

Abstract
The aberrant expression of androgen receptor (AR)-dependent transcriptional programs is a defining pathology of the development and progression of prostate cancers. Transcriptional cofactors that bind AR are critical determinants of prostate tumorigenesis. To gain a deeper understanding of the proteins linked to AR-dependent gene transcription, we performed a DNA-affinity chromatography-based proteomic screen designed to identify proteins involved in AR-mediated gene transcription in prostate tumor cells. Functional experiments validated the coregulator roles of known AR-binding proteins in AR-mediated transcription in prostate tumor cells. More importantly, novel coregulatory functions were detected in components of well-established cell surface receptor-dependent signal transduction pathways. Further experimentation demonstrated that components of the TNF, TGF-β, IL receptor, and epidermal growth factor signaling pathways modulated AR-dependent gene transcription and androgen-dependent proliferation in prostate tumor cells. Collectively, our proteomic dataset demonstrates that the cell surface receptor- and AR-dependent pathways are highly integrated, and provides a molecular framework for understanding how disparate signal-transduction pathways can influence AR-dependent transcriptional programs linked to the development and progression of human prostate cancers.
The application of genomic techniques such as chromatin immunoprecipitation (ChIP) followed by sequencing has been instrumental in defining the androgen receptor (AR) cistrome in prostate epithelial cells, prostate tumor cell lines, and prostatic tissues (1–6). Moreover, the ChIP technology has facilitated identification of transcription factors (TFs), based on the overrepresentation of their binding sites at target androgen-regulated genes (ARGs) (7). TFs that have been shown to influence AR-regulated gene expression programs in prostate tumor cells include members of the forkhead box protein A (FOXA) family of transcriptional factors (eg, FOXA1) (8–13), GATA 2-binding partner 2 (GATA2) (14–16), POU domain, class 2, TF1 (OCT1) (14), homeobox B13 (17, 18), and nuclear factor κB (6, 19–21). The diversity of transcriptional cofactors linked to aberrant AR activity in prostate tumor cells suggests that many TF networks interface with the AR transcriptional program in prostate tumor cells (22).
ChIP-based approaches have been the gold-standard method for studying how androgens mobilize AR to bind androgen-response elements (AREs) at target ARGs (23, 24). Major functional insights into the transcriptional program directed by AR and ancillary TFs in prostate tumor cells and tissues have been obtained through ChIP followed by sequencing experiments (25). However, ChIP-based methods are biased against the discovery of unknown cofactors (26). More importantly, much of the current understanding of how transcriptional and nontranscriptional cofactors that bind AR and either attenuate or potentiate AR-mediated transcription activity as functional coregulators were originally discovered through binary protein-protein interaction (PPI) assays (22, 27). The set of AR-interacting proteins, which represent the “AR-interactome,” continues to grow; more than 350 proteins known to bind AR and potentially modulate AR transcriptional activity in response to androgenic ligands (27–30). The AR-interactome encodes a broad list of functional coregulators that influence AR transcriptional activity at a number of different levels after binding androgenic ligands. AR coregulators can influence AR stability (eg, ubiquitination), intracellular trafficking (eg, ubiquitination, SUMOylation), posttranslational modification (eg, phosphorylation and acetylation), and PPIs (eg, chaperone activity) (22, 31). To date, no single coregulator is known to completely define the aberrant AR activity underlying the development and progression of human prostate cancers. The sheer size of the AR-interactome suggests that aberrant coregulator function (eg, underexpression or overexpression) influences AR transcriptional activity during the development and progression of human prostate cancers (32).
Historically, the proteomic screens carried out to expand the AR-interactome have been restricted to PPI assays designed to detect novel binding proteins through direct or indirect interactions with AR, in the absence of a DNA template (27). In an effort to more completely define the AR-interactome and identify proteins that can bind DNA, either directly or indirectly, we performed a quantitative proteomic screen for androgen-sensitive proteins that copurify with the proximal promoter of the model androgen-regulated rat probasin gene in vitro. Here, we report the identification of novel coregulatory proteins of AR-mediated transcription in prostate tumor cells. The AR-interactome was significantly enriched in the proteomic screen, and the coregulatory functions of these proteins in AR-mediated transcription were verified in prostate tumor cells. More importantly, components of cell surface receptor (CSR)-dependent signaling pathways were identified as androgen-sensitive proteins. Further molecular studies of selected androgen-sensitive adaptor proteins showed that they were functionally linked to the expression to ARGs, and that their expression was required for androgen-dependent proliferation of prostate tumor cells. Our findings demonstrate that AR- and CSR-dependent signaling pathways are biochemically integrated at the molecular level, and demonstrate that perturbations to the expression of components of disparate signal transduction pathways can impact AR transcriptional activity and androgen-mediated proliferation in prostate tumor cells. These findings provide new molecular insights into how CSR-dependent pathways might influence the development and progression of AR-dependent prostate cancers.
Materials and Methods
Reagents
The next reagents were purchased: AR agonist R1881 (methyltrienolone) (PerkinElmer Life Sciences); double-stranded small interfering RNAs (siRNAs) (QIAGEN); Oligofectamine reagent, 4%–12% sodium dodecyl sulfate (SDS)-polyacrylamide gels, and the TOPO TA cloning kit (Invitrogen); prestained Precision Plus protein standards goat antimouse and goat antirabbit horseradish peroxidase-conjugated secondary antibody (Bio-Rad); mouse monoclonal AR antibody (AR441), rabbit polyclonal AR antibody (N-20) (Santa Cruz Biotechnology, Inc); DNA oligonucleotides (Invitrogen); and Fast-start TaqDNA polymerase (Roche Applied Science).
Cell lines
LNCaP, 22Rv1, and PC3 prostate cancer cell lines were purchased from the American Type Culture Collection. LNCaP and 22Rv1 cells were cultured in phenol red-deficient RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (HyClone Laboratories) and penicillin/streptomycin/glutamine (Invitrogen). PC3 cells were cultured in phenol red-deficient DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin/glutamine. Short tandem repeat analysis was used to authenticate the genotype of all human prostate cancer cell lines (August, 2008).
Western blot analysis
For the Western blot analysis shown in Figure 1A, LNCaP nuclear proteins isolated with nuclear extraction buffer (20mM HEPES, 25% glycerol, 1.5mM MgCl2, and 0.2nM ZnCl2; pH 7.9) containing 400mM NaCl or 600mM KCl were quantified using the Pierce Bicinchoninic acid protein assay kit (Thermo Scientific Pierce). A total of 4 μg of nuclear protein extracts were subjected to 4%–12% SDS-PAGE electrophoresis (Invitrogen) and transferred to a polyvinylidene fluoride membrane (Bio-Rad). Membranes were incubated first in Blotto (4% wt/vol nonfat milk in 50mM Tris [pH 7.5] and 150mM NaCl plus 0.1% vol/vol Tween 20 [TBST]) for 1 hour at room temperature and then in TBST containing 5% bovine serum with mouse monoclonal H2A, H2B, H3, or H4 antibody (1:1000 dilution; Cell Signaling Technology). After 3 × 5-minute washes with TBST, the membranes were incubated in TBST containing 5% BSA with goat antimouse horseradish peroxidase-conjugated secondary antibodies (1:10 000; Bio-Rad) for 1 hour at room temperature. After 3 × 5-minute washes with TBST, immunoreactive bands were developed and visualized using the enhanced chemiluminescence reagent kit (Thermo Scientific Pierce), and the membranes were exposed to Hyperfilm ECL film (GE Healthcare) for less than 5 minutes.
Figure 1.
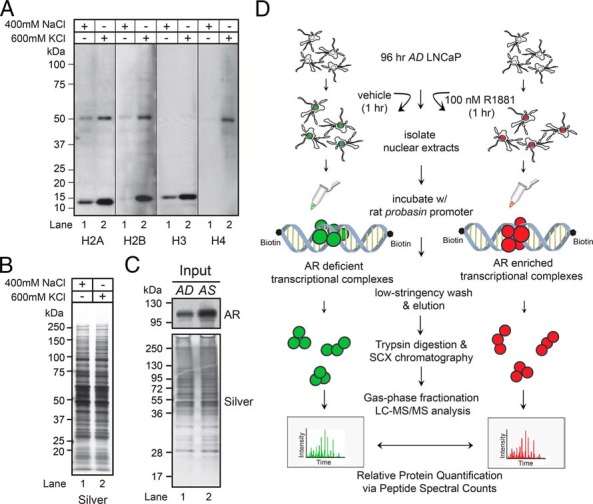
Proteomic workflow to identify DNA-binding proteins. A, Western blot analyses of LNCaP nuclear proteins extracted by the standard (400mM NaCl) or modified (600mM KCl) Dignam method, using antibodies to histones H2A, H2B, H3, and H4. B, Silver-stained gel demonstrated equal protein loading across samples. C, Western blot analysis of AD and AS nuclear protein extracts probed with anti-AR antibody (top panel). Silver-stained gel demonstrated equal protein loading across samples (bottom panel). D, Experimental platform for characterizing AR transcriptional complexes associated with DNA template in LNCaP cells. See Materials and Methods for details of the purification workflow.
The polyclonal and monoclonal antibodies used for all other Western blots are listed here. Rabbit polycolonal antibodies were against: AR (N-20) (1:1000 dilution; Santa Cruz Biotechnology, Inc), poly [ADP-ribose] polymerase 1 (PARP1) (1:1000 dilution; Cell Signaling Technology), α-actinin-4 (ACTN4) (1:1000 dilution; Alexis Biochemicals), transcription intermediary factor 1-β (TRIM28) (1:1000 dilution; Cell Signaling Technology), non-POU domain-containing octamer-binding protein (NONO) (1:1000 dilution; Sigma), soc-2 suppressor of clear homolog (SHOC2) (1:250 dilution; Sigma), and ABL proto-oncogene 1 (ABL1) (1:1000 dilution; Cell Signaling Technology). Rabbit monoclonal antibodies were against: Janus kinase 1 (JAK1) (1:1000 dilution; Cell Signaling Technology) and TGF-β-activated kinase 1/MAP3K7-binding protein 3 (TAB3) (1:1000 dilution; Abcam). Mouse monoclonal antibodies were against: SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1) (1:500 dilution; BD Transduction Laboratories) and filamin A (FLNA) (1:2000 dilution; Santa Cruz Biotechnology, Inc).
siRNA-mediated knockdown (KD) and Western blot analysis
LNCaP cells cultured in androgen-depleted (AD) medium for 24 hours were transfected with 2 sets of validated siRNAs targeting JAK1 (SI00605514, SI00605521), TNF receptor-associated factor 3 (TRAF3) (SI02225622, SI022256269), ABL1 (SI00288316, SI00299082), TAB3 (SI02779504, SI00738318), SHOC2 (SI00096789, SI00096803), AR (SI02757258), and scrambled control siRNA (1027281) (QIAGEN) with Oligofectamine (Life Technologies) for 48 hours. Cells were then treated with vehicle (ethanol) or androgen (1nM R1881) for 18 hours. Isolated protein extracts were probed with JAK1, TRAF3, ABL1, TAB3, SHOC2, AR antibodies. Western blotting results are available upon request.
Generation of the probasin promoter DNA template
The pCMV-myc-probasin vector was PCR amplified using the Advantage GC-2 polymerase (Clonetech) with biotinylated primers, biotinylated dATP, and normal dCTP, dGTP, and dTTP (New England Biolab). The sequence of the 5′ primer is Biotin-gtaatcatacatattatgattatccaataagctttctgg, and that of the 3′ primer is Biotin-agtgtgagcaggagggagggatgaccctcatcgtgtgtg. The DNA was pooled and applied to DNA spin columns to remove excess dNTPs. The DNA was then precipitated with ethanol and quantified using a NanoDrop spectrophotometer. For the DNA-affinity purification of nuclear proteins, equal amounts of DNA template were added to each of the nuclear extracts.
Affinity purification of DNA-binding proteins
LNCaP cells were grown in AD medium in 16 500-cm2 plates to 80% confluency for 96 hours. Eight of the plates were treated with 100nM R1881 for 1 hour (androgen stimulated [AS]), and the other 8 were treated with vehicle (ethanol) for 1 hour (AD). Cells were collected with Dulbecco's Phosphate-Buffered Saline (DPBS), and subjected to hypotonic lysis for subcellular fractionation. The collected cell pellets were resuspended in 25 mL of hypotonic buffer (10mM HEPES, 1.5mM MgCl2, and 10mM KCl [pH 7.9] with 10mM dithiothreitol (DTT) and 1× protease inhibitor cocktail [PIC]), and incubated on ice for 10 minutes. The cells were then subjected to nitrogen cavitation at 100 ψ for 5 minutes, and the nuclei were pelleted by centrifugation at 10 000g for 20 minutes at 4°C. The nuclear proteins were extracted from the pellet using nuclear extraction buffer (20mM HEPES, 600mM KCl, 25% glycerol, 1.5mM MgCl2, and 0.2nM ZnCl2, [pH 7.9] with 10mM DTT and 1× PIC) and rotated end-over-end for 1 hour at 4°C. Insoluble material was removed by centrifugation at 10 000g for 20 minutes at 4°C. Nuclear extracts were dialyzed with buffer A (20mM HEPES, 100mM KCl, 20% glycerol, 1.5mM MgCl2, and 0.2mM ZnCl2; pH 7.9) and analyzed by silver staining to determine protein concentration. 30 mg of AD and AS nuclear extracts in buffer A containing poly(dI-dC) (at a 3:1M excess relative to the molecular weight of the target template), 5mM DTT, 1× PIC, 5mM ATP, and 0.02% NP-40 were incubated with the 15 μg of biotinylated probasin DNA template containing 100 μL of equilibrated magnetic streptavidin beads; 1nM R1881 or vehicle (ethanol) was added to the AS or AD tubes, respectively, and incubated at 4°C with end-over-end rotation overnight. The beads were washed once with buffer A containing 5mM ATP, 0.02% NP-40, and 1nM R1881 or vehicle (ethanol) for the AS or AD tubes, respectively. Bound proteins were eluted with 1% SDS and 50mM Tris (pH 8.5). Samples were analyzed by silver staining to determine total eluted proteins.
Strong cation exchange (SCX) chromatography and tandem mass spectrometry
A total of 300 μg of proteins were reduced with 10mM DTT for 1 hour at 37°C and alkylated in 55mM iodoacetamide for 1 hour at room temperature in the dark. Samples were diluted to 0.05% SDS with 50mM Tris (pH 8.5) and digested with 20-μg trypsin (Promega) overnight at 37°C. The resulting peptide mixture was diluted with buffer C (20mM KH2PO4 and 25% acetonitrile [ACN] [pH 3.0]; final Tris concentration < 15mM). Each sample was individually loaded onto the Integral 100Q HPLC and subjected to SCX chromatography using a 2.1-mm × 20-cm Polysulfoethyl A column (PolyLC, Inc) at a flow rate of 200 μL/min, with a mixture of buffer C and buffer D (20mM KH2PO4, 350mM KCl, and 25% ACN; pH 3.0). The buffer D gradient was set up as follows: 30-minute gradient from 0%–43%, followed by 10-minute gradient from 43%–100%. The gradient was then washed with 100% buffer D for 25 minutes and equilibrated with buffer C for 20 minutes. Fractions 5–26 were collected and desalted with C18 spin columns (The Nest Group, Inc) and subsequently analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS).
LC-MS/MS analysis was performed on the Thermo LTQ mass spectrometer 3 times. Two LC-MS/MS gradients were used in this analysis. Gradient A: the samples were loaded onto a 10-cm Monitor C18 packed 100 inside diameter glass-capillary column for 70 minutes at 2800 nL/min, 180-minute gradient from 5%–35% buffer B (100% ACN, 0.8% acetic acid with a flow rate of 500 nL/min. The column was then washed with 75% buffer B for 45 minutes. Gradient B: the samples were loaded onto a 10-cm Monitor C18 packed 100 inside diameter glass-capillary column for 70 minutes at 2800 nL/min, 5%–28% buffer B (100% ACN, 0.8% acetic acid) with a flow rate of 500 nL/min for 180 minutes and 28%–35% buffer B for 10 minutes. The column was then washed with 75% buffer B for 45 minutes. The samples were analyzed twice using the Pseudo gas-phase fractionation method, with the 2 gradients mentioned before. The data-dependent (DD) settings included mass to charge windows of 400–480, 475–560, 555–640, 635–720, 715–800, and 795–880 m/z (set to top 15 most intense MS/MS ions); mass to charge windows 875–960 and 955–1040 m/z (set to top 10 most intense MS/MS ions); and mass to charge windows 1035–1125 and 1120–1200 m/z (set to top 5 most intense MS/MS ions); minimum signal of 3000; normalized collision energy of 35%; isolation width of 2 atomic mass unit; activation Q of 0.25; and activation time of 30 seconds. The samples were analyzed a third time by the pseudo gas-phase fractionation method, using a higher mass charge window range and gradient B. The MS DD settings were top 15 most intense ions in the mass to charge windows 900–1000, 990–1100, 1090–1200, 1190–1300, 1290–1400, 1390–1500, and 1490–1600 m/z.
Protein identification and data analysis
Raw files were converted to mzXML format using the ReAdW conversion tool. The mzXML files were searched against a forward plus reverse human International Protein Index database, version 3.87) appended with common contaminants (182 934 sequences) using SEQUEST version UW2012.01.6. The next search parameters were applied: peptide mass tolerance 3.0; fragment ion tolerance 0.36; monoisotopic masses; full tryptic search with up to 3 missed cleavages allowed; 57.021464 static modification on C; 15.9949 variable modification on M; 14.01560 variable modification on K and R; 28.0313 variable modification on K and R; 42.046950 variable modification on K and R; 114.042927 variable modification on K; and 79.966331 variable modification on S, T and Y. The search results were subsequently processed with the PeptideProphet and ProphetProphet tools, using Trans-Proteomic Pipeline version 4.6.1., with a less than 1% false discovery rate (FDR). Finally, the data were collated using ProteinProphet files for both the AD and AS samples. The Abacus (33) tool was used to align the 2 ProteinProphet outputs to in a single excel file. The next parameters were used: maxIniProb at 0.80; iniProb at 0.3; E.P.I. at 0.1; combined file probability at 0.3; and minimum protein probability at 0.80. The Abacus output file was then curated and filtered by removing identifications (I.D.s) with multiple International Protein Indexes, less than 2 spectral counts, and no gene names. Multiple MS/MS datasets were analyzed, using Abacus to extract peptide spectra counts and compare protein expression data for subsequent statistical analysis.
Network visualization and accessed public protein interaction databases
Protein interaction networks were visualized using Cytoscape 3.1.0 (http://www.cytoscape.org) (34). The web-based Search Tool for the Retrieval of Interacting Genes (STRING) database (35) was used to assemble functional networks with a medium confidence score more than or equal to 0.4 and default parameters, but with text mining disabled. The STRING network was exported to txt format and imported into Cytoscape for network visualization.
Dual luciferase reporter assay
LNCaP cells were seeded into Falcon (BD Bioscience) 48-well tissue culture dishes at a density of 30 000 cells/cm2. The cells were cultured in AD medium for 24 hours and cotransfected with siRNAs (100nM; QIAGEN), along with pGL4.10-Luc2-probasin (10 ng) and pRLSV40-renilla (25 ng). After transfection was performed using Lipofectamine 2000 (Invitrogen) for 48 hours, the cells were treated with vehicle (ethanol) or androgen (1nM R1881) for 18 hours, and harvested for luciferase activity using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol. Student's t test was used to determine significant differences between experimental and control siRNA-transfected cells (*, P ≤ .05, n = 3). The experiments involving cytokine treatment were carried out as detailed above except that before treatment with vehicle or 1nM R1881 for 18 hours, the transfected cells were treated with the specified cytokine for 30 minutes.
siRNA-mediated KD and real-time qPCR
LNCaP cells cultured in AD medium for 24 hours were transfected with validated siRNAs targeting JAK1, TRAF3, ABL1, TAB3, SHOC2, AR, and scrambled siRNA (control) (QIAGEN) with Oligofectamine (Life Technologies) for 48 hours. Cells were then treated with vehicle (ethanol) or androgen (1nM R1881) for 18 hours. RNA extraction was carried out using the RNeasy Midi kit, following the manufacturer's instructions (QIAGEN). First-strand cDNA synthesis was performed using reverse transcription protocols detailed in the SuperScript III First-Strand Synthesis kit (Invitrogen). Real-time quantitative PCR (qPCR) was carried out in a reaction containing cDNA, respective primer pairs (QIAGEN) and SYBR Green PCR Master Mix (Applied Biosystems). Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control for normalization.
Cytokine treatment and real-time qPCR
LNCaP cells cultured in AD medium for 72 hours were treated with vehicle (BSA), IL-1β (200 pg/mL), IL-6 (50 ng/mL), epidermal growth factor (EGF) (20 ng/mL), TNFα (50 ng/mL), or a cytokine cocktail (CC) (12.5-ng/mL IL-6, 50-pg/mL IL-1β, 12.5-ng/mL TNFα, and 5-ng/mL EGF) for 1 hour and subsequently treated with androgen (1nM R1881) for 18 hours. RNA extraction, reverse transcription, and real-time qPCR were carried out as described above.
Cell growth assays
LNCaP, 22Rv1, and PC3 cells cultured in AD medium for 24 hours were treated with vehicle (0.1% BSA) or a CC (12.5-ng/mL IL-6, 50-pg/mL IL-1β, 12.5-ng/mL TNFα, and 5-ng/mL EGF) for 1 hour and subsequently treated with ethanol (AD) or 1nM R1881 (AS) for 120 hours. The relative cell number was determined using the CyQUANT Cell Proliferation Assay (Life Technologies). Fluorescence measurements were made using a Microplate reader, with excitation at 485 nm and emission detection at 530 nm. For each cell line, asterisks denote differences that were considered statistically significant when cells treated with cytokines were compared with counterparts treated with vehicle (BSA), based on the Student's t test (P ≤ .05, n = 12). For siRNA KD experiments, LNCaP cells cultured in AD medium for 24 hours were transfected with validated siRNAs targeting JAK1, TRAF3, ABL1, TAB3, SHOC2, AR, and scrambled siRNA (control) (QIAGEN) with Oligofectamine (Life Technologies) for 24 hours. Cells were then treated with vehicle (ethanol, 0nM R1881), 0 01nM R1881 or 0.1nM R1881 for 120 hours. The relative cell number was determined using the CyQUANT Cell Proliferation Assay as described above, and ANOVA was used to determine statistical significance between control and experimental siRNA-transfected cells treated with the same dose of androgen (P ≤ .05, n = 6).
Statistics to analyze enrichment of commonly mutated genes in prostate cancer
The lists of commonly mutated genes in localized prostate cancer and castration-resistant prostate cancer (CRPC) were obtained from Grasso et al (99) and Barbieri et al (100), respectively. The contents were compared with the list of proteins identified in our proteomic screen. Fisher's exact test was carried out using the total number of proteins detectable by mass spectrometry (assume ∼10 000), the total number of commonly mutated genes (4295 for localized prostate cancer and 89 for CRPC), the total number of proteins identified in the proteomic screen (2878), and the number of overlapping proteins between the datasets (723 for localized prostate cancer and 38 for CRPC) to determine whether the proteomic screen enriched for commonly mutated genes in clinical prostate cancer samples.
AR-interactome analyses
The annotation of known AR-interacting proteins and androgen-sensitive AR-interacting proteins in the proteomic dataset for results presented below were derived by comparing gene names of known AR-interacting proteins with gene names of identified proteins in the DNA-binding proteomic screen. The raw data files used for these comparative analyses is available upon request.
Probasin promoter analyses
The detection of potential TF binding sites within the probasin promoter was analyzed using the TF database (TRANSFAC version 8.3) interfaced through the web-based PROMO website (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) (36, 37). The 799-bp nucleotide sequence of the probasin promoter (−734 to +65) was uploaded onto the program and the potential TF binding sites were identified based on sequences within a dissimilarity margin less than or equal to 15%.
Data deposition
The mass spectrometry proteomics data have been deposited in the ProteomeExchange repository (PXD002596).
Results
Quantitative mass spectrometry of proteins linked to AR-mediated transcription
The compendium of proteins linked to AR-mediated transcription has yet to be fully defined at the molecular level (22). We thus undertook a large-scale, proteomic approach to the discovery of androgen-sensitive proteins that copurified with a model ARG DNA template in vitro, as established by EMSA (38). The rat probasin promoter was selected as the model ARG DNA template for this screen because the sequence spanning the proximal enhancer and promoter is compact (ie, −705 to +61 bp) relative to the regulatory of the prostate-specific antigen (PSA) gene (spans over 4 kb) (24). The rat probasin promoter also contains 6 well-characterized AREs and directs tissue-specific gene expression to the prostate gland in rodents (39–42). In vitro, the probasin regulatory region exhibits strong mononucleosome phasing (ie, promoter region −268 to −76), and this is speculated to facilitate AR binding to target-gene promoters in vivo (43). Accordingly, the standard Dignam nuclear extraction protocol was modified in preparing EMSA samples, with 600mM KCl substituted for 400mM NaCl to enhance the release of high-affinity DNA-binding proteins such as histones (eg, core histones H2A, H2B, H3, and H4) (44). Western blot analyses confirmed that this protocol was effective when used on crude nuclear protein derived from the LNCaP prostate cancer cell line (Figure 1A, compare lanes 1, 400mM NaCl, and 2, 600mM KCl). Therefore, EMSA nuclear extracts were prepared with this modified protocol.
Next, we developed an experimental workflow (depicted in Figure 1D) to isolate those proteins within nuclear extracts from LNCaP cells that bind to the probasin promoter. To capture acute changes in the composition of proteins linked to AR-mediated transcription, we prepared nuclear extracts from AD (ie, 96-hours exposure to AD growth medium + 1-hour vehicle) or AS (ie, 96-hours exposure to AD growth medium + 1-hour 100nM R1881) LNCaP cells. We purposely challenged androgen-starved LNCaP cells with this superphysiologic dose of androgen so that all unliganded AR would be maximally bound by ligand during the course of the experiment. Androgens promote rapid translocation from the cytoplasm to nucleus in LNCaP cells (45), and, as predicted, AR levels were higher in the nuclear protein extracts from the AS-treated sample than those from the AD-treated sample (Figure 1C). Equal amounts of AD and AS nuclear protein extracts were incubated with the biotinylated probasin promoter and then streptavidin magnetic beads, and subjected to a single low-stringency wash. The remaining proteins were eluted and each sample was processed and subjected to shotgun proteomic analyses (Figure 1D).
To facilitate comprehensive identification and quantification of copurified proteins, we reduced the complexity of the sample peptide before proteomic analysis by subjecting each sample to SCX chromatography. The UV absorbances of the tryptic peptide digests from the eluted AD and AS samples were nearly identical (Supplemental Figure 1), demonstrating the near equal loading and gradient elution of tryptic peptides (∼300 μg). In-depth quantification of proteins based upon peptide spectral counts (PSCs) was facilitated by incorporating a modified gas-phase fractionation (GPFm/z) method; this increased the efficiency of peptide sequencing for all desalted SCX fractions using a 180-minute micro-LC-MS/MS gradient (ie, 5%–35%) (46–48). Eluted peptide ions were interrogated by the GPFm/z protocol, using narrow m/z windows (ie, 400–480, 475–560, 555–640, 635–720, 715–800, 795–880, 875–960, 955–1040, 1035–1125, and 1120–1200 m/z) and DD collision-induced dissociation selection of the top 15, top 10, and top 5 ions for each m/z window spanning the 400–880, 875–1040, and 1035–1200 m/z range, respectively (Supplemental Figure 1B). This proteomic analysis resulted in the acquisition of 784 330 and 791 948 MS/MS spectra in the AD and AS samples, demonstrating the near equal sampling of gas-phase peptide ions across all experiments (Supplemental Table 1). Pep3D and ProteinProphet analysis (eg, AD fraction 12) revealed that many of the peptide I.D.s were likely correct (Supplemental Figure 1, C and D) (49), ie, a single SCX fraction yielded 965 nonredundant protein I.D.s at a FDR of less than or equal to 0.7%, as estimated using the target-decoy approach (ie, ProteinProphet probability cut-off 0.9) (Supplemental Figure 1, C and D) (49, 50). Complete GPFm/z analysis resulted in 2522 and 2502 nonredundant protein I.D.s (FDR = 1%, 2 PSC minimum threshold) for the AD and AS samples, respectively (Supplemental Tables 1–4). These results demonstrated that the GPFm/z method enabled exhaustive peptide sequencing of probasin-copurified proteins.
Identification of androgen-sensitive and androgen-insensitive proteins
The GPFm/z method provided an in-depth proteomic analysis of probasin-copurified proteins detected in the AD and AS samples in vitro. The AD and AS samples shared 2146 nonredundant protein I.D.s (ie, ∼75%, 2146/2878), but also had 376 and 356 nonredundant I.D.s, respectively (Figure 2A). The strong proteomic overlap in probasin-copurified proteins allowed us to define androgen-sensitive proteins (Figure 2A). In classifying the proteins that are differentially expressed in the AD and AS datasets, we considered those for which the difference in expression was more than or equal to 2-fold androgen-sensitive, and those for which the difference was below this threshold. This resulted in the classification of 635 nonredundant I.D.s as androgen-sensitive proteins (ie, 297 AS proteins; 338 AD proteins, total = 635/2146, ∼30%), and 1511 nonredundant proteins I.D.s as androgen-insensitive proteins (ie, 1511/2146 proteins, ∼70%) (Figure 2B). Importantly, the correlation coefficient of PSC values between the AD and AS datasets was relatively high (ie, R2 = 0.80) (Figure 2C), demonstrating that the relative abundance of probasin-copurified proteins was mostly unchanged between the AD and AS samples. Thus, the relative abundance of a subpopulation of probasin-copurified proteins was influenced by androgens in vitro.
Figure 2.

Characterization of androgen-sensitive proteins copurified with the probasin promoter. A, Venn diagram summarizing AD and AS proteins identified in the proteomic screen. B, Natural log AS/AD PSC ratio graph for proteins copurified with the probasin promoter. A total of 635 proteins were defined as androgen sensitive (ln [AS/AD] more than or equal to 0.67 or ln [AS/AD] ≤ −0.67), and 1511 proteins were defined as androgen insensitive (0.67 > ln [AS/AD] > −0.67). C, Correlation coefficient analysis of PSC values between AD and AS samples. D, Western blot analysis of proteins copurified with the probasin promoter under AD and AS conditions and probed with antibodies to AR, SMARCB1, PARP1, ACTN4, TRIM28, NONO, and FLNA. The PSCs quantified in the proteomic screen are labeled below the blots. Silver-stained gel demonstrates equivalent loading across the samples.
Next, the AD and AS samples were subjected to Western blot analyses to validate PSC values and to further verify differences in protein expression between samples. The group of nuclear receptor coregulators selected for Western blot analyses based upon the commercial availability of antibody reagents included the corepressor TRIM28, the ATP-dependent chromatin-remodeling subunit SMARCB1, NONO, FLNA, ACTN4, and PARP1 (51–53). As expected, AR levels were noticeably higher in the AS vs AD sample, concordant with observed PSC values for AR (ie, AD/AS ratio, 2 PSCs:9 PSCs) (Figure 2D). Overall, the Western blot analyses demonstrated that PSC values closely matched the relative abundance of probasin-copurified proteins in the AD and AS samples.
Identification of the AR-interactome
The AR-interactome encodes many coregulators of AR-mediated transcription (22), a finding that prompted us to determine what proportion is detectable in the proteomic dataset. The AR-interactome encodes approximately 351 proteins, based upon well-characterized AR-interacting proteins annotated in the McGill Androgen Receptor Database, Human Proteome Reference Database, Biological General Repository for Interaction Datasets, and STRING database (27–30). A total of 133 members of the AR-interactome were detected in the proteomic dataset (Table 1 and Supplemental Figure 2A), demonstrating that a significant fraction of the AR-interactome (∼37%, 133/351) copurified to the probasin promoter in vitro (Fisher's exact test, 2-sided, right-sided P = 2.4 × 10−4).
Table 1.
AR-Interactome
| Protein Statistics | |
| SwissProt mass spectrometry-detectable human proteins | ∼10 000 |
| Known AR-interacting proteins databases: HRPD/BIOGRID/McGill/STRING | 351 |
| Total nonredundant protein identifications (FDR = 1%) | 2878 |
| Androgen-sensitive proteins; 2-fold threshold | 1367 |
| Androgen-insensitive proteins | 1511 |
| Enrichment of known AR-interacting proteins in the proteomic dataset | 133 out of 351 (Fisher's exact test; P = 1.9 × 10−4) |
| Androgen-sensitivity of known AR interacting proteins. (2-fold threshold, FDR = 1%) | 51 out of 133 (Fisher's exact test; left-tail P = 0.011) |
| Enrichment of androgen-sensitive AR-interacting proteins (T7-phage display, Norris et al, 17 o 57) | 57 out of 156 (Fisher's exact test; P = 1.0 × 10−3) |
| Androgen-sensitivity of the proteomic dataset compared with the T7-phage display study (2-fold threshold) | 18 out of 57 (Fisher's exact test; left-tail value = 0.153) |
BIOGRID, Biological General Repository for Interaction Datasets.
Next, we wanted to determine what proportion of the AR-interactome was influenced by androgens (ie, androgen sensitive; enriched for or depleted of probasin-copurified proteins), because androgens facilitate the coordinated mobilization of AR transcriptional complexes to target ARGs (24, 54, 55). In essence, we wanted to identify proteins that were enriched or depleted after copurification with the probasin promoter in vitro, and thus a 2-fold threshold was applied to the classification androgen-sensitive proteins that included overlapped and unique nonredundant I.D.s. in the AD and AS samples. Using this criterion, we identified 1367 androgen-sensitive and 1511 androgen-insensitive nonredundant I.D.s proteins (Table 1). Interestingly, only 51 of the 133 AR-interacting proteins identified in the proteomic dataset were classified as androgen sensitive; the remaining 82 were considered androgen-insensitive (Table 1). This finding demonstrated no detectable association between AR-interacting proteins and their sensitivity to androgens (ie, Fisher's exact test, right-sided P = 0.989) (Table 1). This was an unexpected finding given current working model(s) of AR-mediated transcription, which posit that AR coregulators coordinate the assembly and/or disassembly of AR-dependent transcriptional complexes at target ARGs in response to androgens (eg, histone acetyltransferase p300) (22, 56).
This finding prompted us to directly test whether or not the AR-interactome discovered by in vitro copurification with the probasin promoter was androgen-sensitive We restricted our statistical analysis to a well-defined group of approximately 156 androgen-sensitive, AR-interacting proteins that had been defined in a landmark, T7-phage display proteomic screen (57). This dataset represents the best annotated group of ligand-dependent, AR-interacting proteins published to date. Among these proteins, 57 were detected in our proteomic screen, demonstrating a significant enrichment of these copurified proteins to the probasin promoter in vitro (ie, Fisher's exact test, 2-sided, right-sided P = 1 × 10−3) (Table 1 and Supplemental Figure 2B). When a 2.0-fold expression threshold was applied to define androgen sensitivity, only 18 of the 57 proteins detected in the proteomic screen were classified as androgen sensitive (ie, androgen-increased, AS/AD ratio ≥ 2.0; or androgen-reduced, AS/AD ratio ≤ 0.50). Thus, T7-phage AR-interacting proteins identified by copurification with the probasin promoter in vitro were not androgen-sensitive (ie, Fisher's exact test, 2-sided, right-sided P = 0.85) (Table 1). This finding suggests that our proteomic screen lacked the power to detect heterogeneous, ligand-dependent protein interactions between AR and T7-phage AR-interacting proteins in the androgen-treated AS sample.
Network topology of androgen-sensitive and androgen-insensitive proteins
To identify novel proteins or protein networks linked to AR-mediated transcription, we examined the molecular connectivity and androgen sensitivity of probasin promoter-copurified proteins in vitro. First, we generated force-directed graphs of androgen-sensitive and androgen-insensitive proteins (using the next definitions: androgen sensitive = AS/AD ratio ≤ 0.5 and AS/AD ratio ≥ 2.0; androgen insensitive = 0.5 < AS/AD ratio < 2.0) to characterize the molecular topology of PPIs in each population of proteins (Supplemental Figure 3). The androgen-sensitive interactome contained 1367 proteins that consisted of 438 hubs, 1336 nodes, and 3830 edges, whereas the androgen-insensitive interactome (ie, 0.5 < PPI < 2.0-fold) contained 1511 proteins that consisted of 910 hubs, 1501 nodes, and 18 511 edges (Supplemental Figure 3). These results demonstrated that the edges in the androgen-insensitive interactome are more highly connected, and highlighted the complexity of PPIs detected in the androgen-sensitive and androgen-insensitive interactomes that copurified with the probasin promoter in vitro.
Next, we determined which biological processes were represented by the top-ranked protein networks for the androgen-sensitive and -insensitive proteomes, using the Genego bioinformatic software (Figure 3A). Notably, the top-ranked protein networks in these proteomes overlapped tremendously. In the androgen-insensitive dataset, the 10 top-ranked networks were 1) transcription mRNA processing; 2) translation initiation; 3) translation, elongation, termination; 4) transcription by RNA polymerase II; 5) cell adhesion/junctions; 6) cell cycle, mitosis; 7) DNA damage, BER-NER repair; 8) transcription, chromatin modification; 9) DNA damage core; and 10) nuclear receptors, transcriptional regulation. Notably, no meaningful difference in these functional themes was observed in the 10 top-ranked protein networks for the androgen-sensitive dataset; only the order was different (Figure 3A). Possible explanations for this finding are that the proteomic screen lacked the power to discern functional differences in protein networks (whether enrichment or depletion) between the 2 proteomes, and that the proteomes act together to coordinate and integrate androgen-dependent transcription. These explanations are not mutually exclusive; both scenarios may have contributed to the protein networks detected in the androgen-sensitive and -insensitive proteomes by our bioinformatic approach.
Figure 3.

A and B, Top differentially ranked process networks of androgen-sensitive (AS/AD ratio ≤ 0.5 and AS/AD ratio ≥ 2.0) vs androgen-insensitive proteins (0.5 < AS/AD ratio < 2.0) (A) and AD (AS/AD ratio ≤ 0.5) vs AS (AS/AD ratio ≥ 2.0) proteins (B), as assessed using Gene Ontology (GO) and analyzed in the GeneGo platform.
To identify more subtle distinctions, we separated the androgen-sensitive proteome into the AD (AS/AD ratio ≤ 0.5) and AS (AS/AD ratio ≥ 2.0) proteomes. This analysis of the proteins that copurified with the probasin promoter under each condition did uncover differences in the networks represented (Figure 3B). Specifically, the AS proteome was enriched for 7 of the top 10 networks with different representation: translational initiation, transcription mRNA processing; cell-cycle mitosis; protein folding response to unfolded proteins; cell-cycle meiosis; protein folding ER and cytoplasm; and protein folding in normal conditions. Although translation might be considered an unexpected finding for our screen given that the probe used is a gene promoter, the concept of nuclear translation, although controversial (58–60), has not been ruled out. Indeed a recent study provided further support for this cellular process taking place in mammalian cells (61). Moreover, the identification of this particular network is consistent with the ability of androgens to elicit rapid changes in protein translation (62). In contrast to the AS proteome, the AD proteome was most highly enriched for the next networks: proteolysis in cell cycle and apoptosis, transcription by RNA polymerase, and inflammation IL-6 signaling. Thus, our findings reveal functional differences in the networks represented by the proteins that can copurify with the probasin promoter in the contexts of androgen depletion and androgen stimulation. Further experimentation will be required to establish whether AR-dependent transcription is coordinated with the physical recruitment and/or dismissal of the androgen-sensitive protein networks reported in this bioinformatic analysis.
Identification of potential TF binding sites on the probasin promoter
The probasin promoter is compact compared with the PSA promoter, but nevertheless represents a relatively long DNA template for the isolation of DNA-binding proteins. This prompted us to examine in greater depth the specificity of TFs that copurified to the DNA template during the proteomic experiment. Specifically, we performed a TRANSFAC analysis against the 799-bp proximal probasin promoter, to identify putative TF binding sites and to validate their copurification to the probasin DNA template in vitro. The TRANSFAC analysis predicted 50 TF binding sites in the probasin promoter (Figure 4), and the proteomic screen validated 12 of these (12/50 = 24%). The TFs for which binding sites were identified are AR and previously annotated TFs in the probasin promoter (FOXA1; JUN; POU domain, class 2, TF1 [OCT1]), and nuclear factor I (63). Applying a threshold of 1.5-fold difference in expression between the 2 proteomes showed that 4 of the 12 TFs were androgen sensitive. These results validated the binding of predicted TFs to the probasin promoter in vitro, and highlighted the androgen-sensitivity of TFs involved AR-dependent transcription.
Figure 4.

Analysis of TF binding sites in the probasin promoter. The probasin promoter (799 bp, −734 to +65) was analyzed using the TF database (TRANSFAC version 8.3) interfaced through the PROMO website. Potential TF binding sites were identified based on sequences with a dissimilarity margin of less than or equal to 15%; 12 of 50 predicted TFs were identified in the proteomic screen, and their potential binding sites are shown. The number in parenthesis indicates the number of predicted binding sites found in the probasin promoter for that TF. Asterisks denote androgen-sensitive proteins identified in the proteomic screen.
Identification of AR transcriptional coregulators
Androgen-bound AR binds to AREs located in the enhancers and/or promoters of ARGs (ie, PSA) to activate or repress gene transcription through the sequential and/or combinatorial recruitment of coregulator complexes, including histone lysine demethylases, histone lysine acetyltransferases, histone lysine deacetylases, histone arginine methyltransferases, nuclear corepressors, SWI/SNF chromatin remodelers (ATPases), mediators (MEDs), and RNA polymerase II preelongation complexes (22, 24, 54, 55, 64–71). We investigated how androgens influenced the relative abundance of known coregulators bound to the probasin promoter in vitro. Functionally related AR coregulators were grouped and color coded based on their abundance in the proteomic dataset. The coregulator groupings included histones, histone lysine demethylases, histone lysine acetyltransferases, KMTs and histone arginine methyltransferases, nuclear corepressors, ATPases, MEDs, the DNA damage response machinery, and polymerase II preelongation (Supplemental Figures 4 and 5) (22). A Genego PPI map identified 25 edges to AR, and 689 edges to known AR coregulators (Supplemental Figure 4). This outcome highlights the extreme connectivity of functionally related AR coregulators within groups, and demonstrates their sensitivity to androgen in binding to the probasin promoter in vitro (Supplemental Figure 5). The findings suggest that the levels of nuclear AR coregulators are influenced by androgens in LNCaP cells. Moreover, they support further investigation into how androgens mobilize (recruitment and/or dismiss) distinct coregulators and coregulator complexes at target ARGs in vivo (54).
Identification of modulators of AR-mediated transcription
Next, we wanted to determine whether the proteomic screen had the power to identify proteins that display “coregulator-like” activity towards AR-mediated transcription in LNCaP cells. We implemented a siRNA-based screen testing for transcription in the context of KD, using AR-mediated probasin luciferase reporter expression as readout (Figure 5). A subpopulation of androgen-sensitive (ie, n = 88, enriched ≥ 2-fold, minimum of 3 PSCs) and -insensitive proteins (ie, n = 26) were targeted for KD using validated Thermo Fisher SMARTpool or QIAGEN FlexiPlate siRNAs (Figure 5B). Androgens facilitate AR-mediated transcription through combinatorial recruitment of coactivators and corepressors of AR-mediated transcription (24, 54, 55). Thus, 2 populations of androgen-sensitive proteins were selected for siRNA-mediated KD. The first consisted of 45 proteins enriched in the AD sample; these proteins were tested on the assumption that they might bind to the probasin promoter in the absence of androgen and facilitate the recruitment of androgen-bound AR to the probasin promoter in response to androgen exposure (Table 2). The second population consisted of 43 proteins enriched in the AS sample; these proteins were tested on the assumption that they might bind the probasin promoter only after ligand-bound AR is recruited, and then facilitate AR-mediated transcription (Table 2). LNCaP cells depleted of androgen for 24 hours were cotransfected with target SMARTpool siRNAs and the probasin luciferase reporter for 48 hours, and luciferase activity was determined after a 24 hours incubation with vehicle (ethanol) or synthetic androgen (1nM R1881) (Figure 5A). Notably, most transfected siRNAs attenuated AR transcriptional activity, as indicated by heat map representations of 8 different KD experiments. In these representations, luciferase activity is normalized to that in cells transfected with a control siRNA. Although a handful of transfected siRNAs potentiated AR activity, most those directed against both androgen-sensitive (ie, 94%, 83/88 target siRNAs) and androgen-insensitive (ie, 96%, 25/26) proteins attenuated AR activity (Table 2). These results suggested that probasin-copurified proteins, both androgen-sensitive and -insensitive, were required for optimal expression of the probasin reporter in LNCaP cells. Moreover, the paucity of androgen-sensitive proteins that displayed corepressor-like activity towards AR-mediated transcription demonstrated that these proteins overwhelmingly possess a coactivator like activity towards the probasin promoter (ie, Fisher's exact test, right-sided P < .05) (Table 2). These findings demonstrated that probasin-copurified proteins are involved in ligand-mediated AR activation or the potentiation of AR-mediated transcription of the probasin reporter in LNCaP cells.
Figure 5.

siRNA luciferase-based screen to identify modulators of AR-mediated transcription. A, Workflow of siRNA KD screen for the identification of proteins capable of modulating AR transcriptional activity. LNCaP cells depleted of androgen for 24 hours were cotransfected with the probasin-luciferase and pRLSV40-renilla vectors, along with control or experimental siRNAs (100nM) for 48 hours. Vehicle (ethanol) or androgen (1nM R1881) was then added to the cells, and luciferase readings were measured 18 hours later. The data are presented as a heatmap. B, Heatmap of the siRNA-luciferase screen results for modulators of AR-mediated transcription in LNCaP cells. LNCaP cells were transfected with siRNAs targeting proteins copurified with the probasin promoter and were tested for AR transcriptional activity. Asterisks denote significant differences between cells transfected with target siRNAs and control siRNAs (Student's t test; *, P < .05, n = 3). Triangles indicate androgen-insensitive proteins (0.67 > ln [AS/AD] > −0.67). Red check marks indicate known AR-interacting proteins.
Table 2.
Statistics of Predicted Modifiers
| Protein Group | Number of Proteins | siRNA KD Modulated AR Transcriptional Activity (P < .05) | Coactivator-Like Activity | Corepressor-Like Activity | No Activity |
|---|---|---|---|---|---|
| Androgen sensitive | 88 | 83 | 81 | 3 | 4 |
| AD | 45 | 44 | 38 | 1 | 1 |
| AS | 43 | 39 | 43 | 2 | 3 |
| Androgen insensitive | 26 | 25 | 21 | 4 | 1 |
| AR-interactome | 17 | 16 | 15 | 1 | 1 |
An association was detected for androgen-sensitive proteins that possessed AR coactivator-like activity (P = 0.047, Fisher's exact test).
Protein kinases as modulators of AR-mediated transcription
Aberrant protein kinase activity is known to initiate and facilitate the progression of human malignancies (72). Thus, protein kinases represent an important class of drug targets in human cancers (73, 74). Several protein kinases encode known coregulators of AR-mediated transcription in prostate tumor cells (22), but their roles in modulating ligand-mediated AR transcription remain unclear (31). Therefore, androgen-sensitive protein kinases were subjected to the probasin-luciferase reporter siRNA screen, and AR activity was assessed. The proteomic screen detected 59 of approximately 500+ protein kinases encoded by the human genome (Supplemental Table 5) (75, 76). Among these, 42 were classified as androgen-sensitive (ie, 2-fold change; minimum of 2 PSCs), including 22 and 20 protein kinases enriched in the AD and AS samples, respectively (Table 3). A schematic illustration of the AR-centric interaction network (Supplemental Figure 6) identified by these data highlights interactions among the 59 protein kinases, shows their sensitivity to androgens, and indicates physical interactions between protein kinases. This interaction network consisted of 44 edges, with 11 directly connected to AR (Supplemental Figure 6 and Table 3). Of the 59 protein kinases found in the screen, 25 were selected for siRNA KD screen based upon the commercial availability of experimentally validated siRNA reagents. The functional siRNA KD screen showed that 20 of 25 transfected siRNAs attenuated probasin-luciferase reporter expression (Figure 6). Notably, AR activity was strongly attenuated in cells transfected with siRNAs directed against glycogen synthase kinase-3β (ie, screen 1), a known AR coregulator in prostate tumor cells (Supplemental Table 5) (77–79). Other protein kinases that attenuated AR activity include CIT (citron rho-interacting serine/threonine kinase) and DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase) (Supplemental Table 5), which are known coregulators of AR-mediated transcription (80, 81). Interestingly, the DNA damage repair protein kinase ATR, which is functionally linked to the formation of transmembrane protease serine 2 (TMPRSS2)-ERG gene fusion transcripts (82), was also required for ligand-mediated AR activity in LNCaP cells. These results show that protein kinases modulate AR-dependent expression of the probasin reporter in LNCaP cells, and provide a molecular rationale for pharmacological inhibition of AR activity in prostate tumor cells.
Table 3.
Kinase Network Statistics
| Total protein kinases identified (2-spectral counts) | 59 |
| 2-Fold or greater | AS: 20 |
| AD: 22 | |
| Less than 2-fold | 17 |
| Total edges in network | 44 |
| Total kinases screened by luciferase | 25 |
Figure 6.
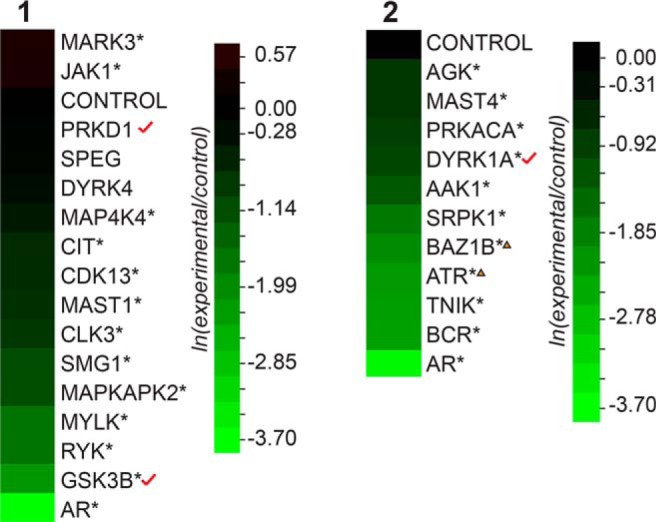
siRNA luciferase screen for the identification of protein kinases that modulate AR-mediated transcription. Dual luciferase assays were performed after cotransfection of the indicated siRNAs, and data were presented as heatmaps. Asterisks denote significant differences between cells transfected with target siRNAs and control siRNAs (Student's t test; *, P < .05, n = 3). Triangles indicate androgen-insensitive proteins (0.67 > ln [AS/AD] > −0.67). Red check marks indicate known AR-interacting proteins.
Because AR transcriptional activity in LNCaP cells was modulated by multiple protein kinases in the siRNA screen, the proteomic dataset was mined for differentially phosphorylated proteins, and androgen-sensitive phosphoproteins linked to AR-mediated transcription were identified (Supplemental Table 6). The proteomic dataset contained 907 unique phosphopeptides representing 365 nonredundant phosphoproteins (Supplemental Tables 7–9). Sixteen of the phosphoproteins encoded TFs and AR coregulators (ie, minimally 2 phospho-PSCs) (Supplemental Table 6). Distinct phosphopeptide residues in these target proteins were differentially phosphorylated in the AD and AS samples (Supplemental Table 6). One of the most notable differentially phosphorylated transcriptional coregulators was FOXA1, a pioneer factor of nuclear receptors (eg, AR, estrogen receptor) (Supplemental Table 6). Other differentially phosphorylated transcriptional coregulators included progesterone receptor, apoptosis antagonizing transcription factor, nuclear receptor corepressor, and trichorhinophalangeal syndrome I (ie, minimally 3 phospho-PSCs) (Supplemental Table 6). These results represent biochemical evidence that androgens influence the phosphorylation status of transcriptional coregulators that may be linked to AR-mediated transcription of the probasin reporter in LNCaP cells.
Adaptors of CSRs modulate AR-mediated transcription
AR coregulators are a functionally diverse group of proteins that modulate AR activity by a number of known mechanisms (ie, influences on trafficking, subcellular localization, stability, PPIs, and posttranslational modifications) (22). Our proteomic screen uncovered a group of androgen-sensitive intracellular adaptors and protein kinases that have been functionally linked to the integration of CSR signaling pathways (Supplemental Table 10). This finding prompted us to explore whether intracellular adaptors and protein kinases could also modulate AR transcriptional activity, because this subpopulation of molecules could facilitate cross talk between the AR- and CSR-signaling pathways in human prostate tumor cells. Thus, a subset of well-studied intracellular adaptors and protein kinases were selected for further study. This included TAB3, SHOC2, JAK1, TRAF3, and nonreceptor tyrosine kinase ABL1. The adaptors TAB3 and TRAF3 are downstream components of the TNF pathway (83–85), TAB3 and the protein kinase JAK1 are major regulators of the IL receptor signaling pathway (85, 86), the adaptor SHOC2 is a known component of the EGF signaling pathway (87, 88), and ABL1 is a component of multiple extracellular and intracellular signal transduction pathways (89). Western blot analysis verified the androgen-sensitivity of all 5 of these proteins in AD and AS nuclear protein extracts (Supplemental Figure 7A). Next, these adaptors and kinases were targeted for siRNA-mediated KD to determine whether their expression was required for AR-dependent expression of the probasin reporter in LNCaP cells. Gene silencing was confirmed by Western blot analysis, demonstrating ≥ 30% reductions in TAB3, SHOC2, JAK1, TRAF3, and ABL1 levels (Figure 7A). Endogenous AR activity was measured in AD LNCaP cells cotransfected with the probasin-luciferase vector and control, AR, TAB3, SHOC2, JAK1, TRAF3, or ABL1 siRNAs for 48 hours, and subsequently challenged with androgen (ie, 1nM R1881) for 24 hours (Figure 7, C and D, and Supplemental Figure 7B). As expected, AR activity was completely abolished in AR siRNA-transfected cells (Figure 7D and Supplemental Figure 7B). AR activity was also attenuated, although to a lesser degree, in TAB3, SHOC2, JAK1, TRAF3, and ABL1 siRNA-transfected cells (Figure 7D and Supplemental Figure 7B). These results showed that siRNAs directed against selected intracellular adaptors and protein kinases attenuated AR-dependent expression of the probasin reporter in LNCaP cells.
Figure 7.

Androgen-sensitive adaptors modulate AR transcriptional activity. A, Western blot analysis of whole-cell extracts (WCLs) isolated from AD (−) or AS (+) LNCaP cells transfected with AR, SHOC2, JAK1, ABL1, TAB3, and TRAF3 siRNAs using indicated antibodies. B, Silver-stained gel demonstrated equal protein loading across samples. C, Workflow of siRNA luciferase screen to identify modulators of AR transcriptional activity; 24-hour AD LNCaP cells were cotransfected with probasin-luciferase and pRLSV40-renilla vectors, along with control or experimental siRNAs (100nM), for 48 hours. Vehicle (ethanol, −) or androgen (1nM R1881, +) was then added to the cells, and luciferase readings were measured 18 hours later. D, LNCaP cells treated as described in C were harvested, and the measured luciferase activities were normalized to the activity of the vehicle control. Error bars indicate SD based on biological triplicates. Asterisks denote significant differences between cells transfected with target and control siRNAs (Student's t test; *, P < .05, n = 3). E, Clustering of qPCR data acquired from control, JAK1, TRAF3, ABL1, TAB3, SHOC2, and AR siRNA-transfected LNCaP cells. LNCaP cells depleted of androgens for 24 hours were transfected with indicated siRNAs for 48 hours and subsequently treated with androgen (1nM R1881). RNAs were extracted from the cells 18 hours later, and RT-qPCR was performed to measure the expression of AR, PSA, TMPRSS2, NKX3.1, and FASN. The values were normalized to cells transfected with control siRNAs.
We extended these findings by exploring whether the same siRNAs had any effect on ARG expression in LNCaP cells (90). The target ARGs included AR, fatty acid synthase (FASN), NK3 homeobox 1 (NKX3.1), PSA, and TMPRSS2 (Figure 7E and Supplemental Figure 7C). qPCR analysis was performed on AD LNCaP cells that were transfected with control, AR, TAB3, SHOC2, JAK1, TRAF3, or ABL1 siRNAs for 72 hours and subsequently challenged with androgen (1nM R1881; AS cells) for 24 hours. As expected, mRNA levels for AR, PSA, TMPRSS2, NKX3.1, and FASN were greatly reduced in AS cells transfected with the AR siRNAs (Figure 7E and Supplemental Figure 7C). Interestingly, transfection of cells with JAK1 siRNA potentiated the expression of most ARGs, excluding TMPRSS2, in AS cells. In contrast, the other siRNAs (ie, targeting TRAF3, ABL1, TAB3, and SHOC2) attenuated the expression of most ARGs, with the exception of FASN (Figure 7E and Supplemental Figure 7C). The luciferase and qPCR results demonstrate that intracellular adaptors and protein kinases modulate AR-dependent gene expression in LNCaP cells.
CSR adaptors modulate androgen-dependent proliferation of prostate tumor cells
Because the KD of androgen-sensitive adaptors and kinases modulated AR-mediated transcription in LNCaP cells, we next investigated whether these proteins were required for androgen-mediated proliferation. To test this, LNCaP cells depleted of androgen for 24 hours were transfected with control, AR, SHOC2, JAK1, TRAF3, ABL1, or TAB3 siRNAs for 24 hours, and then challenged with vehicle, 0.01nM, or 0.1nM of synthetic androgen for 120 hours (Figure 8). As expected, treatment with 0.1nM R1881 resulted in maximal cellular proliferation, and both androgen-dependent and androgen-independent proliferation was attenuated in AR KD cells. In the absence of androgen, cellular proliferation was potentiated in SHOC2 and TAB3 KD cells, whereas it was attenuated in JAK1 and TRAF3 KD cells, and unaffected in ABL1 KD cells. More importantly, at the 0.1nM R1881 dose, androgen-dependent proliferation was antagonized in all cells in which androgen-sensitive adaptors or kinases had been knocked down. These results showed that androgen-sensitive adaptors and protein kinases modulate androgen-mediated proliferation in LNCaP cells.
Figure 8.
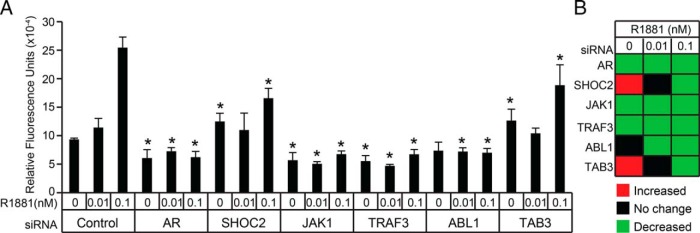
Androgen-sensitive adaptors modulate androgen-dependent proliferation of prostate tumor cells. A, Twenty-four-hour AD LNCaP cells were transfected with control or experimental siRNAs (100nM) for 24 hours. Vehicle (ethanol, 0nM R1881) or androgen (0.01nM or 0.1nM R1881) was then added to the cells, and they were incubated for a further 120 hours. The relative cell number was determined using the CyQUANT Cell Proliferation Assay. ANOVA was only performed between control and target siRNA treated with the same dose of androgen. Asterisks denote statistical significance (ANOVA; *, P < .05, n = 6). B, Heatmap presentation of proliferation assay results. Significant differences between cells transfected with experimental siRNAs and control siRNAs were color coded as indicated in the legend.
CSRs modulate AR-mediated transcription
Many in vitro studies have defined the AR-dependent gene expression program in androgen-starved prostate tumor cells after restimulation with androgens (91, 92). To determine whether CSR-dependent signaling can influence AR-dependent gene expression in LNCaP cells, probasin-luciferase activity was assessed in AD LNCaP cells (ie, androgen starvation for 48 hours) pretreated with cytokines (ie, BSA [0.1%, control], IL-6 [12.5 ng/mL], EGF [5 ng/mL], TNFα [12.5 ng/mL], or IL-1β [50 pg/mL]) for 30 minutes and subsequently challenged with androgen (ie, 1nM R1881) for 24 hours. AR activity was antagonized relative to that in the BSA control by all cytokines tested, in the order: IL-1β > TNFα > EGF > IL-6 (Figure 9A). These findings prompted us to test whether ARG expression was influenced by cytokines. qPCR analysis of a subset of ARGs (ie, AR, PSA, TMPRSS2, NKX3.1, and FASN) showed that cytokines strongly attenuated ARG expression in androgen-treated cells (Figure 9B). ARG expression was further antagonized in cells pretreated with a CC (12.5 ng/mL of IL-6, 5 ng/mL of EGF, 12.5 ng/mL of TNFα, and 50 pg/mL of IL-1β) (Figure 9B), highlighting the synergistic antagonistic effects of cytokines on AR-dependent gene expression in prostate tumor cells. In summary, cytokines antagonized AR-mediated gene expression and mediated cross talk between the CSR- and AR-dependent signaling pathways in LNCaP cells.
Figure 9.

Cytokines modulate AR transcriptional activity of prostate tumor cells. A, Luciferase reporter assay measuring AR activity in the presence of cytokines. LNCaP cells depleted of androgen (AD) for 24 hours were cotransfected with the probasin-luciferase and pRLSV40-renilla vectors. Forty-eight hours after transfection, the cells were treated with indicated cytokines for 30 minutes and then treated with androgen (1nM R1881) for 18 hours. The cells were harvested and the measured luciferase activities were normalized to the activity of the vehicle control (BSA). Asterisks denote significant differences between cells transfected with target and control siRNAs (Student's t test; *, P < .05, n = 3). B, Clustering of qPCR data acquired from cytokine-treated LNCaP cells. Seventy-two-hour AD LNCaP cells were treated with vehicle (BSA) and one or more cytokines for 1 hour before androgen (1nM R1881) treatment. RNAs were extracted from the cells 18 hours later, and RT-qPCR was performed to measure expression of AR and the indicated ARGs. The values were normalized to those in BSA-treated cells. C, Workflow to determine cross talk between cytokines and the roles of intracellular adaptors in modulating AR-mediated transcription. D, Clustering of qPCR data acquired from cytokine-treated LNCaP cells. To determine the effects of cytokines on endogenous ARG expression, 24-hour AD LNCaP cells were transfected with control, AR, SHOC2, or JAK1 siRNAs. Forty-eight hours after transfection, cells were treated with vehicle (BSA), EGF, or IL-6 for 1 hour and subsequently treated with androgen (1nM R1881) for 18 hours. RNAs were extracted from the cells, and RT qPCR was performed to measure AR and indicated ARG expression. The values were normalized to those in BSA-treated cells.
Next, we investigated whether androgen-sensitive adaptors and kinases were required for cytokine-mediated AR antagonism in LNCaP cells, to further test the cross talk between the CSR- and AR-dependent signaling pathways in prostate tumor cells. We selected the androgen-sensitive adaptor SHOC2 and protein kinase JAK1, because they are well-studied components of the EGF and IL-6 signal transduction pathways, respectively. AD LNCaP cells were transfected with a SHOC2 or JAK1 siRNA for 72 hours, pretreated with EGF or IL-6, respectively, for 30 minutes, and subsequently challenged with androgen for 18 hours (Figure 9C). qPCR analysis showed that in BSA-treated cells, the expression of all ARGs except FASN was attenuated in SHOC2 KD cells, and potentiated in JAK1 KD cells (Figure 9D). These results provided independent verification that SHOC2 and JAK1 modulate AR-mediated transcription (Figure 7D). Interestingly, TMPRSS2 and NKX3.1 expression was attenuated in cells transfected with a control siRNA and treated with EGF, suggesting that EGF selectively antagonized ARG expression in LNCaP cells (Figure 9D). Furthermore, the attenuation of TMPRSS2 and NKX3.1 expression was unaffected in SHOC2 KD cells treated with EGF, suggesting that the mechanism(s) of EGF-mediated antagonism was SHOC2 independent. In contrast, EGF slightly potentiated AR and PSA expression in SHOC2 KD cells relative to control cells, suggesting that AR and PSA expression was antagonized by SHOC2 through an EGF-independent mechanism. Similarly, IL-6 treatment led to attenuation of most ARGs, with the exceptions of AR and FASN. Additionally, IL-6 treatment increased the expression of AR, PSA, and NKX3.1 in JAK1 KD cells, suggesting that JAK1 antagonized AR, PSA, and NKX3.1 expression through an IL-6-dependent mechanism. TMPRSS2 was the only ARG that was positively regulated by JAK1 in IL-6-treated cells (Figure 9D). These molecular findings provide further evidence that androgen-sensitive adaptors facilitate cross talk between the CSR- and AR-dependent signaling pathways in LNCaP cells.
Cytokines modulate the proliferation of prostate cancer cells
Previous studies showed that growth factors and cytokines can modulate the proliferation of human prostate cancer cell lines (93–97). Our findings in LNCaP cells further demonstrate that cytokines strongly antagonized AR-mediated transcription in LNCaP cells, prompting us to test whether cytokines can modulate androgen-mediated proliferation, a hallmark pathological feature of early-stage (ie, organ-confined) and late-stage (ie, castrate-resistant) human prostate cancers. Specifically, we investigated whether cytokines influenced the androgen-dependent proliferation of several human prostate cancer cell lines (ie, LNCaP, 22Rv1, PC3, and DU145). Therefore, these cells were grown in AD growth medium for 24 hours, pretreated with a CC (12.5 ng/mL of IL-6, 5 ng/mL of EGF, 12.5 ng/mL of TNFα, and 50 pg/mL of IL-1β) that inhibits AR-mediated transcription in LNCaP cells, and then challenged with vehicle, 0.1nM, or 1nM of synthetic androgen for 120 hours (Figure 10, A–D). Treatment with 0.1nM androgen resulted in maximal cellular proliferation in LNCaP and 22Rv1 cells, but had no effect on proliferation in DU145 and PC3 cells (Figure 10, A–D). To examine the effects of individual cytokines have on the proliferation of prostate tumor cells, 24-hour AD LNCaP cells were pretreated with IL-6, EGF, TNFα, or IL-1β and then challenged with vehicle, 0.1nM, or 1nM of synthetic androgens for 120 hours (Supplemental Figure 7D). Androgen-dependent proliferation of LNCaP cells, to the exception of EGF, was attenuated when treated with individual cytokines (Supplemental Figure 7D). These results demonstrated that cytokines/CSR pathways can antagonize both androgen-dependent and, to a lesser degree, androgen-independent proliferation in prostate tumor cells.
Figure 10.

Cytokines modulate the proliferation of prostate tumor cells. A–D, LNCaP (A), 22Rv1 (B), DU145 (C), and PC3 (D) cells were treated with vehicle (BSA) or CC (12.5-ng/mL IL-6, 50-pg/mL IL-1β, 12.5-ng/mL TNFα, and 5-ng/mL EGF) for 1 hour before incubation with vehicle or androgens (0.1nM or 1nM R1881) for 120 hours. The relative cell number was determined using the CyQUANT Cell Proliferation Assay. Asterisks denote statistical significance (Student's t test; *, P < .05, n = 12).
Bioinformatic analyses of protein-encoding genes that are commonly mutated in human prostate cancers
Whole-exome sequencing analyses of clinical prostate cancers has uncovered a wide spectrum of somatically mutated genes in organ-confined and metastatic, CRPCs (98, 99). This prompted us to determine whether our proteomic screen enriched for the protein-encoding genes that are mutated in a panel of 112 prostate-localized adenocarcinomas (100). Among 4295 genes harboring somatic mutations (ie, somatic point mutations, substitutions, small insertions/deletions), 723 were detected in our screen of 2878 nonredundant I.D.s (Table 4). This finding showed that genes mutated in localized prostate cancer samples were significantly enriched in the proteomic screen (ie, Fisher's exact test, right-sided test P = 0.006) (Table 4). We extended this bioinformatic analysis to genes that are commonly mutated in CRPC. These include AR coregulators, which are thought to be involved in the development of this lethal disease (21). Of 89 AR coregulator genes mutated in CRPC, 38 were detected in the proteomic screen (ie, P = 4.7 × 10−3) (Table 4), demonstrating that AR coregulator genes are frequently mutated in CRPC. Thus, the proteomic screen had sufficient power to detect protein-encoded genes in LNCaP cells that are commonly mutated in organ-confined prostate cancers and CRPC.
Table 4.
Enrichment of Mutations Found in Localized and CRPC
| Total number of proteins detectable by mass spectrometry | ∼10 000 |
| Total number of somatic mutation across 112 prostate cancer samples | 4295 |
| Total number of proteins in proteomic screen | 2878 |
| Total number of overlap proteins | 723 |
| Enrichment of commonly mutated genes in clinical localized prostate cancer patients | 723 out of 4295 (Fisher's exact test; right-sided P = .006) |
| Total number of commonly mutated genes in CRPC | 89 |
| Total number of overlap proteins | 38 |
| Enrichment of commonly mutated genes in clinical CRPC | 38 out of 89 (Fisher's exact test; P = 4.7E-3) |
Discussion
AR-dependent signaling cascades are complex in that they involve both acute “nongenomic” responses and long-term “genomic” responses (101). Binary PPI assays have been crucial to defining the compendium of AR coregulators that bind to AR and modulate AR-dependent transcription (27), identifying 351 AR coregulators. Although this list continues to grow, its limitation is that it has not provided a global understanding of how androgens coordinate AR-dependent transcription. Large-scale proteomic screens/approaches, on the other hand, have gone beyond identifying novel ligand-sensitive AR-interacting proteins (51, 57, 102), also defining subcellular protein networks that are controlled by androgens (103, 104). Such studies are shedding light on novel components and downstream mediators of AR-dependent signaling pathways in prostate tumor cells, at both the molecular and cellular level.
In this study, we have refined the latter approach, applying an unbiased proteomic workflow to identify DNA-binding proteins that are linked to AR-mediated transcription. Our combined use of DNA-affinity chromatography and 2-dimensional LC-MS/MS enabled us to identify nuclear proteins bound directly or indirectly to a naked DNA template encompassing the proximal promoter of the rat probasin gene. We considered using the PSA promoter to isolate DNA-binding proteins because it is one of the most well-studied androgen-regulated genes in prostate cancer (105, 106). However this promoter was ultimately not selected for our biochemical screen because the 4 kb of sequence encompassing the core PSA promoter and upstream enhancer (24) would have been a difficult template for the purification of androgen-sensitive DNA-binding proteins in vitro. We expect that many of the proteins identified in this study will be validated as DNA-binding proteins in future ChIP experiments, and that they will be found to bind, either directly or indirectly, to the probasin gene and other ARGs in prostate tumor cells.
The GPFm/z method used to interrogate probasin-copurified proteins from unstimulated and AS LNCaP nuclear protein extracts offered several advantages. First, acute 1-hour treatment of LNCaP cells with vehicle and androgen facilitated the relative quantification of probasin-copurified proteins under both experimental conditions, and this guided the identification of androgen-sensitive DNA-binding proteins. Second, subjecting the streptavidin-DNA-protein complexes to a low-stringency wash decreased the loss of low-affinity DNA-binding proteins/protein complexes while retaining unspecific DNA-binding proteins. Overall, the advantages gained through in-depth proteomic sequencing by the GPFm/z method appear to have offset any disadvantages of the potential for reduced specificity, because many low-abundance proteins with the ability to regulate transcription were detected in the proteomic screen (eg, MEDs).
We recognize that the single-step affinity-purification protocol for isolating DNA-protein complexes lacked the power to resolve heterogeneous DNA-protein complexes (different molecular weights) that may have formed in vitro, and that such heterogeneity may account for the apparent androgen insensitivity of T7-phage AR-interacting proteins during their copurification. We suspect that because we assessed androgen-sensitivity based solely upon differences in protein abundance between the AD and AS samples, we would not have observed androgen-induced differences in ligand-dependent physical interactions with AR or other binding proteins in the cases of proteins whose relative levels in the 2 samples did not differ. Basically, PSC values lacked the power to distinguish between ligand-dependent protein interactions. In spite of this potential shortcoming of our methodology, the fact that our dataset was enriched for known AR coregulators supports the involvement of the identified proteins in AR-mediated transcription. We also anticipate that the set of identified androgen-sensitive proteins is not only enriched for DNA-binding proteins, but also includes proteins whose levels changed as a consequence of nuclear-cytoplasmic trafficking and/or protein turnover (eg, ubiquitin-mediated proteasome degradation). Transcription-dependent mechanisms are not represented, because a 1-hour exposure to androgens would be too short to allow for such an effect.
Our identification of a subset of androgen-sensitive intracellular adaptors and protein kinases that modulate androgen-mediated proliferation and AR-dependent transcriptional responses in prostate tumor cells is significant because each cofactor (ie, TAB3, SHOC2, JAK1, TRAF3, and ABL1) represents an important signaling node in a CSR-dependent signaling pathway involved in cellular proliferation, differentiation, inflammation, and/or disease (107, 108). Our findings demonstrate that cofactor expression is required for optimal androgen-mediated proliferation of such cells, providing further evidence that they are components of androgen-mediated responses in hormone-responsive prostate tumor cells. Moreover, CSR-dependent pathways modulated AR-mediated gene expression via intracellular adaptors and protein kinases. The modulation of ARGs by these cofactors in response to androgens represents another layer in the modulation of AR-regulated gene expression in prostate tumor cells. Interestingly, the expression of cofactor mRNAs differed significantly between normal prostate epithelial cells and prostate cancers (Table 5) (109–113). Further experimentation will be required to establish whether changes in cofactor expression account for any differences in AR-mediated transcription/signaling responses between normal and cancerous prostate epithelial cells. Although our data were gathered in vitro, this level of gene regulation is expected to be recapitulated in vivo. Early studies showed that CSR-dependent pathways facilitate AR activation at castrate levels of androgens, and that such activity represents one of multiple molecular mechanisms that drive the proliferation of tumor cells in response to ADT (114–116). However, more recently, EGF was found to attenuate AR activity in prostate tumor cells acutely exposed to androgen (castrate levels of androgen) (117), but to potentiate AR activity in cells chronically exposed to castrate levels of androgen (117). Thus, the cellular context of prostate tumor cells in vitro can influence the AR response to signaling by CSR-dependent pathways. A similar observation was made in vivo, for both prostate tumor cells and tissues (6), with the profile of AR-interacting proteins and the AR cistrome differing between in vitro and in xenografted LNCaP prostate tumor cells (6). For example, AR interactions with FOXA1 and STAT5 differed in cultured LNCaP cells and xenografts. Also, the AR-binding site profile of cultured LNCaP cells could be redirected to the in vivo profile when the cells were treated with a CC (eg, SDF1, IL-6). This study clearly demonstrated that CSR-dependent pathways can impact AR-dependent transcriptional responses in prostate tumor cells. Our findings provide further evidence of biochemical cross talk between AR- and CSR-dependent pathways and demonstrate that intracellular adaptors and protein kinases linked to CSR-dependent pathways can influence AR-mediated transcriptional responses. We anticipate that perturbations in the expression (ie, under or overexpression) of intracellular adaptors and protein kinases will also affect AR-mediated transcriptional responses in prostate tumor cells in vivo.
Table 5.
Gene Expression Profiles of Androgen-Sensitive Adaptor Proteins in Human Prostate Cancer Samples
| Gene Name | Cancer vs Normal | References |
|---|---|---|
| ABL1 | ↑ | Tomlins et al (109) |
| Wallace et al (110) | ||
| JAK1 | ↓ | Tomlins et al (109) |
| Arredouani et al (111) | ||
| TRAF3 | ↓ | Tomlins et al (109) |
| SHOC2 | ↓ | Luo et al (112) |
| TAB3 | ↑ | Tomlins et al (109) |
| Varambally et al (113) |
P < .05, 1.5-fold change. ↑ indicates increased expression and ↓ indicates decreased expression.
Our study demonstrates the power of mass spectrometry-based proteomics as a tool for the discovery of hormone-dependent signaling pathways in prostate tumor cells. We successfully followed this proteomic approach up with functional siRNA screening to validate new modifiers of AR-mediated transcription in LNCaP prostate tumor cells. For example, AR activity was attenuated in cells transfected with siRNAs directed against NDUFV2 (encodes NADH dehydrogenase [ubiquinone] flavoprotein 2), and KCTD8 (encodes the potassium channel tetramerization domain containing 8 protein) (Figure 5). We interpret these transcriptional luciferase results with caution because gene-specific siRNA KD could elicit indirect cellular stress-responses (eg, influence cell viability) that might confound biological interpretation. Despite this limitation, our results are important in that they will guide future experiments toward understanding how transcriptional responses, whether direct or indirect, influence AR transcriptional activity. As such, they are expected to reveal why some mutations in tumor cells attenuate and others potentiate AR-mediated transcription, to influence the progression of human prostate cancers. Although further experiments will be required to definitively prove the relevance of interactions of these proteins with AR in prostate tumor cells, our study adds to the growing list of functionally diverse proteins that may modulate AR transcriptional activity in LNCaP prostate tumor cells. Moreover, it suggests that at the molecular level AR signaling is dynamic, and that numerous molecules contribute to aberrant AR signaling in prostate tumor cells. This interpretation can be extended to the protein kinase siRNA screen, because a broad spectrum of protein kinases was found to modulate AR transcriptional activity in LNCaP cells. For example, our siRNA screen validated the findings that cyclin-dependent kinase 5 and glycogen synthase kinase-3 β facilitate androgen-mediated AR activation (77–79, 118) specifically in the context of LNCaP prostate tumor cells. A previous subgenome short hairpin RNA screen of signal transduction molecules and an short hairpin RNA screen directed at protein kinases have identified a subset of kinases that are necessary for AR-dependent gene transcription and the proliferation of human prostate tumor cells after exposure to androgens (80, 119). Our proteomic screen validated several of these protein kinases as androgen-sensitive targets (eg, CIT, TTK), and our siRNA screen validated their involvement in AR-mediated transcription in prostate tumor cells. Recent studies have suggested that AR and aberrant kinase pathways cooperate in the initiation and progression of human prostate cancers. For example, a phosphoprotein profiling experiment in mouse models of prostate cancer uncovered oncogene-specific tyrosine kinase activation (120). The tyrosine kinase pathways included epidermal growth factor receptor, ephrin type A receptor 2, JAK2, ABL1, and SRC. Moreover, phosphoprotein profiling of metastatic CRPC in humans revealed tyrosine kinase activation patterns (eg, SRC, EGFR, anaplastic lymphoma kinase, RET, and MAPK1/3) that are different from those in treatment-naïve prostate cancer (121). It is well established that aberrant kinase activity can modulate AR transcriptional activity and AR-dependent growth responses under castrate levels of androgen in prostate tumor cells and prostate cancer xenografts (31, 122–124). However, the physiological or pathophysiological roles of protein kinases in AR-mediated transcription/signaling in hormone-naïve prostate tumor cells are understudied (125, 126). Unraveling how specific kinases and kinase networks influence AR-dependent gene expression in the presence of androgens is expected to provide novel insights into how AR activity becomes corrupted during the initiation and progression of human prostate cancers. Our siRNA kinase screen suggests a broader role for protein kinases in modulating androgen-dependent AR transcriptional activity in hormone-responsive prostate tumor cells.
In this study, an in vitro proteomic strategy was used to identify novel components of the AR-mediated transcription machinery in prostate tumor cells. These findings will facilitate the development of new hypotheses regarding the proteins and protein networks involved in acute AR signaling in prostate tumor cells. We anticipate that the DNA-affinity purification method detailed here will provide new insight into the mechanisms of action of selective AR modulators, how these drugs modulate the composition of AR coregulators bound to DNA, and how they impact the protein networks that coordinate androgen-regulated processes in prostate tumor cells.
Acknowledgments
This work was supported by University of California Davis and The University of Iowa startup funds (M.E.W.) and by the National Institutes of Health Grant R01 GM094231 (to A.I.N.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ABL1
- ABL proto-oncogene 1
- ACN
- acetonitrile
- ACTN4
- α-actinin-4
- AD
- androgen depleted
- AR
- androgen receptor
- ARE
- androgen-response element
- ARG
- androgen-regulated gene
- AS
- androgen stimulated
- CC
- cytokine cocktail
- ChIP
- chromatin immunoprecipitation
- CRPC
- castration-resistant prostate cancer
- CSR
- cell surface receptor
- DD
- data dependent
- DTT
- dithiothreitol
- EGF
- epidermal growth factor
- FASN
- fatty acid synthase
- FDR
- false discovery rate
- FLNA
- filamin A
- FOXA
- forkhead box protein A
- I.D.s
- identifications
- JAK1
- Janus kinase 1
- KD
- knockdown
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- MED
- mediator
- NKX3.1
- NK3 homeobox 1
- NONO
- non-POU domain-containing octamer-binding protein
- PARP1
- poly [ADP-ribose] polymerase 1
- PIC
- protease inhibitor cocktail
- PPI
- protein-protein interaction
- PSA
- prostate-specific antigen
- PSC
- peptide spectral count
- qPCR
- quantitative PCR
- SCX
- strong cation exchange
- SDS
- sodium dodecyl sulfate
- SHOC2
- soc-2 suppressor of clear homolog
- siRNA
- small interfering RNA
- SMARCB1
- SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1
- STRING
- Search Tool for the Retrieval of Interacting Genes
- TAB3
- TGF-β-activated kinase 1/MAP3K7-binding protein 3
- TBST
- 50mM Tris (pH 7.5) and 150mM NaCl plus 0.1% vol/vol Tween 20
- TF
- transcription factor
- TMPRSS2
- transmembrane protease serine 2
- TRAF3
- TNF receptor-associated factor 3
- TRIM28
- transcription intermediary factor 1-β.
References
- 1. Massie CE, Mills IG. Mapping protein-DNA interactions using ChIP-sequencing. Methods Mol Biol. 2012;809:157–173. [DOI] [PubMed] [Google Scholar]
- 2. Lin B, Wang J, Hong X, et al. Integrated expression profiling and ChIP-seq analyses of the growth inhibition response program of the androgen receptor. PLoS One. 2009;4(8):e6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takayama K, Tsutsumi S, Katayama S, et al. Integration of cap analysis of gene expression and chromatin immunoprecipitation analysis on array reveals genome-wide androgen receptor signaling in prostate cancer cells. Oncogene. 2011;30(5):619–630. [DOI] [PubMed] [Google Scholar]
- 4. Yu J, Mani RS, Cao Q, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17(5):443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Massie CE, Lynch A, Ramos-Montoya A, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30(13):2719–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharma NL, Massie CE, Ramos-Montoya A, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23(1):35–47. [DOI] [PubMed] [Google Scholar]
- 7. Massie CE, Adryan B, Barbosa-Morais NL, et al. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007;8(9):871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao N, Zhang J, Rao MA, et al. The role of hepatocyte nuclear factor-3 α (forkhead box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol. 2003;17(8):1484–1507. [DOI] [PubMed] [Google Scholar]
- 9. Mirosevich J, Gao N, Gupta A, Shappell SB, Jove R, Matusik RJ. Expression and role of Foxa proteins in prostate cancer. Prostate. 2006;66(10):1013–1028. [DOI] [PubMed] [Google Scholar]
- 10. Wang Q, Li W, Zhang Y, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138(2):245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang D, Garcia-Bassets I, Benner C, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474(7351):390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sahu B, Laakso M, Ovaska K, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30(19):3962–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jin HJ, Zhao JC, Ogden I, Bergan RC, Yu J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. 2013;73(12):3725–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Q, Li W, Liu X, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27(3):380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Böhm M, Locke WJ, Sutherland RL, Kench JG, Henshall SM. A role for GATA-2 in transition to an aggressive phenotype in prostate cancer through modulation of key androgen-regulated genes. Oncogene. 2009;28(43):3847–3856. [DOI] [PubMed] [Google Scholar]
- 16. Andreu-Vieyra C, Lai J, Berman BP, et al. Dynamic nucleosome-depleted regions at androgen receptor enhancers in the absence of ligand in prostate cancer cells. Mol Cell Biol. 2011;31(23):4648–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Norris JD, Chang CY, Wittmann BM, et al. The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol Cell. 2009;36(3):405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim YR, Oh KJ, Park RY, et al. HOXB13 promotes androgen independent growth of LNCaP prostate cancer cells by the activation of E2F signaling. Mol Cancer. 2010;9:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nadiminty N, Lou W, Sun M, et al. Aberrant activation of the androgen receptor by NF-κB2/p52 in prostate cancer cells. Cancer Res. 2010;70(8):3309–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCall P, Bennett L, Ahmad I, et al. NF[κ]B signalling is upregulated in a subset of castrate-resistant prostate cancer patients and correlates with disease progression. Br J Cancer. 2012;107(9):1554–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang L, Altuwaijri S, Deng F, et al. NF-κB regulates androgen receptor expression and prostate cancer growth. Am J Pathol. 2009;175(2):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28(7):778–808. [DOI] [PubMed] [Google Scholar]
- 23. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103(6):843–852. [DOI] [PubMed] [Google Scholar]
- 24. Louie MC, Yang HQ, Ma AH, et al. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor-p160 coactivator complex. Proc Natl Acad Sci USA. 2003;100(5):2226–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mills IG. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat Rev Cancer. 2014;14(3):187–198. [DOI] [PubMed] [Google Scholar]
- 26. Goens G, Rusu D, Bultot L, Goval J-J, Magdalena J. Characterization and quality control of antibodies used in ChIP assays. In: Collas P, ed. Chromatin Immunoprecipitation Assays. vol 567 New York City, NY: Humana Press; 2009:27–43. [DOI] [PubMed] [Google Scholar]
- 27. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012;33(5):887–894. [DOI] [PubMed] [Google Scholar]
- 28. Peri S, Navarro JD, Amanchy R, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13(10):2363–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(D1):D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 2013;41(D1):D816–D823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gioeli D, Paschal BM. Post-translational modification of the androgen receptor. Mol Cell Endocrinol. 2012;352(1–2):70–78. [DOI] [PubMed] [Google Scholar]
- 32. Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog. 2011;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fermin D, Basrur V, Yocum AK, Nesvizhskii AI. Abacus: a computational tool for extracting and pre-processing spectral count data for label-free quantitative proteomic analysis. Proteomics. 2011;11(7):1340–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Farré D, Roset R, Huerta M, et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003;31(13):3651–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Messeguer X, Escudero R, Farré D, Núñez O, Martínez J, Albà MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002;18(2):333–334. [DOI] [PubMed] [Google Scholar]
- 38. Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981;9(13):3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson MA, Hernandez I, Wei Y, Greenberg N. Isolation and characterization of mouse probasin: an androgen-regulated protein specifically expressed in the differentiated prostate. Prostate. 2000;43(4):255–262. [DOI] [PubMed] [Google Scholar]
- 40. Yan Y, Sheppard PC, Kasper S, et al. Large fragment of the probasin promoter targets high levels of transgene expression to the prostate of transgenic mice. Prostate. 1997;32(2):129–139. [DOI] [PubMed] [Google Scholar]
- 41. Spence AM, Sheppard PC, Davie JR, et al. Regulation of a bifunctional mRNA results in synthesis of secreted and nuclear probasin. Proc Natl Acad Sci USA. 1989;86(20):7843–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang J, Gao N, Kasper S, Reid K, Nelson C, Matusik RJ. An androgen-dependent upstream enhancer is essential for high levels of probasin gene expression. Endocrinology. 2004;145(1):134–148. [DOI] [PubMed] [Google Scholar]
- 43. Maffey AH, Ishibashi T, He C, et al. Probasin promoter assembles into a strongly positioned nucleosome that permits androgen receptor binding. Mol Cell Endocrinol. 2007;268(1–2):10–19. [DOI] [PubMed] [Google Scholar]
- 44. Dignam JD, Martin PL, Shastry BS, Roeder RG. Eukaryotic gene transcription with purified components. Methods Enzymol. 1983;101:582–598. [DOI] [PubMed] [Google Scholar]
- 45. Veldscholte J, Berrevoets CA, Brinkmann AO, Grootegoed JA, Mulder E. Anti-androgens and the mutated androgen receptor of LNCaP cells: differential effects on binding affinity, heat-shock protein interaction, and transcription activation. Biochemistry. 1992;31(8):2393–2399. [DOI] [PubMed] [Google Scholar]
- 46. Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–4201. [DOI] [PubMed] [Google Scholar]
- 47. Yi EC, Marelli M, Lee H, et al. Approaching complete peroxisome characterization by gas-phase fractionation. Electrophoresis. 2002;23(18):3205–3216. [DOI] [PubMed] [Google Scholar]
- 48. Spahr CS, Davis MT, McGinley MD, et al. Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry. I. Profiling an unfractionated tryptic digest. Proteomics. 2001;1(1):93–107. [DOI] [PubMed] [Google Scholar]
- 49. Li XJ, Pedrioli PG, Eng J, et al. A tool to visualize and evaluate data obtained by liquid chromatography-electrospray ionization-mass spectrometry. Anal Chem. 2004;76(13):3856–3860. [DOI] [PubMed] [Google Scholar]
- 50. Nesvizhskii AI KA, Kelker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–4658. [DOI] [PubMed] [Google Scholar]
- 51. Jasavala R, Martinez H, Thumar J, et al. Identification of putative androgen receptor interaction protein modules: cytoskeleton and endosomes modulate androgen receptor signaling in prostate cancer cells. Mol Cell Proteomics. 2007;6(2):252–271. [DOI] [PubMed] [Google Scholar]
- 52. Loy CJ, Sim KS, Yong EL. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc Natl Acad Sci USA. 2003;100(8):4562–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ozanne DM, Brady ME, Cook S, Gaughan L, Neal DE, Robson CN. Androgen receptor nuclear translocation is facilitated by the f-actin cross-linking protein filamin. Mol Endocrinol. 2000;14(10):1618–1626. [DOI] [PubMed] [Google Scholar]
- 54. Baek SH, Ohgi KA, Nelson CA, et al. Ligand-specific allosteric regulation of coactivator functions of androgen receptor in prostate cancer cells. Proc Natl Acad Sci USA. 2006;103(9):3100–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19(5):631–642. [DOI] [PubMed] [Google Scholar]
- 56. Heemers HV, Sebo TJ, Debes JD, et al. Androgen deprivation increases p300 expression in prostate cancer cells. Cancer Res. 2007;67(7):3422–3430. [DOI] [PubMed] [Google Scholar]
- 57. Norris JD, Joseph JD, Sherk AB, et al. Differential presentation of protein interaction surfaces on the androgen receptor defines the pharmacological actions of bound ligands. Chem Biol. 2009;16(4):452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iborra FJ, Jackson DA, Cook PR. Coupled transcription and translation within nuclei of mammalian cells. Science. 2001;293(5532):1139–1142. [DOI] [PubMed] [Google Scholar]
- 59. Dahlberg JE, Lund E, Goodwin EB. Nuclear translation: what is the evidence? Rna. 2003;9(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nathanson L, Xia T, Deutscher MP. Nuclear protein synthesis: a re-evaluation. Rna. 2003;9(1):9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. David A, Dolan BP, Hickman HD, et al. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol. 2012;197(1):45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liang T, Liao S. A very rapid effect of androgen on initiation of protein synthesis in prostate. Proc Natl Acad Sci USA. 1975;72(2):706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang J, Gao N, DeGraff DJ, et al. Characterization of cis elements of the probasin promoter necessary for prostate-specific gene expression. Prostate. 2010;70(9):934–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kang Z, Jänne OA, Palvimo JJ. Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol Endocrinol. 2004;18(11):2633–2648. [DOI] [PubMed] [Google Scholar]
- 65. Kinyamu HK, Archer TK. Modifying chromatin to permit steroid hormone receptor-dependent transcription. Biochim Biophys Acta. 2004;1677(1–3):30–45. [DOI] [PubMed] [Google Scholar]
- 66. Taatjes DJ. The human mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem Sci. 2010;35(6):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Trotter KW, Archer TK. Nuclear receptors and chromatin remodeling machinery. Mol Cell Endocrinol. 2007;265–266:162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. DeMarzo AM, Nelson WG, Isaacs WB, Epstein JI. Pathological and molecular aspects of prostate cancer. Lancet. 2003;361(9361):955–964. [DOI] [PubMed] [Google Scholar]
- 69. Ding XF, Anderson CM, Ma H, et al. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): multiple motifs with different binding specificities. Mol Endocrinol. 1998;12(2):302–313. [DOI] [PubMed] [Google Scholar]
- 70. Formstecher E, Aresta S, Collura V, et al. Protein interaction mapping: a Drosophila case study. Genome Res. 2005;15(3):376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wissmann M, Yin N, Müller JM, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9(3):347–353. [DOI] [PubMed] [Google Scholar]
- 72. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. [DOI] [PubMed] [Google Scholar]
- 73. Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14(7):455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39. [DOI] [PubMed] [Google Scholar]
- 75. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. [DOI] [PubMed] [Google Scholar]
- 76. Miranda-Saavedra D, Barton GJ. Classification and functional annotation of eukaryotic protein kinases. Proteins. 2007;68(4):893–914. [DOI] [PubMed] [Google Scholar]
- 77. Salas TR, Kim J, Vakar-Lopez F, et al. Glycogen synthase kinase-3β is involved in the phosphorylation and suppression of androgen receptor activity. J Biol Chem. 2004;279(18):19191–19200. [DOI] [PubMed] [Google Scholar]
- 78. Wang L, Lin HK, Hu YC, Xie S, Yang L, Chang C. Suppression of androgen receptor-mediated transactivation and cell growth by the glycogen synthase kinase 3β in prostate cells. J Biol Chem. 2004;279(31):32444–32452. [DOI] [PubMed] [Google Scholar]
- 79. Liao X, Thrasher JB, Holzbeierlein J, Stanley S, Li B. Glycogen synthase kinase-3β activity is required for androgen-stimulated gene expression in prostate cancer. Endocrinology. 2004;145(6):2941–2949. [DOI] [PubMed] [Google Scholar]
- 80. Whitworth H, Bhadel S, Ivey M, et al. Identification of kinases regulating prostate cancer cell growth using an RNAi phenotypic screen. PLoS One. 2012;7(6):e38950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sitz JH, Tigges M, Baumgärtel K, Khaspekov LG, Lutz B. Dyrk1A potentiates steroid hormone-induced transcription via the chromatin remodeling factor Arip4. Mol Cell Biol. 2004;24(13):5821–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chiu YT, Liu J, Tang K, Wong YC, Khanna KK, Ling MT. Inactivation of ATM/ATR DNA damage checkpoint promotes androgen induced chromosomal instability in prostate epithelial cells. PLoS One. 2012;7(12):e51108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-κ B by TNF receptor 2 and CD40. Science. 1995;269(5229):1424–1427. [DOI] [PubMed] [Google Scholar]
- 84. Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78(4):681–692. [DOI] [PubMed] [Google Scholar]
- 85. Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, Matsumoto K. Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J. 2003;22(23):6277–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lütticken C, Wegenka UM, Yuan J, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 1994;263(5143):89–92. [DOI] [PubMed] [Google Scholar]
- 87. Galperin E, Abdelmoti L, Sorkin A. Shoc2 is targeted to late endosomes and required for Erk1/2 activation in EGF-stimulated cells. PLoS One. 2012;7(5):e36469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jeoung M, Abdelmoti L, Jang ER, Vander Kooi CW, Galperin E. Functional integration of the conserved domains of Shoc2 scaffold. PLoS One. 2013;8(6):e66067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pendergast AM, Quilliam LA, Cripe LD, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75(1):175–185. [PubMed] [Google Scholar]
- 90. Lamont KR, Tindall DJ. Androgen regulation of gene expression. Adv Cancer Res. 2010;107:137–162. [DOI] [PubMed] [Google Scholar]
- 91. Nelson PS, Clegg N, Arnold H, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci USA. 2002;99(18):11890–11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. DePrimo SE, Diehn M, Nelson JB, et al. Transcriptional programs activated by exposure of human prostate cancer cells to androgen. Genome Biol. 2002;3(7):RESEARCH0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wagley Y, Yoo YC, Seo HG, et al. The IL-6/sIL-6R treatment of a malignant melanoma cell line enhances susceptibility to TNF-α-induced apoptosis. Biochem Biophys Res Commun. 2007;354(4):985–991. [DOI] [PubMed] [Google Scholar]
- 94. Malinowska K, Neuwirt H, Cavarretta IT, et al. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr Relat Cancer. 2009;16(1):155–169. [DOI] [PubMed] [Google Scholar]
- 95. Schuurmans AL, Bolt J, Mulder E. Androgens and transforming growth factor β modulate the growth response to epidermal growth factor in human prostatic tumor cells (LNCaP). Mol Cell Endocrinol. 1988;60(1):101–104. [DOI] [PubMed] [Google Scholar]
- 96. Bouraoui Y, Ben Rais N, Culig Z, Oueslati R. Involvement of interleukin-1β mediated nuclear factor κB signalling pathways to down-regulate prostate-specific antigen and cell proliferation in LNCaP prostate cancer cells. Cell Biol Int. 2012;36(5):449–454. [DOI] [PubMed] [Google Scholar]
- 97. Nakajima Y, DelliPizzi A, Mallouh C, Ferreri NR. Effect of tumor necrosis factor-α and interferon-γ on the growth of human prostate cancer cell lines. Urol Res. 1995;23(4):205–210. [DOI] [PubMed] [Google Scholar]
- 98. Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470(7333):214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sen A, Prizant H, Hammes SR. Understanding extranuclear (nongenomic) androgen signaling: what a frog oocyte can tell us about human biology. Steroids. 2011;76(9):822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Paliouras M, Zaman N, Lumbroso R, et al. Dynamic rewiring of the androgen receptor protein interaction network correlates with prostate cancer clinical outcomes. Integr Biol (Camb). 2011;3(10):1020–1032. [DOI] [PubMed] [Google Scholar]
- 103. Wright ME, Eng J, Sherman J, et al. Identification of androgen-coregulated protein networks from the microsomes of human prostate cancer cells. Genome Biol. 2003;5(1):R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Martinez HD, Hsiao JJ, Jasavala RJ, Hinkson IV, Eng JK, Wright ME. Androgen-sensitive microsomal signaling networks coupled to the proliferation and differentiation of human prostate cancer cells. Gene Cancer. 2011;2(10):956–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Young CY, Montgomery BT, Andrews PE, Qui SD, Bilhartz DL, Tindall DJ. Hormonal regulation of prostate-specific antigen messenger RNA in human prostatic adenocarcinoma cell line LNCaP. Cancer Res. 1991;51(14):3748–3752. [PubMed] [Google Scholar]
- 106. Balk SP, Ko YJ, Bubley GJ. Biology of prostate-specific antigen. J Clin Oncol. 2003;21(2):383–391. [DOI] [PubMed] [Google Scholar]
- 107. Jin G, Klika A, Callahan M, et al. Identification of a human NF-κB-activating protein, TAB3. Proc Natl Acad Sci USA. 2004;101(7):2028–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Matsunaga-Udagawa R, Fujita Y, Yoshiki S, et al. The scaffold protein Shoc2/SUR-8 accelerates the interaction of Ras and Raf. J Biol Chem. 2010;285(10):7818–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tomlins SA, Mehra R, Rhodes DR, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39(1):41–51. [DOI] [PubMed] [Google Scholar]
- 110. Wallace TA, Prueitt RL, Yi M, et al. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68(3):927–936. [DOI] [PubMed] [Google Scholar]
- 111. Arredouani MS, Lu B, Bhasin M, et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin Cancer Res. 2009;15(18):5794–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Luo JH, Yu YP, Cieply K, et al. Gene expression analysis of prostate cancers. Mol Carcinog. 2002;33(1):25–35. [DOI] [PubMed] [Google Scholar]
- 113. Varambally S, Yu J, Laxman B, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8(5):393–406. [DOI] [PubMed] [Google Scholar]
- 114. Culig Z, Hobisch A, Cronauer MV, et al. Androgen receptor activation in prostatic tumor cell lines by insulin- like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54(20):5474–5478. [PubMed] [Google Scholar]
- 115. Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5(3):280–285. [DOI] [PubMed] [Google Scholar]
- 116. Ueda T. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem. 2002;277:7076–7085. [DOI] [PubMed] [Google Scholar]
- 117. Cai C, Portnoy DC, Wang H, Jiang X, Chen S, Balk SP. Androgen receptor expression in prostate cancer cells is suppressed by activation of epidermal growth factor receptor and ErbB2. Cancer Res. 2009;69(12):5202–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hsu FN, Chen MC, Chiang MC, et al. Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. J Biol Chem. 2011;286(38):33141–33149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Dahlman KB, Parker JS, Shamu T, et al. Modulators of prostate cancer cell proliferation and viability identified by short-hairpin RNA library screening. PLoS One. 2012;7(4):e34414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Drake JM, Graham NA, Stoyanova T, et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc Natl Acad Sci USA. 2012;109(5):1643–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Drake JM, Graham NA, Lee JK, et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci USA. 2013;110(49):E4762–E4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Guo Z, Dai B, Jiang T, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10(4):309–319. [DOI] [PubMed] [Google Scholar]
- 123. Dai B, Chen H, Guo S, et al. Compensatory upregulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res. 2010;70(13):5587–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Mahajan NP, Liu Y, Majumder S, et al. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci USA. 2007;104(20):8438–8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gioeli D, Ficarro SB, Kwiek JJ, et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J Biol Chem. 2002;277(32):29304–29314. [DOI] [PubMed] [Google Scholar]
- 126. Gordon V, Bhadel S, Wunderlich W, et al. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol Endocrinol. 2010;24(12):2267–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]