

Abstract
Pancreatic β-cells possess a highly active protein synthetic and export machinery in the endoplasmic reticulum (ER) to accommodate the massive production of proinsulin. ER homeostasis is vital for β-cell functions and is maintained by the delicate balance between protein synthesis, folding, export, and degradation. Disruption of ER homeostasis by diabetes-causing factors leads to β-cell death. Among the 4 components to maintain ER homeostasis in β-cells, the role of ER export in insulin biogenesis is the least understood. To address this knowledge gap, the present study investigated the molecular mechanism of proinsulin ER export in MIN6 cells and primary islets. Two inhibitory mutants of the secretion-associated RAS-related protein (Sar)1 small GTPase, known to specifically block coat protein complex II (COPII)-dependent ER export, were overexpressed in β-cells using recombinant adenoviruses. Results from this approach, as well as small interfering RNA-mediated Sar1 knockdown, demonstrated that defective Sar1 function blocked proinsulin ER export and abolished its conversion to mature insulin in MIN6 cells, isolated mouse, and human islets. It is further revealed, using an in vitro vesicle formation assay, that proinsulin was packaged into COPII vesicles in a GTP- and Sar1-dependent manner. Blockage of COPII-dependent ER exit by Sar1 mutants strongly induced ER morphology change, ER stress response, and β-cell apoptosis. These responses were mediated by the PKR (double-stranded RNA-dependent kinase)-like ER kinase (PERK)/eukaryotic translation initiation factor 2α (p-eIF2α) and inositol-requiring protein 1 (IRE1)/x-box binding protein 1 (Xbp1) pathways but not via activating transcription factor 6 (ATF6). Collectively, results from the study demonstrate that COPII-dependent ER export plays a vital role in insulin biogenesis, ER homeostasis, and β-cell survival.
Insulin plays a crucial role in the regulation of blood glucose homeostasis. In pancreatic β-cells, the well-developed endoplasmic reticulum (ER) is responsible for the synthesis, folding, and export of proinsulin. Newly synthesized preproinsulin polypeptide chain enters ER lumen where its signal peptide is cleaved to produce proinsulin. Proinsulin undergoes folding in the ER lumen, facilitated by molecular chaperones and protein disulfide isomerases (1, 2), to form 3 correctly paired disulfide bonds. Properly folded proinsulin is exported from ER to the Golgi apparatus and then packaged into immature secretory (Sec) granules where proinsulin is converted into insulin via prohormone convertase 1/3, prohormone convertase 2 (PC2), and carboxypeptidase E (3, 4). Mature insulin is exocytosed upon glucose stimulation (5). In β-cells, proinsulin biosynthesis dominates the ER activities even under fasting conditions (6). Therefore, ER homeostasis, namely the delicate balance between protein synthesis, folding, export, and degradation, is vital for normal β-cell functions and survival. The disruption of the ER homeostasis induces ER stress. Chronically elevated ER stress contributes to β-cell dysfunction and death in both type 1 and type 2 diabetes (7–9). Compared with our knowledge in protein synthesis and folding in β-cells, the role of ER export in insulin biogenesis and ER homeostasis in β-cells is much less understood.
Coat protein complex II (COPII)-coated vesicles have been shown to mediate cargo proteins to exit ER from yeast to mammalian cells (10–12). The 5 coat proteins, secretion-associatiated RAS-related protein (Sar)1, Sec23, Sec24, Sec13 and Sec31, are the minimal machinery to drive COPII vesicle formation (13). The assembly of the COPII coat on the ER membrane is initiated through the activation and subsequent membrane insertion of the small GTPase Sar1 (13). Upon activation by its guanine nucleotide exchange factor Sec12, Sar1 recruits Sec23-Sec24 heterodimers, which forms the inner COPII coat, and subsequently the Sec13-Sec31 heterotetramers, which forms the outer coat, to promote vesicle fission (14–16). Due to the essential role of Sar1 in COPII coat assembly, its GDP/GTP exchange and GTP hydrolysis are crucial steps in regulating COPII vesicle biogenesis. Sar1 mutants, which block Sar1 activation (Sar1 T39N) or GTP hydrolysis (Sar1 H79G), have been widely used to specifically inhibit COPII-dependent ER exit of cargo molecules (17–19). Although the COPII-coated vesicles is considered a conserved pathway for ER export, evidence does exist for COPII-independent ER exit (20–23). Proinsulin is the major soluble cargo in pancreatic β-cells. However, the molecular mechanism mediating its ER export remains uncharacterized (4, 24). Furthermore, the role of the COPII-dependent export pathway in maintaining normal β-cell ER functions has not yet been examined. To elucidate the molecular mechanism by which proinsulin exits ER, we utilized inhibitory Sar1 mutants as well as Sar1 knockdown together with an in vitro vesicle formation assay and demonstrated that COPII-dependent ER export plays a vital role in insulin biogenesis, maintenance of ER homeostasis, and β-cell survival.
Materials and Methods
Materials
Antibodies against insulin (H-86), Sec22B, C/EBP-homologous protein (CHOP), ATF4, and x-box binding protein 1 (XBP1) were from Santa Cruz Biotechnology, Inc; insulin B (L6B10), eukaryotic translation initiation factor 2 α (p-eIF2α), and cleaved caspase-3 (Asp175) antibodies from Cell Signaling Technology; lectin mannose binding 1 (LMAN1), ribophorin 1, and Sar1 antibodies were generous gifts from Dr Randy Schekman at the University of California, Berkeley; mCherry, protein disulfide-isomerase A3, and β-actin antibodies from GeneTex. Mouse monoclonal anti-insulin antibody was a gift from Dr Ming Liu at University of Michigan. Collagenase P was from Roche. Tunicamycin (Tm), thapsigargin (TG), cycloheximide (CHX), and Histopaque-1077 were purchased from Sigma-Aldrich. ER-Tracker Blue-White DPX was purchased from Life Technologies. Red fluorescent protein (RFP) ELISA kit was from Cell Biolabs. BJ5183-AD-1 electroporation competent cells were from Agilent Technologies. Halt protease inhibitors cocktail were from Thermo Scientific. Other chemicals were purchased from Sigma.
Construction of plasmids encoding mCherry, preproinsulin-mCherry, wild-type, and mutant Sar1A
The full-length mCherry in PEN-DD-N-IES-mCherry (a gift from Dr Stuenkel at the University of Michigan) was amplified by PCR (forward primer, 5′-ggctcgagatggtgagcaagggcgaggagg-3′ and reverse primer, 5′-cctctagattacttgtacagctcgtccatg-3′), digested with XhoI and XbaI, and subcloned into adenoviral shuttle vector, pShuttle-cytomegalovirus (CMV), to generate pShuttle-CMV-mCherry. For generating the preproinsulin-mCherry fusion construct, human preproinsulin was amplified by PCR (forward primer, 5′-ccggtaccaccatggccctgtggatgcgcc-3′ and reverse primer, 5′-ggctcgagcggccgcgtacgcgagttgcagtagttctccagc-3′) with the stop codon replaced. The PCR product was then digested with KpnI and XhoI and inserted into corresponding sites in pShuttle-CMV-mCherry. This construct is designated as pShuttle-CMV-preproinsulin-mCherry where the preproinsulin is inserted upstream of the mCherry coding sequence with a 7-amino acid linker (SRTRPLE) in between. The human full-length Sar1A, Sar1A T39N, and Sar1A H79G in pGEX-2T (kind gifts from Dr Schekman at the University of California, Berkeley) were PCR-amplified using forward primer (5′-cgctcgagccaccatgtctttcatctttgagtgg-3′) and reverse primer (5′-gctctagattagtcaatatactgggagag-3′). The PCR products were digested with XhoI and XbaI and then ligated into another adenoviral shuttle vector, pAdTrack-CMV. The sequences of all the above constructs were confirmed by restriction mapping and direct DNA sequencing.
Generation of recombinant adenoviruses
The recombinant adenoviruses expressing the wild-type and mutant Sar1A, mCherry, and preproinsulin-mCherry were produced using the pAdEASY-1 system as previously described (25, 26). Briefly, the shuttle vectors with genes of interest (the wild-type and mutant Sar1A in pAdTrack-CMV; mCherry and preproinsulin-mCherrry in pShuttle-CMV) were linearized and transformed into Escherichia coli BJ5183-AD-1 cells containing the adenoviral backbone vector pAdEasy-1. Appropriate recombinants were selected and confirmed, then linearized and transfected into HEK293 cells for adenovirus packaging. Adenoviral titers were estimated by counting the number of green fluorescent cells 48 hours after infection using serial dilution of the viral stocks.
Culture and adenoviral infection of MIN6 cells, dispersed mouse islet cells, and isolated human islets
MIN6 mouse insulinoma cells were cultured in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS), 50μM β-mercaptoethanol, and antibotics. Before experiments, MIN6 cells were seeded in 6-well plates for biochemical studies or in 35-mm dish with glass coverslip bottom (MatTek) for imaging analyses. The next day, cells were infected with control or experimental adenoviruses. Infected MIN6 cells were imaged or harvested for Western blotting 48 hours after viral infection. For coinfection experiments, MIN6 cells were first infected with wild-type or mutant Sar1A virus 1 hour before proinsulin-mCherry viral infection. Infected cells were then used for imaging or Western blotting. ICR mouse islets were isolated using collagenase P at 1-mg/mL concentration, purified with Histopaque-1077 and handpicked under a stereomicroscope. Isolated mouse islets were then dispersed into single islet cells by trypsin digestion. Single islet cells were cultured on poly-D-lysine treated glass coverslips in 6-well plates using RPMI 1640 medium with 10% FBS, 50μM β-mercaptoethanol, 10mM HEPES, 2mM L-glutamine, 1mM sodium-pyruvate, and antibiotics. Human islets from healthy donors were isolated in University of Minnesota and distributed by LONZA. After arrival, the human islets were cultured in the same medium as used for the single mouse islet cells. Isolated human islets and dispersed mouse islet cells were infected with Sar1 and proinsulin-mCherry viruses using the same procedure as used in the MIN6 cells.
Glucose stimulation of insulin-mCherry secretion
INS-1 832/13 cells (from Dr Christopher Newgard at Duke University) were cultured in RPMI 1640, 10% FBS, 10mM HEPES, 2mM L-glutamine, 1mM sodium-pyruvate, and 50μM β-mercaptoethanol. INS-1 832/13 cells were infected with proinsulin-mCherry adenovirus overnight. To study glucose stimulated insulin-mCherry secretion, infected INS-1 832/13 cells were first incubated for 2 hours in Hank's Balanced Salt Solution buffer containing 2.5mM glucose and then for 2 hours in new Hank's Balanced Salt Solution media containing either 2.5mM or 20mM glucose. At the end of the incubation, the media were collected and centrifuged. The supernatants were stored at −80°C for subsequent analysis. The cells were lysed to determine the total cellular contents of insulin-mCherry. The amount of insulin-mCherry secreted into the medium was determined by RFP ELISA kit following the manufacturer's instruction.
In vitro vesicle formation assay in MIN6 cells
The procedure for the in vitro vesicle formation assay was adopted from Dr Schekman's lab as described previously (28, 29). Briefly, MIN6 cells were removed from 100-mm plates with trypsin, washed once, and then the cell membranes were permeabilized with digitonin. After confirming the permeabilization by trypan blue staining, these semi-intact cells were used as donor membranes in subsequent budding assays. Vesicle budding reactions were set up to contain GTP, an ATP regeneration system, semi-intact MIN6 cells, and rat liver cytosol which serves as a source of COPII proteins. After 1 hour of incubation at 30°C, the newly formed COPII vesicles were separated from the donor membranes by a medium speed centrifugation at 17 000g for 15 minutes. Vesicles were then collected by ultracentrifugation. The donor membrane and the vesicle pellets were solubilized in sodium dodecyl sulfate sample buffer and analyzed by SDS-PAGE and Western blotting. One microgram of Sar1A H79G protein, purified as previously described (28), was used in the assay to inhibit the COPII vesicle budding.
Quantitative real-time RT-PCR analysis of ER stress markers
MIN 6 cells cultured in 6-well plates were infected with green fluorescent protein (GFP), wild-type, or mutant Sar1A adenoviruses for 48 hours, or treated with Tm (5 μg/mL) for 6 hours. The treated cells in each well were then harvested for further analysis. For real-time RT-PCR, total RNA was isolated using TRIzol reagent (Invitrogen) and then synthesized to cDNA using a random primer. The cDNA (0.5 μg) was used as a template for real-time quantitative PCR analysis on a 7500 Fast Real-Time PCR System (Applied Biosystems) with primers and SYBR Green PCR Master Mix (Invitrogen). The sequences information of the real-time PCR primers in this study was described previously (30). The internal control was mouse β-actin gene. Relative quantities of mRNA levels were normalized against relative quantities of the β-actin.
SDS-PAGE and Western blot analysis
MIN6 cell total lysates were prepared in lysis buffer supplemented with the Halt protease inhibitors cocktail and the protein concentrations were determined using bicinchoninic acid assay protein assay (Thermo Scientific). Protein samples were separated on NuPAGE 4%–12% Bis Tris or Novex 10%–20% Tris-Glycine mini gels from Invitrogen. For Western blotting, the membranes were incubated with the next primary antibodies overnight at 4°C: anti-mCherry (1:2500), antiactin (1:3000), anti-Sar1 (1:1000), anti-insulin (1:1000), anti-immunoglobulin heaving chanin-binding protein (BiP) (1:7000), anti-CHOP (1:1000), anti-ATF4 (1:1000), anti-p-eIF2α (1:1000), anti-XBP1 (1:200), and anticleaved caspase-3 (1:500). Densitometry analyses of protein bands on the appropriately exposed films which provided a linear dynamic range were performed using ImageJ software. The quantification was normalized with the protein level of β-actin.
Fluorescent imaging using confocal and total internal reflection fluorescent (TIRF) microscopy
Live cell imaging was performed with confocal microscopy on a Zeiss LSM-510 Meta system using a 63/1.4 oil immersion objective with appropriate filters for mCherry and FITC. The laser scanning confocal microscope was equipped with a controlled environmental chamber that maintains a 5% CO2/humidified atmosphere at 37°C. For the ER staining, MIN6 cells were labeled with 200nM of ER-Tracker Blue-White DPX (Invitrogen) for 30 minutes at 37°C. After incubation, cells were washed with PBS to remove free dye and the stained cells were imaged. For the TIRF analysis, infected MIN6 cells were imaged in 35-mm dishes with No. 1 coverslip bottom. TIRF imaging was performed on an Olympus IX81 inverted microscope equipped with TIRF objective lens (1.49 NA, ×60, Apo N) and a ×4 relay lens (U-TVCAC; Olympus). Digital images were captured on an electron multiplying charge-coupled device camera (Image-EM, Model C9100–13; Hamamatsu). The red fluorescence intensity profiles for proinsulin-mCherry were determined from a line scan analysis of images using MetaMorph software (Molecular Devices, Inc).
Electron microscopy (EM)
Transmission EM of MIN6 cells was performed as described previously (31). Briefly, 48 hours after adenoviral infection, MIN6 cells were washed in ice-cold PBS and then fixed in 2% glutaraldehyde/2% paraformaldehyde in ice-cold PBS for 24 hours. After fixation, MIN6 cells were washed, embedded in 2% SeaPrep agarose, and then followed by postfixation for 1 hour at 4°C using 1% OsO4 in 0.1M cacodylate buffer. Finally, the samples were dehydrated in a graded series of ethanol, through propylene oxide, and infiltrated and embedded in Spurr's resin. Ultrathin sections were cut with a diamond knife, retrieved onto 200 mesh nickel thin-bar grids, and contrasted with alcoholic uranyl acetate and lead citrate. Grids were viewed with a JEOL 1400 transmission EM (JEOL USA, Inc) operating at 60 or 80 kV, and digital images were acquired with an AMT-XR611 11 megapixel charge-coupled device camera (Advanced Microscopy Techniques).
Sar1 knockdown by small interfering RNA (siRNA)
A mixture of siRNAs against mouse Sar1A and Sar1B (Smartpool ON-TARGETplus Sar1A siRNA, L-042764-00 and Sar1B siRNA, L-057585-01, from GE Dharmacon) or a scrambled siRNA (ID, SIC001; Sigma-Aldrich) was transfected into MIN6 cells, at a final concentration of 75nM, using Lipofectamine RNAiMAX (Invitrogen) in accordance with the manufacturer's instructions. Total cell lysates were collected 72 hours after transfection to analyze various protein expression levels by Western blotting.
Statistical analysis
Experimental data (quantitative real-time RT-PCR analyses and quantitative Western blot analyses) were reported as mean ± SD. The mean values in this study from the experimental groups were compared using a paired 2-tailed Student's t test. P < .05 was considered statistically significant.
Results
The proinsulin-mCherry fusion protein was correctly targeted and processed
To study the molecular mechanism by which proinsulin exits ER, we first generated a red fluorescent fusion protein, human preproinsulin-mCherry, to monitor proinsulin transport from ER to the Sec granules. In this construct, mCherry was fused in frame with human preproinsulin at its C terminus with a 7-amino acid linker in between (Figure 1A, top). A recombinant adenovirus was thereafter generated to express this fusion protein in β-cell lines and islets. Another recombinant adenovirus only expressing mCherry was also created as a control. When expressed in MIN6 cells, this fusion protein displayed punctuate red fluorescence consistent with its primary localization in the insulin Sec granules (ISGs). In contrast, the mCherry alone showed a homogenous distribution in the cytoplasmic region (Figure 1A, bottom).
Figure 1.
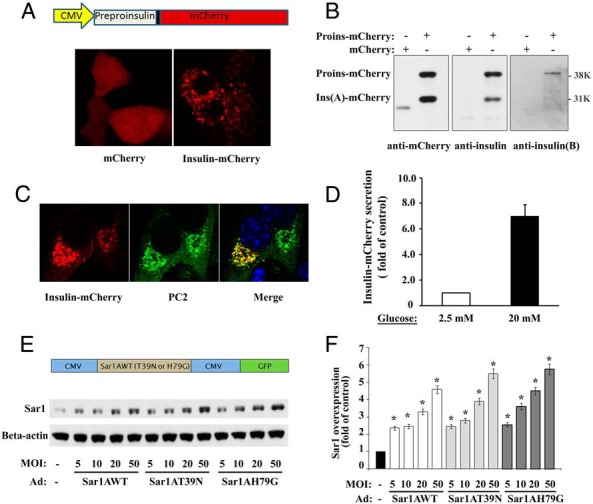
Adenoviral-mediated expression of proinsulin-mCherry and Sar1 proteins in MIN6 cells. A, top, The construction of preproinsulin-mCherry fusion protein is shown. Bottom, The cellular localization of overexpressed mCherry and proinsulin-mCherry was examined 48 hours after adenoviral infection by confocal fluorescent microscopy. B, Western blot results, using anti-mCherry or anti-insulin antibodies, indicated that a significant portion of the proinsulin-mCherry was converted into insulin (A-chain)-mCherry. This was further confirmed by Western blotting using an anti-insulin B-chain antibody, which only detected the upper band corresponding to proinsulin-mCherry. Notice that no truncated fragment was detected in the proinsulin-mCherry-infected MIN6 cells corresponding to mCherry alone. C, MIN6 cells infected with proinsulin-mCherry adenovirus were fixed and immunostained for insulin granule marker, PC2. Insulin-mCherry (left panel, red) colocalizes with PC2 (middle panel, green) and displays punctuated, yellow spots in the merged image (right panel). DAPI staining (blue) was also shown in the merged image. D, Secretion of insulin-mCherry under unstimulated or glucose-stimulated conditions was measured by RFP ELISA and expressed as averaged fold change (mean ± SD, n = 3). E, top, Construction of wild-type and mutant Sar1A in a shuttle vector, which also expresses GFP under a separate promotor. Bottom, Adenoviral titer-dependent expressions of the wild-type and mutants (T39N and H79G) Sar1A proteins in MIN6 cells were examined by Western blotting 48 hours after adenoviral infection with specified MOIs. β-Actin was used as the internal loading control. The WB image shown was a representative of 4 experiments. F, After normalized with β-actin, the overexpressions of Sar1 protein were expressed as fold increased (mean ± SD, n = 4) relative to that of the control uninfected MIN6 cells. Student's t test was used for the statistical analysis; *, P < .05.
To examine whether the proinsulin-mCherry was properly processed by the converting enzymes as the endogenous proinsulin, Western blot analyses were carried out in MIN6 cells using anti-mCherry, anti-insulin, and anti-insulin B-chain antibodies, respectively (Figure 1B). Both mCherry and insulin antibodies detected 2 bands related to the exogenous proinsulin-mCherry: the full-length proinsulin-mCherry at approximately 39 kDa and the A-chain-mCherry at approximately 31 kDa under the reducing condition. In contrast, when an anti-insulin B-chain antibody was used, only the upper full-length proinsulin-mCherry band could be detected (Figure 1B). The correct targeting of insulin-mCherry to the ISGs was further confirmed by its colocalization with ISG marker, PC2 (Figure 1C). Furthermore, overexpressed insulin-mCherry displayed a robust glucose stimulated secretion as determined by mCherry ELISA in the culture media (Figure 1D). The glucose-stimulated secretion of insulin-mCherry was expressed as averaged fold change over unstimulated condition after normalizing with the proinsulin-mCherry contents in the total cell lysates. INS-1 832/13 cells were used in this study to obtain the most robust glucose-stimulated insulin secretion (32). Taken together, these results indicated that the mCherry-tagged proinsulin was correctly targeted to, processed, and stored in the ISGs. The proinsulin-mCherry behaved like the endogenous proinsulin and can be used as a reliable reporter to monitor intracellular transport of proinsulin as well as glucose-stimulated insulin secretion.
The recombinant adenoviruses were generated to mediate the expression of wild-type or mutant Sar1A
In order to investigate ER exit of proinsulin using both morphological and biochemical approaches, recombinant adenoviruses were generated for highly efficient overexpression of mutant Sar1 proteins in MIN6 cells and in isolated islets to inhibit endogenous Sar1 activity and therefore the COPII-dependent ER export. The adenoviral constructs are described in Figure 1E, top. The adenovirus-mediated overexpression of wild-type and mutant Sar1A proteins were readily detectable in MIN6 cells, showing viral titer-dependent increase of Sar1 proteins (Figure 1E, bottom). When compared with the endogenous Sar1 protein level in control MIN6 cells, the total Sar1 protein level was increased to over 200% at multiplicity of infections (MOIs) of 5 and to around 500% at MOIs of 50 (Figure 1F). Furthermore, the levels of wild-type and mutant Sar1A proteins were comparable at the same viral titers (Figure 1, E and F).
Sar1 mutants blocked the ER export of proinsulin-mCherry
Because proinsulin is processed into insulin in the immature granules (3, 4), the level of mature insulin is expected to decrease if the ER export of proinsulin can be blocked by Sar1 mutants. To test this scenario, MIN6 cells were coinfected with proinsulin-mCherry adenovirus and 1 of the Sar1A-expressing adenoviruses, either wild-type Sar1A, Sar1AT39N, or Sar1AH79G. Forty-eight hours after infection, Western blot analyses were carried out to examine the levels of proinsulin-mCherry and insulin-mCherry as an indicator of proinsulin processing. Compared with control MIN6 cells, neither proinsulin-mCherry nor insulin-mCherry level was altered by the wild-type Sar1A at any viral titer (Figure 2A). In contrast, the protein level of insulin-mCherry showed a dramatic decrease in MIN6 cells expressing Sar1AT39N or H79G (Figure 2A). The reduction was in a viral titer-dependent manner ranging from approximately 10% at MOI of 5 to greater than 90% at MOI of 50 (Figure 2B). These results indicated that Sar1A mutants almost completely blocked proinsulin to insulin conversion presumably because the proinsulin-mCherry was unable to enter the Sec granules. It is worth noting that this blockage was confirmed using siRNA-mediated Sar1 knockdown (Supplemental Figure 1B, middle), demonstrating that these findings were not due to any nonspecific effect of the overexpressed Sar1A mutants. This blockage was also demonstrated independently using the TIRF microscopy (Figure 2C). Although docked red fluorescent insulin granules were readily detectable in MIN6 cells expressing proinsulin-mCherry and wild-type Sar1A, no such granule could be detected in cells expressing proinsulin-mCherry and Sar1AT39N (Figure 2C) or H79G (data not shown).
Figure 2.

Sar1 mutants inhibited proinsulin-mCherry processing and granule targeting. A, The expression and processing of proinsulin-mCherry was examined in MIN6 cells coinfected with proinsulin-mCherry and wild-type or mutant (T39N or H79G) Sar1A adenoviruses. β-Actin was used as the internal loading control. B, After normalized with β-actin, the protein levels of mature insulin (A-chain)-mCherry were expressed as the percentage (mean ± SD, n = 4) of that of the control uninfected MIN6 cells. Student's t test was used for the statistical analysis; *, P < .05. C, top left, TIRF microscope image of a representative MIN6 cell coinfected with proinsulin-mCherry and wild-type Sar1 viruses. Top right, TIRF image of a representative MIN6 cell coinfected with proinsulin-mCherry and Sar1 T39N viruses. Red lines in both images indicate the line scans. The intensity profiles of the corresponding red lines are shown below each image. Note that the scales of y-axis of the 2 profiles are different. D, Subcellular localizations of proinsulin-mCherry were examined by confocal microscopy in MIN6 cells coinfected with proinsulin-mCherry and either wild-type, T39N, or H79G Sar1 adenovirus. Red, Proinsulin-mCherry fluorescence. Blue, ER tracker fluorescent dye (0.5μM), added 30 minutes before imaging. Colocalization of proinsulin-mCherry with ER marker was displayed in the merged images in the rightmost column.
To examine whether the deficient proinsulin processing was due to blockage of ER export of proinsulin, confocal microscopy analyses were performed in MIN6 cells coexpressing proinsulin-mCherry with either wild-type or mutant Sar1A. In MIN6 cells expressing wild-type Sar1A, red fluorescence corresponding to insulin-mCherry showed punctuate staining consistent with ISG localization, whereas cells expressing Sar1A mutants displayed a homogenous distribution of red fluorescence, indicating that the proinsulin-mCherry was blocked in the ER due to an inhibition of the COPII-dependent ER exit (Figure 2D). The homogenous distribution of proinsulin-mCherry was further confirmed to be in the ER by colocalization of the red fluorescence with a live cell ER marker, ER-Tracker (Figure 2D).
Sar1 mutants blocked ER export of the endogenous proinsulin
Next, we examined the effect of Sar1 mutants on the ER export of the endogenous proinsulin in MIN6 cells. To maximize the detection of endogenous insulin, a custom insulin antibody, which can recognize both proinsulin and insulin B chain efficiently, was utilized. On the SDS-PAGE gels, mature insulin (A chain and B chain together) could be readily detected by immunoblotting as a band of around 6 kDa from nonreduced MIN6 cell lysates (data not shown), whereas proinsulin of approximately 12 kDa and insulin B chain of approximately 3.5 kDa were detected from reduced MIN6 cell lysate (Figure 3A). Under reducing conditions, the amounts of mature insulin were compared among MIN6 cells infected for 48 hours with GFP, Sar1A, Sar1AT39N, and Sar1AT79G adenovirus, respectively (Figure 3A). It was found that both Sar1A mutants dramatically reduced the level of endogenous mature insulin to approximately 10% of that of control (Figure 3C). This finding is consistent with the inhibitory roles of the Sar1 mutants in ER exit of proinsulin. In addition, it was found that in Sar1A mutants treated MIN6 cells proinsulin levels were also significantly reduced to approximately 60% of that of control (Figure 3B). At earlier time points, we detected a similar decrease of proinsulin level at 36 hours after viral infection (data not shown) and no significant change of proinsulin levels at 24 hours after viral infection (Figure 3D). Because we expected to detect an accumulation of proinsulin in the ER, this decrease of proinsulin level was unexpected. CHX has been used to block new proinsulin synthesis and then monitor the existing proinsulin level decrease as they transit/mature through the Sec pathway (33). In our experiments, when the MIN6 cells were treated briefly with CHX, proinsulin levels were found significantly higher in the Sar1A mutants expressing MIN6 cells (Figure 3D). This effect is consistent with an accumulation of proinsulin in the ER due to a blockage of its ER exit. The same effect was also observed at a longer time after viral infection (data not shown). The effect of defective Sar1 function on endogenous proinsulin and mature insulin was also confirmed by siRNA-mediated Sar1 knockdown (Supplemental Figure 1B, bottom). These results together indicated that blockage of COPII-dependent ER export has a compound effect on proinsulin processing: a primary effect is to block proinsulin ER exit which by itself should cause an accumulation of proinsulin and a reduction of mature insulin; and a secondary effect seems to inhibit proinsulin synthesis likely through the induction of ER stress response, which will be further discussed in later sections. In addition, defective ER exit may also impact ER-associated degradation of proinsulin. This issue requires future investigation.
Figure 3.

Sar1 mutants blocked ER exit of endogenous proinsulin. A, MIN6 cells were infected with either GFP, wild-type Sar1A, Sar1AT39N, or Sar1AH79G adenovirus. After 48 hours, total protein lysates were harvested and analyzed by Western blot analyses with anti-insulin, anti-Sar1A, and antiactin antibodies. After normalized with β-actin, the protein levels of proinsulin (B) as well as insulin (B-chain) (C) were expressed as the percentage (mean ± SD, n = 4) of that of the control GFP virus infected MIN6 cells. *, P < .05. D, MIN6 cells were infected with either GFP, wild-type Sar1A, Sar1AT39N, or Sar1AH79G adenovirus. After 22 hours, infected cells were treated with or without CHX at 15 μg/mL for 2 hours. Then, the total protein lysates were harvested and analyzed by Western blotting with anti-insulin, anti-Sar1, and antiactin antibodies. A representative image of 3 separate experiments is shown.
Proinsulin is packaged into COPII vesicles using an in vitro budding assay
The above results demonstrated that ER export of proinsulin required COPII-dependent vesicle transport. To further elucidate the COPII dependence of proinsulin ER export, we tested whether proinsulin could be packaged into COPII vesicles using an in vitro COPII vesicle budding assay which has been well established in yeast (34) as well as in multiple mammalian cell systems (28, 29, 35–37) but not yet in β-cells. To apply this approach to pancreatic β-cells in this study, permeabilized MIN6 cells were incubated with rat liver cytosol (served as the donor for COPII proteins), ATP-regenerating system and GTP. The budded vesicles were then collected by ultracentrifugation. When the budding samples were analyzed by immunoblotting, the well-characterized COPII cargo proteins, LMAN1 and Sec22B, were efficiently packed into these vesicles (Figure 4, A and B). In contrast, ribophorin 1, a known ER-resident protein, was excluded from the COPII vesicles, indicating a minimal contamination of the ER membrane in the isolated COPII vesicles (Figure 4A). Moreover, consistent with previously published results in other mammalian cell lines, the formation of the COPII vesicles were ATP and GTP dependent and could be inhibited by GTPγS (28, 36). Particularly, the COPII vesicle formation could be inhibited by the purified Sar1A mutant protein, Sar1A H79G (Figure 4, A and B). Most importantly, it was found that proinsulin was able to enter the budded COPII vesicles with the same ATP, GTP, and Sar1 dependence as other well-characterized COPII cargo proteins (Figure 4, A and B).
Figure 4.

Proinsulin was packaged into COPII vesicles in MIN6 cells. A, In vitro COPII budding reactions were performed using rat liver cytosol, ATP-regenerating system, permeabilized MIN6 cells, and GTP (or GTPrS as the negative control). Purified Sar1A H79G mutant protein was also included in one of the budding reactions to demonstrate the vesicle budding is COPII dependent. COPII vesicles were then isolated by ultracentrifugation and analyzed by Western blotting. Different conditions were tested in the reactions to demonstrate the specificity of the COPII in vitro budding assay. The vesicle fractions were compared on the same gel (vesicle fractions, lanes 2–6) together with 5% of the permeabilized MIN6 cells total lysate on lane 1. The presence or absence of resident ER protein, ribophorin 1, COPII markers, LMAN1 and Sec22B, and proinsulin in each reaction was examined by Western blotting. A representative image of 3 separate experiments is shown. B, The averaged quantitative results (mean ± SD, n = 3) were expressed as the percentage of input to indicate the budding efficiency of the corresponding cargo protein.
Blockage of COPII-dependent ER export altered ER morphology and induced ER stress response in MIN6 cells
To evaluate the effect of defective COPII-dependent ER export on ER morphology, MIN6 cell ultrastructure was examined by transmission EM. Compared with the normal flattened cisternae structure of the rough ER in GFP and wild-type Sar1A-infected MIN6 cells (Figure 5, A and B), drastically distended ER was observed in MIN6 cells infected with both Sar1A mutants (Figure 5, C and D). This type of dramatic ER distension is usually an indicative of severe ER stress (38). It is known that accumulation of unfolded or misfolded proteins in the ER lumen causes ER stress and activate unfolded protein response (UPR) in pancreatic β-cells. ER stress can be induced by a variety of conditions, including pharmacological ER stress inducers Tm, TG, as well as proinsulin mutants (6, 39–41). However, whether defective COPII-dependent ER export in β-cells induces ER stress response has not been investigated previously. Next, we examined the effect of adenovirus-mediated expression of Sar1A mutants on the induction of ER stress markers. In these experiments, Tm or TG, 2 well-studied pharmacological ER stress inducers, was included as a positive control.
Figure 5.

Sar1 mutants induced abnormal ER morphology and accumulation of proteins in the ER lumen. Electron micrograph of MIN6 cells infected with either GFP (A), wild-type Sar1A (B), Sar1A T39N (C), or Sar1A H79G (D) adenovirus for 48 hours. Note, compared with the GFP (A) and Sar1 wild-type (B) expressing MIN6 cells, Sar1 mutant T39N (C) and H79G (D) expressing MIN6 cells display grossly distended ER lumen with protein contents accumulated within the lumen. Arrows indicate ER. N, nucleus. All images are presented at ×8000 magnification.
Although wild-type Sar1A did not alter the expression of any of the ER stress markers tested, Sar1A mutant T39N or H79G increased the levels of ER chaperones, protein folding enzymes, and UPR mediators in MIN6 cells. In particular, Sar1A mutants significantly induced the expression of ER chaperone BiP, a prominent ER stress marker, at both the mRNA (Figure 6) and protein (Figure 7, A and B) levels. Moreover, inhibition of the Sar1 activity through dominant negative strategy also led to the activation of the UPR pathways mediated through inositol-requiring protein 1 (IRE1)α/XBP1 and PKR (double-stranded RNA-dependent kinase)-like endoplasmic kinase (PERK)/eIF2α, respectively, in MIN6 cells. Activation of the IRE1α/XBP1 UPR pathway in Sar1-defective MIN6 cells was evidenced by increased levels of IRE1α-targets, including spliced XBP1 mRNA, ER-associated degradation mediators ER degradation-enhancing alpha-mannosidase-like protein 1 and HMG-CoA reductase degradation protein 1, and ER oxidoreductin 1 (Figures 6 and 7, A and B) (42). Activation of the PERK/eIF2α UPR pathway in Sar1-defective MIN6 cells was reflected by increased expression of phosphorylated eIF2α, ATF4, growth arrest and DNA damage-inducible protein, and ER stress-induced proapoptotic factor CHOP (Figures 6 and 7, A and B).
Figure 6.

Sar1 mutants induced ER stress; real-time PCR analyses. MIN6 cells were infected with no virus (control), GFP virus (GFP), wild-type Sar1A (WT), Sar1AT39N (T39N), or Sar1AH79G (H79G) adenovirus for 48 hours. As a positive control of ER stress inducer, MIN6 cells were also treated with Tm at 5 μg/mL for 6 hours. RNA was extracted from treated MIN6 cells, and quantitative real-time PCR was performed as described in Materials and Methods. Results were expressed as fold changes of corresponding mRNA levels relative to that of the control cells (mean ± SE from 4 separate biological experiments). Student's t test was used for the statistical analysis; *, P < .05.
Figure 7.

Sar1 mutants induced ER stress; Western blot analyses. A, MIN6 cells were infected with no virus (control), GFP virus (GFP), wild-type Sar1A (WT), Sar1AT39N (T39N), or Sar1AH79G (H79G) adenovirus for 48 hours. As a positive control of ER stress inducer, MIN6 cells were also treated with Tm at 5 μg/mL for 6 hours. Total protein lysates were extracted from the treated cells and analyzed by Western blotting using corresponding antibodies. B, The averaged quantitative results from 4 experiments were expressed as fold changes of corresponding protein levels relative to that of the control cells (mean ± SD, n = 4). Student's t test was used for the statistical analysis; *, P < .05. C, To test whether Sar1 mutants induced ER stress via activating ATF6, MIN6 cells were infected with GFP, wild-type, or mutant Sar1A adenoviruses and then transfected with HA-ATF6 (3 μg/well) for 48 hours in duplicate groups, one group as controls and the other groups were treated with Tm at 5 μg/mL for 6 hours before harvest. MIN6 cells transfected with only empty vector or HA-ATF6 active form were used as negative and positive controls. Protein samples were analyzed by Western blotting using HA-HRP antibody. A representative WB image from 4 independent experiments is shown. D, Diagram indicates that Sar1 mutants induced β-cell ER stress via the PERK/pEIF2α and IRE1/Xbp1 pathways but not by ATF6. E, MIN6 cells were infected with either GFP, or various Sar1 adenoviruses for 48 hours. As a positive control, MIN6 cells were also treated with TG at 1μM for 16 hours. Total protein lysates from control and treated cells were analyzed by Western blotting with anticleaved caspase-3 and anti-β-actin antibodies.
Although Sar1A mutants and classic ER stress inducers such as Tm shared common mechanisms to induce ER stress response, there was a very important distinction between them: Sar1A mutants induced ER stress response did not activate the activating transcription factor 6 (ATF6) pathway. This was indicated by the absence of the active form of hemagglutinin (HA)-tagged ATF6, ATF6 p50, in MIN6 cells expressing Sar1A mutants (Figure 7C). A lighter exposed WB image is also shown in Supplemental Figure 2. This is because ATF6 activation requires its translocation from the ER to the Golgi, which was blocked by the Sar1A mutants. Furthermore, Sar1A mutants were also able to block ATF6 activation induced by Tm (Figure 7C). A diagram was included to indicate the unique mechanism by which Sar1 mutants induced β-cell ER stress, namely, it is through the PERK/p-eIF2α and IRE1α/XBP1 pathways but not the ATF6 pathway (Figure 7D). Notably, induction of CHOP in Sar1-defective MIN6 cells implicates that impaired COPII activity may cause accumulation of proinsulin in the ER and eventually lead to β-cell death through ER stress-induced apoptosis. This was indeed the case as demonstrated by the detection of apoptosis marker, cleaved caspase-3, only in MIN6 cells expressing Sar1A mutants as well as in MIN6 cells treated with TG (Figure 7E). The degree of caspase-3 activation between TG and Sar1A mutants seemed fairly comparable.
Blockage of COPII vesicle formation inhibited proinsulin ER export and induced ER stress in mouse and human islets
The major findings in β-cell lines were reproduced in dispersed mouse islet cells and isolated human islets. Same as in MIN6 cells (Figure 2D), confocal microscopy analyses were performed in dispersed mouse islet cells coexpressing proinsulin-mCherry with either wild-type or Sar1A H79G (Figure 8A) or T39N (data not shown). In islet cells expressing wild-type Sar1A, red fluorescence corresponding to insulin-mCherry showed punctuate staining consistent with ISG localization, whereas islet cells expressing Sar1A mutants displayed a homogenous distribution of red fluorescence, indicating that the proinsulin-mCherry was blocked in the ER due to defective COPII-dependent ER export (Figure 8A). Furthermore, in isolated human islets infected with Sar1A viruses, it was found that Sar1A mutants inhibited insulin biogenesis, reduced proinsulin level and induced ER stress markers BiP, ATF4, and CHOP (Figure 8B).
Figure 8.
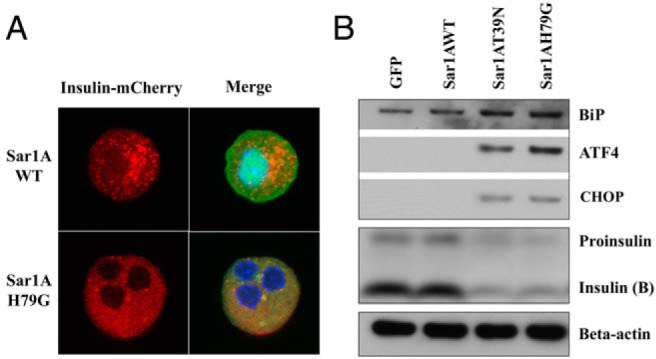
Sar1 mutants blocked ER export of proinsulin and induced ER stress in isolated islets. A, Subcellular localizations of proinsulin-mCherry were examined by confocal microscopy in dispersed mouse islet cells coinfected with proinsulin-mCherry and either wild-type or H79G Sar1A adenovirus. Red, Proinsulin-mCherry fluorescence. Blue, DAPI staining for nuclei. Green, GFP coexpressed with Sar1A under a separate CMV promotor. Representative images from 3 separate experiments are shown. B, Western blot analyses of ER stress markers and proinsulin processing in isolated human islets (50 islets handpicked for each group) treated with specified viruses for 48 hours. Human islets from a healthy donor were isolated at Schulze Diabetes Institute in University of Minnesota and distributed by LONZA.
Discussion
To monitor insulin granule trafficking and proinsulin processing in living β-cells, 2 general strategies have been used to construct fluorescent protein-tagged proinsulin reporters. One strategy is to fuse the fluorescent protein in frame at the C terminus of proinsulin (43–45). The second strategy is to insert the fluorescent proteins into the C peptide of proinsulin, therefore to track the fluorescent protein as a stoichiometric reporter without being part of the mature insulin (46, 47). Multiple labs have previously reported technical challenges of proper and efficient targeting of insulin-fluorescent protein chimers to the ISGs (45, 48, 49). Here, we constructed a recombinant adenovirus expressing proinsulin-mCherry, which has shown normal insulin granule targeting, processing, and regulated secretion. This fusion protein behaved like the endogenous proinsulin and represents an additional tool to monitor insulin biogenesis and secretion not only in β-cell lines but also in isolated islets. Sar1 T39N and H79G mutants have been widely used to demonstrate COPII-dependent ER exit of a variety of cargo proteins (17–19). However, the vast majority of these studies focused on cellular imaging analyses. The Sar1 recombinant adenoviruses generated here have allowed us not only in the imaging studies of individual cells but also in the biochemical analyses of the entire cell population, particularly the studies in isolated islets, which are very difficult to transfect. An earlier study has reported the adenovirus-mediated overexpression of Sar1 mutants (50). However, because the Sar1 mutant proteins seemed highly toxic to viral packing cells, their inducible expression required the coinfection of the Cre adenovirus. We did not encounter such a problem in the current study when packaging and amplifying the recombinant viruses in HEK293 cells. This is likely because a newer adenoviral system was used here which does not require recombination to take place in HEK293 cells. In this study, we were also able to reproduce major findings obtained with Sar1 mutants by using a combination of Sar1A and Sar1B siRNAs. Although the inhibitory Sar1 mutants likely abolish the overall activity of the endogenous Sar1 regardless of its specific isoform, siRNA based Sar1 knockdown approach has the potential to determine the relative contribution of each Sar1 isoform, Sar1A or Sar1B, to insulin biogenesis in future studies.
Proinsulin is the major soluble cargo in pancreatic β-cells. Properly folded proinsulin is exported from the ER. However, the molecular mechanism of this process has not yet been thoroughly investigated before this study. Although COPII vesicle is generally considered the conserved route for cargo proteins to exit the ER, evidence does exist for the presence of COPII-independent ER exit pathways (20–23). Whether ER export of proinsulin is COPII dependent has not been determined experimentally before our study. Here, we provided definitive evidence that the ER exit of proinsulin requires COPII-dependent pathway. First, when a proinsulin-mCherry reporter was used in MIN6 cells and dispersed mouse islet cells, it was found that mutant but not wild-type Sar1 blocked the exogenous proinsulin in the ER and abolished its conversion to mature insulin. Subsequently, the inhibitory effect of the Sar1 mutants on endogenous proinsulin processing was confirmed in both β-cell lines and isolated human islets. Last, by applying an in vitro COPII vesicle formation assay to a pancreatic β-cell line, it was demonstrated, for the first time, that proinsulin was packaged into COPII vesicles in a GTP and Sar1-dependent manner. Two distinct molecular mechanisms have been shown by which soluble cargo proteins to exit ER via COPII-coated vesicles. One mechanism is by receptor-mediated sorting. Such examples include ER-derived vesicle protein-mediated ER export of glycosylated pro-α-factor (51, 52) and LMAN1-mediated ER export of clogging factors V and VIII (53, 54) as well as α1-antitrypsin (55). The other mechanism involves a so-called bulk flow process in which soluble cargo enter COPII vesicles without active sorting. It has been postulated that digestive enzymes in the exocrine pancreatic cells might exit ER in such a manner (56). As for proinsulin, the predominant soluble cargo in pancreatic β-cells, it will be of great interest to elucidate in future studies whether it exits ER via any active sorting mechanism, and if so, via what transmembrane receptor(s). In addition, it is well known that under normal physiological conditions proinsulin synthesis can fluctuate dramatically (>20-fold) in response to blood glucose (9). It will be of great interest to elucidate how COPII machinery is regulated to adapt to such a wide range of demand in ER export.
ER homeostasis is vital for normal β-cell functions and is maintained by the delicate balance between protein synthesis, folding, export, and degradation. Disruption of the ER homeostasis plays a critical role in the pathogenesis of both type 1 and type 2 diabetes (8, 9). Compared with our knowledge in protein synthesis, folding and degradation in the β-cell ER, the exact molecular mechanism of protein export from the ER and how this process is regulated is much less understood. In the current study, we showed for the first time that Sar1 mutants blocked proinsulin in the ER, reduced β-cell insulin content and caused severe ER stress. This represents a new way to disrupt the ER homeostasis, namely via the defective COPII-dependent ER export. Misfolded cargo proteins, such as proinsulin mutants, have been shown to accumulate in the ER, induce ER stress and cause diabetes in mice and human (6, 40, 41, 57, 58). Different from the above situation where the defect in ER export was primarily restricted to individual cargo mutants, in this study, we have demonstrated a scenario where impaired COPII budding led to a global defect in ER export. In this case, wild-type proinsulin was retained in the ER and induced severe ER stress, a situation likely more reminiscent of the ER stress and β-cell dysfunction that occurs in human type 2 diabetes. These findings suggested that impaired COPII-dependent ER export could potentially contribute to β-cell failure in some forms of diabetes. In fact, emerging evidence suggests that reduced ER-Golgi transport was at least one of the primary causes for lipotoxicity-induced ER stress and β-cell failure (27, 59, 60). Because our study has established for the first time the critical role of COPII-dependent pathway in β-cell ER export and insulin biogenesis, now we have a working model for subsequent mechanistic studies on how various diabetes-causing factors, such as gluco- and lipidtoxicity, may reduce ER export through altering COPII coat assembly and vesicle budding on the ER membrane. The COPII machinery and related molecules could become new therapeutic targets to alleviate chronic ER stress and preserve β-cell mass in diabetes.
Acknowledgments
We thank Dr Randy Schekman at the University of California, Berkeley and Dr Kanika Bajaj Pahuja in his lab for training us on the in vitro vesicle budding assay in Hela cells and providing us the Sar1A vectors and antibodies to Sar1, LMAN1, and ribophorin 1.
This work was supported by the start-up fund from Wayne State University and by a Pilot and Feasibility grant from the Michigan Diabetes Research and Training Center (National Institutes of Health Grant 5P60-DK020572) (to X.C.). A.K. is supported by the National Institutes of Health Grant EY022230 and the Department of Veterans Affairs Grant 1BX000469. A.K. is also the recipient of the Department of Veterans Affairs Senior Research Career Scientist Award 13S-RCS-006.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ATF6
- activating transcription factor 6
- BiP
- immunoglobulin heavy chain-binding protein
- CHOP
- C/EBP-homologous protein
- CHX
- cycloheximide
- CMV
- cytomegalovirus
- COPII
- coat protein complex II
- EM
- electron microscopy
- ER
- endoplasmic reticulum
- FBS
- fetal bovine serum
- GFP
- green fluorescent protein
- HA
- hemagglutinin
- IRE1
- inositol-requiring protein 1
- ISG
- insulin secretory granule
- LMAN1
- lectin mannose-binding 1
- MOI
- multiplicity of infection
- PC2
- prohormone convertase 2
- p-eIF2α
- eukaryotic translation initiation factor 2 α
- PERK
- PKR (double-stranded RNA-dependent kinase)-like ER kinase
- RFP
- red fluorescent protein
- Sar
- secretion-associated RAS-related protein
- Sec
- secretory
- siRNA
- small interfering RNA
- TG
- thapsigargin
- TIRF
- total internal reflection fluorescent
- Tm
- tunicamycin
- UPR
- unfolded protein response
- Xbp1
- x-box binding protein 1.
References
- 1. Zito E, Chin KT, Blais J, Harding HP, Ron D. ERO1-β, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J Cell Biol. 2010;188(6):821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajpal G, Schuiki I, Liu M, Volchuk A, Arvan P. Action of protein disulfide isomerase on proinsulin exit from endoplasmic reticulum of pancreatic β-cells. J Biol Chem. 2012;287(1):43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arvan P. Secretory protein trafficking: genetic and biochemical analysis. Cell Biochem Biophys. 2004;40(3 suppl):169–178. [DOI] [PubMed] [Google Scholar]
- 4. Steiner DF. Adventures with insulin in the islets of Langerhans. J Biol Chem. 2011;286(20):17399–17421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leung YM, Kwan EP, Ng B, Kang Y, Gaisano HY. SNAREing voltage-gated K+ and ATP-sensitive K+ channels: tuning β-cell excitability with syntaxin-1A and other exocytotic proteins. Endocr Rev. 2007;28(6):653–663. [DOI] [PubMed] [Google Scholar]
- 6. Liu M, Hodish I, Haataja L, et al. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab. 2010;21(11):652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocr Rev. 2008;29(3):317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Volchuk A, Ron D. The endoplasmic reticulum stress response in the pancreatic β-cell. Diabetes Obes Metab. 2010;12(suppl 2):48–57. [DOI] [PubMed] [Google Scholar]
- 9. Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dancourt J, Barlowe C. Protein sorting receptors in the early secretory pathway. Annu Rev Biochem. 2010;79:777–802. [DOI] [PubMed] [Google Scholar]
- 11. Routledge KE, Gupta V, Balch WE. Emergent properties of proteostasis-COPII coupled systems in human health and disease. Mol Membr Biol. 2010;27(8):385–397. [DOI] [PubMed] [Google Scholar]
- 12. Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. COPII and the regulation of protein sorting in mammals. Nat Cell Biol. 2012;14(1):20–28. [DOI] [PubMed] [Google Scholar]
- 13. Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122(4):605–617. [DOI] [PubMed] [Google Scholar]
- 14. Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419(6904):271–277. [DOI] [PubMed] [Google Scholar]
- 15. Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Dev Cell. 2007;13(5):635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129(7):1325–1336. [DOI] [PubMed] [Google Scholar]
- 17. Aridor M, Fish KN, Bannykh S, et al. The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J Cell Biol. 2001;152(1):213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taneja TK, Mankouri J, Karnik R, et al. Sar1-GTPase-dependent ER exit of KATP channels revealed by a mutation causing congenital hyperinsulinism. Hum Mol Genet. 2009;18(13):2400–2413. [DOI] [PubMed] [Google Scholar]
- 19. Venditti R, Scanu T, Santoro M, et al. Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science. 2012;337(6102):1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009;10(2):148–155. [DOI] [PubMed] [Google Scholar]
- 21. Rabouille C, Malhotra V, Nickel W. Diversity in unconventional protein secretion. J Cell Sci. 2012;125(pt 22):5251–5255. [DOI] [PubMed] [Google Scholar]
- 22. Steringer JP, Bleicken S, Andreas H, et al. Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J Biol Chem. 2012;287(33):27659–27669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cutrona MB, Beznoussenko GV, Fusella A, Martella O, Moral P, Mironov AA. Silencing of mammalian Sar1 isoforms reveals COPII-independent protein sorting and transport. Traffic. 2013;14(6):691–708. [DOI] [PubMed] [Google Scholar]
- 24. Weiss MA. Proinsulin and the genetics of diabetes mellitus. J Biol Chem. 2009;284(29):19159–19163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X, Edwards JA, Logsdon CD, Ernst SA, Williams JA. Dominant negative Rab3D inhibits amylase release from mouse pancreatic acini. J Biol Chem. 2002;277(20):18002–18009. [DOI] [PubMed] [Google Scholar]
- 26. Chen X, Ernst SA, Williams JA. Dominant negative Rab3D mutants reduce GTP-bound endogenous Rab3D in pancreatic acini. J Biol Chem. 2003;278(50):50053–50060. [DOI] [PubMed] [Google Scholar]
- 27. Boslem E, Weir JM, MacIntosh G, et al. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic β-cells. J Biol Chem. 2013;288(37):26569–26582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim J, Hamamoto S, Ravazzola M, Orci L, Schekman R. Uncoupled packaging of amyloid precursor protein and presenilin 1 into coat protein complex II vesicles. J Biol Chem. 2005;280(9):7758–7768. [DOI] [PubMed] [Google Scholar]
- 29. Merte J, Jensen D, Wright K, et al. Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat Cell Biol. 2010;12(1):41–46; suppp 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee JS, Zheng Z, Mendez R, Ha SW, Xie Y, Zhang K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol Lett. 2012;211(1):29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang S, Lee JS, Bishop N, et al. 3D organization and function of the cell: Golgi budding and vesicle biogenesis to docking at the porosome complex. Histochem Cell Biol. 2012;137(6):703–718. [DOI] [PubMed] [Google Scholar]
- 32. Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49(3):424–430. [DOI] [PubMed] [Google Scholar]
- 33. Haataja L, Snapp E, Wright J, et al. Proinsulin intermolecular interactions during secretory trafficking in pancreatic β cells. J Biol Chem. 2013;288(3):1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barlowe C, Orci L, Yeung T, et al. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77(6):895–907. [DOI] [PubMed] [Google Scholar]
- 35. Kim J, Kleizen B, Choy R, Thinakaran G, Sisodia SS, Schekman RW. Biogenesis of γ-secretase early in the secretory pathway. J Cell Biol. 2007;179(5):951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saito K, Chen M, Bard F, et al. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. 2009;136(5):891–902. [DOI] [PubMed] [Google Scholar]
- 37. Zheng C, Liu HH, Yuan S, Zhou J, Zhang B. Molecular basis of LMAN1 in coordinating LMAN1-MCFD2 cargo receptor formation and ER-to-Golgi transport of FV/FVIII. Blood. 2010;116(25):5698–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wikstrom JD, Israeli T, Bachar-Wikstrom E, et al. AMPK regulates ER morphology and function in stressed pancreatic β-cells via phosphorylation of DRP1. Mol Endocrinol. 2013;27(10):1706–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13(10):1211–1233. [DOI] [PubMed] [Google Scholar]
- 40. Støy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA. 2007;104(38):15040–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meur G, Simon A, Harun N, et al. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes. 2010;59(3):653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang K, Wang S, Malhotra J, et al. The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30(7):1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kasai K, Fujita T, Gomi H, Izumi T. Docking is not a prerequisite but a temporal constraint for fusion of secretory granules. Traffic. 2008;9(7):1191–1203. [DOI] [PubMed] [Google Scholar]
- 44. Ohara-Imaizumi M, Nishiwaki C, Kikuta T, Nagai S, Nakamichi Y, Nagamatsu S. TIRF imaging of docking and fusion of single insulin granule motion in primary rat pancreatic β-cells: different behaviour of granule motion between normal and Goto-Kakizaki diabetic rat β-cells. Biochem J. 2004;381(pt 1):13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsuboi T, Ravier MA, Parton LE, Rutter GA. Sustained exposure to high glucose concentrations modifies glucose signaling and the mechanics of secretory vesicle fusion in primary rat pancreatic β-cells. Diabetes. 2006;55(4):1057–1065. [DOI] [PubMed] [Google Scholar]
- 46. Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci USA. 2007;104(40):15841–15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Watkins S, Geng X, Li L, Papworth G, Robbins PD, Drain P. Imaging secretory vesicles by fluorescent protein insertion in propeptide rather than mature secreted peptide. Traffic. 2002;3(7):461–471. [DOI] [PubMed] [Google Scholar]
- 48. Jain RK, Joyce PB, Molinete M, Halban PA, Gorr SU. Oligomerization of green fluorescent protein in the secretory pathway of endocrine cells. Biochem J. 2001;360(pt 3):645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pouli AE, Kennedy HJ, Schofield JG, Rutter GA. Insulin targeting to the regulated secretory pathway after fusion with green fluorescent protein and firefly luciferase. Biochem J. 1998;331(pt 2):669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoshimura S, Yamamoto A, Misumi Y, et al. Dynamics of Golgi matrix proteins after the blockage of ER to Golgi transport. J Biochem. 2004;135(2):201–216. [DOI] [PubMed] [Google Scholar]
- 51. Otte S, Barlowe C. Sorting signals can direct receptor-mediated export of soluble proteins into COPII vesicles. Nat Cell Biol. 2004;6(12):1189–1194. [DOI] [PubMed] [Google Scholar]
- 52. Belden WJ, Barlowe C. Role of Erv29p in collecting soluble secretory proteins into ER-derived transport vesicles. Science. 2001;294(5546):1528–1531. [DOI] [PubMed] [Google Scholar]
- 53. Zhang B, Kaufman RJ, Ginsburg D. LMAN1 and MCFD2 form a cargo receptor complex and interact with coagulation factor VIII in the early secretory pathway. J Biol Chem. 2005;280(27):25881–25886. [DOI] [PubMed] [Google Scholar]
- 54. Zhang B, Cunningham MA, Nichols WC, et al. Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nat Genet. 2003;34(2):220–225. [DOI] [PubMed] [Google Scholar]
- 55. Nyfeler B, Reiterer V, Wendeler MW, et al. Identification of ERGIC-53 as an intracellular transport receptor of α1-antitrypsin. J Cell Biol. 2008;180(4):705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martínez-Menárguez JA, Geuze HJ, Slot JW, Klumperman J. Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell. 1999;98(1):81–90. [DOI] [PubMed] [Google Scholar]
- 57. Park SY, Ye H, Steiner DF, Bell GI. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun. 2010;391(3):1449–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rajan S, Eames SC, Park SY, et al. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am J Physiol Endocrinol Metab. 2010;298(3):E403–E410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pétremand J, Puyal J, Chatton JY, et al. HDLs protect pancreatic β-cells against ER stress by restoring protein folding and trafficking. Diabetes. 2012;61(5):1100–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Preston AM, Gurisik E, Bartley C, Laybutt DR, Biden TJ. Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse β cells by promoting protein overload. Diabetologia. 2009;52(11):2369–2373. [DOI] [PubMed] [Google Scholar]