329 bacterial strains cultured from feces of 12 isolated Amerindians.
Table S12. Typing of 24 E. coli strains from 11 isolated Amerindians.
Fig. S1. Meta-analysis of 16S V4 region fecal data.
Fig. S2. Microbiome diversity between different human groups.
Fig. S3. Microbiota diversity in fecal, oral, and skin samples from uncontacted Yanomami in relation to U.S. subjects using the V2 region of the 16S rRNA gene.
Fig. S4. Taxonomic distribution across gut samples sorted by the dominant genera within each population.
Fig. S5. Prevalence/abundance curves of representative bacterial OTUs in feces (A), oral (B), and skin (C).
Fig. S6. ClonalFrame analysis of Escherichia coli strains based on the sequence of seven housekeeping genes.
Fig. S7. PCoA plot of Yanomami, Malawian, and Guahibo fecal samples.
Fig. S8. Fecal microbiome of non-Bacteroides OTUs in Yanomami, Malawian,Guahibo, and U.S. samples.
Fig. S9. Bacterial diversity as a function of age and body site.
Fig. S10. Procrustes analyses of shotgun metagenomic and 16S rRNA data for different subsets of samples.
Fig. S11. Comparison of PICRUSt versus sequenced shotgun metagenomes of fecal and oral samples with at least 10,000 mapped reads.
Fig. S12. Bacterial genome assembly.
REFERENCES AND NOTES
- 1.Henao-Mejia J., Elinav E., Jin C., Hao L., Mehal W. Z., Strowig T., Thaiss C. A., Kau A. L., Eisenbarth S. C., Jurczak M. J., Camporez J. P., Shulman G. I., Gordon J. I., Hoffman H. M., Flavell R. A., Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho I., Yamanishi S., Cox L., Methé B. A., Zavadil J., Li K., Gao Z., Mahana D., Raju K., Teitler I., Li H., Alekseyenko A. V., Blaser M. J., Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turnbaugh P. J., Hamady M., Yatsunenko T., Cantarel B. L., Duncan A., Ley R. E., Sogin M. L., Jones W. J., Roe B. A., Affourtit J. P., Egholm M., Henrissat B., Heath A. C., Knight R., Gordon J. I., A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diaz H. R., Wang S., Anuar F., Qian Y., Björkholm B., Samuelsson A., Hibberd M. L., Forssberg H., Pettersson S., Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. U.S.A. 108, 3047–3052 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turnbaugh P. J., Ley R. E., Mahowald M. A., Magrini V., Mardis E. R., Gordon J. I., An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Koren O., Goodrich J. K., Cullender T. C., Spor A., Laitinen K., Bäckhed H. K., Gonzalez A., Werner J. J., Angenent L. T., Knight R., Bäckhed F., Isolauri E., Salminen S., Ley R. E., Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150, 470–480 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ministerio del Poder Popular para la Salud. Memoria 2008–2011 (Caracas, Venezuela, 2009–2012).
- 8.Sommer M. O., Dantas G., Church G. M., Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325, 1128–1131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartoloni A., Pallecchi L., Rodríguez H., Fernandez C., Mantela A., Bartalesi F., Strohmeyer M., Kristiansson C., Gotuzzo E., Paradisi F., Rossolini G. M., Antibiotic resistance in a very remote Amazonas community. Int. J. Antimicrob. Agents 33, 125–129 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Warinner C., Rodrigues J. F., Vyas R., Trachsel C., Shved N., Grossmann J., Radini A., Hancock Y., Tito R. Y., Fiddyment S., Speller C., Hendy J., Charlton S., Luder H. U., Salazar-García D. C., Eppler E., Seiler R., Hansen L. H., Castruita J. A., Barkow-Oesterreicher S., Teoh K. Y., Kelstrup C. D., Olsen J. V., Nanni P., Kawai T., Willerslev E., von Mering C., Lewis C. M. Jr, Collins M. J., Gilbert M. T., Rühli F., Cappellini E., Pathogens and host immunity in the ancient human oral cavity. Nat. Genet. 46, 336–344 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yatsunenko T., Rey F. E., Manary M. J., Trehan I., Dominguez-Bello M. G., Contreras M., Magris M., Hidalgo G., Baldassano R. N., Anokhin A. P., Heath A. C., Warner B., Reeder J., Kuczynski J., Caporaso J. G., Lozupone C. A., Lauber C., Clemente J. C., Knights D., Knight R., Gordon J. I., Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.American Gut, http://github.com/biocore/American-Gut
- 13.Pontzer H., Raichlen D. A., Wood B. M., Mabulla A. Z., Racette S. B., Marlowe F. W., Hunter-gatherer energetics and human obesity. PLOS One 7, e40503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., Huttenhower C., Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groth I., Schumann P., Schutze B., Augsten K., Stackebrandt E., Knoellia sinensis gen. nov., sp. nov. and Knoellia subterranea sp. nov., two novel actinobacteria isolated from a cave. Int. J. Syst. Evol. Microbiol. 52, 77–84 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Shin N. R., Roh S. W., Kim M. S., Jung M. J., Whon T. W., Bae J. W., Knoellia locipacati sp. nov., from soil of the Demilitarized Zone in South Korea. Int. J. Syst. Evol. Microbiol. 62, 342–346 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Ward N. L., Challacombe J. F., Janssen P. H., Henrissat B., Coutinho P. M., Wu M., Xie G., Haft D. H., Sait M., Badger J., Barabote R. D., Bradley B., Brettin T. S., Brinkac L. M., Bruce D., Creasy T., Daugherty S. C., Davidsen T. M., DeBoy R. T., Detter J. C., Dodson R. J., Durkin A. S., Ganapathy A., Gwinn-Giglio M., Han C. S., Khouri H., Kiss H., Kothari S. P., Madupu R., Nelson K. E., Nelson W. C., Paulsen I., Penn K., Ren Q., Rosovitz M. J., Selengut J. D., Shrivastava S., Sullivan S. A., Tapia R., Thompson L. S., Watkins K. L., Yang Q., Yu C., Zafar N., Zhou L., Kuske C. R., Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl. Environ. Microbiol. 75, 2046–2056 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langille M. G., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., Clemente J. C., Burkepile D. E., RL Vega T., Knight R., Beiko R. G., Huttenhower C., Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wirth T., Falush D., Lan R., Colles F., Mensa P., Wieler L. H., Karch H., Reeves P. R., Maiden M. C., Ochman H., Achtman M., Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 60, 1136–1151 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forsberg K. J., Patel S., Gibson M. K., Lauber C. L., Knight R., Fierer N., Dantas G., Bacterial phylogeny structures soil resistomes across habitats. Nature 509, 612–616 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Contreras M., Costello E. K., Hidalgo G., Magris M., Knight R., Dominguez-Bello M. G., The bacterial microbiota in the oral mucosa of rural Amerindians. Microbiology 156, 3282–3287 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Lester C. H., Frimodt-Møller N., Sørensen T. L., Monnet D. L., Hammerum A. M., In vivo transfer of the vanA resistance gene from an Enterococcus faecium isolate of animal origin to an E. faecium isolate of human origin in the intestines of human volunteers. Antimicrob. Agents Chemother. 50, 596–599 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cox L. M., Yamanishi S., Sohn J., Alekseyenko A. V., Leung J. M., Cho I., Kim S. G., Li H., Gao Z., Mahana D., Zárate Rodriguez J. G., Rogers A. B., Robine N., Loke P., Blaser M. J., Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158, 705–721 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dominguez-Bello M. G., Costello E. K., Contreras M., Magris M., Hidalgo G., Fierer N., Knight R., Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U.S.A. 107, 11971–11975 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chesson P., Mechanisms of maintenance of species diversity. Annu. Rev. Ecol. Syst. 31, 343–366 (2000). [Google Scholar]
- 26.Leibold M. A., Holyoak M., Mouquet N., Amarasekare P., Chase J. M., Hoopes M. F., Holt R. D., Shurin J. B., Law R., Tilman D., Loreau M., Gonzalez A., The metacommunity concept: A framework for multi-scale community ecology. Ecol. Lett. 7, 601–613 (2004). [Google Scholar]
- 27.Faith J. J., Guruge J. L., Charbonneau M., Subramanian S., Seedorf H., Goodman A. L., Clemente J. C., Knight R., Heath A. C., Leibel R. L., Rosenbaum M., Gordon J. I., The long-term stability of the human gut microbiota. Science 341, 1237439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charlton A., Medicinal uses of tobacco in history. J. R. Soc. Med. 97, 292–296 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.F. M. Salzano, S. M. Callegari-Jacques, South American Indians: A Case Study in Evolution (Clarendon Press, Oxford, 1988). [Google Scholar]
- 30.D’Costa V. M., King C. E., Kalan L., Morar M., Sung W. W., Schwarz C., Froese D., Zazula G., Calmels F., Debruyne R., Golding G. B., Poinar H. N., Wright G. D., Antibiotic resistance is ancient. Nature 477, 457–461 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Forsberg K. J., Reyes A., Wang B., Selleck E. M., Sommer M. O., Dantas G., The shared antibiotic resistome of soil bacteria and human pathogens. Science 337, 1107–1111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dantas G., Sommer M. O., Context matters—The complex interplay between resistome genotypes and resistance phenotypes. Curr. Opin. Microbiol. 15, 577–582 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Martínez J. L., Coque T. M., Baquero F., What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 13, 116–123 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Tracz D. M., Boyd D. A., Hizon R., Bryce E., McGeer A., Ofner-Agostini M., Simor A. E., Paton S., MR; Canadian Nosocomial Infection Surveillance Program Mulvey. ampC gene expression in promoter mutants of cefoxitin-resistant Escherichia coli clinical isolates. FEMS Microbiol. Lett. 270, 265–271 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Houndt T., Ochman H., Long-term shifts in patterns of antibiotic resistance in enteric bacteria. Appl. Environ. Microbiol. 66, 5406–5409 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walson J. L., Marshall B., Pokhrel B. M., Kafle K. K., Levy S. B., Carriage of antibiotic-resistant fecal bacteria in Nepal reflects proximity to Kathmandu. J. Infect. Dis. 184, 1163–1169 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Bartoloni A., Bartalesi F., Mantela A., Dell’Amico E., Roselli M., Strohmeyer M., Barahona H. G., Barrón V. P., Paradisi F., Rossolini G. M., High prevalence of acquired antimicrobial resistance unrelated to heavy antimicrobial consumption. J. Infect. Dis. 189, 1291–1294 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Huntley J., Fierer N., Owens S. M., Betley J., Fraser L., Bauer M., Gormley N., Gilbert J. A., Smith G., Knight R., Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faith D. P., Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10 (1992). [Google Scholar]
- 40.Lozupone C., Knight R., UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song S. J., Lauber C., Costello E. K., Lozupone C. A., Humphrey G., Berg-Lyons D., Caporaso J. G., Knights D., Clemente J. C., Nakielny S., Gordon J. I., Fierer N., Knight R., Cohabiting family members share microbiota with one another and with their dogs. eLife 2, e00458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schnorr S. L., Candela M., Rampelli S., Centanni M., Consolandi C., Basaglia G., Turroni S., Biagi E., Peano C., Severgnini M., Fiori J., Gotti R., De Bellis G., Luiselli D., Brigidi P., Mabula A., Marlowe F., Henry A. G., Crittedenn A. N., Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5, 3654 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parks D. H., Tyson G. W., Hugenholtz P., Beiko R. G., STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fierer N., Ladau J., Clemente J. C., Leff J. W., Owens S. M., Pollard K. S., Knight R., Gilbert J. A., McCulley R. L., Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342, 621–624 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Roggenbuck M., Bærholm S. I., Blom N., Bælum J., Bertelsen M. F., Pontén T. S., Sørensen S. J., Gilbert M. T., Graves G. R., Hansen L. H., The microbiome of New World vultures. Nat. Commun. 5, 5498 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Muegge B. D., Kuczynski J., Knights D., Clemente J. C., González A., Fontana L., Henrissat B., Knight R., Gordon J. I., Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zerbino D. R., Birney E., Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurtz S., Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., Salzberg S. L., Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alneberg J., Bjarnason B. S., de Bruijn I., Schirmer M., Quick J., Ijaz U. Z., Lahti L., Loman N. J., Andersson A. F., Quince C., Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Feil E. J., Li B. C., Aanensen D. M., Hanage W. P., Spratt B. G., eBURST: Inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jolley K. A., Feil E. J., Chan M. S., Maiden M. C., Sequence type analysis and recombinational tests (START). Bioinformatics 17, 1230–1231 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Ochman H., Selander R. K., Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 157, 690–693 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O., New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Didelot X., Falush D., Inference of bacterial microevolution using multilocus sequence data. Genetics 175, 1251–1266 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S., MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moore A. M., Patel S., Forsberg K. J., Wang B., Bentley G., Razia Y., Qin X., Tarr P. I., Dantas G., Pediatric fecal microbiota harbor diverse and novel antibiotic resistance genes. PLOS One 8, e78822 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gibson M. K., Forsberg K. J., Dantas G., Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 9, 207–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blainey P. C., Quake S. R., Digital MDA for enumeration of total nucleic acid contamination. Nucleic Acids Res. 39, e19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fu L., Niu B., Zhu Z., Wu S., Li W., CD-HIT: Accelerated for clustering the next generation sequencing data. Bioinformatics 28, 3150–3152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rice P., Longden I., Bleasby A., EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R., A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V., Lescot M., Claverie J. M., Gascuel O., Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anisimova M., Gascuel O., Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qin J., Li R., Raes J., Arumugam M., Burgdorf K. S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., Mende D. R., Li J., Xu J., Li S. , Li D., Cao J., Wang B., Liang H., Zheng H., Xie Y., Tap J., Lepage P., Bertalan M., Batto J. M., Hansen T., Le Paslier D., Linneberg A., Nielsen H. B., Pelletier E., Renault P., Sicheritz-Ponten T., Turner K., Zhu H., Yu C., Li S., Jian M., Zhou Y., Li Y., Zhang X., Li S., Qin N., Yang H., Wang J., Brunak S., Doré J., Guarner F., Kristiansen K., Pedersen O., Parkhill J., Weissenbach J. MetaHIT Consortium Bork P., Ehrlich S. D., Wang J., A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmieder R., Edwards R., Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLOS One 6, e17288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.vegan: Community Ecology Package. R package version 2.0-10 (2013).
- 70.RColorBrewer: ColorBrewer palettes. R package version 1.0-5 (2011).
- 71.Gaujoux R., Seoighe C., A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11 367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rideout J. R., He Y., Navas-Molina J. A., Walters W. A., Ursell L. K., Gibbons S. M., Chase J., McDonald D., Gonzalez A., Robbins-Pianka A., Clemente J. C., Gilbert J. A., Huse S. M., Zhou H. W., Knight R., Caporaso J. G., Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2, e545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McDonald D., Price M. N., Goodrich J., Nawrocki E. P., DeSantis T. Z., Probst A., Andersen G. L., Knight R., Hugenholtz P., An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kazimierczak K. A., Flint H. J., Scott K. P., Comparative analysis of sequences flanking tet(W) resistance genes in multiple species of gut bacteria. Antimicrob. Agents Chemother. 50, 2632–2639 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beaber J. W., Hochhut B., Waldor M. K., SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427, 72–74 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/3/e1500183/DC1
Text
Table S1. Samples obtained from the 34 Amerindian subjects included in this study: skin from right (BD) or left (BI) forearm (n = 28), oral (n = 28), and fecal (n = 12) specimens.
Table S2. Composition of Amerindian E. coli isolate genomic libraries.
Table S3. Functional capture of AR genes from Amerindian E. coli isolate genomic libraries.
Table S4. AR genes identified from functional selections of Amerindian E. coli isolate genomic libraries and Amerindian and Puerto Rican metagenomic libraries.
Table S5. Composition of metagenomic libraries from Amerindian and Puerto Rican fecal and oral microbiota.
Table S6. Antibiotics used in genomic and metagenomic library selections for AR genes.
Table S7. List of all taxa significantly associated with each population in fecal samples as determined by LEfSe analysis.
Table S8. List of all taxa significantly associated with each population in oral samples as determined by LEfSe analysis.
Table S9. List of all taxa significantly associated with each population in skin samples as determined by LEfSe analysis.
Table S10. List of clusters at different k-mer and minimum contig length and their associated taxonomy.
Table S11. Taxa of 329 bacterial strains cultured from feces of 12 isolated Amerindians.
Table S12. Typing of 24 E. coli strains from 11 isolated Amerindians.
Fig. S1. Meta-analysis of 16S V4 region fecal data.
Fig. S2. Microbiome diversity between different human groups.
Fig. S3. Microbiota diversity in fecal, oral, and skin samples from uncontacted Yanomami in relation to U.S. subjects using the V2 region of the 16S rRNA gene.
Fig. S4. Taxonomic distribution across gut samples sorted by the dominant genera within each population.
Fig. S5. Prevalence/abundance curves of representative bacterial OTUs in feces (A), oral (B), and skin (C).
Fig. S6. ClonalFrame analysis of Escherichia coli strains based on the sequence of seven housekeeping genes.
Fig. S7. PCoA plot of Yanomami, Malawian, and Guahibo fecal samples.
Fig. S8. Fecal microbiome of non-Bacteroides OTUs in Yanomami, Malawian,Guahibo, and U.S. samples.
Fig. S9. Bacterial diversity as a function of age and body site.
Fig. S10. Procrustes analyses of shotgun metagenomic and 16S rRNA data for different subsets of samples.
Fig. S11. Comparison of PICRUSt versus sequenced shotgun metagenomes of fecal and oral samples with at least 10,000 mapped reads.
Fig. S12. Bacterial genome assembly.