

Abstract
Parkinson's disease (PD) is characterized by the progressive loss of select neuronal populations, but the prodeath genes mediating the neurodegenerative processes remain to be fully elucidated. Trib3 (tribbles pseudokinase 3) is a stress-induced gene with proapoptotic activity that was previously described as highly activated at the transcriptional level in a 6-hydroxydopamine (6-OHDA) cellular model of PD. Here, we report that Trib3 immunostaining is elevated in dopaminergic neurons of the substantia nigra pars compacta (SNpc) of human PD patients. Trib3 protein is also upregulated in cellular models of PD, including neuronal PC12 cells and rat dopaminergic ventral midbrain neurons treated with 6-OHDA, 1-methyl-4-phenylpyridinium (MPP+), or α-synuclein fibrils (αSYN). In the toxin models, Trib3 induction is substantially mediated by the transcription factors CHOP and ATF4. Trib3 overexpression is sufficient to promote neuronal death; conversely, Trib3 knockdown protects neuronal PC12 cells as well as ventral midbrain dopaminergic neurons from 6-OHDA, MPP+, or αSYN. Mechanism studies revealed that Trib3 physically interacts with Parkin, a prosurvival protein whose loss of function is associated with PD. Elevated Trib3 reduces Parkin expression in cultured cells; and in the SNpc of PD patients, Parkin levels are reduced in a subset of dopaminergic neurons expressing high levels of Trib3. Loss of Parkin at least partially mediates the prodeath actions of Trib3 in that Parkin knockdown in cellular PD models abolishes the protective effect of Trib3 downregulation. Together, these findings identify Trib3 and its regulatory pathways as potential targets to suppress the progression of neuron death and degeneration in PD.
SIGNIFICANCE STATEMENT Parkinson's disease (PD) is the most common neurodegenerative movement disorder. Current treatments ameliorate symptoms, but not the underlying neuronal death. Understanding the core neurodegenerative processes in PD is a prerequisite for identifying new therapeutic targets and, ultimately, curing this disease. Here, we describe a novel pathway involving the proapoptotic protein Trib3 in neuronal death associated with PD. These findings are supported by data from multiple cellular models of PD and by immunostaining of postmortem PD brains. Upstream, Trib3 is induced by the transcription factors ATF4 and CHOP; and downstream, Trib3 interferes with the PD-associated prosurvival protein Parkin to mediate death. These findings establish this new pathway as a potential and promising therapeutic target for treatment of PD.
Keywords: ATF4, cell death, CHOP, Parkin, Parkinson's disease, Trib3
Introduction
Parkinson's disease (PD), the most common neurodegenerative movement disorder, is characterized by the progressive loss of several neuronal populations, including dopaminergic neurons of the substantia nigra pars compacta (SNpc) (Fahn and Sulzer, 2004; Davie, 2008). The current treatments available for PD temporarily ameliorate some of the clinical symptoms but do not stop or slow down the underlying degenerative processes (Olanow et al., 2009; Schapira, 2009). Although studies of familial forms of PD have provided important insights on initiating causes of the disease, much remains to be learned about the downstream pathways and effectors of neurodegeneration in this disorder (Levy et al., 2009; Schapira et al., 2014). Cellular models of PD represent potentially powerful systems to study such downstream pathways and effectors, and these have revealed that neuronal death requires the transcriptional induction of specific prodeath genes. A previous study identified Trib3 (tribbles pseudokinase 3) transcripts as among the most highly induced in neuronal PC12 cells treated with the dopaminergic toxin 6-hydroxydopamine (6-OHDA) (Ryu et al., 2005). Converging evidence indicates that Trib3 is a pseudokinase with scaffold-like regulatory functions for a number of signaling pathways (Hegedus et al., 2006, 2007). Trib3 is induced by a wide variety of stresses with potential relevance to PD pathophysiology, including metabolic stress (Du et al., 2003; Bi et al., 2008; Carraro et al., 2010; Liu et al., 2010), endoplasmic reticulum stress (Corcoran et al., 2005; Ohoka et al., 2005; Ord and Ord, 2005; Salazar et al., 2009; Zou et al., 2009), oxidative stress (Lange et al., 2008), mitochondrial stress (Ishikawa et al., 2009), and neurotrophic factor deprivation (Mayumi-Matsuda et al., 1999; Kristiansen et al., 2011; Zareen et al., 2013). Both proapoptotic (Ohoka et al., 2005; Shang et al., 2009) and antiapoptotic (Ord et al., 2007; Zhou et al., 2013) actions have been attributed to Trib3. Relatively little is known about the actions and role of Trib3 in neurons. Trib3 is upregulated in sympathetic neurons and neuronal PC12 cells following NGF deprivation (Mayumi-Matsuda et al., 1999; Kristiansen et al., 2011; Zareen et al., 2013) and in cortical neurons by oxidative stress (Lange et al., 2008). Overexpression of Trib3 is sufficient to promote death of sympathetic neurons and neuronal PC12 cells, whereas knockdown of Trib3 protects these neuronal cells from apoptotic death induced by NGF withdrawal (Zareen et al., 2013).
In the present study, because of Trib3's high induction in a PD model and known proapoptotic activity in neuronal cells, we examined its expression in cellular models of PD and in postmortem substantia nigrae from human PD patients. We also used a variety of cellular models of PD to define the role of Trib3 in neuronal death as well as to identify its upstream transcriptional regulators and to describe one of its relevant downstream targets.
Materials and Methods
Cell culture.
PC12 cells were cultured as described previously (Greene and Tischler, 1976). Cells were cultured on plastic cell culture dishes coated with rat tail collagen (Roche). Nondifferentiated PC12 cells were grown in RPMI 1640 cell culture medium supplemented with 10% heat inactivated horse serum (Sigma), 5% FBS, and penicillin/streptomycin. For neuronal differentiation, cells were grown in RPMI 1640 cell culture medium supplemented with 1% horse serum, penicillin/streptomycin, and a 100 ng/ml final concentration of human recombinant NGF (kind gift of Genentech). Cell culture medium was changed every other day. HEK293T/17 cells were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin.
Ventral midbrain dopaminergic neurons from P0-P3 rats and mice were dissected, dissociated, and plated on a confluent glial monolayer following the protocol kindly provided by Dr. David Sulzer (Columbia University) and as described previously (Rayport et al., 1992).
PD toxins and α-synuclein-preformed fibrils.
For PC12 cells, 10 mm stock solutions of 6-OHDA or 1-methyl-4-phenylpyridinium (MPP+) (Sigma) diluted in water were freshly prepared just before each experiment. 6-OHDA was used at final concentrations ranging from 100 to 150 μm, and MPP+ was used at a final concentration of 1 mm, for the indicated times. For ventral midbrain dopaminergic neurons, the 10 mm stock solution of 6-OHDA was prepared in MEM supplemented with ascorbic acid (Sigma) to prevent 6-OHDA oxidation and degradation (Ding et al., 2004). 6-OHDA was used at a final concentration of 40 μm in 0.015% ascorbic acid. MPP+ was diluted in water and used at a final concentration of 40 μm.
α-Synuclein-preformed fibrils were prepared from recombinant human wild-type α-synuclein as described previously (Volpicelli-Daley et al., 2011). Briefly, α-synuclein-preformed fibrils were generated by shaking purified α-synuclein (5 mg/ml in PBS) at 1000 rpm and 37°C for 7 d. α-Synuclein-preformed fibrils were diluted in PBS at 0.1 mg/ml and sonicated with 65 pulses over 40 s. α-Synuclein-preformed fibrils were added to ventral midbrain neuron cultures at a final concentration of 5 μg/ml for 10–14 d.
Plasmids and lentiviral preparations.
The DDK-tagged rat parkin cDNA cloned in a pCMV6-entry vector was obtained from Origene (#RR212553). The plasmid used for Trib3 and ATF4 overexpression was pWPI (AddGene; https://www.addgene.org/12254/), a bicistronic lentiviral vector allowing the simultaneous expression of the transgene and EGFP under the control of the EF1-α promoter. Trib3 and ATF4 cDNAs were generated and cloned into the pWPI vector, as described previously (Sun et al., 2013; Zareen et al., 2013). The following sequences were used for shRNA-mediated downregulation of Trib3: shTRIB3#1 5′-CGAGTGAGAGATGAGCCTG-3′ and shTRIB3#2 5′-CCTGGAGGATGCCTGTGTG-3′. The corresponding scrambled shRNAs with no specific rat targets were used as control shRNAs for Trib3 (and Parkin) shRNAs: shTRIB3#1 SCRAMBLED 5′-GCGACATGAGACGAGTGGT-3′; shTRIB3#2 SCRAMBLED 5′-GCGGTCCGGTGCAGATTGT-3′. An shRNA targeted against DSRED 5′-GCAGCGTCGTTCGATACTA-3′ was used as a control in two sets of experiments. The following sequences were used for shRNA-mediated downregulation of Parkin: sh-Parkin 5′-GGACACATCAGTAGCTTTG-3′ and ATF4: shATF4, 5′-GCCTGACTCTGCTGCTTATAT-3′. For shRNA-mediated ATF4 downregulation experiments, a mutated version of shATF4 (mutated bases are italicized; shATF4mutant 5′-GCCAGATTCAGCGGCCTACAT-3′) was used as a control shRNA. The transfer plasmid used for Trib3, ATF4, and Parkin shRNA expression was pLVTHM (AddGene; https://www.addgene.org/12247/) expressing shRNA from the H1 promoter along with GFP from the EF1-α promoter. The pLL3.7 plasmid was used for Trib3 shRNA and shDSRED expression in two sets of experiments (AddGene; https://www.addgene.org/11795/). The pGIPZ lentivector expressing CHOP (DDIT3) shRNA (shCHOP#1 5′-CGATTTCCTGCTTGAGCCG-3′) and TurboGFP under the control of the hCMV promoter was obtained from Open Biosystems (#RHS4430-200227707). The empty pGIPZ vector was use as a control in shRNA-mediated CHOP downregulation experiments.
Lentiviruses were prepared in HEK293T/17 cells by cotransfecting pWPI, pLVTHM, or pGIPZ expression plasmids along with second-generation lentiviral packaging plasmids (obtained from AddGene) using the calcium phosphate transfection method. pLL3.7 expression plasmids were cotransfected with Rsv/Rev, pMDLg/pRRE, and CMV-VSVG. Lentiviral particles were collected twice (48 and 72 h after transfection) and concentrated using Lenti-X concentrator (Clontech, #631231) following the manufacturer's protocol, resuspended in PBS, and stored at −80°C.
Transfection and lentiviral infection.
For the transfection procedure, after 3 d in vitro, PC12 cells were washed and placed in RPMI 1640 without serum or antibiotics. Cells were transfected with a total of 0.6 μg of DNA per well of a 48-well plate with Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. After 3–5 h, the medium was replaced with fresh RPMI 1640 medium containing 1% horse serum, penicillin/streptomycin, and 100 ng/ml NGF. The transfected PC12 cells were analyzed by immunofluorescence after 5–7 d.
For lentiviral infection, 0.1 up to 5 × 107 viral particles were added per cm2 of culture area, directly in the medium of PC12 cells or ventral midbrain dopaminergic neurons. The transduced neurons were analyzed by qPCR, Western blot, or immunofluorescence after 5–15 d.
qPCR.
PC12 cells were lysed and total RNA was extracted using TRI reagent (Molecular Research Center) following the manufacturer's protocol. RNA concentration and purity were assessed by measuring the optical density at 260 and 280 nm with a NanoDrop (Thermo Scientific). cDNA was synthesized using the first-strand cDNA synthesis kit (Origene) with 1 μg of total RNA, following the manufacturer's instructions. Quantitative real-time PCR was performed using FastStart SYBR Green Master Mix (Roche) and an Eppendorf Realplex Mastercyler with the following settings: 1 cycle at 95°C for 10 min and 40 cycles of amplification, 95°C for 15 s, 58–60°C for 30–60 s, 72°C for 30–60 s. The amounts of Trib3, CHOP, ATF4, and Parkin mRNAs were quantified and normalized to α-tubulin mRNA or 18S rRNA using the following primer pairs: Trib3 forward 5′-GTTGCGTCGATTTGTCTTCA-3′ and reverse 5′-CGGGAGCTGAGTATCTCTGG-3′; ATF4 forward 5′- CCTTCGACCAGTCGGGTTTG-3′ and reverse 5′-CTGTCCCGGAAAAGGCATCC-3′; CHOP forward 5′-CTGGAAGCCTGGTATGAGGA-3′ and reverse 5′-AGGTGCTTGTGACCTCTGCT-3′; Parkin forward 5′-CGGATGAGTGGAGAGTGC-3′ and reverse 5′-TGGCGGTGGTTACATTGG-3′; α-tubulin forward 5′-TACACCATTGGCAAGGAGAT-3′ and reverse 5′-GGCTGGGTAAATGGAGAACT-3′; 18S forward 5′- TTGATTAAGTCCCTGCCCTTTGT-3′ and reverse 5′- CGATCCGAGGGCCTCACTA-3′. The threshold cycles were determined for each gene of interest and normalized to the threshold cycles of a housekeeping gene. Relative mRNAs levels for our genes of interest were expressed as a fold induction in an experimental condition compared with a control condition.
Immunoprecipitation.
Neuronal PC12 cells were collected and homogenized with IP lysis buffer (Thermo Scientific, #87787) supplemented with protease and phosphatase inhibitor (Thermo Scientific, #1861281). Cell extracts were incubated overnight at 4°C on a rotator with 10 μg/ml of mouse anti-parkin antibody (Cell Signaling Technology, #4211) or mouse IgG (Cell Signaling Technology, #5415) as a negative control. Immunocomplexes were incubated with magnetic beads covalently coated with protein G (Invitrogen, #10007D) on a rotator for 2 h at 4°C. The beads were then concentrated with a magnet and washed 3 times with 1× cell lysis buffer (Cell Signaling Technology, #9803) supplemented with complete mini protease inhibitor mixture (Roche, #11836170001). The immunocomplexes were resuspended in cell lysis buffer, LDS-sample buffer (Invitrogen), 50 mm dithiothreitol, and analyzed by Western blotting.
Western immunoblotting.
Cells and brain tissue were homogenized in 1× cell lysis buffer (Cell Signaling Technology, #9803) supplemented with complete mini protease inhibitor mixture (Roche, #11836170001). Samples were sonicated, and protein concentrations were determined by BCA assay according to the manufacturer's protocol (Thermo Scientific, #23225). Protein samples were prepared for loading with LDS-sample buffer (Invitrogen) supplemented with 50 mm dithiothreitol. A total of 20 μg of proteins was loaded per well of 10% Bis-Tris polyacrylamide gels (Invitrogen) and separated by electrophoresis for 1 h at 110 V. Proteins were then transferred onto a PVDF membrane (Bio-Rad) for 1 h 30 min at 40 V. Membranes were blocked with 5% powdered milk in TBS containing 0.1% of Tween 20 (TBST) and incubated overnight at 4°C with primary antibodies. The following primary antibodies were used for Western blotting: rabbit anti-Trib3 (Calbiochem, #ST1032), mouse anti-Parkin (Santa Cruz Biotechnology, #sc-32282), and rabbit anti-Erk1/2 (Santa Cruz Biotechnology, #sc-93). Membranes were washed 3 times with TBST and incubated with HRP-conjugated secondary antibodies. After 3 final washes with TBST, blots were incubated with ECL reagents (GE Healthcare), and chemiluminescent signals were detected by exposure to autoradiography film. Films were scanned using a desktop scanner, and band intensities were determined using ImageJ.
Immunofluorescence.
Cells were fixed for 12–15 min in 4% PFA and washed 3 times with PBS. Cells were blocked with Superblock (Thermo Scientific) supplemented with 0.3% Triton-X for 1 h at room temperature and incubated overnight at 4°C with primary antibodies. The following primary antibodies were used for immunofluorescence: goat anti-Trib3 (Santa Cruz Biotechnology, #sc-34214), mouse anti-tyrosine hydroxylase (Millipore, #MAB318), rabbit anti-tyrosine hydroxylase (Millipore, #AB152), mouse anti-Parkin (Cell Signaling Technology, #4211), rabbit anti-GFP (Invitrogen, #A11122), chicken anti-GFP (Invitrogen, #A10262), and rabbit anti-phosphoS129-α-synuclein (Sigma, #SAB4300139). Cells were washed 3 times with PBS and incubated with fluorescent secondary antibodies for 2 h at room temperature: AlexaFluor-568 anti-mouse, anti-rabbit, or anti-goat, AlexaFluor-488 anti-chicken, anti-mouse, or anti-rabbit, and AlexaFluor-350 anti-mouse or anti-rabbit (Invitrogen). For PC12 cells grown in multiwell dishes, Hoechst 33328 was added to the secondary antibody solution, cells were washed in PBS and observed with an inverted fluorescence microscope. For ventral midbrain dopaminergic neurons grown on glass coverslips, after 3 final washes with PBS, coverslips were mounted on slides with Vectashield mounting medium containing DAPI for nuclear staining (Vector Laboratories). Images were acquired using a Zeiss epifluorescence microscope equipped with a digital camera and Axiovision software.
Immunohistochemistry.
Two independent sets of paraffin-embedded postmortem midbrain samples from PD patients and age-matched controls were obtained from the brain bank at Columbia University. The first set was used for Trib3 staining only (controls: n = 8, 4 males, 4 females; PD patients: n = 7, 6 males, 1 female). The second set was used for Trib3 and Parkin double immunostaining (controls: n = 5, 2 males, 3 females; PD patients, n = 6, 5 males, 1 female). The 5 μm sections were deparaffinized in xylene and rehydrated in an ethanol series. For antigen retrieval, tissue sections were placed in citrate buffer (10 mm, pH 6.0) for 45 min in a rice cooker at 100°C. Sections were stained for Trib3 and Parkin using Elite Vectastain ABC kits (rabbit IgG, mouse IgG, respectively) from Vector Laboratories according to the manufacturer's instructions. Sections were stained using rabbit anti-Trib3 (human) polyclonal antibody (Abcam, # 84174; final concentration of 1.0 μg/ml) and mouse anti-Parkin (Santa Cruz Biotechnology, #sc-32282; final concentration 0.4 μg/ml) overnight at 4°C. To test the specificity of the Trib3 antibody, some sections were incubated with the antibody that was mixed with Trib3-immunizing peptide (Abcam, #93788, lot #941648; final concentration of 1.0 μg/ml). To test the specificity of the Parkin antibody, gallbladder sections were incubated with or without primary antibody. Sections were then incubated with biotinylated anti-rabbit and anti-mouse secondary antibodies. ImmPACT SG Peroxidase HRP (blue/gray, for Trib3 staining) and ImmPACT VIP Peroxidase HRP (violet/purple, for Parkin staining) from Vector Laboratories were used as a substrates and left on the slides for 15 min, after which slides were rinsed for 10 min under running tap water. Sections with single Trib3 staining were also counterstained with Nuclear Fast Red (Vector Laboratories) for 5–10 min. Finally, sections were dehydrated and mounted with coverslips with VectaMount Permanent Mounting Medium (Vector Laboratories) and examined under light microscopy.
Survival assays.
For PC12 cells infected with lentiviral particles (typically achieving an 80%–90% transduction rate in PC12 cells) and/or treated with PD toxins, cell survival was assessed on the total cell population by incubating the cell cultures with a detergent solution that lyses the plasma membrane and leaves the nuclei intact (10× counting lysis buffer: 5 g of cetyldimethyl-ethanolammonium bromide, 0.165 g of NaCl, 2.8 ml of glacial acetic acid, 50 ml of 10% Triton-X, 2 ml of 1 M MgCl2, 10 ml of 10 × PBS, 35.2 ml of H2O); 250 μl of 1 × counting lysis buffer was added per cm2 of culture dish area, and the suspended nuclei were counted into a hemacytometer.
For transfected PC12 cells and transduced ventral midbrain dopaminergic neurons, cell survival was assessed by performing immunofluorescence and counting GFP+ (PC12 cells) or TH+ and GFP+ cells (ventral midbrain dopaminergic neurons).
Statistical analysis.
All statistical analyses were performed with GraphPad Prism software. Simple comparisons of two experimental groups were performed using t tests. Multiple comparisons of more than two experimental groups were performed using one-way ANOVA and Student-Newman-Keuls or Dunnett's post hoc tests. The threshold of significance was set at α = 0.05 for all experiments.
Results
Trib3 is induced in cellular models of PD
To study the role of Trib3 in neuron death in the context of PD, we first conducted experiments with two different cellular models: neuronally differentiated (i.e., NGF-treated) PC12 cells (Greene and Tischler, 1976), and rat and mouse postnatally derived ventral midbrain dopaminergic neurons (Rayport et al., 1992). Both culture types have been used extensively for cellular models of PD (Walkinshaw and Waters, 1994; Mosharov et al., 2009; Malagelada et al., 2010; Sun et al., 2013). A previous study with PC12 cells (Ryu et al., 2005) revealed that Trib3 transcripts were among the most elevated following 8 h of treatment with 6-OHDA, a toxin that mimics PD in vitro and in vivo. We confirmed and extended these results by measuring cell death as well as Trib3 mRNA and protein levels in neuronal PC12 cells treated with 6-OHDA or MPP+, an additional toxin used to model PD (Fig. 1). These toxins induced 30%–50% cell death starting at ∼16 h (Fig. 1A; ANOVA with Student-Newman-Keuls tests, p < 0.005, p < 0.0005). Under these conditions, Trib3 mRNA was induced by ∼4.5 fold with 6-OHDA after 8 h of treatment, well before cell death became evident, and Trib3 mRNA levels were maintained at 16 and 24 h (Fig. 1B; ANOVA with Student-Newman-Keuls tests, p < 0.0005). In MPP+-treated cells, Trib3 mRNA started rising at 8 h and was significantly upregulated by ∼3-fold and ∼4-fold at 16 and 24 h, respectively (Fig. 1B; ANOVA with Student-Newman-Keuls tests, p < 0.005, p < 0.0005). The increase of Trib3 mRNA was paralleled by an elevation of Trib3 protein: MPP+ induced a significant 1.5 fold-increase at 8 h, and 6-OHDA induced a significant 1.7-fold increase at 8 h that was maintained at both 16 and 24 h (Fig. 1C; ANOVA with Student-Newman-Keuls tests, p < 0.05).
Figure 1.

Trib3 mRNA and protein are induced before cell death in cellular models of PD. A, Time course of cell death elicited by 6-OHDA and MPP+. Survival assay shows the remaining viable nuclei in neuronal PC12 cell cultures untreated (control) or treated with 100–150 μm 6-OHDA (left) or 1000 μm MPP+ (right) for 2, 8, 16, or 24 h. Cell death becomes evident by 16 h. **p < 0.005 (ANOVA with Student-Newman-Keuls tests). ***p < 0.0005 (ANOVA with Student-Newman-Keuls tests). Values are mean ± SEM from five independent experiments performed in triplicate. B, Trib3 mRNA is upregulated in cellular PD models before cell death. qPCR analysis of Trib3 mRNA levels in neuronal PC12 cells either untreated (control) or treated with 100–150 μm 6-OHDA (left) or 1000 μm MPP+ (right) for 2, 8, 16, and 24 h. 6-OHDA induces an increase in Trib3 mRNA starting at 8 h that is maintained at 16 and 24 h. ***p < 0.0005 (ANOVA with Student-Newman-Keuls tests). Trib3 mRNA level starts rising at 8 h and keeps increasing at 16 and 24 h with MPP+. **p < 0.005 (ANOVA with Student-Newman-Keuls tests). ***p < 0.0005 (ANOVA with Student-Newman-Keuls tests). Trib3 mRNA levels are normalized against those of α-tubulin and are mean ± SEM from at least three independent experiments performed in triplicate. C, Trib3 protein levels are elevated before cell death in cellular models of PD. Western blot quantification of Trib3 protein levels in neuronal PC12 cells untreated (control) or treated with 100–150 μm 6-OHDA or 1000 μm MPP+ for the indicated times. *p < 0.05 (ANOVA with Student-Newman-Keuls tests). Top portion of the panel shows representative Western blots. Trib3 corresponds to the top band; the bottom band noted with an asterisk is nonspecific. Densitometric quantification of Trib3 protein levels is shown at the bottom of the panel. Trib3 values are normalized against ERK1 and are mean ± SEM from at least six independent experiments.
To extend the findings obtained with PC12 cells, we also examined the induction and cellular localization of Trib3 protein in cultured rat ventral midbrain dopaminergic neurons treated with either 6-OHDA or MPP+. After exposure for 8 and 24 h, the cultures were immunostained for the dopaminergic neuron marker TH and for Trib3 to assess cell death (Fig. 2) and Trib3 protein induction (Fig. 3). Consistent with previous literature (Gearan et al., 2001; Ding et al., 2004; Ganser et al., 2010), there was extensive death with only 55% of dopaminergic neurons (TH+) remaining after 24 h of 6-OHDA treatment, and 44% after 24 h of MPP+ treatment (Fig. 2A,B; t tests, p < 0.05). Although basal levels of Trib3, as detected by immunostaining, were extremely low in control cultures, treatment with 6-OHDA or MPP+ triggered an induction of Trib3 protein in dopaminergic neurons detectable mostly in the cytoplasm and proximal processes (Fig. 3A,B). The toxins also induced Trib3 immunostaining in nondopaminergic neurons and other cell types present in the mixed cultures. Densitometric quantification in a random set of TH+ neurons in each culture condition revealed a 1.9- or 1.5-fold increase in Trib3 protein immunostaining levels after 8 h of 6-OHDA or MPP+ treatment, respectively (Fig. 3A,B; t tests, p < 0.005, p < 0.005) (i.e., at a time when cell death was not yet significant). Although elevated Trib3 protein was no longer detectable after 24 h of MPP+ treatment, Trib3 immunostaining was still strongly elevated at 24 h in 6-OHDA-treated dopaminergic neurons (1.9-fold, t test, p < 0.005). These observations of an early transient induction of Trib3 protein immunostaining with MPP+ and a sustained elevation with 6-OHDA in dopaminergic neurons are very similar to the results obtained by Western immunoblotting of PC12 cells (Fig. 1C).
Figure 2.

6-OHDA, MPP+, and preformed α-synuclein fibrils induce death of cultured postnatally derived ventral midbrain dopaminergic neurons. A–C, Representative overview images of dopaminergic cells (positive for tyrosine hydroxylase (TH, white, left panels) and quantifications (right panels) show a decrease in the numbers of surviving dopaminergic (TH+) neurons after treatment with (A) 6-OHDA, (B) MPP+, or (C) preformed α-synuclein fibrils. *p < 0.05 (t tests). **p < 0.005 (t tests). A, B, Cultures were either untreated (control) or treated as indicated with 40 μm 6-OHDA in 0.015% ascorbic acid (AA + 6-OHDA) or 0.015% AA for 8 or 24 h, or with 40 μm MPP+ for 8 or 24 h and then immunostained for TH. C, Cultures were treated with PBS or 5 μg/ml of human WT α-synuclein-preformed fibrils (α-synuclein pffs) for 10–14 d and then immunostained for TH. Survival assay values are expressed as the mean ± SEM of the percentage of remaining TH+ neurons in cultures treated with 6-OHDA, MPP+, or α-synuclein, compared with the corresponding control conditions. Values were analyzed from at least three independent experiments performed in triplicate.
Figure 3.
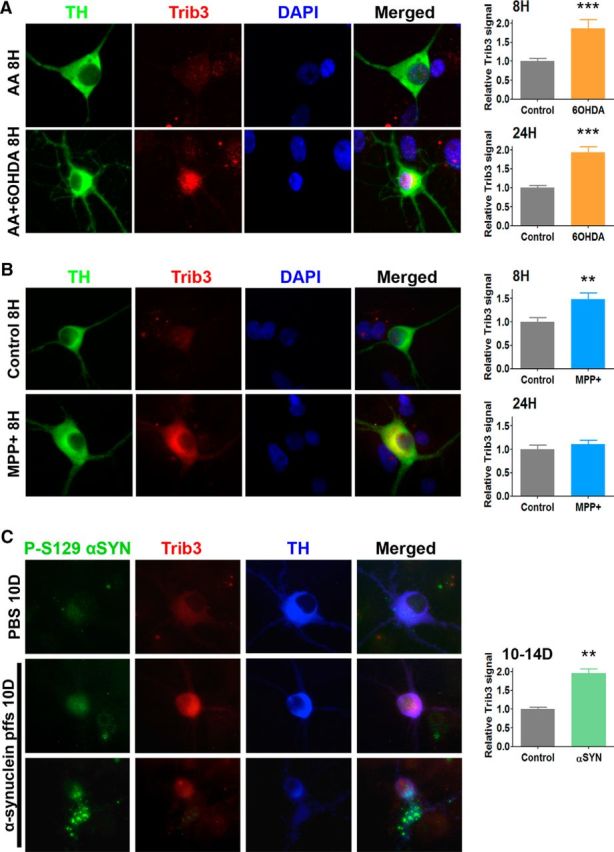
6-OHDA, MPP+, and preformed α-synuclein fibrils induce Trib3 expression in cultured postnatally derived ventral midbrain dopaminergic neurons. A–C, Representative images (left panels) and corresponding quantification (right panels) show an increase of Trib3 signal in dopaminergic cells (positive for TH) after treatment with (A) 6-OHDA, (B) MPP+, or (C) preformed α-synuclein fibrils. **p < 0.005 (t tests). ***p < 0.0005 (t tests). A, B, Cultures were either untreated (control) or treated as indicated with 40 μm MPP+ for 8 or 24 h; 0.015% ascorbic acid (AA) or 40 μm 6-OHDA in 0.015% AA (AA + 6-OHDA) for 8 or 24 h, and immunostained for TH (green) and Trib3 (red) expression. DAPI staining (blue) is shown as a nuclear marker. C, Cultures were treated with PBS or 5 μg/ml of human WT α-synuclein-preformed fibrils (α-synuclein pffs) for 10–14 d and then immunostained for phosphorylated α-synuclein (P-S129 α-syn, green) to show pathogenic intracellular α-synuclein aggregates, Trib3 (red) and TH (blue). To exemplify the diversity of α-synuclein aggregates induced by α-synuclein pffs treatment, images of two morphologically distinct dopaminergic neurons are shown in the two bottom rows. Trib3 relative protein levels correspond to the mean ± SEM of Trib3 densitometric signals measured in 40–85 individual neurons and expressed relative to the corresponding control conditions. Values were analyzed from 2 to 4 independent experiments performed in triplicate.
To extend our results beyond toxin-based models of PD, we evaluated cell death (Fig. 2) and Trib3 induction (Fig. 3) in an α-synuclein-based cellular model. Recent studies have shown that exogenous preformed fibrils from recombinant human WT α-synuclein are taken up into cultured cells by endocytosis and induce endogenous α-synuclein aggregation into Lewy neurite- and Lewy body-like intracellular inclusions (Luk et al., 2009; Volpicelli-Daley et al., 2011). α-Synuclein recruited into intracellular synucleinopathy lesions undergoes extensive phosphorylation at Ser129 (Fujiwara et al., 2002). The intracellular accumulation of aggregated α-synuclein leads to synaptic dysfunction, impairments in connectivity and excitability, and, ultimately, neuron death (Volpicelli-Daley et al., 2011; Luk et al., 2012). As expected, we found that fibril-treated cultured dopaminergic neurons were positive for phosphorylated, thus pathogenic, α-synuclein (P-S129 αSYN) inclusions. Interestingly, these neurons also displayed a marked Trib3 induction (Fig. 3C). Densitometric quantification of Trib3 protein immunostaining levels revealed that α-synuclein fibril treatment induced a concomitant twofold increase in Trib3 levels in the cultured midbrain dopaminergic neurons (t test, p < 0.005). The magnitude of induction and cellular distribution of Trib3 in the fibril-treated cultures were thus highly similar to that observed with 6-OHDA and MPP+. Under these conditions, we found that endogenous α-synuclein aggregation induced death of dopaminergic neurons (Fig. 2C) with loss of 30% of cultured ventral midbrain dopaminergic neurons after 10–14 d of treatment with 5 μg/ml α-synuclein fibrils (t test, p < 0.005).
Trib3 expression is elevated in the substantia nigra of PD patients
The results obtained in vitro prompted us to compare the levels and cellular localization of Trib3 protein in postmortem midbrains of PD and age-matched control patients (control: 81.9 ± 2.9 years, PD: 79.4 ± 1.1 years, t test, p > 0.05; N = 8 for controls and 7 for PD patients). At low magnification, Trib3 staining (blue) was evident in the substantia nigra of both PD and control patients (Fig. 4A). Staining of the substantia nigra was completely absent when the Trib3 antiserum was preincubated with the corresponding Trib3 peptide immunogen, thus supporting the specificity of the antibody and staining. At higher magnification (Fig. 4B), Trib3 staining was found in the neuropil of both control and PD patients. Examination of the dopaminergic neurons, identified by the presence of the brown pigment neuromelanin, revealed an intracytoplasmic granular Trib3 staining pattern in a subset of the population. A proportion of nearby neuromelanin-negative neurons also displayed a similar pattern of Trib3 staining. On a random set of sections, the proportions of dopaminergic neurons positive for Trib3 in the substantia nigra were quantified in a blinded manner for each group. Consistent with the considerable loss of dopaminergic neurons classically described to occur in the substantia nigra of PD patients (Braak et al., 2006), we found that the PD brains were massively depleted of dopaminergic neurons compared with control brains. Significantly, in the remaining dopaminergic neurons of PD patients, there was on average a >2-fold increase in the proportion of nigral dopaminergic neuron cell bodies that were positive for Trib3 immunostaining compared with control patients (Fig. 4C; t test, p < 0.0005).
Figure 4.

The proportion of Trib3+ dopaminergic neurons is increased in the substantia nigra of PD patients. A, Representative low-magnification images (4×) of postmortem midbrains from age-matched human control and PD patients immunostained for Trib3 (blue) and counterstained with Fast Red (pink) show basal expression of Trib3 in the substantia nigra (black dashed lines). Specificity of the Trib3 antibody was verified by a competition experiment performed by incubating the antibody with the corresponding immunizing peptide. Virtually no staining was observed in this control experiment. B, Representative high-magnification (40×) images of sections from the substantia nigra of a control and a PD patient brain. Dopaminergic neurons are identified by the presence of neuromelanin (NM) inclusions (brown). Examples of neurons with granular cytoplasmic Trib3 immunostaining are shown in the right panel (including insets). Black bar represents 25 μm. C, Quantification shows an increase in the percentage of NM+ neurons with Trib3 immunostaining in the substantia nigra of PD patients compared with control cases. ***p < 0.0005 (t test). These data are based on the observation of 8 control and 7 PD patients' brains. A total of 3923 NM+ neurons were scored in controls, and 1489 NM+ neurons were scored in PD cases.
Together, these results show that Trib3 is induced in multiple cellular models of PD, that this induction consistently precedes cell death, and that there is an increase in the proportion of Trib3-immunostained dopaminergic neurons in PD patients. Because Trib3 has proapoptotic activity, we next conducted a series of gain- and loss-of-function experiments to assess its potential role in PD-associated neuron degeneration and death.
Trib3 overexpression is sufficient to induce death of cultured neuronal PC12 cells and ventral midbrain dopaminergic neurons
We recently reported that Trib3 overexpression induces death of cultured neuronal PC12 cells and superior cervical ganglion neurons (Zareen et al., 2013). To confirm and extend these results, we evaluated the effect of lentiviral-mediated Trib3 overexpression on survival of neuronal PC12 cells and ventral midbrain dopaminergic neurons (Fig. 5). Trib3 overexpression was sufficient to induce death of neuronal PC12 cells: 1 and 3 d after transduction, there was loss of 16% of cells and significant 42% loss by 7 d (Fig. 5A; ANOVA with Dunnett's test, p < 0.0005). Similarly, by 6–9 d of Trib3 overexpression, there was loss of 57% of cultured ventral midbrain dopaminergic neurons (Fig. 5B; t tests, p < 0.0005).
Figure 5.

Trib3 induces death of neuronal PC12 cells and postnatally derived ventral midbrain dopaminergic neurons. A, Left, Survival assay shows a decrease in the proportion of surviving neuronal PC12 cells after infection with a lentivirus carrying a Trib3-expressing vector (pWPI Trib3) for the indicated times (1, 3, or 7 d), compared with cells transduced with an empty control vector (pWPI empty). ***p < 0.005 (ANOVA with Dunnett's test). Right, Representative Western blot images show the level of Trib3 expression typically obtained during such experiments. B, Trib3 overexpression decreases survival of cultured postnatally derived ventral midbrain dopaminergic neurons. Left, Representative images of cultures infected with lentivirus expressing Trib3 and GFP (pWPI Trib3) or GFP alone (pWPI empty) and immunostained for TH (red) or GFP (green, lentiviral-infected cells) and stained with DAPI (blue, nuclear marker). Right, Corresponding quantifications of TH+-infected cells showing a decrease in the number of dopaminergic neurons (TH+/GFP+) infected with lentivirus carrying Trib3 for 6–9 d compared with dopaminergic neurons transduced with an empty control vector. ***p < 0.0005 (t test). Values are mean ± SEM and were obtained from at least three independent experiments done in triplicate.
Trib3 downregulation and Trib3 knock-out protect from cell death in PD cellular models
We next assessed whether Trib3 was not only sufficient, but also necessary, for the death of neuronal PC12 cells and ventral midbrain dopaminergic neurons in cellular models of PD. To achieve this, we first used two sets of shRNAs targeted against different regions of Trib3 mRNA that have been shown to effectively and specifically knock down Trib3 protein expression (Zareen et al., 2013). Figure 6A shows a representative Western immunoblot demonstrating the efficacy of shTrib3#1 in knocking down endogenous Trib3 in control and 6-OHDA-treated neuronal PC12 cells. Comparable results were achieved with shTrib3#2 (data not shown). We analyzed the effects of Trib3 knockdown on the survival of neuronal PC12 cells and rat ventral midbrain dopaminergic neurons treated with 6-OHDA, MPP+, or α-synuclein fibrils (Fig. 6B,C). In cultures of neuronal PC12 cells, lentivirus carrying shTrib#1 completely blocked 6-OHDA-induced cell death and significantly reduced MPP+-induced cell death in comparison with control shRNAs (Fig. 6B; ANOVA with Student-Newman-Keuls tests, p < 0.05, p < 0.0005). Similar results were obtained with shTrib3#2 (data not shown). In rat ventral midbrain dopaminergic neuron cultures, Trib3 knockdown also provided significant protection from 6-OHDA and MPP+, as reflected by the percentage of TH+ neurons (Fig. 6C; ANOVA with Student-Newman-Keuls tests, p < 0.05, p < 0.005). As an alternative to the toxin models, we further examined cultured ventral midbrain dopaminergic neurons exposed to α-synuclein fibrils after treatment with virus expressing either scrambled or shTrib3#1 (Fig. 6C). Only approximately half of the dopaminergic neurons survived 10 d of fibril treatment (ANOVA with Student-Newman-Keuls tests, p < 0.0005), and there was significant protection by Trib3 knockdown (ANOVA with Student-Newman-Keuls tests, p < 0.005).
Figure 6.
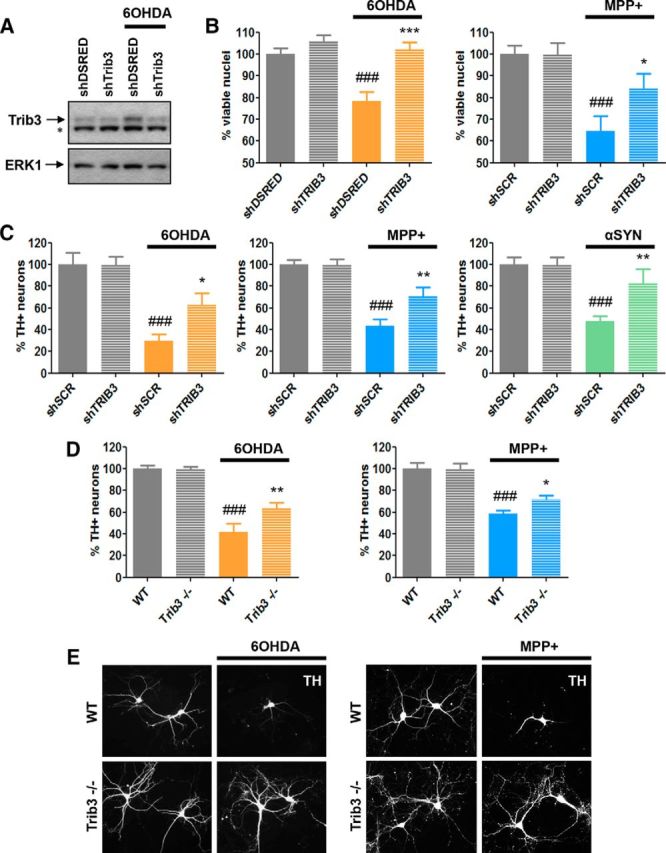
Trib3 downregulation and Trib3 knock-out protect from cell death in PD cellular models. A, Representative Western blot images showing the extent of Trib3 knockdown obtained in neuronal PC12 cells treated with 150 μm 6-OHDA. Top band represents Trib3. Bottom band noted with an asterisk is nonspecific. B, Trib3 knockdown protects neuronal PC12 cells from death elicited by 6-OHDA and MPP+. Survival assay shows the proportion of surviving cells after treatment with 125–150 μm 6-OHDA (left) or 1000 μm MPP+ (right) for 24 h. Cells were infected as indicated with a control shRNA (against either DSRED, shDSRED) or a scrambled version of the shTRIB3 sequence (shSCR) or with an shRNA targeted against Trib3 (shTRIB3). C, Trib3 knockdown protects cultured rat ventral midbrain dopaminergic neurons from death elicited by 6-OHDA, MPP+, or preformed α-synuclein fibrils. Survival assays show the proportion of remaining rat dopaminergic (TH+) ventral midbrain neurons after treatment with 40 μm 6-OHDA for 24 h (left), 40 μm MPP+ for 24 h (middle), or 5 μg/ml of α-synuclein fibrils for 10 d (right). D, E, Dopaminergic (TH+) neurons cultured from Trib3−/− mouse ventral midbrain show decreased sensitivity to 6-OHDA and MPP+. D, Survival assays show the proportion of surviving wild-type (WT) and Trib3−/− mouse dopaminergic (TH+) ventral midbrain neurons after treatment for 24 h with 40 μm 6-OHDA (left) or 40 μm MPP+ (right). E, Representative images of TH+ neurons in cultures of WT and Trib3−/− ventral midbrain show that the absence of Trib3 protects processes from degeneration elicited by 6-OHDA and MPP+. A–D, Values are mean ± SEM from three or four independent experiments performed in triplicate. Multiple comparisons were performed using ANOVA with Student-Newman-Keuls post hoc tests: #Compared with untreated cells expressing a control shRNA or wild-type cells. *Compared with toxin-treated cells expressing a control shRNA or wild-type cells. *p < 0.05; **p < 0.005; ***p < 0.0005; ###p < 0.0005.
To extend these results and to assess the effect of a complete deletion of the Trib3 gene in cellular models of PD, we evaluated the survival of dopaminergic neurons cultured from the midbrains of wild-type as well as Trib3-null mice after treatment with 6-OHDA or MPP+ (Fig. 6D,E). Consistent with reports that Trib3 deletion has no metabolic or behavioral effects under basal conditions (Okamoto et al., 2007; Örd et al., 2014), we did not observe an obvious altered phenotype in Trib3-null animals, nor did examination of the anatomy and cellular content of cortical and subcortical regions, including the ventral midbrain, reveal any evident anomalies or defects (data not shown). Also, under basal conditions, cultured Trib3-null ventral midbrain dopaminergic neurons appeared healthy and were morphologically indistinguishable from wild-type ventral midbrain dopaminergic neurons. However, we found that the Trib3-null neurons were significantly more resistant than wild-type neurons to 6-OHDA and MPP+ (Fig. 6D; ANOVA with Student-Newman-Keuls tests, p < 0.05, p < 0.005). In addition, microscopic examination of the cultures revealed that TH+ neurites of Trib3-null neurons consistently appeared more robust and with more elaborate neuritic trees than those of their wild-type counterparts after 24 h of neurotoxin treatment (Fig. 6E).
The data presented thus far demonstrate that Trib3 mediates, at least in part, death and degeneration in multiple cellular models of PD. We next undertook characterization of the upstream regulators and downstream effectors of Trib3 to better understand the molecular pathways in which it is involved and potentially to identify means to target its induction or proapoptotic actions.
Trib3 induction in Parkinson's cellular models is regulated in part by ATF4 and CHOP
Multiple transcription factors have been implicated in activation of Trib3 in response to stress. Among these are the forkhead box, class O (FoxO) transcription factors, activating transcription factor 4 (ATF4), and C/EBP homologous protein (CHOP; product of the Ddit3 gene), all of which have been shown to activate and/or bind the Trib3 promoter under stress conditions in a variety of cellular models (Ohoka et al., 2005; Ord and Ord, 2005; Carraro et al., 2010; Bromati et al., 2011; Han et al., 2013; Zareen et al., 2013). To investigate the potential roles of these factors in the context of PD, we used lentiviral-delivery of commercially available (shCHOP) or previously validated shRNAs (shFoxO) (Zareen et al., 2013) or shATF4 (Sun et al., 2013) to knock each of them down in toxin-treated neuronal PC12 cells and then assessed induction of Trib3 mRNA by qPCR (Fig. 7). A previous study showed that dephosphorylated FoxO transcription factors are required for induction of Trib3 in neuronal PC12 cells and sympathetic neurons in response to NGF deprivation and that knockdown of either FoxO or Trib3 protects these cells from death caused by such treatment (Zareen et al., 2013). In contrast, however, we found that shRNA-mediated knockdown of FoxO family proteins failed to protect neuronal PC12 cells in PD cellular models (data not shown) or to block Trib3 mRNA induction by 6-OHDA (Fig. 7E; ANOVA with Student-Newman-Keuls tests, p > 0.05). Next, we assessed ATF4 and CHOP; these proteins and their corresponding transcripts are highly upregulated in cellular models of PD, and ATF4 protein expression is elevated in SN dopaminergic neurons in a significant proportion of PD patients (Ryu et al., 2002, 2005; Holtz and O'Malley, 2003; Sun et al., 2013). We confirmed that 6-OHDA and MPP+ induce ATF4 and CHOP transcripts in our models and that this was effectively reduced by the corresponding shRNAs (Fig. 7A–C). Knockdown of ATF4 (Fig. 7A) or CHOP (Fig. 7B) significantly blocked induction of Trib3 mRNA in both the 6-OHDA and MPP+ models (ANOVA with Student-Newman-Keuls tests, p < 0.05, p < 0.005, p < 0.0005). Moreover, CHOP knockdown conferred significant protection in both of these toxin models (Fig. 7D; ANOVA with Student-Newman-Keuls tests, p < 0.05, p < 0.005). We also tested concurrent knockdown of both ATF4 and CHOP in the MPP+ model, and this provided no greater reduction of Trib3 induction than achieved with ATF4 or CHOP alone (Fig. 7C; ANOVA with Student-Newman-Keuls tests, p < 0.005, p < 0.0005). Together, these findings identify ATF4 and CHOP as major regulators of Trib3 induction in cellular toxin models of PD.
Figure 7.

ATF4 and CHOP contribute to Trib3 induction by 6-OHDA and MPP+ in neuronal PC12 cells. A, ATF4 knockdown reduces induction of Trib3 mRNA in response to 6-OHDA and MPP+. Real-time qPCR analyses show the effects of lentivirally delivered ATF4 shRNA on levels of ATF4 mRNA (left) and Trib3 mRNA (right) after treatment with 150 μm 6-OHDA or 1000 μm MPP+ for 8 h. Control cultures were infected with a control shRNA (sh-MUTANT). B, CHOP knockdown reduces induction of Trib3 mRNA in response to 6-OHDA and MPP+. Real-time qPCR analyses show the effects of lentivirally delivered CHOP shRNA on levels of CHOP mRNA (left) and Trib3 mRNA (right) after treatment with 150 μm 6-OHDA or 1000 μm MPP+ for 8 h. Control cultures were infected with a control empty vector (empty). C, Combined knockdown of both ATF4 and CHOP does not enhance suppression of Trib3 mRNA induction by MPP+ over that achieved with knockdown of either alone. Experimental details are given in A and B, except that cells were infected with a mixture of both control vectors (control, empty vector, and sh-MUTANT), sh-ATF4, shCHOP, or both shRNAs (shATF4/shCHOP) as indicated. D, CHOP knockdown protects neuronal PC12 cells from 6-OHDA and MPP+. E, Knockdown of FOXO transcription factors does not significantly block Trib3 induction in PC12 cells treated with 6-OHDA. Neuronal PC12 cells were transduced with lentiviruses expressing either a control shRNA (shDSRED) or an shRNA targeted against FOXO family transcription factors (shFOXO) and treated with 6-OHDA. A–C, E, mRNA levels are normalized against α-tubulin or 18S rRNA. Values are mean ± SEM and were analyzed from at least three independent experiments done in duplicate or triplicate. Comparisons were performed using ANOVA with Student-Newman-Keuls post hoc tests: #Compared with untreated cells expressing a control shRNA. *Compared with toxin-treated cells expressing a control shRNA. *p < 0.05; **p < 0.005; ##p < 0.005; ***p < 0.0005; ###p < 0.0005.
Trib3 interacts with and decreases in vitro and in vivo expression of Parkin
Trib3 appears to have scaffold-like properties and interacts with a number of protein partners (Hegedus et al., 2006, 2007). One example is Akt; Trib3 binds Akt and inhibits its activation (Du et al., 2003). However, we were unable to find consistent effects of Trib3 manipulation on levels of activated Akt in the context of our PD models (data not shown). Trib3 also binds the E3 ubiquitin-protein ligases Smurf1 and Smurf2 and promotes their degradation (Chan et al., 2007; Hua et al., 2011). Parkin (product of the PARK2 gene) is an E3 ubiquitin-protein ligase that plays a key role in pathogenic substrate clearance and mitochondrial homeostasis and that appears to be involved in both familial and sporadic forms of PD (Dawson and Dawson, 2014). Mutations leading to Parkin loss of function cause an autosomal recessive form of juvenile parkinsonism (Kitada et al., 1998). Decreases in Parkin protein levels have been reported in in vitro and in vivo toxin-based models of PD (Kühn et al., 2003; Sonia Angeline et al., 2012; Sun et al., 2013). We therefore explored whether Trib3 might bind to and affect Parkin expression in the context of our cellular PD models. First, we assessed whether Trib3 interacts with Parkin (Fig. 8). Because Trib3 protein levels are low in unstressed cells, we used Trib3 overexpression in these experiments. After 48 h of lentivirus-mediated expression of Trib3 in neuronal PC12 cells, immunoprecipitation with an anti-Parkin antibody specifically coimmunoprecipitated Trib3, indicating that Trib3 and Parkin physically interact in this context.
Figure 8.

Trib3 and Parkin physically interact in neuronal PC12 cells. Top, Short exposures. Bottom, Long exposures. Left panels, Western immunoblot images showing Trib3. Right panels, Parkin and ERK protein levels. Neuronal PC12 cells were transduced with an empty control vector (pWPI empty) or a Trib3-expressing vector (pWPI Trib3) for 48 h, and an immunoprecipitation (IP) experiment was performed with anti-Parkin antibody or control IgG.
Next, we assessed whether Trib3 overexpression is sufficient to affect Parkin levels. In neuronal PC12 cells, Trib3 overexpression led to an ∼40% fall in Parkin levels within 24 h (Fig. 9A,B; ANOVA with Student-Newman-Keuls tests, p < 0.005). At that time, Trib3 overexpression was ∼2-fold to 3-fold, similar to what occurs with toxin and α-synuclein treatment. Interestingly, there was only a minor additional decrease in parkin expression beyond 1 d at times when Trib3 overexpression reaches >10-fold, suggesting a possible threshold effect. Similarly, a densitometric quantification of Parkin immunostaining signal in dopaminergic neurons infected with a Trib3-expressing lentivirus revealed a 33% decrease in Parkin protein levels compared with neurons infected with control empty virus (Fig. 9D,E; t test, p < 0.0005). To analyze a possible transcriptional effect of Trib3 overexpression on Parkin levels, a similar experiment was conducted by qPCR to measure both Trib3 and Parkin mRNA levels after 1–2 d of Trib3 overexpression in neuronal PC12 cells (Fig. 9C). Although lentiviral-mediated Trib3 overexpression led to a massive increase in Trib3 mRNA level (∼70 fold, t test, p < 0.0005), Parkin mRNA levels remained unaffected, thus ruling out a transcriptional regulation of Parkin by Trib3.
Figure 9.

Trib3 overexpression decreases Parkin levels. A, B, Trib3 overexpression decreases Parkin protein levels in neuronal PC12 cells. Representative Western blot images (A) and corresponding quantifications (B) of Trib3 (top) and Parkin (bottom) protein levels in neuronal PC12 cells transduced with an empty control vector (pWPI empty) or a Trib3-expressing vector (pWPI Trib3) for the indicated times (1, 3, or 7 d). Protein levels were quantified and normalized to ERK protein levels, and values are mean ± SEM from five independent experiments. Multiple comparisons were performed using ANOVA with Student-Newman-Keuls post hoc tests: **p < 0.005; ***p < 0.0005. C, qPCR analyses of Trib3 (left) and Parkin (right) mRNA levels in neuronal PC12 cells, transduced with pWPI empty or pWPI Trib3 for 1–2 d, shows that Trib3 overexpression does not affect Parkin mRNA levels. Trib3 and Parkin mRNA levels are normalized against 18S rRNA, and values are mean ± SEM and were analyzed from three independent experiments performed in triplicate. ***p < 0.0005 (t test). D, E, Trib3 overexpression reduces Parkin expression in cultured postnatally derived ventral midbrain dopaminergic neurons. D, Representative immunofluorescence images of cultured postnatally derived ventral midbrain dopaminergic neurons immunostained for TH (blue), GFP (green, lentiviral-infected cells), and Parkin (red) after 2 d of infection with pWPI empty or pWPI Trib3. E, Quantification of relative Parkin immunostaining signals in dopaminergic neurons expressing pWPI Trib3 for 1–4 d, compared with dopaminergic neurons expressing pWPI-empty. ***p < 0.0005 (t test). Trib3 relative protein levels correspond to the mean ± SEM of Trib3 densitometric signals measured in 45–58 individual neurons and normalized to the corresponding control conditions. Values were analyzed from three independent experiments done in triplicate.
Next, we determined whether Trib3 deletion affects Parkin levels in vivo. By comparing Parkin levels in whole-brain extracts of Trib3-null and wild-type mice, we found that complete suppression of Trib3 led to a significant increase (by ∼50%) of basal Parkin protein expression (Fig. 10A,B; t test, p < 0.05). We then studied the potential role of Trib3 induction in the fall in Parkin levels that occurs in response to 6-OHDA (Fig. 10C,D). As anticipated, for neuronal PC12 cells expressing a control shRNA, 6-OHDA elevated Trib3 protein levels and concomitantly decreased Parkin protein expression. However, when Trib3 induction was blocked by infection with shTrib3 lentivirus, there was a partial rescue of Parkin protein levels in the presence of toxin (Fig. 10C,D; ANOVA with Student-Newman-Keuls tests, p < 0.05, p < 0.005, p < 0.0005).
Figure 10.

Trib3 downregulation or deletion increases Parkin levels. A, B, Parkin level is increased in brains of Trib3-null mice. Representative Western immunoblot images (A) and corresponding quantifications (B) of Parkin protein levels in whole-brain lysates from postnatal day 1 (P1) wild-type and P1 Trib3-null mice. Values are normalized to ERK and expressed as mean ± SEM from 3 animals of each genotype. *p < 0.05 (t test). C, D, Trib3 knockdown suppresses the fall in Parkin levels that occurs in neuronal PC12 cells in response to 6-OHDA. C, Representative Western blot image showing Parkin and Trib3 protein levels in cells infected with lentivirus expressing either a control shRNA (shSCR) or shTrib3 and then treated for 8 h with 150 μm 6-OHDA. Top band represents Trib3. Bottom band noted with an asterisk is nonspecific. D, Quantification of relative Trib3 (left) and Parkin (right) protein levels under the conditions corresponding to those in C. Values are normalized to ERK and expressed as mean ± SEM from five independent experiments. #Compared with untreated cells expressing shControl. *Compared with cells treated with 6-OHDA and expressing shControl. ANOVA with Student-Newman-Keuls tests: *p < 0.05; ##p < 0.005; *** p < 0.0005; ###p < 0.0005.
Parkin overexpression protects from Trib3 overexpression and Parkin knockdown reverses the protective effects of Trib3 knockdown in a cellular toxin model
Converging evidence indicates a protective role for Parkin overexpression in multiple in vitro (Jiang et al., 2004; Sun et al., 2013) and in vivo (Haque et al., 2012) models of PD. We therefore assessed whether Parkin overexpression could prevent death induced by Trib3 overexpression. As shown in Figure 11A, Parkin significantly protected neuronal PC12 cells from death caused by Trib3 overexpression (ANOVA with Student-Newman-Keuls tests, p < 0.0005). Next, to ascertain the extent to which the protective actions of Trib3 knockdown are mediated through maintenance of Parkin levels, we examined whether Trib3 silencing would still be protective when Parkin was also knocked down (Fig. 11B,C). Indeed, knocking down Parkin completely abolished the protective effect of Trib3 knockdown on 6-OHDA-treated neuronal PC12 cells (Fig. 11B; ANOVA with Student-Newman-Keuls tests, p < 0.0005).
Figure 11.
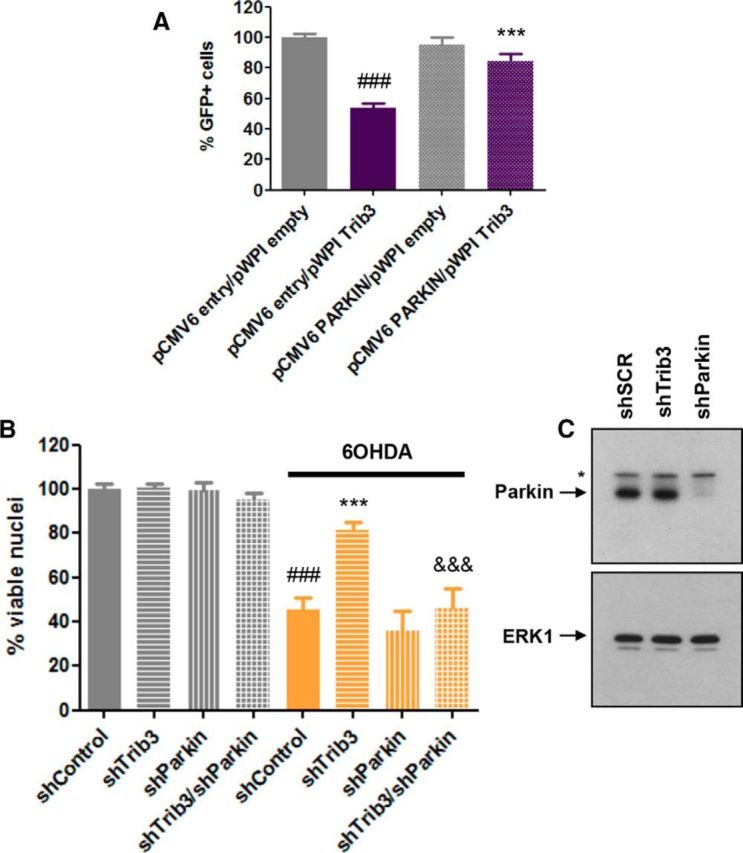
Parkin overexpression protects from death caused by Trib3 overexpression, and Parkin knockdown abolishes the protective effect of Trib3 knockdown. A, Parkin overexpression protects neuronal PC12 cells from death caused by Trib3 overexpression. Data show the proportion of surviving GFP+ neuronal PC12 cells cotransfected with a Trib3-expressing vector (pWPI Trib3) and either a Parkin-expressing vector (pCMV6 Parkin) or an empty control vector (pWPI empty, pCMV6 entry). Values are mean ± SEM and were obtained from two independent experiments performed in quadruplicate. Multiple comparisons were performed using ANOVA with Student-Newman-Keuls post hoc tests. #Compared with cells cotransfected with pCMV6 entry and pWPI empty. *Compared with cells cotransfected with pCMV6 entry and pWPI TRIB3. ***p < 0.0005. ###p < 0.0005. B, Parkin knockdown abolishes the protective effect of Trib3 knockdown on 6-OHDA-treated neuronal PC12 cells. Replicate cultures of neuronal PC12 cells were infected with lentiviruses expressing a control shRNA (shControl), shTrib3, shParkin, or both shTrib3 and shParkin (shTrib3/shParkin) and then treated with 150 μm 6-OHDA for 24 h. Values show the relative proportion of viable cell nuclei after treatment for each condition and are mean ± SEM and were derived from three independent experiments performed in triplicate. Multiple comparisons were performed using ANOVA with Student-Newman-Keuls post hoc tests. #Compared with untreated cells expressing shControl. *Compared with cells treated with 6-OHDA and expressing shControl. &Compared with cells treated with 6-OHDA and expressing shTrib3. ***p < 0.0005. ###p < 0.0005. &&&p < 0.0005. C, Representative Western blot images showing the efficiency of Parkin knockdown in neuronal PC12 cells typically achieved by lentiviral-mediated delivery of shParkin used for these experiments. Parkin corresponds to the bottom band; the top band noted with an asterisk is nonspecific.
Costaining of Parkin and Trib3 in SNpc reveals a large increase in proportion of dopaminergic neurons expressing high levels of Trib3 and low levels of Parkin in PD patients
We next analyzed the potential PD-associated relevance of the effects of Trib3 on Parkin levels by assessing double immunostaining of Trib3 and Parkin proteins in dopaminergic neurons of postmortem midbrains of PD and age-matched control patients (control: 82.8 ± 6.3 years, PD: 75.8 ± 4.5 years, t test, p > 0.05; N = 5 for controls and 6 for PD patients). As a positive control for Parkin expression, we immunostained gallbladder sections, a tissue known to contain high levels of Parkin protein (the human protein atlas; http://www.proteinatlas.org) (Uhlén et al., 2015). This revealed strong Parkin cytoplasmic immunoreactivity in glandular cells lining the gallbladder lumen, which was absent when primary antibody was omitted (data not shown). We then proceeded to assess, in a blinded manner, the coexpression of Trib3 and Parkin in a random set of neuromelanin neurons in postmortem substantia nigrae. Images of single neurons were presented to the blinded observer so that PD and control brains could not be distinguished on the basis of neuronal density.
For each neuron examined, expression of each protein was scored as either high or low (Fig. 12A,B). In agreement with the findings in Figure 4, for this new set of midbrains, we found a subset of neuromelanin-positive neurons highly positive for Trib3 granular cytoplasmic immunostaining (blue staining, Fig. 12A, arrows, B) and confirmed that the proportion of these was substantially increased in the midbrain of PD patients (Fig. 12C,D). In this sample set, the mean proportion of neuromelanin-positive neurons with high Trib3 immunostaining was elevated by ∼5-fold in PD brains compared with controls (Fig. 12D; t test, p < 0.05). The higher proportion of highly Trib3-positive neurons detected in the PD brains (compared with that in Fig. 4) might be attributable to a great interindividual variability seen in this new sample set as well as technical variations and independent experimental conditions.
Figure 12.
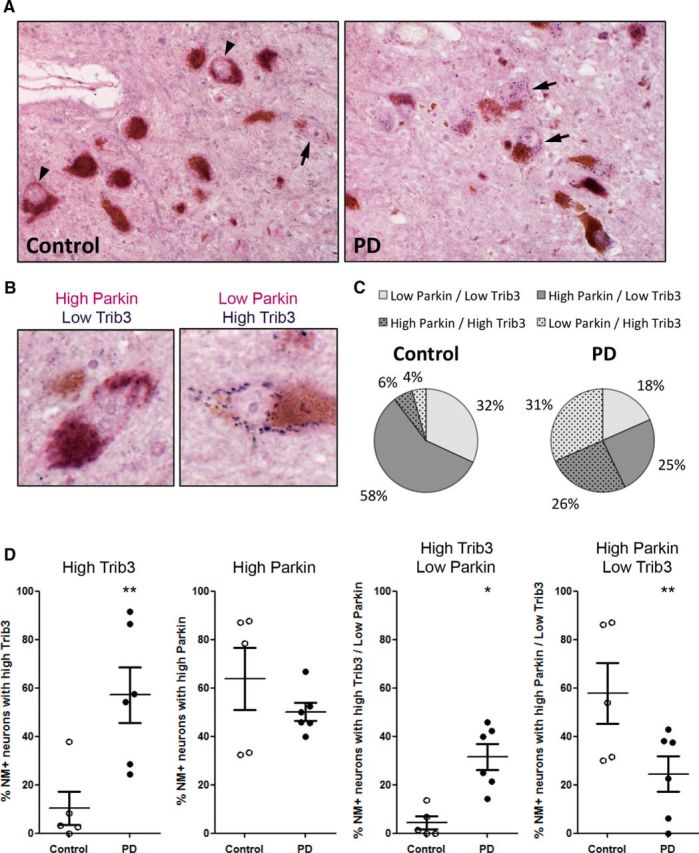
Trib3 and Parkin coexpression in the substantia nigra of control and PD patients. A, Representative images of dopaminergic neurons in postmortem midbrains from age-matched human control and PD patients immunostained for Trib3 (blue) and Parkin (violet). Dopaminergic neurons are identified by the presence of neuromelanin (NM) inclusions (brown). Arrows indicate NM+ neurons highly positive for Trib3 staining. Arrowheads indicate NM+ neurons highly positive for Parkin immunostaining. B, High-magnification images of two examples of dopaminergic neurons displaying high Parkin and low Trib3 levels (left) and low Parkin and high Trib3 levels (right). C, Pie charts showing the proportion of NM+ neurons with low Parkin/low Trib3, high Parkin/low Trib3, low Parkin/high Trib3, and high Parkin/high Trib3 staining observed in controls (left) and PD cases (right). D, Scatter plots showing, from left to right: the proportion of NM+ neurons with high Trib3, high Parkin, high Trib3/low Parkin, high Parkin/low Trib3 staining in controls and PD patients. *p < 0.05 (t tests). **p < 0.005 (t tests). These data are based on the blinded scoring of 5 control and 6 PD patients' brains. A total of 225 NM+ neurons were scored in controls, and 233 NM+ neurons were scored in PD cases.
We also observed a subset of dopaminergic neurons displaying high Parkin levels (homogeneous purple staining, Fig. 12A, arrowheads, B). The proportion of dopaminergic neurons with substantial levels of Parkin was decreased in the midbrain of PD patients (Fig. 12C,D). The mean proportion of neuromelanin-positive neurons with high levels of Parkin corresponded to 64% of neuromelanin-positive neurons in controls, and to 50% of neuromelanin-positive neurons in PD patients. However, this difference did not reach statistical significance (Fig. 12D; t test, p = 0.29).
Finally, analysis of Parkin and Trib3 costaining revealed a marked distinction between neuromelanin-positive neurons in control and PD midbrains (Fig. 12C,D). The most prominent neuronal population in control cases was that with high Parkin and low Trib3 levels; and, by contrast, the most prominent neuronal population in PD cases was that with high Trib3 and low Parkin levels (Fig. 12C). The mean proportion of neuromelanin-positive neurons with high Trib3 and low Parkin staining was elevated by approximately sevenfold in PD brains compared with controls (Fig. 12D; t test, p < 0.05). By contrast, the mean proportion of dopaminergic neurons with high Parkin and low Trib3 staining was decreased by approximately twofold in PD brains compared with controls (Fig. 12D; t test, p < 0.005).
Discussion
Together, the present results indicate that Trib3 is a potential mediator of neuronal death and degeneration in PD. We found that Trib3 is induced in multiple cellular models of PD, including those using PD-mimetic toxins and α-synuclein fibrils and that the SNpc of PD patients contains a significantly higher number of dopaminergic neurons with detectable or high Trib3 expression compared with non-PD patients. Trib3 induction in cellular models occurred before cell death was detectable, consistent with it being a cause of, rather than consequence of, cell degeneration. These results establish Trib3 as an early mediator of cell death, and its potential as a therapeutic target is of particular interest in an effort to intervene in early events of neurodegeneration. In line with this, Trib3 overexpression was sufficient to promote neuronal death. Moreover, knockdown or knock-out of Trib3 repressed death of neuronal PC12 cells and dopaminergic ventral midbrain neurons evoked by either PD-mimicking toxins or α-synuclein fibrils. In addition, Trib3-null neurons were protected from neurite degeneration induced by 6-OHDA and MPP+.
Trib3 is a member of a small family consisting of Tribs 1–3 (Hegedus et al., 2006, 2007; Dobens and Bouyain, 2012; Lohan and Keeshan, 2013). Among their multiple activities, Trib1 and Trib2 have been described as having either context-dependent proapoptotic or antiapoptotic activities in non-neuronal cells (Jin et al., 2007; Gilby et al., 2010; Rishi et al., 2014). It remains to be determined whether these proteins play roles in neurons or in PD. We have reported that Trib1 and 2 are upregulated in brains and superior cervical sympathetic ganglia of Trib3-null mice (Zareen et al., 2013). Compensation by these proteins could explain why dopaminergic neurons from Trib3-null mice, although resistant to 6-OHDA and MPP+, were less protected compared with those in which Trib3 was acutely knocked down with shRNA. Such a consideration would also suggest that mice null only for Trib3 may not be optimal for testing the role of this gene in PD-associated neuron death.
Numerous transcription factors have been implicated in stress-induced Trib3 induction, including ATF4 (Ohoka et al., 2005; Ord and Ord, 2005; Rzymski et al., 2008), CHOP (product of the Ddit3 gene) (Ohoka et al., 2005; Shang et al., 2010), NFkB (Rzymski et al., 2008), FOXO family genes (Zareen et al., 2013), Nrf2 (Juknat et al., 2013), C/EBPβ (Ishikawa et al., 2009), and myb and c-Jun (Kristiansen et al., 2011; Zareen et al., 2013). In contrast to the induction of Trib3 that occurs in response to NGF-deprivation, Trib3 induction in the 6-OHDA model was not significantly affected by FoxO knockdown. Instead, we found that knocking down CHOP or ATF4 reduced induction of Trib3 mRNA in response to 6-OHDA or MPP+ by approximately half. We found that CHOP and ATF4 were strongly induced by 6-OHDA and MPP+ in neuronal PC12 cells. This is consistent with previous studies that have shown induction of ATF4 and CHOP in in vitro and in vivo toxin models of PD (Ryu et al., 2002; Holtz and O'Malley, 2003; Silva et al., 2005) and of ATF4 in dopaminergic neurons of PD patients (Sun et al., 2013). We also observed that combined silencing of ATF4 and CHOP did not lead to additional blockade of Trib3 induction. This suggests that the two work in the same pathway and is consistent with the report that ATF4 and CHOP form heterodimers that synergistically cooperate to activate the Trib3 promoter (Ohoka et al., 2005). The importance of CHOP in regulating Trib3 and death in PD models is supported by our observation that CHOP knockdown was protective in both the MPP+ and 6-OHDA models and the report that dopaminergic neurons in CHOP null mice are resistant to 6-OHDA (Silva et al., 2005). Although our studies identify CHOP and ATF4 as major upstream regulators of Trib3 in cellular PD models, our data also indicate that additional transcription factors remain to be identified that participate in Trib3 expression in the context of PD. Several reports point to p8 (also known as Nupr1) as a major upstream activator of the ATF4/CHOP-TRIB3 pathway leading to the apoptosis of cancer cells (Carracedo et al., 2006; Salazar et al., 2009). Because p8 is present in the nervous system (Khalyfa et al., 2007; Sárvári et al., 2010), it would be interesting to test in the future whether p8 knockdown prevents induction of ATF4, CHOP, and Trib3 and cell death in PD cellular models.
Although ATF4 plays a major role in induction of proapoptotic Trib3, several studies indicate that this transcription factor has a net protective function in PD models, at least in part by maintaining levels of the antiapoptotic protein Parkin (Sun et al., 2013; Wu et al., 2014). Thus, knocking down ATF4 sensitizes neuronal cells to PD-mimicking toxins while overexpressing it provides partial protection (Sun et al., 2013). It therefore appears that ATF4 may activate both proapoptotic and antiapoptotic genes in the context of PD. Trib3 has been shown to bind to and inactivate ATF4 (Ord and Ord, 2003, 2005; Jousse et al., 2007; Liew et al., 2010). Once Trib3 has been sufficiently induced, this negative feedback might shut down the protective pathway orchestrated by ATF4 upon sustained stress, therefore participating in the prodeath action of Trib3. This suggests that reducing Trib3 expression may not only relieve the proapoptotic activities of this protein in PD but also enhance the protective actions of ATF4.
Our studies revealed at least one potential mechanism by which Trib3 mediates neuronal death in cellular models of PD, namely, by regulating levels of the prosurvival Parkin protein. In humans, Parkin loss of function via mutation is associated with autosomal recessive early-onset PD (Kitada et al., 1998). Loss of Parkin activity also has been suggested to play a role in sporadic PD (Dawson and Dawson, 2014). As confirmed here, Parkin protein levels fall by ∼50% in response to 6-OHDA and MPP+, and this decrease appears to sensitize cells to these toxins (Kühn et al., 2003; Sonia Angeline et al., 2012; Sun et al., 2013). In our studies, we found that Trib3 physically interacts with Parkin and that Trib3 overexpression was sufficient to decrease Parkin levels by ∼40%. This decrease in Parkin protein was near maximal after just one day of Trib3 overexpression, when levels of Trib3 protein were increased by approximately twofold, an elevation comparable with that achieved with 6-OHDA, MPP+, or αSYN treatments. Conversely, Parkin levels were elevated in brains of Trib3 knock-out animals and Trib3 knockdown in culture substantially protected Parkin levels during treatment with 6-OHDA. Significantly, the protective effect of Trib3 knockdown was abolished when Parkin was also downregulated.
Our findings indicate that Trib3 overexpression does not change Parkin mRNA levels. Therefore, it seems unlikely that Trib3 regulates Parkin transcription, thus suggesting that Trib3 rather affects Parkin turnover. This is consistent with findings that Trib3 can target other proteins for degradation (Chan et al., 2007; Hua et al., 2011; Aynaud et al., 2012). Parkin has been reported to be degraded by both proteasomal (Yu and Zhou, 2008; Sun et al., 2013) and autophagic (Durcan and Fon, 2011) pathways as well as by caspases (Kahns et al., 2002, 2003), so the exact means by which Trib3 might affect Parkin stability remains to be established.
In agreement with previous studies in human postmortem substantia nigra, we found that Parkin is localized in the cytoplasm and the neurites of dopaminergic neurons (Shimura et al., 1999; Huynh et al., 2000; Zarate-Lagunes et al., 2001). In addition, we found substantial heterogeneity of Parkin staining in both control and PD patients. This is consistent with Zarate-Lagunes et al. (2001), who reported observing some neuromelanin-positive substantia nigral neurons without detectable Parkin immunostaining. By evaluating costaining of Trib3 and Parkin in dopaminergic neurons, we found that high Parkin and low Trib3 levels were the most highly represented in control cases. Conversely, high Trib3 and low Parkin levels were the most frequent in PD cases. These findings demonstrate a clear redistribution of Parkin and Trib3 coexpression in PD. Although these data do not show a strictly inverse relationship between high Trib3 and low Parkin expression (neurons with low Parkin and low Trib3 expression were observed in both control and PD patients; as were those with high Trib3 and high Parkin), they do suggest that, in a substantial number of neurons, increased Trib3 correlates with a decrease in Parkin protein levels. Thus, these findings suggest that Trib3 may influence Parkin levels in PD and that additional factors also contribute to regulation of Parkin expression.
Together, our observations support the idea that Trib3 expression is elevated in both PD and in PD models and that this contributes to neuron degeneration and death, at least in part, by diminishing the expression of Parkin protein. Targeting Trib3 and/or the pathways that lead to its induction in PD (such as CHOP) may therefore represent a novel means to delay or prevent the progressive degeneration and loss of neurons that occurs in this and potentially additional neurodegenerative diseases associated with activation of cellular stress pathways.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants NS072050 and 5P50NS038370 to L.A.G. and Grant K08NS070608 to O.A.L., Parkinson's Disease Foundation (to L.A.G. and O.A.L.), National Research Service Award 5F31AG033477 to N.Z., American Parkinson's Disease Association (to L.V.-D.), William and Bernice E. Bumpus Foundation Fellowship to X.S., and Columbia's College of Dental Medicine Pre-Doctoral Summer Research Fellowship to Z.B. We thank Dr. David Sulzer, Dr. Eugene Mosharov, and Ellen Kanter for sharing their expertise and providing help for setting up postnatally derived dopaminergic ventral midbrain cultures in our laboratory.
The authors declare no competing financial interests.
References
- Aynaud MM, Suspène R, Vidalain PO, Mussil B, Guétard D, Tangy F, Wain-Hobson S, Vartanian JP. Human Tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J Biol Chem. 2012;287:39182–39192. doi: 10.1074/jbc.M112.372722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi XP, Tan HW, Xing SS, Wang ZH, Tang MX, Zhang Y, Zhang W. Overexpression of TRB3 gene in adipose tissue of rats with high fructose-induced metabolic syndrome. Endocr J. 2008;55:747–752. doi: 10.1507/endocrj.K08E-049. [DOI] [PubMed] [Google Scholar]
- Braak H, Bohl JR, Müller CM, Rüb U, de Vos RA, Del Tredici K. Stanley Fahn Lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- Bromati CR, Lellis-Santos C, Yamanaka TS, Nogueira TC, Leonelli M, Caperuto LC, Gorjão R, Leite AR, Anhê GF, Bordin S. UPR induces transient burst of apoptosis in islets of early lactating rats through reduced AKT phosphorylation via ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr Comp Physiol. 2011;300:R92-R100. doi: 10.1152/ajpregu.00169.2010. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Gironella M, Lorente M, Garcia S, Guzmán M, Velasco G, Iovanna JL. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006;66:6748–6755. doi: 10.1158/0008-5472.CAN-06-0169. [DOI] [PubMed] [Google Scholar]
- Carraro V, Maurin AC, Lambert-Langlais S, Averous J, Chaveroux C, Parry L, Jousse C, Ord D, Ord T, Fafournoux P, Bruhat A. Amino acid availability controls TRB3 transcription in liver through the GCN2/eIF2α/ATF4 pathway. PLoS One. 2010;5:e15716. doi: 10.1371/journal.pone.0015716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MC, Nguyen PH, Davis BN, Ohoka N, Hayashi H, Du K, Lagna G, Hata A. A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol Cell Biol. 2007;27:5776–5789. doi: 10.1128/MCB.00218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran CA, Luo X, He Q, Jiang C, Huang Y, Sheikh MS. Genotoxic and endoplasmic reticulum stresses differentially regulate TRB3 expression. Cancer Biol Ther. 2005;4:1063–1067. doi: 10.4161/cbt.4.10.2205. [DOI] [PubMed] [Google Scholar]
- Davie CA. A review of Parkinson's disease. Br Med Bull. 2008;86:109–127. doi: 10.1093/bmb/ldn013. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Parkin plays a role in sporadic Parkinson's disease. Neurodegener Dis. 2014;13:69–71. doi: 10.1159/000354307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding YM, Jaumotte JD, Signore AP, Zigmond MJ. Effects of 6-hydroxydopamine on primary cultures of substantia nigra: specific damage to dopamine neurons and the impact of glial cell line-derived neurotrophic factor. J Neurochem. 2004;89:776–787. doi: 10.1111/j.1471-4159.2004.02415.x. [DOI] [PubMed] [Google Scholar]
- Dobens LL, Jr, Bouyain S. Developmental roles of tribbles protein family members. Dev Dyn. 2012;241:1239–1248. doi: 10.1002/dvdy.23822. [DOI] [PubMed] [Google Scholar]
- Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- Durcan TM, Fon EA. Mutant ataxin-3 promotes the autophagic degradation of parkin. Autophagy. 2011;7:233–234. doi: 10.4161/auto.7.2.14224. [DOI] [PubMed] [Google Scholar]
- Fahn S, Sulzer D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx. 2004;1:139–154. doi: 10.1602/neurorx.1.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Ganser C, Papazoglou A, Just L, Nikkhah G. Neuroprotective effects of erythropoietin on 6-hydroxydopamine-treated ventral mesencephalic dopamine-rich cultures. Exp Cell Res. 2010;316:737–746. doi: 10.1016/j.yexcr.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Gearan T, Castillo OA, Schwarzschild MA. The parkinsonian neurotoxin, MPP+ induces phosphorylated c-Jun in dopaminergric neurons of mesencephalic cultures. Parkinsonism Relat Disord. 2001;8:19–22. doi: 10.1016/S1353-8020(00)00078-X. [DOI] [PubMed] [Google Scholar]
- Gilby DC, Sung HY, Winship PR, Goodeve AC, Reilly JT, Kiss-Toth E. Tribbles-1 and -2 are tumour suppressors, down-regulated in human acute myeloid leukaemia. Immunol Lett. 2010;130:115–124. doi: 10.1016/j.imlet.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque ME, Mount MP, Safarpour F, Abdel-Messih E, Callaghan S, Mazerolle C, Kitada T, Slack RS, Wallace V, Shen J, Anisman H, Park DS. Inactivation of Pink1 gene in vivo sensitizes dopamine-producing neurons to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and can be rescued by autosomal recessive Parkinson disease genes, Parkin or DJ-1. J Biol Chem. 2012;287:23162–23170. doi: 10.1074/jbc.M112.346437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus Z, Czibula A, Kiss-Toth E. Tribbles: novel regulators of cell function; evolutionary aspects. Cell Mol Life Sci. 2006;63:1632–1641. doi: 10.1007/s00018-006-6007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus Z, Czibula A, Kiss-Toth E. Tribbles: a family of kinase-like proteins with potent signalling regulatory function. Cell Signal. 2007;19:238–250. doi: 10.1016/j.cellsig.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H, Yang H, Chen X, Hu Z. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J Cell Sci. 2011;124:3235–3246. doi: 10.1242/jcs.082875. [DOI] [PubMed] [Google Scholar]
- Huynh DP, Scoles DR, Ho TH, Del Bigio MR, Pulst SM. Parkin is associated with actin filaments in neuronal and nonneural cells. Ann Neurol. 2000;48:737–744. doi: 10.1002/1531-8249(200011)48:5<737::AID-ANA7>3.3.CO%3B2-4. [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Akimoto T, Yamamoto H, Araki Y, Yoshie T, Mori K, Hayashi H, Nose K, Shibanuma M. Gene expression profiling identifies a role for CHOP during inhibition of the mitochondrial respiratory chain. J Biochem. 2009;146:123–132. doi: 10.1093/jb/mvp052. [DOI] [PubMed] [Google Scholar]
- Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–1754. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- Jin G, Yamazaki Y, Takuwa M, Takahara T, Kaneko K, Kuwata T, Miyata S, Nakamura T. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood. 2007;109:3998–4005. doi: 10.1182/blood-2006-08-041202. [DOI] [PubMed] [Google Scholar]
- Jousse C, Deval C, Maurin AC, Parry L, Chérasse Y, Chaveroux C, Lefloch R, Lenormand P, Bruhat A, Fafournoux P. TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J Biol Chem. 2007;282:15851–15861. doi: 10.1074/jbc.M611723200. [DOI] [PubMed] [Google Scholar]
- Juknat A, Pietr M, Kozela E, Rimmerman N, Levy R, Gao F, Coppola G, Geschwind D, Vogel Z. Microarray and pathway analysis reveal distinct mechanisms underlying cannabinoid-mediated modulation of LPS-induced activation of BV-2 microglial cells. PLoS One. 2013;8:e61462. doi: 10.1371/journal.pone.0061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahns S, Lykkebo S, Jakobsen LD, Nielsen MS, Jensen PH. Caspase-mediated parkin cleavage in apoptotic cell death. J Biol Chem. 2002;277:15303–15308. doi: 10.1074/jbc.M111534200. [DOI] [PubMed] [Google Scholar]
- Kahns S, Kalai M, Jakobsen LD, Clark BF, Vandenabeele P, Jensen PH. Caspase-1 and caspase-8 cleave and inactivate cellular parkin. J Biol Chem. 2003;278:23376–23380. doi: 10.1074/jbc.M300495200. [DOI] [PubMed] [Google Scholar]
- Khalyfa A, Chlon T, Qiang H, Agarwal N, Cooper NG. Microarray reveals complement components are regulated in the serum-deprived rat retinal ganglion cell line. Mol Vis. 2007;13:293–308. [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kristiansen M, Menghi F, Hughes R, Hubank M, Ham J. Global analysis of gene expression in NGF-deprived sympathetic neurons identifies molecular pathways associated with cell death. BMC Genomics. 2011;12:551. doi: 10.1186/1471-2164-12-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn K, Wellen J, Link N, Maskri L, Lübbert H, Stichel CC. The mouse MPTP model: gene expression changes in dopaminergic neurons. Eur J Neurosci. 2003;17:1–12. doi: 10.1046/j.1460-9568.2003.02408.x. [DOI] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson's disease: proximal triggers, distal effectors, and final steps. Apoptosis. 2009;14:478–500. doi: 10.1007/s10495-008-0309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew CW, Bochenski J, Kawamori D, Hu J, Leech CA, Wanic K, Malecki M, Warram JH, Qi L, Krolewski AS, Kulkarni RN. The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse beta cells. J Clin Invest. 2010;120:2876–2888. doi: 10.1172/JCI36849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wu X, Franklin JL, Messina JL, Hill HS, Moellering DR, Walton RG, Martin M, Garvey WT. Mammalian Tribbles homolog 3 impairs insulin action in skeletal muscle: role in glucose-induced insulin resistance. Am J Physiol Endocrinol Metab. 2010;298:E565–E576. doi: 10.1152/ajpendo.00467.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohan F, Keeshan K. The functionally diverse roles of tribbles. Biochem Soc Trans. 2013;41:1096–1100. doi: 10.1042/BST20130105. [DOI] [PubMed] [Google Scholar]
- Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayumi-Matsuda K, Kojima S, Suzuki H, Sakata T. Identification of a novel kinase-like gene induced during neuronal cell death. Biochem Biophys Res Commun. 1999;258:260–264. doi: 10.1006/bbrc.1999.0576. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Latres E, Liu R, Thabet K, Murphy A, Valenzeula D, Yancopoulos GD, Stitt TN, Glass DJ, Sleeman MW. Genetic deletion of Trb3, the mammalian Drosophila tribbles homolog, displays normal hepatic insulin signaling and glucose homeostasis. Diabetes. 2007;56:1350–1356. doi: 10.2337/db06-1448. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease. Neurology. 2009;72(Suppl):S1–S136. doi: 10.1212/WNL.0b013e3181a1d44c. [DOI] [PubMed] [Google Scholar]
- Ord D, Ord T. Mouse NIPK interacts with ATF4 and affects its transcriptional activity. Exp Cell Res. 2003;286:308–320. doi: 10.1016/S0014-4827(03)00070-3. [DOI] [PubMed] [Google Scholar]
- Ord D, Ord T. Characterization of human NIPK (TRB3, SKIP3) gene activation in stressful conditions. Biochem Biophys Res Commun. 2005;330:210–218. doi: 10.1016/j.bbrc.2005.02.149. [DOI] [PubMed] [Google Scholar]
- Ord D, Meerits K, Ord T. TRB3 protects cells against the growth inhibitory and cytotoxic effect of ATF4. Exp Cell Res. 2007;313:3556–3567. doi: 10.1016/j.yexcr.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Örd T, Innos J, Lilleväli K, Tekko T, Sütt S, Örd D, Kõks S, Vasar E, Örd T. Trib3 is developmentally and nutritionally regulated in the brain but is dispensable for spatial memory, fear conditioning and sensing of amino acid-imbalanced diet. PLoS One. 2014;9:e94691. doi: 10.1371/journal.pone.0094691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayport S, Sulzer D, Shi WX, Sawasdikosol S, Monaco J, Batson D, Rajendran G. Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology. J Neurosci. 1992;12:4264–4280. doi: 10.1523/JNEUROSCI.12-11-04264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishi L, Hannon M, Salomè M, Hasemann M, Frank AK, Campos J, Timoney J, O'Connor C, Cahill MR, Porse B, Keeshan K. Regulation of Trib2 by an E2F1-C/EBPα feedback loop in AML cell proliferation. Blood. 2014;123:2389–2400. doi: 10.1182/blood-2013-07-511683. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Angelastro JM, Greene LA. Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol Dis. 2005;18:54–74. doi: 10.1016/j.nbd.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Rzymski T, Paantjens A, Bod J, Harris AL. Multiple pathways are involved in the anoxia response of SKIP3 including HuR-regulated RNA stability, NF-kappaB and ATF4. Oncogene. 2008;27:4532–4543. doi: 10.1038/onc.2008.100. [DOI] [PubMed] [Google Scholar]
- Salazar M, Carracedo A, Salanueva IJ, Hernández-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C, Torres S, García S, Nowak J, Fimia GM, Piacentini M, Cecconi F, Pandolfi PP, González-Feria L, Iovanna JL, Guzmán M, Boya P, Velasco G. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119:1359–1372. doi: 10.1172/JCI37948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sárvári M, Kalló I, Hrabovszky E, Solymosi N, Tóth K, Likó I, Molnár B, Tihanyi K, Liposits Z. Estradiol replacement alters expression of genes related to neurotransmission and immune surveillance in the frontal cortex of middle-aged, ovariectomized rats. Endocrinology. 2010;151:3847–3862. doi: 10.1210/en.2010-0375. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Neuroprotection in Parkinson's disease. Parkinsonism Relat Disord. 2009;15(Suppl 4):S41–S43. doi: 10.1016/S1353-8020(09)70834-X. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet. 2014;384:545–555. doi: 10.1016/S0140-6736(14)61010-2. [DOI] [PubMed] [Google Scholar]
- Shang YY, Zhong M, Zhang LP, Guo ZX, Wang ZH, Zhang Y, Deng JT, Zhang W. Tribble 3, a novel oxidized low-density lipoprotein-inducible gene, is induced via the activating transcription factor 4-C/EBP homologous protein pathway. Clin Exp Pharmacol Physiol. 2010;37:51–55. doi: 10.1111/j.1440-1681.2009.05229.x. [DOI] [PubMed] [Google Scholar]
- Shang YY, Wang ZH, Zhang LP, Zhong M, Zhang Y, Deng JT, Zhang W. TRB3, upregulated by ox-LDL, mediates human monocyte-derived macrophage apoptosis. FEBS J. 2009;276:2752–2761. doi: 10.1111/j.1742-4658.2009.06998.x. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, Mizuno Y. Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann Neurol. 1999;45:668–672. doi: 10.1002/1531-8249(199905)45:5<668::AID-ANA19>3.0.CO%3B2-Z. [DOI] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonia Angeline M, Chaterjee P, Anand K, Ambasta RK, Kumar P. Rotenone-induced parkinsonism elicits behavioral impairments and differential expression of parkin, heat shock proteins and caspases in the rat. Neuroscience. 2012;220:291–301. doi: 10.1016/j.neuroscience.2012.06.021. [DOI] [PubMed] [Google Scholar]
- Sun X, Liu J, Crary JF, Malagelada C, Sulzer D, Greene LA, Levy OA. ATF4 protects against neuronal death in cellular Parkinson's disease models by maintaining levels of parkin. J Neurosci. 2013;33:2398–2407. doi: 10.1523/JNEUROSCI.2292-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, et al. Proteomics: tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkinshaw G, Waters CM. Neurotoxin-induced cell death in neuronal PC12 cells is mediated by induction of apoptosis. Neuroscience. 1994;63:975–987. doi: 10.1016/0306-4522(94)90566-5. [DOI] [PubMed] [Google Scholar]
- Wu L, Luo N, Zhao HR, Gao Q, Lu J, Pan Y, Shi JP, Tian YY, Zhang YD. Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain Res. 2014;1549:52–62. doi: 10.1016/j.brainres.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Yu F, Zhou J. Parkin is ubiquitinated by Nrdp1 and abrogates Nrdp1-induced oxidative stress. Neurosci Lett. 2008;440:4–8. doi: 10.1016/j.neulet.2008.05.052. [DOI] [PubMed] [Google Scholar]
- Zarate-Lagunes M, Gu WJ, Blanchard V, Francois C, Muriel MP, Mouatt-Prigent A, Bonici B, Parent A, Hartmann A, Yelnik J, Boehme GA, Pradier L, Moussaoui S, Faucheux B, Raisman-Vozari R, Agid Y, Brice A, Hirsch EC. Parkin immunoreactivity in the brain of human and non-human primates: an immunohistochemical analysis in normal conditions and in Parkinsonian syndromes. J Comp Neurol. 2001;432:184–196. doi: 10.1002/cne.1096. [DOI] [PubMed] [Google Scholar]
- Zareen N, Biswas SC, Greene LA. A feed-forward loop involving Trib3, Akt and FoxO mediates death of NGF-deprived neurons. Cell Death Differ. 2013;20:1719–1730. doi: 10.1038/cdd.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Luo Y, Chen JH, Hu J, Luo YZ, Wang W, Zeng Y, Xiao L. Knockdown of TRB3 induces apoptosis in human lung adenocarcinoma cells through regulation of Notch 1 expression. Mol Med Rep. 2013;8:47–52. doi: 10.3892/mmr.2013.1453. [DOI] [PubMed] [Google Scholar]
- Zou CG, Cao XZ, Zhao YS, Gao SY, Li SD, Liu XY, Zhang Y, Zhang KQ. The molecular mechanism of endoplasmic reticulum stress-induced apoptosis in PC-12 neuronal cells: the protective effect of insulin-like growth factor I. Endocrinology. 2009;150:277–285. doi: 10.1210/en.2008-0794. [DOI] [PubMed] [Google Scholar]