

Abstract
A new series of potent potent aryl/alkylated (bis)urea- and (bis)thiourea polyamine analogues were synthesized and evaluated in vitro for their antiplasmodial activity. Altering the carbon backbone and terminal substituents increased the potency of analogues in the compound library 3-fold, with the most active compounds, 15 and 16, showing half-maximal inhibitory concentrations (IC50 values) of 28 and 30 nM, respectively, against various Plasmodium falciparum parasite strains without any cross-resistance. In vitro evaluation of the cytotoxicity of these analogues revealed marked selectivity towards targeting malaria parasites compared to mammalian HepG2 cells (>5000-fold lower IC50 against the parasite). Preliminary biological evaluation of the polyamine analogue antiplasmodial phenotype revealed that (bis)urea compounds target parasite asexual proliferation, whereas (bis)thiourea compounds of the same series have the unique ability to block transmissible gametocyte forms of the parasite, indicating pluripharmacology against proliferative and non-proliferative forms of the parasite. In this manuscript, we describe these results and postulate a refined structure-activity relationship (SAR) model for antiplasmodial polyamine analogues. The terminally aryl/alkylated (bis)urea- and (bis)thiourea-polyamine analogues featuring a 3-5-3 or 3-6-3 carbon backbone represent a structurally novel and distinct class of potential antiplasmodials with activities in the low nanomolar range, and high selectivity against various lifecycle forms of P. falciparum parasites.
Keywords: Malaria, Plasmodium, Polyamine analogue, Antimalarial drugs, (Bis)urea, (Bis)thiourea
1. Introduction
Malaria remains a deadly parasitic disease transmitted to humans by the female Anopheles mosquito in the tropical and sub-tropical parts of the world and still causes approximately 600,000 deaths each year, mostly of African children. At present, the primary component of parasite control remains the use of antimalarial drugs; however, these have to remain effective against a background of continuous development of drug resistant parasite strains. As a result, global efforts are focused on the development of novel antimalarial chemotypes with unique mechanisms of action compared to currently used antimalarial drugs. The antimalarial drug pipeline is currently populated by, for instance, imidazolopiperazines,1 spiroindolones,2 synthetic peroxides,3 dihydroorotate dehydrogenase inhibitors4 and 3,5-diaryl-2-aminopyridines5(www.mmv.org). However, several other chemotypes are effective antiproliferative agents against both cancer and parasitic diseases, and we have previously explored some of these as starting points to assess their efficacy against malaria parasites. Although cancer and parasitic diseases have different etiologies, they share pathophysiological features including uncontrolled, rapid proliferation; enhanced metabolic activities; some signal transduction pathways and immune evasion strategies.
Biogenic amines and polyamines (the naturally occurring spermidine and spermine, and their precursor putrescine) are well recognized for their pleiotropic biological activities during cell growth and development, making them essential constituents of highly proliferative cell types including cancerous cells and parasites.6 While the pKa values of the amine groups within the polyamine chain vary, these groups are all essentially protonated, and are positively charged at physiological pH.7 Polyamines are therefore able to electrostatically interact with RNA, DNA, dTNPs, proteins, and other anionic substances, thereby modulating their functions. The polyamines therefore play essential roles in many of the following processes: cell growth, proliferation, differentiation, synthesis of nucleic acids and proteins, cell adhesion, signaling processes and in the repair of the extracellular matrix.
Inhibition of polyamine biosynthesis is well recognized for its therapeutic efficacy against parasitic diseases including Trypanosoma brucei gambiense infections (West African sleeping sickness).8 Moreover, analogues of the naturally occurring polyamines are structurally similar to the natural polyamines but act as either antimetabolites or polyamine mimetics, thereby resulting in the non-functional replacement of the natural polyamines from their specific effector sites.9,10 These chemotypes were originally studied for their antiproliferative properties against various forms of cancer,11 but terminally (bis)aryl/alkylated polyamine analogues such as compounds 1–5 (Fig. 1) and (bis)biguanides such as verlindamycin (compound 6; Fig. 1) have previously been reported as highly effective antiparasitic agents against various parasites.8,12,13 In particular, compound 4 was found to be curative for the microsporidia Encephalitozoan cuniculi in a murine model.8 Compound 1 demonstrated impressive antileishmanial activity, eliminating Leishmania donovani amastigotes from mouse macrophages at 1 µM concentrations.14 This analogue was also an effective antitrypanosomal agent, inhibiting the In vitro growth of various drug-sensitive and -resistant strains of T. brucei parasites.15 Compound 5 had an IC50 of 110 nM against the intraerythrocytic P. falciparum drug-sensitive 3D7 strain and an IC50 of 120 nM against the P. falciparum Dd2 drug-resistant strain and was as effective as pyrimethamine at reducing parasitemia and extending life span in a murine model of Plasmodium yoelii (unpublished observations). Compound 1 also presented remarkable antimalarial activity and was curative of disease in a P. berghei malaria model.16 Additionally, mice treated with compound 1 combined with a polyamine biosynthesis inhibitor were resistant to Plasmodium berghei re-infections.16 This finding demonstrated the prospective value of the polyamine pathway as a drug target in Plasmodia.17
Figure 1.

Structures of (bis)alkylpolyamines (compounds 1–5), verlindamycin (compound 6) and 1,14-bis-(phenylurea)-tetradecane (compound 7).
As part of our continued search for structurally novel antimalarials, potent terminally substituted aryl/alkylated (bis)urea and (bis)thiourea polyamine analogues based on the lead compound verlindamycin (6) were synthesized and evaluated for their antimalarial properties.18 These compounds contained 3-3-3, 3-4-3, 3-6-3 or 3-7-3 carbon backbone architecture, combined with a variety of terminal substituents. The majority of these compounds possessed IC50 values below 500 nM against intraerythrocytic P. falciparum parasites. The leading compound in this series, (bis)urea 7 (IC50 = 88 nM) contained a 3-6-3 carbon backbone architecture and terminal phenyl substitutions. Importantly, these polyamine analogues represent a chemotype of highly selective antiplasmodial agents (>7000-fold In vitro selectivity compared to mammalian HepG2 cells) and produce a significant irreversible, cytotoxic response in the parasite.18
In an effort to complete the characterization and interrogate potential structure-activity relationships for this chemotype, we report here additional isosteres based on the structure of compound 6 that feature a 3-5-3 carbon backbone architecture (8–16). (Bis)urea- and (bis)thiourea analogues with the 3-5-3 backbone architecture have not yet been reported or evaluated in any antiparasitic assay systems. These compounds were evaluated here for their ability to inhibit the In vitro proliferation of both drug-sensitive and drug-resistant strains of P. falciparum parasites. With this data, we now have a structurally complete polyamine library, spanning the backbone pharmacophore from the 3-3-3 norspermine scaffold to an extended 3-7-3 scaffold, all with various terminal substitutions. This library has facilitated the development of a preliminary structure activity relationship model for these novel antiplasmodial agents.
2. Results and discussion
2.1. Chemistry
Polyamine analogues exemplify a class of compounds that are structurally distinct from any existing antimalarial drug or new chemotype under investigation. To enable comparative analysis of the complete chemotype based on variant polyamine backbones, compounds 8–16 were designed as a novel series with 3-5-3 polyamine backbones. These compounds were synthesized using adaptations of our previously reported synthesis, as shown in Scheme 1.18,19 A reaction of N4,N10-(bis)-[(tert-butyloxy) carbonyl]-1,14-aminotridecane 17 with the appropriate isothiocyanate or isocyanate gave the corresponding protected (bis)thiourea analogues 18–21, or the protected (bis)urea analogues 22–26, respectively. Removal of the N-Boc protection groups (1.0 M HCl in EtOAc) provided the desired (bis)thiourea polyamines 8–11, and the (bis)urea polyamines 12–16.
Scheme 1.

2.2. Biological activity
2.2.1. In vitro antiplasmodial activity against asexual parasites
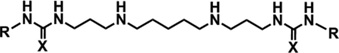
To interrogate the antiplasmodial activity of the 3-5-3 polyamine backbone series, compounds 8–16 were evaluated for their antiplasmodial activities against drug-sensitive and -resistant strains of intraerythrocytic P. falciparum parasites In vitro. In the In vitro assay, compounds with IC50 values ⩽100 nM were considered potently active, while compounds with IC50 values between 100 and 900 nM were considered active. Thus 9–16 were identified as antiplasmodial agents against the 3D7 strain of intraerythrocytic P. falciparum parasites, with activities between 28 and 793 nM (Table 1). The two most potent 3-5-3 compounds, (bis)ureas 15 and 16, had IC50 values of 28 and 30 nM, respectively. Compounds 8 and 12 had moderate activities of 249 and 246 nM, respectively, while compound 13 was found to be the least active against the 3D7 strain of P. falciparum, with an IC50 of 793 nM.
Table 1.
In vitro antiplasmodial activity of 3-5-3 carbon backbone aryl/alkylated polyamine analogues against various strains of intraerythrocytic P. falciparum parasites
| Compd |
 |
P. falciparum IC50 (nM) |
||||
|---|---|---|---|---|---|---|
| R | X | 3D7a | W2b | HBc | RId | |
| 8 | Benzyl | S | 249 ± 30 | 42 ±6 | 178 ±2 | 0.17 |
| 9 | Diphenylmethyl | S | 68 ±13 | 35 ±3 | 75 ±9 | 0.51 |
| 10 | Diphenylethyl | S | 40 ±9 | 47 ±1 | 56 ±3 | 1.18 |
| 11 | Diphenylpropyl | S | 76 ±24 | 41 ±3 | 62 ±7 | 0.54 |
| 12 | Phenyl | O | 246 ± 34 | 49 ±13 | 177±4 | 0.20 |
| 13 | Benzyl | O | 793 ± 78 | 171 ±25 | 593 ± 20 | 0.22 |
| 14 | Diphenylmethyl | O | 47 ±7 | 20 ±3 | 65 ±2 | 0.43 |
| 15 | Diphenylethyl | O | 28 ±4 | 18±3 | 40 ±3 | 0.64 |
| 16 | Diphenylpropyl | O | 30 ±5 | 36 ±7 | 56 ±7 | 1.20 |
| CQ | Chloroquine | - | 9 ± 0.04 | 70 ±1 | - | 7.78 |
All data points are the average of 3 or more determinations ± SE.
P. falciparum 3D7, chloroquine-sensitive, (n = 9).
P. falciparum W2, chloroquine-resistant (n = 4).
P. falciparum HB3, antifolate-resistant (n = 3).
Resistance index (RI) defined as the ratio of the IC50 values of the resistant to sensitive strains, W2/3D7.
Parallel cross-reactivity screening against drug resistant strains of intraerythrocytic P. falciparum parasites indicated resistance indices (RI, ratio of the IC50 values of resistant to sensitive strain) from 0.17 to 1.2, compared to the calculated chloroquine value of 7.8 (Table 1). This strongly suggests that these compounds would not share the same resistance mechanism as chloroquine or that their activity is independent of such resistance mechanisms. The two most active 3-5-3 carbon backbone compounds against the 3D7 strain, compounds 15 and 16, did not show any cross-resistance and both were highly active against both the antifolate-resistant HB3 strain (compound 15 IC50 = 40 nM; compound 16 IC50 = 56 nM, Table 1) and the chloroquine-resistant W2 strains of intraerythrocytic P. falciparum (compound 15 IC50 = 18 nM; compound 16 IC50 = 36 nM, Table 1). In addition, compound 10 proved to be equipotent with 16, with an IC50 = 56 nM against the P. falciparum HB3 strain. In the chloroquine-resistant W2 strain of intraerythrocytic P. falciparum parasites, all compounds except compound 13 had IC50 values below 50 nM, and compounds 14 and 15 showed the greatest activity (14 IC50 = 20 nM; 15 IC50 = 18 nM). Interestingly, this strain also seemed more sensitive overall to polyamine perturbation.
The antiproliferative effect of the polyamine analogues may, in addition to the polyamine functionality, also be due to the effect on other metabolic activities of the parasite as both (bis)urea and bis(thiourea) derivatives have been reported to be β-hematin inhibitors, similar to the action of chloroquine.20–22 To further investigate the activity of the series on the chloroquine resistant W2 parasites, compound 10 (the most active thiourea), 16 (one of the leading urea compounds) and 13 (a urea and the least active compound) were screened for their ability to inhibit hemozoin (β-hematin) formation (Fig. 2). Due to the catabolism of erythrocyte hemoglobin, the intraerythrocytic P. falciparum parasite is exposed to cytotoxic oxidative stress caused by the free heme formation.23 Aggregating the heme into hemozoin crystals circumvents this and interference with this process has been ascribed as the mode of action of quinoline type antimalarials. All compounds tested had showed significantly poor abilities to inhibit β-hematin formation compared to chloroquine at an IC50 of 10.1 ± 1.2 µM (Fig. 2). Even though compound 10 was able to inhibit β-hematin formation with an IC50 of 16.7 ± 0.07 µM, this was still significantly less (P = 0.0054, n = 3) than that of CQ. More dramatically, compound 16, with a RI of 1.2 only showed activity at >400 µM and compound 13 showed no activity even at 1 mM, indicating that β-hematin inhibition is not the primary mechanism of action for these two compounds.
Figure 2.

Investigating similarities to chloroquine action. (A) Summary of the activity of a selection of the 3-5-3 series to drug sensitive (3D7) and chloroquine (CQ) resistant (W2) P. falciparum parasites. (B) Dose-response curves (µM) of CQ. (circle), compound 10 (square) and compound 16 (triangle) with the NP-40 mediated β-hematin formation assay. Absorbance was measured at 405 nm. In each instance, data are of three independent biological repeats performed in triplicate, ±SE.
2.2.2. In vitro antiplasmodial evaluation: cytotoxicity evaluation
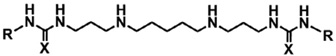
All polyamine analogues from the 3-5-3 series was found to be highly selective towards intraerythrocytic P. falciparum parasites, as concurrent counter-screening for In vitro cytotoxicity against mammalian HepG2 cells determined the selectivity index (SI, activity against mammalian cells to that against P. falciparum; Table 2). With the exception of compounds 9 (SI = 659 fold) and 11 (SI = 666 fold), the majority of the compounds showed >1000-fold selectivity toward the parasite. Moreover, compounds 15 and 16 are not only the most active against the P. falciparum 3D7 parasite, but also highly selective for the parasite, with SI values of 3375 and 5090, respectively. The polyamine parent molecule from which the 3-5-3 series was derived has previously been shown to produce a low level of general cytotoxicity, and the observed antitumor effect is primarily through re-expression of aberrantly silenced tumor suppressor genes. As such, this series is not inherently cytotoxic to non-cancerous mammalian cells when administered alone.18 Interestingly, the enhanced sensitivity of P. falciparum parasites compared to mammalian cells has also been observed with phenylacetamido-polyamines having nM activity against the parasite with low general cytotoxicity.24,25
Table 2.
In vitro antiplasmodial activity of 3-5-3 carbon backbone aryl/alkylated polyamine analogues against various strains of intraerythrocytic P. falciparum parasites
| Compd |
 |
P. falciparum 3D7 IC50 (nM) |
HepG2 GI50 (µM) |
SIe | |
|---|---|---|---|---|---|
| R | X | ||||
| 8 | Benzyl | S | 249 ± 30 | 100% viability* | >5000 |
| 9 | Diphenylmethyl | s | 68 ±13 | 44.8 ± 0.8 | 659 |
| 10 | Diphenylethyl | s | 40 ±9 | 52.20 ± 7 | 1305 |
| 11 | Diphenylpropyl | s | 76 ±24 | 50.6 ± 2 | 666 |
| 12 | Phenyl | O | 246 ± 34 | 100% viability* | >5000 |
| 13 | Benzyl | O | 793 ± 78 | 1947.33 ±43 | 2456 |
| 14 | Diphenylmethyl | O | 47 ±7 | 240.3 ± 11 | 5113 |
| 15 | Diphenylethyl | O | 28 ±4 | 94.5 ± 9 | 3375 |
| 16 | Diphenylpropyl | O | 30 ±5 | 152.7 ±8 | 5090 |
| CQ | Chloroquine | - | 9 ± 0.04 | 43.09 ± 2 | 4788 |
All data points are the average of 3 or more determinations ± SE.
Selectivity index (SI) was determined as the compound GI50 mammalian cell/IC50 P. falciparum.
No inhibition of HepG2 viability at 5000 × IC50 (3D7 value).
2.2.3. In vitro antiplasmodial activity: stage-specificity and kinetics
A major goal of antimalarial discovery programs is to define the cidal nature of compounds, in addition to descriptors of stage-specificity and kinetics of killing. Ideally, new antimalarials should be fast acting on all stages of the P. falciparum parasite’s life cycle to fulfill target candidate profile (TCP) 1 criteria, or alternatively, be relatively slow acting but still affecting all lifecycle stages to be used in combination treatments, fulfilling TCP 2.26 Treatment of intraerythrocytic P. falciparum 3D7 parasites with the 3-5-3 series of polyamine analogues resulted in a non-progressive pyknotic phenotype with a apparently dead or dying morphology corresponding to the stage of parasite affected as early trophozoites (Fig. 3A). Early ring stage parasites were not affected by treatment with these compounds. The phenotype corresponds moreover to a similar phenotype observed with polyamine biosynthesis inhibitors (such as difluoromethylornithine, Fig. 3A), indicating a stage-specific preference of action of these polyamine analogues correlating to the requirement of polyamines for biological function in the parasites.18,27
Figure 3.

Stage-specificity and kinetics evaluation of antiplasmodial action. (A) Morphological monitoring (Giemsa stained microscopy) of intraerythrocytic P. falciparum parasites treated with lead compound 16 (IC90) over a single 48 h life cycle compared to a polyamine biosynthesis inhibitor, difluoromethylornithine (DFMO) or to untreated (DMSO) control parasites. (B) Flow cytometric analysis of nuclear content of intraerythrocytic P. falciparum parasites treated with compound 15 and 16 (1 × IC90) using SYBR Green-I in the FITC channel. Parasite population (%) illustrates rings and early trophozoite stage parasites based on their 1 N nuclear content. After nuclear division the trophozoites contain 2 N and the multi-nucleated schizonts contain >2 N. Nuclear content was measured with flow cytometric analysis. (C) Intraerythrocytic P. falciparum parasites were treated with compounds 15 and 16 (1 × IC90) for 96 h and parasitemia monitored by flow cytometry compared to untreated, DMSO control parasites. (D) Dose-dependent cidal action of compound 16 was indicated as follows: ring stage parasites were treated with 1× (diamond) and 10x (square) IC90 of compound 16 and chloroquine (triangle, control) for 12 h, washed and allowed to proliferate for 36 h. Proliferation was measured at different time points with the SYBR Green-I fluorescence.
Quantification of this phenotype confirmed that the parasites treated with compounds 15 and 16 could not progress beyond early trophozoite stages (Fig. 3B) and this effect was observed within the first 24 h of drug treatment. Due to the nature of the flow cytometric measurement (detection of DNA content through SYBR Green I fluorescence), it was also clear that these analogues enforce their inhibitory activity through preventing DNA synthesis. The dramatic halt in schizogony distinctly correlated to a cessation of nuclear division with parasites containing ∼88% rings/trophozoites (1 N) with only ∼10% 2 N and ∼2% >2 N schizonts at 96 h after treatment (compound 16, Fig. 3B). These results are similar to previous reports where the utter requirement of P. falciparum parasites of polyamines for nuclear division and cell cycle progression was indicated.18 Again, the data supports the requirement of these metabolites for DNA synthesis and nuclear division in the schizont stages.27
With the activity of the lead compounds from this series clearly established on early trophozoite stage parasites, we subsequently evaluated the kinetics thereof. Parasite proliferation ceased entirely within the first 24 h of drug treatment for both compounds 15 and 16, continuously for two lifecycles (96 h) at 1 × IC90 (Fig. 3C). To fully investigate the long-term effects of exposure to compound 16 (as example) including the ability of parasites to overcome drug pressure if only dormant after treatment, parasite proliferation was monitored for 18, 24, 36 and 48 h after a single 12 h pulse with the compound (1× IC90; Fig. 3D). An irreversible decrease in intraerythrocytic P. falciparum 3D7 parasite viability (>40%, as measured by parasite proliferation over 36 h) was seen after this single 12 h pulse with no outgrowth observed even at 10× IC90, confirming the rapid and cidal nature of this perturbation. Previous polyamine analogues (e.g., compound 7) were also characterized as targeting early trophozoite forms of the parasite with rapid action (within the first 24 h of drug pressure),18 indicating that these compounds have a relatively fast and cidal mode of action.28
Antimalarial drug discovery programs currently focus not only on novel descriptors of chemotherapies against asexual, blood stage parasites but also, to support malaria elimination endeavors, need to additionally identify compounds with action against the sexual, gametocyte forms of the parasites to be used as malaria transmission blocking drugs.26,29 For the first time, we also show that polyamine analogues not only target the asexual intraerythrocytic development of P. falciparum parasites but have the ability to also affect the viability of sexual stages of P. falciparum parasites. Compounds 8–16 were analyzed for their gametocytocidal activity at 5 µM (48 h) against early (>90% stage II/III) and late (>90% stage IV/V) gametocytes of P. falciparum using a luciferase reporter assay30 (Fig. 4). Thioureated compounds (except for 8) were particularly interesting as they had a preference for late-stage gametocytes, with 9 and 11 fully inhibitory at the concentration tested. This finding is significant given the current focus of malaria drug discovery efforts on identification of potential gametocytocidal compounds, with killing of mature gametocytes seen as highly desirable for transmission blocking compounds.26 The urea-based compounds all had relatively poor activity against both early-and late-stage gametocytes but overall showed a preference towards early gametocytes, correlating with their significant activity against the asexual forms of the parasite. Nonetheless, the polyamine chemotype does not have any chemical signatures corresponding to any other compound series that have been described before for gametocytocidal activity (e.g.,31,32) and this therefore extends their potential applications as antiplasmodials useful as either chemotherapeutics or transmission blocking drugs.
Figure 4.

Gametocytocidal activity of the 3-5-3 series against P. falciparum parasites. NF54-luc reporter strains of the parasite was used to induce gametocytogenesis and activity (% inhibition of luciferase readout) of the compounds assayed at 5 µM after 48 h exposure. Both early gametocytes (>90% stage II/II gametocytes) as well as mature gametocytes (>90% stage IV/V gametocytes) were assayed independently. Data are of three independent biological repeats performed in triplicate, ±SE.
2.3. Structure–activity relationship analysis
The data presented above, together with a number of series previously tested,18 allowed for completion of the polyamine backbone to a full series (3-3-3, 3-4-3, 3-5-3, 3-6-3 and 3-7-3) tested for activity against P. falciparum parasites. The antiplasmodial chemotype is considered complete as previous have shown that extending the central carbon beyond 7 carbons results in greater toxicity in a malaria mouse model.36 The analogues containing a 3-3-3, 3-4-3, 3-6-3 or 3-7-3 carbon backbone architecture, combined with a variety of terminal substituents (compounds 7, 27–57, Fig. 5,18), as well as the new (bis)urea- and (bis)thiourea analogues of 7 with 3-5-3 carbon backbone architecture (8–16), were considered.
Figure 5.

Evaluation of the (bis)urea- and (bis)thiourea polyamine chemotype. Data from this study (3-5-3 series, compounds 8–16) as well as previous studies18 (compounds 7, 27–57) were compiled and compared for their antiplasmodial activity (IC50 values, various strains), selectivity (HepG2 and SI), physicochemical properties (solubility, permeability and HIA). Data were ranked according to 3D7 potency (IC50 values) with a color gradient scale indicating more potent activity / desirable features in darker squares. Solubility was either obtained physically at pH 7.4 or predicted with Discovery Studio. Permeability was indicated with a PAMPA assay. HIA predictors were obtained through Discovery Studio analysis.
By applying a primary filter of 100 nM for activity, the majority of the 3-5-3 series (with the exception of compounds 8, 12, 13) passed these selection criteria. However, among previously reported (bis)urea compounds (Fig. 5), only the 3-6-3 phenyl substituted analogue (compound 7) had an IC50 value below 100 nM, but compound 7 was as selective towards the parasite as compounds 8, 10, 14, 15, 16, 28, 29 all with SI >1000, indicative of little to no hepatotoxicity at least In vitro. Regrettably, compound 1 previously showed hepatotoxicity in preclinical trials;33 this prompted preliminary predictions of ADME and druggability properties of the compounds tested in silico. Overall, the compounds show moderate solubility and permeability with lead compounds 15, 16 (3-5-3 series) and 7 (3-6-3 series) displaying QED values of 0.21, suggesting possible druggability.
Based upon the compiled data for the chemotype (Fig. 5), the structure-activity relationship (SAR) correlations for symmetrically aryl/alkylated (bis)urea- and (bis)thiourea-polyamine analogues could be refined. Further objective chemi-informatic analysis of SAR was employed to define the contributions of the carbon backbone and terminal substituents to antimalarial activity (Fig. 6). Activity cliff analyses34,35 were created analyzing either the pairwise structural similarity and associated activities based on the carbon backbone (Fig. 6) or on the terminal substituents, in each case with an 80% structural similarity threshold.
Figure 6.

Structure activity landscape analysis for the symmetrically substituted (bis)urea- and (bis)thiourea polyamines. Pairwise activity to structural feature analysis was performed with Activity Cliff Analysis (Osiris DataWarrior V 3.12.1) on the complete series from Figure 5 at a stringency of 80% similarly in structural features. Cluster analysis based on backbone structural features. In all cases, two structurally similar isosteres were compared and a single bar acts as identifier of such correlations based on the respective activities of the two isosteres as indicated on the two axes (decreasing potency from crosspoint). Activity values are based on the In vitro IC50 values in nM (asexual P. falciparum parasites 3D7). Isostere pairs are identified numerically based on compound numbers in Figure 5 for example, 15–16 with activity 1-activity 2 desciptors.
A distinct correlation was observed between antiplasmodial activity and length of the central carbon spacers. Our data suggests that, in the (bis)urea series, a central carbon spacer of 5 or 6 carbons is optimal for antiplasmodial activity In vitro (Fig. 6). Backbone clustering clearly confirmed that a 3-5-3 backbone shows preferred activity below the 100 nM filter. Additionally, all (except for one 3-4-3 backbone pair, compounds 36-34) the additional compounds below a 500 nM activity filter are from either the 3-7-3 (63%) or 3-6-3 (21%) series. This corroborates previous work, which showed that extending the central carbon beyond 7 carbons results in greater toxicity in the malaria mouse model.36 The spacing of these nitrogens37 and the pKa of the terminal nitrogens38 have a significant effect on normal polyamine biological activity. The nitrogens in the polyamines spermidine and spermine are fully protonated at physiological pH, and it is thus generally assumed that they interact with 3 or 4 anionic sites at a biological target.39 However, the pKa of the terminal nitrogen equivalent does not appear to be as critical, particularly in polyamine analogues. Compounds such as 1 have terminal secondary nitrogens with pKa values near 10 that are protonated at physiological pH. However, since substituted ureas and thioureas do not have acid-base characteristics at physiological pH, it would appear that the ability to hydrogen bond at the terminal urea or thiourea moiety is sufficient for retaining activity. Terminal guanidines or biguanides produce enhanced antiproliferative effects as these moieties would result in an even larger charge perturbation than the natural polyamines.40 Substituted guanidines and biguanides, possessing pKa values of 13.5341 and 13.0342 respectively, are more basic than the terminal nitrogens of polyamines with pKa values approximating 1043 and are therefore likely to interfere with polyamine metabolism.11
Within the 3-5-3 backbone, structural changes from (bis)urea to (bis)thiourea were associated with relative loss in activity to asexual forms of P. falciparum parasites (Fig. 6). Additionally, the (bis)urea analogues in the 3-5-3 series (compounds 14, 15, 16) proved to be more selective towards the parasite compared to mammalian cells than their thiourea isosteres (compounds 9, 10, 11). However, the preference for (bis)urea analogues in this series does not hold true when activity is evaluated against the non-replicating gametocyte forms of the parasite, where clear preference seen towards (bis)thiourea analogues. This could indicate pluripharmacology for this chemotype as either antimalarial chemotherapies or transmission blocking drugs. Further exploitation of the potential transmission blocking activity of this series is underway.
However, the activities described above are completely dependent on symmetrical terminal nitrogen substituents; size, flexibility and electronic character appear to affect antiplasmodial activity, with clear preference towards diphenylaryl constituents compared to monoarylalkylation. Here, the diphenylethyl substituent appears to be optimal (among analogues examined to date), possibly due to favorable hydrophobic contacts with the biological target. In (bis)urea analogues with the 3-5-3 backbone, the phenyl and benzyl substituted compounds 12 and 13 exhibited a dramatic decrease in activity, exhibiting IC50 values of 246 and 793 nM, respectively (pairs 12–13 and 8–13, Fig. 6). Conversely, compounds with diphenylaryl substitutions such as compounds 14–16 retained the antiplasmodial activity, with the diphenylethyl substituted analogue 15 being the most potent compound IC50 = 28 nM (3D7) and 18 nM (W2). In all but 3 examples in Figure 5, compounds featuring the diphenylethyl substituent are more potent than the corresponding diphenylmethyl or diphenylpropyl homologues, possibly due to a combination of steric and electronic effects including pi stacking.
In the (bis)thiourea series, a different SAR emerged. The 3-6-3 (bis)benzyl analogue 28 (IC50 106 nM) and the 3-6-3 (bis)diphenyl-methyl analogue 31 (IC50 211 nM) had activity below 250 nM. In the corresponding 3-5-3 series, the (bis)benzyl analogue 8 (IC50 249 nM) was significantly less potent than the corresponding 3-6-3 homologue 28, while the diphenylaryl compounds 9–11 all had IC50 values below 80 nM. The diphenylethyl compound 10 was the most potent among these three (IC50 40 nM). Compounds with simple alkyl substituents such as compounds 53 and 54 were inactive. Analogues that featured a diphenylalkyl terminal substitution were not significantly more potent than 7.
In summary, the preliminary SAR for symmetrically substituted (bis)urea- and (bis)thiourea-polyamines against intraerythrocytic P. falciparum parasites could be delineated based on the mono-or diarylalkyl constituency as follows (Fig. 7): In all cases, the best linker is an alkylation, preferably ethyl and X = O. In the (bis)mono-arylalkyl compounds, there is a preference for phenyl terminal substituents and n1 = 4 (3-6-3), with n1 = 3 (3-5-3) significantly reducing activity for this series. These compounds are most like the lead compound 1, and as such these data extend the SAR from previous studies.12,36 In the more potent (bis)diarylalkyl series, preference exist for diphenylethyl and where n1 = 3 (3-5-3) compared to n1 = 4 (3-6-3), the reduction of the central carbon chain by 1 carbon increases antiplasmodial activity by as much as 10-fold compared to the (bis)monoarylalkyl compounds.
Figure 7.

Antiplasmodial structure activity relationship model for symmetrically substituted (bis)urea- and (bis)thioureapolyamines.
3. Conclusions
The terminally aryl/alkylated (bis)urea- and (bis)thiourea-polyamine analogues of the 3-5-3 and 3-6-3 carbon backbone represent a structurally novel and distinct class of potential antiplasmodials with activities in the low nanomolar range and high selectivity against intraerythrocytic P. falciparum parasites. Further mechanistic studies and in vivo activity determinations are currently underway.
4. Materials and methods
4.1. Chemistry
4.1.1. Reagents
All reagents and dry solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI), Sigma Chemical Co. (St. Louis, MO) Fisher Scientific (Chicago, IL) or VWR Scientific (Atlanta, GA) and were used without further purification except where noted below. Triethylamine was distilled from potassium hydroxide and stored in a nitrogen atmosphere. Dry methanol, ethyl acetate, tetrahydrofuran, dimethyl formamide and hexane were prepared using a Glass Contour Solvent Purification System (Pure Process Technology, LLC, Nashua, NH). Methanol was distilled from magnesium and iodine under a nitrogen atmosphere and stored over molecular sieves. Routine chromatographic purification on silica gel was performed on a Teledyne Isco CombiFlash Rf200. Preparative scale chromatographic procedures were carried out using E. Merck silica gel 60, 230–440 mesh. Thin layer chromatography was conducted on Merck precoated silica gel 60 F-254. All 1H and 13C NMR spectra were recorded on a Varian Mercury 400 MHz spectrometer, and all chemical shifts are reported as δ values referenced to TMS. In all cases, 1H NMR, 13C NMR and mass spectra were consistent with assigned structures. Mass spectra were recorded on a Kratos MS 80 RFA (EI and CI) or Kratos MS 50 TC (FAB) mass spectrometer. N4,N10-(Bis)-[(tert-butyloxy)carbonyl]-1,14-aminotridecane 17 was synthesized from 1,5-diaminopentane as previously described.19,44 Prior to biological testing, previously unreported target molecules 8–16 were determined to be 95% pure or greater by UPLC chromatography using a Waters Acquity system with a photodiode array (PDA) detector (190–500 nm) fitted with a C18 reversed-phase column.
4.2. General procedure for the synthesis of N-Boc protected (bis)ureas and (bis)thioureas
4.2.1. 1,13-Bis-{3-[1-(benzyl)thiourea]}-4,10-di(tert-butyloxycarbonyl)-4,10-diazatridecane, 18
A 203 mg (1.361 mmol) portion of benzyl isothiocyanate dissolved 10 mL of anhydrous dichloromethane was added to 270 mg (0.648 mmol) of 17 in 20 mL of anhydrous dichloromethane in a drop wise fashion. The reaction mixture was allowed to stir at room temperature for 24 h. During this time, the formation of product was monitored by TLC (75% ethyl acetate in hexane). After the completion of the reaction, the solvent was removed in vacuo and the crude product was purified on silica gel eluted with 75% ethyl acetate in hexane to afford 315 mg of pure 18 as a white solid (68%). 1H NMR (CDCl3): δ 7.32–7.25 (m, 10H), 4.57 (s, 4H), 3.90 (m, 4H), 4.10 (m, 4H), 1.71 (m, 4H), 1.56–1.47 (m, 4H), 1.40 (s, 18H), 1.23–1.20 (m, 2H). 13C NMR (CDCl3): δ 181.01, 156.70, 136.97, 128.68, 127.64, 79.98, 47.51, 47.01, 43.25, 41.14, 28.38, 27.09, 24.18. M+ = 714.4, [M+1]+ = 715.3.
4.2.2. 1,13-Bis-{3-[1-(1′,1′-diphenylmethyl)thiourea]}-4,10-di- (tert-butyloxycarbonyl)-4,10-diazatridecane, 19
Compound 19 was prepared from 17 (218 mg, 0.523 mmol) and 2,2-diphylmethylisothiocyanate (236 mg, 1.047 mmol) using the general procedure described above. Yield 348 mg (71%). 1H NMR (CDCl3): δ 7.43–7.26 (m, 20H), 6.50 (s, 2H), 3.53 (m, 4H), 3.075 (m, 8H), 1.67 (m, 4H), 1.51–1.48 (m, 4H), 1.38 (s, 18H), 1.24–1.18 (m, 2H). 13C NMR (CDCl3): δ 180.07, 156.64, 140.25, 128.75, 127.84, 127.58, 79.89, 61.42, 47, 42.93, 41.31, 28.32, 26.95, 24.17; M+ = 866.46, [M+1]+ = 867.2.
4.2.3. 1,13-Bis-{3-[1-(2′,2′-diphenylethyl)thiourea]}-4,10-di- (tert-butyloxycarbonyl)-4,10-diazatridecane, 20
Compound 20 was prepared from 17 (200 mg, 0.48 mmol) and 2,2-diphenylethylisothiocyanate (230 mg, 0.96 mmol) using the general procedure described above. Yield 292 mg (68%). 1H NMR (CDCl3): δ 7.33–7.21 (m, 20H), 5.9 (s, 2H), 4.14–4.05 (m, 4H), 3.55 (m, 4H), 3.26 (m, 4H), 3.12 (t, 4H), 1.71 (m, 4H), 1.54 (m, 4H), 1.29 (s, 18H), 1.27 (t, 4H, J = 6.8 Hz); 13C NMR (CDCl3): δ 181.22, 156.78, 141.55, 128.78, 128.08, 126.94, 80.08, 49.85, 47, 43.29, 40.99, 28.41, 26.96, 24.22. M+ = 894.49, [M+1]+ = 895.3.
4.2.4. 1,13-Bis-{3-[1-(3′,3′-diphenylpropyl)thiourea]}-4,10-di- (tert-butyloxycarbonyl)-4,10-diazatridecane, 21
Compound 21 was prepared from 17 (200 mg, 0.48 mmol) and 3,3-diphenylpropylisothiocyanate (255 mg, 1.008 mmol) using the general procedure described above. Yield 328 mg (74%). 1H NMR (CDCl3): δ 7.31–7.17 (m, 20H), 4.05 (t, 2H, J = 8.32 Hz), 3.55 (m, 4H), 3.3–3.25 (m, 8H), 3.12 (m, 4H), 2.41-2.36 (m, 4H), 1.71 (m, 4H), 1.56–1.53 (m, 4H), 1.43 (s, 18H), 1.29–1.26 (m, 2H); 13C NMR (CDCl3): δ 171.19, 156.77, 143.95, 128.63, 127.75, 126.46, 80.01, 48.69, 46.99, 43.22, 40.94, 34.48, 31.59, 27.05, 24.22, 22.66. M+ = 922.52, [M+1]+ = 923.4.
4.2.5. 1,13-Bis-{3-[1-(phenyl)urea]}-4,10-di(tert-butyloxycarbonyl)-4,10-diazatridecane, 22
Compound 22 was prepared from 17 (268 mg, 0.643 mmol) and phenylisocyanate (153 mg, 1.287 mmol) using the general procedure described above. Yield 282 mg (67%). 1H NMR (CH3OD): δ 7.34–7.32 (m, 8H), 7.23 (t, 2H, J = 7.6 Hz), 6.99 (t, 2H, J = 7.24 Hz), 3.19-3.10 (m, 12H), 1.65 (m, 4H), 1.44 (m, 22H), 1.19 (m, 2H). 13C NMR (CDCl3): δ 156.49, 156.16, 139.27, 128.96, 122.76, 119.82, 79.80, 46, 44, 36.38, 28.45, 24.09. M+ = 654.41, [M+1]+ = 655.2.
4.2.6. 1,13-Bis-{3-[1-(benzyl)urea]}-4,10-di(tert-butyloxycarbonyl)-4,10-diazatridecane, 23
Compound 23 was prepared from 17 (282 mg, 0.677 mmol) and benzylisocyanate (189 mg, 1.422 mmol) using the general procedure described above. Yield 324 mg (70%). 1H NMR (CDCl3): δ 7.28 (m, 10H), 4.32 (s, 4H), 3.21–3.11 (m, 12H), 1.61 (m, 4H), 1.52–1.47 (m, 22H), 1.24–1.20 (m, 2H). 13C NMR (CDCl3): δ 158.59, 156.27, 139.53, 128.49, 127.37, 127.08, 79.56, 46.99, 44.32, 43.6, 36.8, 28.44, 24.11. M+ = 682.44, [M+1]+ = 683.3.
4.2.7. 1,13-Bis-{3-[1-(1′,1′-diphenylmethyl)urea]}-4,10-di(tert-butyloxycarbonyl)-4,10-diazatridecane, 24
Compound 24 was prepared from 17 (230 mg, 0.552 mmol) and 2,2-diphenylmethylisothiocyanate (243 mg, 1.159 mmol) using the general procedure described above. Yield 332 mg (72%). 1H NMR (CDCl3): δ 7.40–7.10 (m, 20H), 5.99 (d, 2H), 3.3–2.9 (m, 12H), 1.54 (m, 4H), 1.43 (m, 22H), 1.2 (m, 2H); 13C NMR (CDCl3): δ 157.78, 156.25, 142.62, 128.51, 127.36, 127.14, 58.13, 47.01, 45.09, 43.67, 36.73, 28.46, 25.30, 24.15. M+= 834.5, [M+1]+ = 835.3.
4.2.8. 1,13-Bis-{3-[1-(2′,2′-diphenylethyl)urea]}-4,10-di(tert-butyloxycarbonyl)-4,10-diazatridecane 25
Compound 25 was prepared from 17 (200 mg, 0.48 mmol) and 2,2-diphenylethylisocyanate (225 mg, 1.008 mmol) using the general procedure described above. Yield 290 mg (70%). 1H NMR (CDCl3): δ 7.31–7.19 (m, 20H), 4.19 (t, 2H, J = 7.8 Hz), 3.81 (t, 2H, J = 7.1 Hz), 3.22 (m, 4H), 3.10 (m, 8H), 1.61 (m, 4H), 1.51 (m, 4H), 1.43 (s, 18H), 1.23 (m, 2H); 13C NMR (CDCl3): δ 158.16, 142.22, 128.63, 128.16, 126.66, 79.61, 51.21, 46.91, 44.98, 44.46, 36.5, 28.45, 28.24. Exact mass = 862.54, [M+1]+ = 863.3.
4.2.9. 1,13-Bis-{3-[1-(3′,3′-diphenylpropyl)urea]}-4,10-di(tert-butyloxycarbonyl)-4,10-diazatridecane 26
Compound 26 was prepared from 17 (217 mg, 0.521 mmol) and 3,3-diphenylpropylisocyanate (260 mg, 1.094 mmol) using the general procedure described above. Yield 348 mg (75%). 1H NMR (CDCl3): δ 7.30–7.16 (m, 20H), 3.99 (t, 2H, J =7.5 Hz), 3.30–3.26 (m, 4H), 3.15–3.10 (m, 12H), 2.30–2.24 (m, 4H), 1.67 (m, 4H), 1.54–1.51 (m, 4H), 1.46 (s, 18H), 1.25–1.22 (m, 2H); 13C NMR (CDCl3): δ 158.48, 156.5, 144.41, 128.54, 127.81, 126.29, 79.59, 48.8, 46.94, 39.21, 36.01, 28.47. M+ = 890.57, [M+1]+ = 891.3.
4.3. General procedure for the deprotection of the N-Boc protecting group
4.3.1. 1,13-Bis-{3-[1-(benzyl)thiourea]}-4,10-diazatridecane dihydrochloride, 8
A stirred solution of 0.1 g (0.140 mmol) of the (bis)-N-Boc protected (bis)thiourea derivative 18 in 25 mL of 1.0 M HCl in ethyl acetate was allowed to stir overnight at room temperature, and the formation of the product was monitored by TLC. The product precipitated as a crystalline solid during the course of the reaction. After the completion of the reaction, the solvent was removed under reduced pressure followed by the addition of fresh ethyl acetate, and the mixture was stirred for 15 min and decanted. The solid thus obtained was dried in vacuo as above to afford the final product 8 in 85% yield (69.9 mg). 1H NMR (CD3OD): δ 7.4–7.26 (m, 10H), 4.34 (s, 4H), 3.31 (t, 4H, J = 6.2 Hz), 3.01–2.98 (m, 4H), 2.91 (m, 4H), 1.88–1.85 (m, 4H), 1.72–1.68 (m, 4H). 1.48–1.46 (m, 2H); 13C NMR (CD3OD): δ 128.21, 127.09, 126.99, 47.06, 44.51, 40.20, 26.39, 25.31, 25.28, 23.15.
4.3.2. 1,13-Bis-{3-[1-(1’,1’-diphenylmethyl)thiourea]}-4,10-diazatridecane dihydrochloride, 9
Compound 9 was synthesized from 19 (100 mg, 0.115 mmol) as per the procedure described above in 85% yield (72.5 mg). 1H NMR (CD3OD): δ 7.45–7.27 (m, 20H), 6.67 (s, 2H), 3.78 (t, 4H, J =5.76 Hz), 3.00–2.94 (m, 8H), 1.72–1.69 (m, 4H), 1.55 (br, 4H), 1.48–1.46 (m, 2H); 13C NMR (CD3OD): δ 141.72, 132.48, 129.62, 128.23, 127.27, 127.07, 61.38, 47.04, 44.50, 40.12, 26.43, 25.83, 25.22, 23.10. M+ (2HCl) = 666.35; calculated = 667.2.
4.3.3. 1,13-Bis-{3-[1-(2′,2′-diphenylethyl)thiourea]}-4,10-diazatridecane dihydrochloride, 10
Compound 10 was synthesized from 20 (100 mg, 0.112 mmol) as per the procedure described above in 90% yield (77 mg). 1H NMR (CD3OD): δ 7.32–7.22 (m, 20H), 4.45 (s, 2H), 4.13 (t, 4H, J =7.08 Hz), 3.67 (m, 4H), 3.04–2.97 (m, 8H), 1.92-1.89 (m, 4H), 1.80–1.77 (m, 4H), 1.56–1.53 (m, 2H); 13C NMR (CD3OD): δ 142.22, 128.29, 127.86, 126.39, 50.22, 47.09, 44.04, 39.87, 26.41, 25.31, 23.17. M+ (2HCl) = 694.39; calculated = 695.3.
4.3.4. 1,13-Bis-{3-[1-(3′,3′-diphenylpropyl)thiourea]}-4,10-diazatridecane dihydrochloride, 11
Compound 11 was synthesized from 21 (100 mg, 0.108 mmol) as per the procedure described above in 87% yield (75 mg). 1H NMR (CD3OD): δ 7.30–7.16 (m, 20H), 4.05 (t, 2H, J =7.6 Hz), 3.70 (br, 4H), 3.41 (br, 4H), 3.04–2.99 (m, 8H), 2.4–2.34 (m, 4H), 1.99–1.92 (m, 4H), 1.8–1.72 (m, 4H), 1.54–1.51 (m, 2H); 13C NMR (CD3-OD): δ 144.43, 128.17, 127.50, 125.97, 48.59, 44.48, 40.04, 34.41, 26.46, 25.26, 23.12. M+ (2HCl) = 722.42; calculated = 723.3.
4.3.5. 1,13-Bis-{3-[1-(phenyl)urea]}-4,10-diazatridecane dihydrochloride, 12
Compound 12 was synthesized from 22 (100 mg, 0.153 mmol) as per the procedure described above in 85% yield (68.5 mg). 1H NMR (CD3OD): δ 7.39 (d, 4H, J = 7.92 Hz), 7.28 (t, 4H, J = 7.6 Hz), 7.02 (t, 2H, 7.28 Hz), 3.36 (t, 4H, J = 6.2), 3.10–3.03 (m, 8H), 1.96–1.91 (m, 4H), 1.83–1.76 (m, 4H), 1.60–1.56 (m, 2H); 13C NMR (CD3-OD): δ 157.76, 139.20, 128.51, 122.46, 119.25, 119.12, 47.13, 44.91, 35.83, 26.99, 25.3, 23.06; M+ (2HCl) = 454.31; calculated = 455.2.
4.3.6. 1,13-Bis-{3-[1-(benzyl)urea]}-4,10-diazatridecane dihydrochloride, 13
Compound 13 was synthesized from 23 (100 mg, 0.146 mmol) as per the procedure described above in 90% yield (73.2 mg). 1H NMR (CD3OD): δ 7.47–7.28 (m, 10H), 4.7 (s, 4H), 3.75 (br, 4H), 3.04–3.01 (m, 8H), 1.99–1.96 (m, 4H), 1.81–1.75 (m, 4H), 1.55 (m, 2H); 13C NMR (CD3OD): δ 160.47, 139.87, 128.48, 128.16, 126.69, 47.09, 44.71, 43.42, 35.72, 27.12, 25.32, 22.99. M+ (2HCl) = 482.34; calculated = 483.2.
4.3.7. 1,13-Bis-{3-[1-(1′,1′-diphenylmethyl)urea]}-4,10-diazatridecane dihydrochloride, 14
Compound 14 was synthesized from 24 (100 mg, 0.12 mmol) as per the procedure described above in 88% yield (74.6 mg). 1H NMR (CD3OD): δ 7.37–7.25 (m, 20H), 5.98 (s, 2H), 3.3 (m, 4H), 2.94 (m, 4H), 2.76 (m, 4H), 1.87 (m, 4H), 1.59 (m, 4H), 1.34 (m, 2H). 13C NMR (CD3OD): δ 159.72, 142.67, 129.63, 128.27, 126.93, 58.07, 47.12, 44.63, 35.52, 27.11, 25.27, 22.94. M+ (2HCl) = 634.4; calculated = 635.3.
4.3.8. 1,13-Bis-{3-[1-(2′,2′-diphenylethyl)urea]}-4,10-diazat-ridecane dihydrochloride, 15
Compound 15 was synthesized from 25 (100 mg, 0.116 mmol) as per the procedure described above 89% yield (76 mg). 1H NMR (CD3OD): δ 7.33–7.21 (m, 20H), 4.2 (t, 2H, J = 8.0 Hz), 3.81 (d, 4H), 3.23 (t, 4H, 5.8 Hz), 2.98 (t, 4H, J = 7.1 Hz), 2.92 (t, 4H, J = 6.3 Hz), 1.8 (m, 8H), 1.57 (m, 2H); 13C NMR (CD3OD): δ 160.3, 142.54, 128.25, 127.82, 126.29, 51.37, 44.66, 44.33, 35.59, 27.12, 25.43, 23.09. M+ (2HCl) = 662.43; observed calculated = 663.3.
4.3.9. 1,13-Bis-{3-[1-(3′,3′-diphenylpropyl)urea]}-4,10-diazatridecane dihydrochloride, 16
Compound 16 was synthesized from 26(100 mg, 0.112 mmol) as per the procedure described above 82% yield. 1HNMR (CD3OD): δ 7.3–7.17 (m, 20H), 4.02 (t, 2H, J = 7.8 Hz), 3.27 (t, 4H, J = 6.08 Hz), 3.09 (t, 4H, 7 Hz), 3.02–2.94 (m, 8H), 2.28–2.26 (m, 4H), 1.88–1.85 (m, 4H), 1.76–1.72 (m, 4H), 1.50–1.48 (m, 2H); 13C NMR (CD3OD): δ 160.51, 144.59, 128.16, 127.47, 125.93, 48.48, 47.04, 44.71, 38.52, 35.69, 35.46, 27.15, 25.34, 23.0. M+ (2HCl) = 690.46; calculated = 691.4.
4.4. Biological activity evaluations
4.4.1. In vitro cultivation of intraerythrocytic P. falciparum parasites
Intraerythrocytic P. falciparum parasites (strains 3D7, W2 and HB3) were cultivated in RPMI-1640 culture medium (25 mM HEPES pH 7.5, 0.2 mM hypoxanthine, 0.024 mg/mL gentamycin, 5 g/L Albumax II, 23.81 mM sodium bicarbonate, 0.2% D-glucose)45 in human erythrocytes (O+, suspended at 5% hematocrit) under hypoxic conditions (5% O2, 5% CO2, 90% N2) with shaking at 37 °C. Cultures were synchronized to >95% in the ring stage with three consecutive rounds of 5% (w/v) D-Sorbitol treatment.46
4.4.2. In vitro assessment of antiplasmodial activity against asexual parasites
Intraerythrocytic P. falciparum parasites (3D7, W2 and HB3) were used to assess the In vitro antiplasmodial efficacy of the polyamine analogues using a SYBR Green I-based fluorescence assay as previously described.18,47 Compounds were dissolved in a nonlethal DMSO concentration (<0.025%), serially diluted in culture medium and added to ring stage intraerythrocytic P. falciparum parasites (1% parasitemia, 2% hematocrit), incubated at 37 °C for 96 h in a N2 atmosphere. Chloroquine disulfate was used as positive drug control (0.5 µM; IC50 9 ± 0.04 nM) for complete inhibition of parasite proliferation. Parasite proliferation was normalized to the percentage of untreated control, after subtracting the background fluorescence (chloroquine treated samples). Nonlinear regression curves were generated using GraphPad Prism 5, from which the half-maximal inhibitory concentrations (IC50) could be determined.
4.4.2.1. Kinetics and stage-specificity
Highly synchronized (>95% ring stage) intraerythrocytic P. falciparum parasites (2% parasitemia; 5% hematocrit) were treated with each of the polyamine analogues for 12 h at 1× and 10× IC90, respectively. After treatment, the parasites were washed once with RPMI 1640 culture medium, diluted (1:40) before further monitoring proliferation at set times over a total period of 36 h by assessing DNA content with SYBR Green I as described above.48
4.4.2.2. Flow cytometric evaluation
The effect of the aryl/alkylated polyamine analogues on intraerythrocytic P. falciparum parasite’ DNA replication and nuclear division was determined using flow cytometric analysis as described previously.18 Parasites (2% parasitemia, 2% hematocrit) were treated in duplicate with 2× IC90 of the polyamine analogues Following drug treatment, samples (50 µL) were taken at specific time intervals (24, 48, 72 and 96 h) and nuclear content detected through SYBR Green I fluorescence (FITC channel; excitation wavelength of 488 nm with 502 nm long-band-pass and 530 nm band-pass emission filters) with a BD FACS Aria I flow cytometer, analysing 106 cells for each sample. Data was analyzed using FlowJo 7.6.5 and Cyflogic.
4.4.2.3. β-Hematin inhibition assays
The parasite rids itself of toxic levels of heme released during the degradation of hemoglobin through the lipid-mediated production of hemozoin. The water-lipid interface can be mimicked In vitro to measure the formation of β-hematin (a synthetic analogue of hemozoin). β-Hematin formation was assayed as described previously using the pyridine-ferrichrome method.23,49 Briefly, a mixture consisting of NP-40 detergent (305.5 µM) water and DMSO was added to polyamine analogues at various concentrations (0–1 M). The samples were then incubated at 37 °C for 5 h with 25 mM hematin (Fluka; Biochemika) in a 1 M acetate buffer (pH 4.8) with occasional shaking.
4.4.3. Cytotoxicity determinations in mammalian cells
Cytotoxicity against human hepatocellular liver carcinoma cells (HepG2) was determined using the lactate dehydrogenase (LDH) assay as previously described.18 In brief, HepG2 cells (±200,000 cells) were seeded into 96-well plates and grown for 24 h at 37 °C (5% CO2, 90% humidity) in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum and 1% penicillin/streptomycin. Subsequently, the HepG2 cells were treated with various concentrations of the polyamine compounds for 48 h, following which the cells were pelleted at 600 g for 10 min and the LDH activity in the supernatant (10 µL) measured by adding 100 µL LDH reaction mix (Biovision LDH-Cytotoxicity Assay Kit II). The production of the colored water-soluble tetrazolium salt was measured spectrophotometrically at 450 nm.
4.4.4. Induction of P. falciparum gametocytogenesis and maintenance of gametocyte cultures
Gametocytogenesis was induced from asexual P. falciparum parasites by a combination of nutrient starvation and a drop in hematocrit. Synchronized asexual parasites cultivated as above at 10% parasitemia were diluted to 0.5% parasitemia (6% hematocrit) into glucose-free medium. Cultures were then maintained under hypoxic conditions without shaking. After 72 h, the hematocrit was dropped to 3% (day 0 of induction). Gametocytogenesis was subsequently monitored microscopically with daily medium (glucose-free) changes. Residual asexual parasites were eliminated by continuous 50 mM N-acetyl glucosamine NAG, from day 1–4 in the presence of 0.2% glucose.
4.4.5. In vitro assessment of antiplasmodial activity against sexual gametocytes
Luciferase reporter lines NF54-PfS16-GFP-Luc (early gametocyte marker) and NF54-Mal8p1.16-GFP-Luc (late gametocyte marker) (kind gift from David Fidock, Columbia University, USA)50 enabled stage-specific descriptors of gametocytocidal activities. Assays were performed as described elsewhere (Reader et al., in press). Drug assays were set up on day 5 and 10 (representing early stage I/II/III and mature stage IV/V gametocytes, respectively). In each instance, assays were set up in triplicate using a 2% gametocytemia, 2% hematocrit culture and 48 h drug pressure (5 µM) under hypoxic conditions at 37 °C. Luciferase activity was determined in 20 µL parasite lysates by adding 50 µL luciferin substrate (Promega Luciferase Assay System) at room temperature and detecting resultant bioluminescence at an integration constant of 10 s with the GloMax®-Multi+ Detection System with Instinct® Software.
4.4.6. Solubility
Solubility assay was carried out on Biomek FX lab automation workstation (Beckman Coulter, Inc., Fullerton, CA) using mSOL Evolution software (pION Inc., Woburn, MA). The detailed method is described as follows: 10 mL of 10 mM compound stock (in DMSO) was added to 190 mL 1-propanol to make a reference stock plate. 5 mL from this reference stock plate was mixed with 70 mL 1-propanol and 75 mL citrate phosphate buffered saline (isotonic) to make the reference plate, and the UV spectrum (250–500 nm) of the reference plate was read. 6 mL of 10 mM test compound stock was added to 594 mL buffer (at pH 3 or 5 or 7.4) in a 96-well storage plate and mixed. The storage plate was sealed and incubated at room temperature for 18 h. The suspension was then filtered through a 96-well filter plate (pION Inc., Woburn, MA). 75 mL filtrate was mixed with 75 mL 1-propanol to make the sample plate, and the UV spectrum of the sample plate was read. Calculations were carried out by mSOL Evolution software based on the AUC (area under curve) of UV spectrum of the sample plate and the reference plate. All compounds were tested in triplicates.
4.4.7. Permeability
Parallel Artificial Membrane Permeability Assay (PAMPA) was conducted by Biomek FX lab automation workstation (Beckman Coulter, Inc., Fullerton, CA) and PAMPA evolution 96 command software (pION Inc., Woburn, MA). The detailed method is described as following. 3 mL 10 mM test compound stock in DMSO was mixed with 597 mL of citrate phosphate buffered saline (isotonic) to make diluted test compound. 150 mL of diluted test compound was transferred to a UV plate (pION Inc., Woburn, MA) and the UV spectrum was read as the reference plate. The membrane on pre-loaded PAMPA sandwich (pION Inc., Woburn, MA) was painted with 4 mL GIT lipid (pION Inc., Woburn, MA). The acceptor chamber was then filled with 200 mL ASB (acceptor solution buffer, pION Inc., Woburn, MA), and the donor chamber was filled with 180 mL diluted test compound. The PAMPA sandwich was assembled, placed on the Gut-box and stirred for 30 min. Aqueous Boundary Layer was set to 40 mm for stirring. The UV spectrum (250–500 nm) of the donor and the acceptor were read. The permeability coefficient was calculated using PAMPA evolution 96 command software (pION Inc., Woburn, MA) based on the AUC of the reference plate, the donor plate and the acceptor plate. All compounds were tested in triplicates.
4.5. Chemi-informatic analyses
The analysis of structure activity relationships was performed in Osiris Data Warrior V 3.12.1(Actelion Pharmaceuticals Ltd.) freely available at (http://www.openmolecules.org/).34,35 Activity Cliff Analysis was performed with similarity criterion as Skeleton-Spheres descriptor, which considers stereochemistry, counts duplicate fragments and encodes hetero-atom depleted skeletons as the most accurate descriptor for calculating similarities of chemical graphs. The similarity limit threshold was set to 80%.
Physiochemical properties descriptors and Quantitative Estimate of Drug-likeness (QED) were predicted with Discovery Studio (Accelrys Software Inc., V4.0) using CHARMm force field simulations on all structures (mol files in supplementary information). An energy minimization cascade was performed using an energy tolerance of 0.001 kcal/mol. ADMET descriptors were predicted using the small-molecule protocols for intestinal absorption, aqueous solubility, blood-brain barrier penetration, plasma protein binding, cytochrome P450 2D6 inhibition, and hepatotoxicity. The QED was determined for each structure using small-molecule protocols predicting molecular weight, lipophilicity, hydrogen bond donors, hydrogen bond acceptors, rotatable bonds aromatic rings, violations of Lipinski’s rules and the quantitative estimate of drug-likeness value.
4.6. Statistical analyses
Unless otherwise stated, all data are of at least three independent biological experiments (n ⩾ 3), each performed in triplicate. Statistical analysis was performed using either paired or unpaired Student t-tests to indicate statistical significance. Data were analyzed using GraphPad Prism 5.0.
Supplementary Material
Acknowledgements
We thank Kathryn Wicht for assistance with the β-hematin inhibition assays at the University of Cape Town; Colin Dubick, Ethan Marrow, Youxuan Li for assistance with chemical synthesis at University of South Carolina and Gloria Holbrook and Fangyi Zhu for assistance with physicochemical properties at St Judes. This work was supported by the South African National Research Foundation (FA2007050300003 & UID: 84627), the University of Pretoria and the South African Medical Research Council Strategic Health Initiatives Partnerships with the Medicines for Malaria Venture for funding. Any opinion, findings and conclusion exqpressed in this paper are those of the author(s) and therefore the NRF does not accept any liability in regard hereto.
Abbreviations
- BW-1
1,15-[N-(2-phenyl)benzyl)]amino-4,12-diazapentadecane
- DFMO
α-difluoromethylornithine
- FIC
fractional inhibitory concentrations
- MDL 27695
1,15-(N-benzyl)amino-4,12-diazapentadecane
- RI
resistance indices
- SAR
structure-activity relationship
- SI
selectivity index
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.Org/10.1016/j.bmc.2015.01.036. These data include MOL files and InChiKeys of the most important compounds described in this article.
References and notes
- 1.Meister S, Plouffe DM, Kuhen KL, Bonamy GM, Wu T, Barnes SW, Bopp SE, Borboa R, Bright AT, Che J, Cohen S, Dharia NV, Gagaring K, Gettayacamin M, Gordon P, Groessl T, Kato N, Lee MC, McNamara CW, Fidock DA, Nagle A, Nam TG, Richmond W, Roland J, Rottmann M, Zhou B, Froissard P, Glynne RJ, Mazier D, Sattabongkot J, Schultz PG, Tuntland T, Walker JR, Zhou Y, Chatterjee A, Diagana TT, Winzeler EA. Science. 2011;334:1372. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Science. 2010;329:1175. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FC, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, Sriraghavan K, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4400. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen L, Ferrer SB, Jimenez-Diaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst L, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. J. Med. Chem. 2011;54:5540. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Younis Y, Douelle F, Feng TS, Cabrera DG, Manach CL, Nchinda AT, Duffy S, White KL, Shackleford DM, Morizzi JJ. Med. Chem. 2012;55:3479. doi: 10.1021/jm3001373. [DOI] [PubMed] [Google Scholar]
- 6.Wallace HM, Fraser AV, Hughes A. Biochem. J. 2003;15:1. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallace HM, Fraser AV. Biochem. Soc. Trans. 2003;31:393. doi: 10.1042/bst0310393. [DOI] [PubMed] [Google Scholar]
- 8.Zou Y, Wu Z, Sirisoma N, Woster PM, Casero RA, Weiss LM, Rattendi D, Lane S, Bacchi CJ. Bioorg. Med. Chem. Lett. 2001;11:1613. doi: 10.1016/s0960-894x(01)00315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seiler N, Atanassov CL, Raul F. Int. J. Oncol. 1998;13:993. doi: 10.3892/ijo.13.5.993. [DOI] [PubMed] [Google Scholar]
- 10.Fraser AV, Woster PM, Wallace HM. Biochem. J. 2002;367:307. doi: 10.1042/BJ20020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casero RA, Marton LJ. Nat. Rev. Drug Disc. 2007;6:373. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 12.Bitonti A, Dumont J, Bush T, Edwards M, Stemerick D, McCann P, Sjoerdsma A. Proc. Natl. Acad. Sci. U.S.A. 1989;86:651. doi: 10.1073/pnas.86.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woster PM. Ann. Rep. Med. Chem. 2003;5:203. [Google Scholar]
- 14.Hanson W, Baumann RJ, McCann PP, Sjoerdsma A, Bitonti AJ. Antimicrob. Agents Chemother. 1990;34:722. doi: 10.1128/aac.34.5.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baohbedaso M, Bellevue FH, Wu R, Woster PM, Casero RA, Jr, Rattendi D, Lane S, Bacchi CJ. Bioorg. Med. Chem. Lett. 1996;6:2765. [Google Scholar]
- 16.Dumont J, Bitonti AJ, Bush TL, Edwards ML, Stemerick DM, McCann PP, Sjoerdsma A. Proc. Natl. Acad. Sci. U.S.A. 1989;86:651. doi: 10.1073/pnas.86.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.William M, Birkholtz LM, Niemand J, Louw AL, Persson L, Heby O. Biochem. J. 2011;438:229. doi: 10.1042/BJ20110362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verlinden BK, Niemand J, Snyman J, Sharma SK, Beattie RJ, Woster PMa, Birkholtz L. J. Med. Chem. 2011;54:1. doi: 10.1021/jm200463z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma SK, Wu Y, Steinbergs N, Crowley ML, Hanson AS, Casero RAa, Woster PM. J. Med. Chem. 2010;53:5197. doi: 10.1021/jm100217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominguez JN, Leon C, Rodrigues J, De Dominguez GN, Gut Ja, Rosenthal PJ. J. Med. Chem. 2005;48:3654. doi: 10.1021/jm058208o. [DOI] [PubMed] [Google Scholar]
- 21.Mei Z, Wang L, Lu W, Pang C, Maeda T, Peng W, Kaiser M, Sayed IEa, Inokuchi T. J. Med. Chem. 2013;56:1431. doi: 10.1021/jm300887b. [DOI] [PubMed] [Google Scholar]
- 22.Egan TJ, Koch KR, Swan PL, Clarkson C, Van Schalkwyk DAa, Smith PJ. J. Med. Chem. 2004;47:2926. doi: 10.1021/jm031132g. [DOI] [PubMed] [Google Scholar]
- 23.Carter MD, Phelan VV, Sandlin RD, Bachmann BO, Wright DW. Comb. Chem. HTS. 2010;13:285. doi: 10.2174/138620710790980496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liew LP, Kaiser M, Copp BR. Bioorg. Med. Chem. Lett. 2013;23:452. doi: 10.1016/j.bmcl.2012.11.072. [DOI] [PubMed] [Google Scholar]
- 25.Liew LP, Pearce AN, Kaiser M, Copp BR. Eur. J. Med. Chem. 2013;69:22. doi: 10.1016/j.ejmech.2013.07.055. [DOI] [PubMed] [Google Scholar]
- 26.Burrows JN, van Huijsduijnen RH, Mohrle JJ, Oeuvray C, Wells TN. Malar. J. 2013;12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Assaraf YG, Abu-Elheiga L, Spira DT, Desser Ha, Bachrach U. Biochem. J. 1987;242:221. doi: 10.1042/bj2420221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Manach C, Scheurer C, Sax S, Schleiferbock S, Cabrera DG, Younis Y, Paquet T, Street L, Smith P, Ding XC, Waterson D, Witty MJ, Leroy D, Chibale K, Wittlin S. Malar. J. 2013;12:424. doi: 10.1186/1475-2875-12-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leroy D, Campo B, Ding XC, Burrows JN, Cherbuin S. Trends Parasitol. 2014;30:478. doi: 10.1016/j.pt.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Adjalley SH, Johnston GL, Li T, Eastman RT, Ekland EH, Eappen AG, Richman A, Sim BK, Lee MC, Hoffman SL, Fidock DA. Proc. Natl. Acad. Sci. U.S.A. 2011;108:E1214. doi: 10.1073/pnas.1112037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun W, Tanaka TQ, Magle CT, Huang W, Southall N, Huang R, Dehdashti SJ, McKew JC, Williamson KC, Zheng W. Sci. Rep. 2014;4:3743. doi: 10.1038/srep03743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duffy S, Avery VM. Malar. J. 2013;12:408. doi: 10.1186/1475-2875-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birkholtz L. Encyclopedia of Malaria. In: Ginsburg H, editor. Polyamine Metabolism in Plasmodia. Springer; 2013. Chapter 2. [Google Scholar]
- 34.Guha R, Van Drie JHJ. Chem. Inf. Model. 2008;48:1716. doi: 10.1021/ci8001414. [DOI] [PubMed] [Google Scholar]
- 35.Guha R, Van Drie JHJ. Chem. Inf. Model. 2008;48:646. doi: 10.1021/ci7004093. [DOI] [PubMed] [Google Scholar]
- 36.Edwards ML, Stemerick DM, Bitonti AJ, Dumont JA, McCann PP, Bey P, Sjoerdsma AJ. Med. Chem. 1991;34:569. doi: 10.1021/jm00106a015. [DOI] [PubMed] [Google Scholar]
- 37.Bergeron R, Hawthorne T, Vinson J, Beck DJ, Ingeno M. Cancer Res. 1989;1:2959. [PubMed] [Google Scholar]
- 38.Bergeron RJ, McManis JS, Weimar WR, Schreier KM, Gao F, Wu Q, Ortiz-Ocasio J, Luchetta GR, Porter C, Vinson JR. J. Med. Chem. 1995;38:2278. doi: 10.1021/jm00013a003. [DOI] [PubMed] [Google Scholar]
- 39.Melchoirre C, Bolognesi ML, Minarini A, Rosini Ma, Tumiatti VJ. Med. Chem. 2010;53:5906. doi: 10.1021/jm100293f. [DOI] [PubMed] [Google Scholar]
- 40.Lopez C, Bi X, Bacchi CJ, Rattendi D, Woster PM. Bioorg. Med. Chem. Lett. 2006;16:3229. doi: 10.1016/j.bmcl.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 41.Angyal WW, Warburton W. J. Chem. Soc. 1951;1951:2492. [Google Scholar]
- 42.Rodima T, Kaljurand I, Leito I, Koppel IA, Schwesinger R. J. Org. Chem. 2000;65:6202. doi: 10.1021/jo005521j. [DOI] [PubMed] [Google Scholar]
- 43.Wallace AV, Wallace HM, Hughes A. Biochem. J. 2003;376:1. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bi X, Lopez C, Bacchi CJ, Rattendi D, Woster PM. Bioorg. Med. Chem. Lett. 2006;16:3229. doi: 10.1016/j.bmcl.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 45.Trager WA, Jensen JB. Science. 1976;193:673. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 46.Lambros C, Vanderberg JP. J. Parasitol. 1979;65:418. [PubMed] [Google Scholar]
- 47.Smilkstein M, Sriwilaijaroen N, Kelly J, Wilairat P, Riscoe M. Antimicrob Agents Chemother. 2004;48:1803. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malmquist NA, Moss TA, Mecheri S, Scherf Aa, Fuchter MJ. Proc. Natl. Acad. Sci. U.S.A. 2012:1. doi: 10.1073/pnas.1205414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ncokazi KK, Egan TJ. Anal. Biochem. 2005;338:306. doi: 10.1016/j.ab.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 50.Lucantoni L, Duffy S, Adjalley SH, Fidock DA, Avery VM. Antimicrob. Agents Chemother. 2013;57:6050. doi: 10.1128/AAC.00870-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.