

Abstract
Eight G protein–coupled P2Y receptor (P2YR) subtypes are important physiologic mediators. The human P2YRs are fully activated by ATP (P2Y2 and P2Y11), ADP (P2Y1, P2Y12, and P2Y13), UTP (P2Y2 and P2Y4), UDP (P2Y6 and P2Y14), and UDP glucose (P2Y14). Their structural elucidation is progressing rapidly. The X-ray structures of three ligand complexes of the Gi-coupled P2Y12R and two of the Gq-coupled P2Y1Rs were recently determined and will be especially useful in structure-based ligand design at two P2YR subfamilies. These high-resolution structures, which display unusual binding site features, complement mutagenesis studies for probing ligand recognition and activation. The structural requirements for nucleotide agonist recognition at P2YRs are relatively permissive with respect to the length of the phosphate moiety, but less so with respect to base recognition. Nucleotide-like antagonists and partial agonists are also known for P2Y1, P2Y2, P2Y4, and P2Y12Rs. Each P2YR subtype has the ability to be activated by structurally bifunctional agonists, such as dinucleotides, typically, dinucleoside triphosphates or tetraphosphates, and nucleoside polyphosphate sugars (e.g., UDP glucose) as well as the more conventional mononucleotide agonists. A range of dinucleoside polyphosphates, from triphosphates to higher homologs, occurs naturally. Earlier modeling predictions of the P2YRs were not very accurate, but recent findings have provided much detailed structural insight into this receptor family to aid in the rational design of new drugs.
Introduction
The discovery and cloning of the P2Y family of G protein–coupled receptors (GPCRs), which respond to a range of extracellular nucleotides, has spawned a vast array of biologic studies (Webb et al., 1993; Abbracchio et al., 2006). These eight receptors can be divided into two subfamilies based on sequence homology and second messengers: five Gq-coupled P2Y1-like (P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11) and three Gi-coupled P2Y12-like (P2Y12–P2Y14) receptors. The first native agonists of P2Y receptors (P2YRs) with recognized biologic effects were ATP and ADP, and later, UTP, UDP, and UDP glucose (UDPG) were found to activate various P2YRs (Fig. 1A). Thus, the diversity of purine and pyrimidine nucleotide agonists of this family is broader than for most other GPCR families, which typically respond to a single molecule endogenous agonist. The correspondence of the principal native agonists to human P2YR subtypes is ATP (P2Y2 and P2Y11), ADP (P2Y1, P2Y12, and P2Y13), UTP (P2Y2 and P2Y4), UDP (P2Y6 and P2Y14), and UDPG (P2Y14). At increased concentrations, there are some additional crossovers in the activation patterns, such as UDPG acting as a full agonist at P2Y2R (Ko et al., 2009). ATP may act as an antagonist or partial agonist at several P2YR subtypes, including antagonism at the human (but not rat) P2Y4R (Kennedy et al., 2000).
Fig. 1.

(A) Action at P2YRs of nucleotides released from cells (e.g., ATP, UTP, and UDPG) and their conversion outside the cell to 5′-diphosphates, which act at different P2YRs, and/or to 5′-monophosphates, which are inactive (inact.). 5′-nucleotidase catalyzes the final conversion of AMP to adenosine, which acts at its own set of four GPCRs (adenosine A1, A2A, A2B, and A3 receptors). There is a redundancy of ligands that activate various P2YR subtypes. The nucleotides may act as full agonists (green arrows) or variably partial agonists and antagonists (orange arrow). EC50 or IC50 values (µM) at human P2YRs from measurement of adenylate cyclase or phospholipase C activity are indicated in italics. Weaker interactions, such as UDP, as an agonist of P2Y2R (∼10) or P2Y6R (16) are not shown. The enzymatic conversions are catalyzed by ecto-nucleotidases (blue arrows): (a) ecto-nucleoside triphosphate diphosphohydrolases (CD39s) act on either 5′-triphosphates or 5′-diphosphates; (b) ecto-nucleotide pyrophosphatase/phosphodiesterases convert 5′-trimonophosphates to 5′-monophosphates; (c) NPP1 and NPP3 hydrolyze UDPG to produce UMP. (B) Naturally occurring dinucleotides (n = 2–7; B is a nucleobase) are shown schematically and described in detail later in the text. The dinucleotides, such as Up4A and Ap4A, may either act directly on P2YRs, in some cases, or be converted by ecto-nucleotide pyrophosphatase/phosphodiesterases to active mononucleotides, such as ADP. P2YR potencies of simple dinucleotides are reported (Shaver et al., 2005).
P2YRs are widespread in the body and involved in the regulation of nearly all systems, notably, immune, skeletomuscular, digestive, nervous, endocrine, cardiovascular, pulmonary, gastrointestinal, and renal systems (Abbracchio et al., 2006). The broad distribution of P2YRs and the multiplicity of effects of each subtype throughout the body, which are often both protective and damaging, make this system both highly attractive and challenging for drug discovery and development (Jacobson and Boeynaems, 2010).
In addition to the conventional mononucleotide (i.e., nucleoside 5′-polyphosphate) agonists, each of the eight P2YR subtypes has the ability to be activated by structurally bifunctional nucleotides, principally, dinucleotides (Jankowski et al., 2009). They are bifunctional in the respect that the receptor binding site would have to accommodate two nonphosphate end groups, such as nucleoside moieties, linked through a phosphate or polyphosphate moiety (Fig. 1B). Such bifunctional nucleotides typically would include dinucleoside triphosphates or tetraphosphates and nucleoside polyphosphate sugars (e.g., UDPG). The concentrations of dinucleotides achieved in the extracellular medium are often sufficient to activate a range of P2YRs; thus, this is a physiologically relevant component of the purinergic system (Rapaport and Zamecnik, 1976). For example, the endogenous levels of P1-(5′-adenosinyl)-P4-(5′-adenosinyl)-tetraphosphate (Ap4A) and related dinucleotides vary in response to stress, and they participate in extracellular signaling in many tissues and cells, ranging from bacterial to human (Schlüter et al., 1994; Monds et al., 2010).
This phenomenon of broader agonist recognition beyond the mononucleotide agonists was not discovered in a systematic manner for each P2YR subtype, but rather stemmed from the observation that dinucleotides are naturally occurring substances having considerable biologic activity (Miras-Portugal et al., 1999). A range of dinucleoside polyphosphates, from triphosphates to higher homologs, occurs naturally. For example, P1-(5′-adenosinyl)-P4-(5′-uridinyl)-tetraphosphate (Up4A) is released from the vascular endothelium to induce vasoconstriction and has been explored in various biologic contexts, such as P2Y2R-induced migration of smooth muscle cells and activation of enteric neuronal P2Y1R (Wiedon et al., 2012; Durnin et al., 2014). Diadenosine polyphosphates, such as Ap4A, are plentiful in platelet granules and secretory granules of nerve terminals. They contribute either directly or after cleavage to ADP/ATP to thrombus formation and participate in synaptic transmission (Zamecnik et al., 1992; Pintor et al., 2000).
Now with the availability of structural information on the P2Y family and mutagenesis data on the role of specific amino acid residues in ligand binding and/or receptor activation for a few of the P2YRs (Fig. 2), it is feasible to compare the recognition pattern for the various types of agonists more systematically within the P2YR family. Until recently, the empirically detected dual recognition at the P2YRs of mononucleotides and dinucleotides has lacked a structural explanation.
Fig. 2.

Sequence alignment of the human P2YRs. Residues that have been identified using site-directed mutagenesis, as involved in ligand binding and/or receptor activation at P2Y1R (Abbracchio et al., 2006; Zhang et al., 2015), P2Y2R (Erb et al., 1995; Hillmann et al., 2009), P2Y4R (Herold et al., 2004), P2Y11R (Zylberg et al., 2007), and P2Y12R (Hoffmann et al., 2008; Mao et al., 2010; Ignatovica et al., 2012; Zhang et al., 2014a,b) are highlighted with different colors: residues whose mutation can have a major effect on ligand binding and/or receptor activation (red); residues whose mutation modulates ligand binding and/or receptor activation (orange); and residues whose mutation has a minor or no effect on ligand binding and/or receptor activation (yellow). Residues within 3 Å from the crystallographic pose of 2MeSADP at P2Y12R or within 3 Å from the crystallographic pose of MRS2500 at P2Y1R are circled in green. The most highly conserved residue among GPCRs of each helix is highlighted in gray. Cysteine residues involved in disulfide bridges are highlighted in cyan.
Medicinal Chemistry of P2YRs: Focus on Nucleotides
This review emphasizes the action of nucleotides, most of which in this context are P2YR agonists. The characterization of nucleotides as receptor ligands is challenging due to their pharmacological lability, low bioavailability, nonselectivity in activating specific P2YRs, and difficulties in chemical synthesis. Potency values in medicinal chemical studies often reflect either activation of phospholipase C within the P2Y1-like receptor subfamily or other second messengers, such as cAMP, rather than binding affinity because only three of the P2YRs (P2Y1, P2Y12, and P2Y14 receptors) have radioligands available.
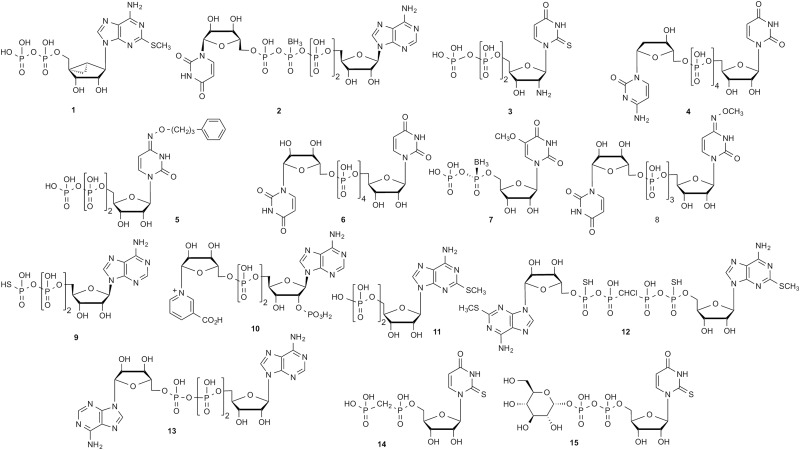
Ecto-nucleotidases and other enzymes are involved in the interconversion of nucleotides that act as P2YR ligands (Fig. 1A) and finally by 5′-nucleotidase for the conversion to adenosine, which acts at its own set of four GPCRs. Recently, the structures of 5′-nucleotidase and ecto-nucleoside triphosphate diphosphohydrolase (CD39) were determined using X-ray crystallography (Heuts et al., 2012; Zimmermann et al., 2012). The structures of some of the other enzymes involved in processing purine receptor ligands, such as ecto-nucleotide pyrophosphatase/phosphodiesterase-1, have also been determined (Jansen et al., 2012). Inhibition or activation of these enzymes is an appealing means of indirectly modulating the activation of the receptors at which the nucleotides and the nucleoside adenosine act. This is an alternative approach to the design of directly acting receptor ligands, either orthosteric or allosteric. The polyphosphate moiety of synthetic nucleotide ligands may contain substitution at limited positions: methylene or halomethylene bridges or P-thio or P-borano substitution (Table 1), all of which can reduce their enzymatic degradation.
TABLE 1.
Representative examples of different types of synthetic or natural ligands (agonists, unless noted), either mononucleotide or dinucleotide (or nucleotide sugar), for each of the P2YRs
The potency (nM) was measured in functional assays at the human P2YRs.
 | |||
|---|---|---|---|
| P2YR |
Synthetic Agonist (Mononucleotide), Potency |
Bifunctional Ligand, Potency |
References |
| nM | |||
| P2Y1 | 1, MRS2365, 0.4 | 2, Up4(β-B)A, A isomer, 500 | Houston et al., 2008; Yelovitch et al., 2012 |
| P2Y2 | 3, MRS2698, 8.0 | 4, INS37217, 220 | Houston et al., 2008; Yerxa et al., 2002 |
| P2Y4 | 5, MRS4062, 26 | 6, INS365 (Up4U)a, 130 | Ko et al., 2008; Maruoka et al., 2011 |
| P2Y6 | 7, 5-OMe-UDPαB, 8 | 8, MRS2957, 12 | Maruoka et al., 2010; Haas et al., 2014 |
| P2Y11 | 9, ATP-γ-S, 24,000 | 10, NAADP, 64,000 | Djerada and Millart, 2013 |
| P2Y12 | 11, 2MeSADPb, 5 | 12, compound 17 (R/S)c, 13 | Zhang et al., 2002; Yanachkov and Wright, 2010 |
| P2Y13 | 11, 2MeSADPb, 19 | 13, Ap3A, 72 | Zhang et al., 2002 |
| P2Y14 | 14, MRS2905, 2.0 | 15, MRS2690, 70 | Das et al., 2010 |
NAADP, nicotinic acid adenine dinucleotide phosphate.
INS365 also activates P2Y2R (EC50 = 210 nM).
2MeSADP activates P2Y1R (EC50 = 6.6 nM), P2Y12R, and P2Y13R (also used as a high affinity 3H- or 33P-radioligand) (Takasaki et al., 2001).
Antagonist.
The only P2YR subtypes that are currently targeted by pharmaceutical agents are P2Y12 (antithrombotic antagonists) (Ferri et al., 2013) and P2Y2 (agonist treatment of dry eye, which is approved in Japan) (Lau et al., 2014). Two of the three P2Y12R antagonists in use as antithrombotics (thienopyridines) are actually prodrugs of irreversibly receptor-binding thiols and therefore have clinical limitations. The attempt to use P2Y2 agonists in the treatment of cystic fibrosis unfortunately failed in clinical trials (Deterding et al., 2007).
Most of the P2YRs still lack uncharged, drug-like antagonists. However, recent extensive exploration of the structure activity relationship (SAR) at P2Y1R has provided such agents, which are also being evaluated as potential antithrombotics (Yang et al., 2014). Also, the SAR of nucleotide antagonists is particularly advanced for the platelet ADP receptors P2Y1 and P2Y12. Several reviews have cataloged the variety of nucleotide and non-nucleotide ligands of P2YRs in detail (Brunschweiger and Müller, 2006; Houston et al., 2008; Jacobson et al., 2012).
The structural requirements for P2YR nucleotide recognition are relatively permissive with respect to the length of the phosphate moiety, but less so with respect to base recognition. Nucleoside polyphosphates beyond 5′-triphosphates, e.g., uridine 5′-tetraphosphate, are also reported to activate various P2YR subtypes (Ko et al., 2008). Bifunctional agonist analogs of Up4U [P1-(5′-uridinyl)-P4-(5′-uridinyl)-tetraphosphate] and uridine 5′-tetraphosphate glucose are tolerated at P2Y2, P2Y4, and P2Y6 receptors. Only P2Y2R readily accepts either A or U as the nucleobase in 5′-triphosphate agonists. ATP binds to human P2Y4R as an antagonist. Alternate nucleobases are sometimes recognized at P2YRs, but at much higher concentrations, for example, IDP as an agonist at P2Y12R (EC50 = 3.18 µM) and P2Y13R (EC50 = 0.552 µM) (Lazarowski et al., 1995). ITP and GTP act as agonists at P2Y4R, with EC50 values of 7.38 and 6.59 µM (intracellular Ca+2), respectively (Kennedy et al., 2000). Some 2-alkylthio derivatives of AMP interact with P2Y1 and P2Y12 receptors as agonists (Boyer et al., 1996b), whereas 2MeSAMP [2-methylthioadenosine 5′-monophosphate] is a P2Y12R antagonist (Zhang et al., 2002).
At P2Y2 and P2Y4 receptors, UMP and UDP are inactive, but some of their analogs activate these receptors as well as UMP analogs at P2Y6R (El-Tayeb et al., 2011). Steric constraint of the ribose ring using a bicyclo[3.1.0]hexane (methanocarba) ring system has demonstrated a strong preference for P2Y6R for the south (S) conformation over the north (N) (Maruoka et al., 2010). Substitution of the uracil 5 position, e.g., with iodo or methoxy, is tolerated at P2Y6, but not P2Y2 and P2Y4 receptors (Haas et al., 2014). Thiocarbonyl substitution of the uracil 2 or 4 position is variably tolerated at the P2Y2, P2Y4, and P2Y14 receptors. 2′- or 3′-deoxynucleotides are not well tolerated as P2YR agonists. Methylene or halomethylene bridges, such as in antagonist 12 or agonist 14, are tolerated at some of the P2YRs (Das et al., 2010; Yelovitch et al., 2012). Boronation of the α-phosphate of ADP derivatives is conducive to activity at the P2Y1R; a pure stereoisomer of the 2-Cl member of that series displayed an EC50 of 7 nM (Azran et al., 2013). Although P2Y6R prefers UDP over UTP, various 5′-triphosphate analogs have proven to be potent (Maruoka et al., 2010). 4-Alkoxyimino groups on the pyrimidine ring, which preserve a double bond character in a C=O substitution, are tolerated at P2Y2, P2Y4, and P2Y6 receptors, and this has allowed the attachment of long-chain fluorophores through that linkage (Jayasekara et al., 2014).
Furthermore, nucleotide-like antagonists and partial agonists are also known for P2Y1, P2Y2, P2Y4, and P2Y12 receptors. Some of these structures are shown in Fig. 3. A3p5p 16 was identified as a partial agonist of human P2Y1R, a key finding that was later optimized by extensive structural modification (Boyer et al., 1996a). Thus, the separation of the two phosphate moieties of ADP and attachment to ribose as bisphosphates (either 3′,5′ or 2′,5′) reduced its efficacy at P2Y1R. N1 was not required for recognition (e.g., 1-deaza analog 19), and several other modifications, N6-methylation, and removal of the 2′-OH, further reduced the efficacy, leading to antagonists, such as MRS2179 18 (Houston et al., 2008). N6-dimethylation or the addition of N-alkyl groups larger than ethyl greatly reduced affinity at P2Y1R, suggesting the presence of a small hydrophobic pocket in the receptor, with a requirement for NH as an H-bond donor. Replacement of the 3′,5′-bisphosphates with bisthiophosphates also greatly reduced affinity. Halogen 20 or small thioethers 21 were tolerated at the C2 position. Substitution of the ribose ring with an (N)-methanocarba ring system, as in 22-24, greatly enhanced potency in the antagonist series by maintaining a P2Y1R-preferred conformation (Kim et al., 2003). Halo (by IC50; I < Cl < F), methyl, methythio, and methylseleno substitution at the C2 position preserved high affinity (Costanzi et al., 2007). The presence of the N6-methyl group in (1′R,2′S,4′S,5′S)-4-(2-iodo-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane (MRS2500) 23 (also used as a high affinity 3H- or 125I-radioligand) enhanced the antagonist affinity by 16-fold. The same (N)-bicyclic ring in P2Y1R agonists, such as 1, was also greatly potency enhancing, which suggests a common binding site for nucleotide antagonists and agonists at this receptor, along with other SAR parallels. Curiously, although rigidity of the ribose enhanced pharmacological properties, acyclic ribose substitutes (25, 26) were also tolerated with micromolar affinities as long as two charged phosphate or phosphonate groups were present. Thus, the binding site for the anionic moieties in P2Y1R must have some flexibility. The uracil phosphonate 27 appears to be an allosteric partial agonist, with selectivity for P2Y2R, but additional characterization of this compound is required (Cosyn et al., 2009).
Fig. 3.

Structures of nucleotide and nucleotide-like antagonists and partial agonists of P2YRs (IC50 values in micromolar at the human P2Y1R are shown in italics).
At P2Y12R, 5′-triphosphates were found to be partial agonists or, in some cases, antagonists (Kauffenstein et al., 2004; Springthorpe et al., 2007). In platelets, triphosphates and triphosphate mimics, such as 28-30, inhibit ADP-induced aggregation, which is consistent with P2Y12R antagonism. 29 has been used as a high affinity radioligand, [3H]PSB-0413 (Ohlmann et al., 2013). Conversely, there are studies showing that ATP seems to be a full P2Y12R agonist (Schmidt et al., 2013). Simplifications of the unwieldy triphosphate group are possible. Monophosphate derivative 31 (Douglass et al., 2008) and carboxyl derivative 32, which was used as a 125I-radioligand (van Giezen et al., 2009), are P2Y12R antagonists. Even uncharged nucleotide-like derivatives, such as acyclic diester 33 and carbocyclic 8-aza derivative 34 (ticagrelor, now approved as an antithrombotic), act as reversibly binding P2Y12R antagonists.
Toward a Systematic Characterization of the SAR of Dinucleotides at P2YRs
Distinct biologic activities are associated with dinucleotides acting at P2YRs, and both P2Y1- and P2Y12-like subfamilies are represented. Zamecnik et al. published early reports on both the chemistry and biology of dinucleoside polyphosphates (Zamecnik et al., 1992). Using recombinant P2YRs, the actions of dinucleotides have been studied systematically at individual molecular targets. For example, Ap4A was found to activate the recombinant human P2Y4R (Lazarowski et al., 1995). At P2Y12R, which is involved in ADP-induced platelet aggregation, the series of ApnA has been studied. In certain conditions, Ap4A appears to be either an agonist, antagonist, or partial agonist (Chang et al., 2010). Diadenosine polyphosphates are also known to activate P2X ion channels. For example, diadenosine pentaphosphate (Ap5A) activates P2X receptors on human cerebrocortical synaptic terminals (Delicado et al., 2006).
Dinucleoside polyphosphates tend to be more stable than mononucleotides at the cell surface because they are not substrates of the ecto-nucleotidases, such as CD39, which cleaves the terminal P-O-P bond from nucleoside 5′-polyphosphates (Kukulski et al., 2011). However, dinucleoside polyphosphates are hydrolyzed by NPP4, which is expressed on the surface of vascular endothelial cells and elsewhere (Albright et al., 2012). Fischer et al. studied diadenosine polyphosphates as inhibitors of nucleotide pyrophosphatase/phosphodiesterases and agonists of various P2YRs (Yelovitch et al., 2012). The inclusion of a borano group in place of OH at a specific location on the polyphosphate moiety was found to have a major enhancing effect on potency and enzymatic stability. The borano substitution of an asymmetric phosphate may also create a new chiral center, e.g., 7, which necessitates separation of diastereomers, and 31P and 1H NMR can be used to determine the relative configuration.
We include in the scope of this review terminal sugar derivatives, which are related structurally to dinucleotides. The first recognized native ligand of what is now designated P2Y14R (originally called GPR105) was UDPG (Chambers et al., 2000). Other related UDP sugars have considerable potency at P2Y14R, and UDP itself is now known to be one of the cognate ligands of this receptor (Carter et al., 2009). There are other reported examples of nucleoside polyphosphates as potent P2YR ligands, in which the terminal phosphate is blocked with a simple aromatic or aliphatic moiety (Das et al., 2010). However, in some cases, blocking the terminal phosphate moiety of a nucleoside 5′-diphosphate or 5′-triphosphate can lead to a great reduction in activity. For example, if the β-phosphate of the P2Y1/P2Y12 agonist 2-methylthioadenosine 5′-diphosphate (2MeSADP) is esterified with a photocleavable o-nitrobenzyl alcohol, receptor activity is lost (Gao et al., 2008).
The pharmacological properties within the series of NpnN (dinucleotides with base N and polyphosphate length n) also vary considerably with the value of n. The potency of various dinucleoside polyphosphates to induce a rise in intracellular calcium in 1321N1 astrocytoma cells heterologously expressing P2YRs of the P2Y1-like subfamily was studied systematically (Shaver et al., 2005). Using highly purified analogs, the rank order of agonist potencies in general was Np3N > > Np4N, Np2N at P2Y1 and P2Y6 receptors, and Np4N > > Np3N > Np2N at P2Y2 and P2Y4 receptors. However, the results are inconsistent with earlier reports that may not purely reflect their potency in activating a given P2YR, and species differences may exist. For example, Ap4A was reported to be either inactive (Patel et al., 2001) or active at P2Y1R. Recent studies have expanded the SAR of dinucleotide analogs, including boranophosphates, at P2YRs (Maruoka et al., 2011; Yelovitch et al., 2012).
The length of the polyphosphate chain required for activation of each P2YR in some cases is highly limited, i.e., with narrow SAR requirements, suggesting that specific interactions with the receptor are involved. Thus, the distal terminal moiety, i.e., either a nucleoside or sugar, with respect to the primary pharmacophore, is not likely to be disassociated from the constraints of the receptor protein. Rather than have complete conformational freedom in the extracellular space, this terminal moiety appears to occupy a secondary binding region that reflects specific interactions with amino acid residues on the receptor.
Representative dinucleotides and related bifunctional compounds (i.e., blocked on both ends of the polyphosphate chain with a phosphodiester) that potently interact with each of the P2YRs are shown in Table 1. The dinucleotides found to modulate P2YRs are often symmetric tail-to-tail dimers of the principal native ligands, such as Ap4A, a dimer of ADP, at P2Y12R. Analogs of Up4A and Ap4A have been studied at the recombinant P2Y1R (e.g., 2) and platelet P2Y12R (e.g., 12), whereas analogs of Up4U (e.g., 3 and 4) have been studied at P2Y2R and P2Y4R. Up3U [P1-(5′-uridinyl)-P3-(5′-uridinyl)-triphosphate] and its derivatives (e.g., 8) have demonstrated high potency at P2Y6R. Ap3A 13 clearly activates P2Y13R, whereas higher diadenosine polyphosphate homologs are inactive (Zhang et al., 2002). Nicotinic acid adenine dinucleotide phosphate (10) is an endogenous agonist of P2Y11R. β-Nicotinamide adenine dinucleotide is released from sympathetic nerve terminals and appears to activate P2Y1R and P2Y11R (Moreschi et al., 2006; Mutafova-Yambolieva et al., 2007; Klein et al., 2009). The production and enzymatic stability of an endogenous P2Y14R agonist, UDPG 14, was studied (Lazarowski et al., 2003). UDPG was also used as a 3H-radioligand (Brunschweiger and Müller, 2006). It is cleaved by nucleotide pyrophosphatase/phosphodiesterases but is stable to the action of several ecto-nucleotidases, such as CD39, which hydrolyze mononucleotides. UDPG release accompanies trafficking of proteins to the cell surface.
Each P2YR subtype has a characteristic SAR for the nucleoside moiety that is not necessarily in parallel between the mononucleotide and dinucleotide series. SAR analysis of mononucleotide pharmacophores at P2YRs is better characterized than for the terminal ends of P2YR-active dinucleotides. In some cases, there is freedom of substitution, and in other cases, the activity is highly dependent on subtle structural changes. For example, if a terminal glucose or other sugar is present on the β-phosphate of UDP, the P2Y14R potency is highly sensitive to changes in sugar functional groups and stereochemistry (Ko et al., 2009). Thus, at P2Y14R, UDP sugars seem to have a different SAR from 5′-diphosphates. Many of the simple UDP analogs are equipotent or more potent than UDPG. However, when present, the terminal β-sugar has specific structural requirements that can greatly reduce potency, and when absent, there is no detrimental effect in general on potency, which suggests a defined binding site for the distal end of UDPG on the receptor. Also, uridine 5′-tetraphosphate sugars and uridine triphosphates each have distinct SAR patterns at P2Y4R (Maruoka et al., 2011) and uridine 5′-diphosphate sugars, such as 15, and uridine 5′-diphosphates, such as 14, each have distinct patterns at P2Y14R (Das et al., 2010).
Dinucleotides have been the focus of pharmaceutical development. Diuridine polyphosphates have been explored as drug candidates by virtue of activating P2Y2R (tetraphosphates) or P2Y6R (triphosphates). Inspire Pharmaceuticals introduced the former as a candidate for the treatment of cystic fibrosis (4, INS37217) (Deterding et al., 2007), which displayed exceptional stability to nucleotidases but later lacked efficacy in clinical trials, and reported selective dinucleotide antagonists of P2Y12R (Douglass et al., 2008). A simple diuridine tetraphosphate (6, INS365) is approved for the treatment of dry eye disease in Japan but not in the United States and is roughly equipotent at P2Y2R and P2Y4R (Lau et al., 2014). A diuridine triphosphate P2Y6R agonist (8, MRS2957) has been shown to increase insulin release from mouse β-islet cell cultures in a glucose-dependent manner and protect against apoptosis induced by TNFα (Balasubramanian et al., 2013). Both actions of P2Y6R might be favorable in cases of diabetes.
Miras-Portugal, Pintor, and colleagues studied the effects of dinucleoside polyphosphates in the nervous system and eye (Castany et al., 2011). Diinosine polyphosphates have been proposed as antiglaucoma agents based on their activation of P2YRs when applied to the corneal surface (Guzman-Aranguez et al., 2012). Ip4I [P1-(5′-inosyl)-P4-(5′-inosyl)-tetraphosphate] was the most efficacious in the inosine series, with a 26% reduction in intraocular pressure and an EC50 value of 0.63 μM, but the P2YR subtype involved was not determined. One complication in interpreting the biologic activity in this series is that diinosine polyphosphates also act as antagonists of P2X receptors, with Ip5I [P1-(5′-inosyl)-P5-(5′-inosyl)-pentaphosphate] as the most potent at P2X1R (North and Jarvis, 2013).
Freilinger et al. have recently reported analogs of Ap4A that are potent and selective antagonists of the platelet P2Y12R and have an antithrombotic action (Yanachkov and Wright, 2010; Chang et al., 2012). Some of the analogs have enhanced stability in biologic systems due to the inclusion of methylene or halomethylene bridges between several phosphorus atoms. Because the thiophosphate group of 12 is a stereocenter, clarification of the biologic implications of this stereochemistry was needed. Pure diastereomers of a monochloromethylene diphosphonate derivative of Ap4A were separated chromatographically and characterized biologically (Chang et al., 2014). One of the isomers was clearly the most potent in inhibiting platelet aggregation through antagonism of P2Y12R without action at P2Y1R or P2X1R.
Structural Characterization of P2YRs
Mutagenesis studies have identified residues in various P2YRs that are likely involved in ligand recognition, regulation, and/or receptor activation (Fig. 2) (Brinson and Harden, 2001; Costanzi et al., 2004; Abbracchio et al., 2006). Given the requirement for negatively charged groups in all agonists thus far identified, the presence of many positively charged amino acid residues in the outer regions of P2YRs suggests direct interaction with nucleotide ligands. The requirement for specific positively charged residues in the recognition of agonists of P2YRs has been well documented (see Fig. 2).
The role of extracellular loops (ELs) in recognition at P2YRs was indicated in several mutagenesis studies (Jacobson et al., 2012). Single amino acid replacements of P2Y1R and P2Y2R led to this conclusion. For example, a single Asp residue (D204) in P2Y1R was found by Ala mutation to be essential for activation by ADP analogs, and even Glu, Asn, and Gln replacement of this residue failed to restore recognition (Moro et al., 1999). Meta-binding sites, which refer to transient complexes of nucleotide ligands as they approach the principal binding site, have been proposed for P2Y1R. Chimeric P2Y1/P2Y6Rs, both of which respond to nucleoside 5′-diphosphates, indicated the role of several ELs in ligand selectivity (Hoffmann et al., 2004). The relatively low structural homology of P2YRs to the available GPCR structural templates, until recently, has impeded the effort to understand these mutagenesis findings in a three-dimensional structural context.
The high-resolution X-ray structures of the P2Y12R complexes with the non-nucleotide antagonist ethyl 6-(4-[(benzylsulfonyl)carbamoyl]piperidin-1-yl)-5-cyano-2-methylnicotinate (AZD1283), nucleotide full agonist 2MeSADP 11, and nucleotide partial agonist 2MeSATP [2-methylthioadenosine 5′-triphosphate] were reported (Fig. 4A) (Zhang et al., 2014a,b), providing major structural insight into this receptor family. The shape of the binding site suggests two distinct subpockets, which are more prominent in the antagonist-bound structure than in the nucleotide complexes. Both AZD1283 and 2MeSADP bind in pocket 1, which is delimited by transmembrane helices (TMs) 3–7, whereas pocket 2, which is delimited by TMs 1, 2, 3, and 7, is mainly empty. The structures revealed major conformational changes in the binding pocket between nucleotides and AZD1283-bound complexes of P2Y12R. Thus, the negatively charged phosphate groups of the nucleotide ligands attract positively charged (R19, R933.21, R2566.55, and K2807.35) and hydrogen-bonding groups (NHα of C175, Y1053.33, Y2596.58, and Q2636.62), and the extracellular regions of TMs 6 and 7 are bent inward toward the bound ligand, further enclosing the ligand (Fig. 5A). The entrance to this pocket is completely blocked by the ELs, suggesting that ligand access to its binding site requires high plasticity of the extracellular region. In contrast, the non-nucleotide antagonist AZD1283 stabilizes a wide-open structure of the pocket by pushing TM6 and TM7 outward with its phenyl group. The long EL2 appears to be flexible and lacking a well defined three-dimensional conformation in the AZD1283 complex. Moreover, the disulfide bond connecting TM3 with EL2, which is highly conserved among family A GPCRs, is missing, contributing to the open conformation of the binding site in the AZD1283-bound structure of P2Y12R. Formation of this disulfide bond in the nucleotide complexes requires an ∼60° rotation of TM3 as compared with the AZD1283 structure. Adenine of 2MeSADP and the pyridine ring of AZD1283 form a π-π interaction with Y1053.33. The methylthio group is situated between TMs 3–5.
Fig. 4.

(A) Human P2Y12R X-ray structures in complex with AZD1283 (the non-nucleotide antagonist is shown in green carbon sticks, and the receptor is shown in orange ribbons) and 2MeSADP (the nucleotide full agonist is shown in orange carbon sticks, and the receptor is shown in cyan ribbons) (Zhang et al., 2014a,b). (B) Human P2Y1R X-ray structures in complex with the antagonists MRS2500 (the nucleotide antagonist is shown in pink carbon sticks, and the receptor is shown in cyan ribbons) and BPTU (the allosteric antagonist is shown in green carbon sticks, and the receptor is shown in orange ribbons) (Zhang et al., 2015).
Fig. 5.

P2Y12R structures and binding of mononucleotides and dinucleotides. (A) Top view of the crystallographic pose of 2MeSADP (green carbon sticks) at P2Y12R (Zhang et al., 2014b). Side chains of some residues important for ligand recognition are displayed (cyan carbon sticks). H-bonds and ionic interactions are pictured as red dotted lines. (B) Top view of the theoretical docking pose of Ap4A (pink carbon sticks) at the antagonist-bound P2Y12R structure (Zhang et al., 2014a). Side chains of some residues in contact with the ligand are displayed (cyan carbon sticks). Semitransparent surface of binding site’s residues is displayed in pale cyan.
The classic ionic lock of many GPCRs, which is a pair of oppositely charged residues in TMs 3 and 6 and holds the receptor in an inactive conformation, is absent in P2Y12R. The potential sodium-binding site, which is associated with a highly conserved Asp residue in TM2 of class A GPCRs (Katritch et al., 2014), is conserved in P2Y12R. Similar to the most closely related PAR1 structure, which contains a sodium ion in the crystal structure, P2YRs have a second Asp residue in TM7 that can participate in the coordination of cations. The crystallized P2Y12R construct has an Asp-to-Asn mutation in this TM7 position, which improved the purified yield of the protein but apparently reduced sodium ion binding.
The crystal structures of the P2Y1R complexes with the nucleotide antagonist MRS2500 23 and with a non-nucleotide allosteric antagonist 1-(2-[2-(tert-butyl)phenoxy]pyridin-3-yl)-3-[4-(trifluoromethoxy)phenyl]urea (BPTU) (Chao et al., 2013) were recently determined (Fig. 4B) (Zhang et al., 2015). The two binding sites were dramatically different from each other and from the P2Y12R structures. Thus, the modeling of the two subfamilies of P2YRs requires distinct structural approaches and assumptions. In both P2Y1R structures, two disulfide bonds were present, connecting the N-terminus to TM7 and TM3 to EL2. The nucleotide-binding site is situated in the EL region, above the region corresponding to P2Y12R-bound nucleotides. This position is reminiscent of an allosteric site of the muscarinic m2 receptor that is above the orthosteric site (Kruse et al., 2013). The 5′-phosphate of MRS2500 is coordinated by R3107.39 and makes hydrogen bonds with T205 in EL2 and Y3067.35. The 3′-phosphate is coordinated by the N-terminus and EL2 and by the phenol groups of Y2.63 and Y7.32. N6.58 coordinates the N6 and N7 groups of the adenine moiety, and a small 2-iodo group is complementary to a small subpocket in the N-terminal segment, including the main chain carbonyl of C42. R2876.62 and L44 were on opposite sides of the adenine moiety, and a π-π stacking, as in P2Y12R, was lacking. The N6 methyl group of MRS2500 was inserted between TMs 6 and 7, forming hydrophobic interactions with A2866.61 and N2997.28. The (N)-methanocarba ring contacts the phenyl group of Y203 in ECL2, which is essential for binding of 23. Although the two antagonist-bound P2Y1R protein structures were very similar, the non-nucleotide antagonist BPTU binds to a novel site on the exterior of the 7TM bundle, and its allosteric antagonism was shown by dissociation kinetics of [3H]2MeSADP. The increase in the dissociation rate of this agonist induced by BPTU was lost when this unusual allosteric site, which is located outside of the TM region and in contact with the phospholipid bilayer, was blocked by a mutation that sterically interfered with this binding region. Mutagenesis indicated that mutually exclusive residues are essential for the two antagonists and suggested that the agonist 2MeSADP may be bound in a similar fashion as 23. The Y306F mutant receptor lost affinity for both agonist and antagonist nucleotide ligands but not BPTU.
Although an unequivocal orientation for dinucleotides at P2Y12R and other P2YRs is still undetermined, the recently solved crystallographic structures suggest the accommodation of both nucleoside moieties of dinucleotides in the unusual bifurcated cavity at P2Y12R and possibly at other P2YRs (Trujillo et al., 2015). This is consistent with docking studies showing dinucleotide ligands reaching both subpockets in the bound state (Zhang et al., 2014a). Figure 5B shows a hypothetical docking of Ap4A in P2Y12R based on the structure of its AZD1283 complex. In particular, the docking pose of Ap4A at the antagonist-bound P2Y12R shows one nucleotide moiety accommodated in pocket 1 and the other one in pocket 2, with the phosphate groups interacting with positively charged residues on TMs 6 and 7. The subsequent resolution of the agonist-bound P2Y12R structure revealed that the proposed orientation of the nucleotide moiety of Ap4A in pocket 1 is different from the one observed for 2MeSADP in the crystal (Fig. 5A). In fact, residues stabilizing the orientation of the ribose and base have different conformations in the two P2Y12R structures: Lys179 in EL2; Cys97 in TM3 because of the missing disulfide bond; and His187 and Asn191 in TM5. This indicates that the receptor conformation that binds dinucleotides may probably combine some of the structural features observed in the two available crystal structures, and further molecular modeling studies can help to obtain a more realistic orientation of dinucleotides in the binding site. The 5′-phosphate of P2Y1R-bound MRS2500 points downward toward a sterically limited region. Thus, the mode of dinucleotide binding to P2Y1R is not apparent from the currently available structure.
Conclusions
The structural understanding of the interaction of nucleotide ligands with P2YRs has been greatly advanced with the resolution of two antagonist-bound structures of P2Y1R and three structures of P2Y12R, which will be especially useful in characterizing recognition at the subfamilies of P2Y1R-like and P2Y12R-like receptors. With new structural data on the P2YR family currently available, it will be possible to use more rational design processes to explore the SAR of different classes of nucleotides at P2YRs.
At each of the P2YR subtypes, both mononucleotides and dinucleotides can act; thus, both naturally occurring and synthetically optimized dinucleoside polyphosphates can serve as agonist or antagonist ligands at various P2YRs. There are several preclinical drug candidates based on this phenomenon. The empirical observation of very specific patterns of SAR of dinucleotides or nucleoside phosphosugars at each of the P2YR subtypes is now partly understandable structurally with the observation that P2Y12R, and potentially other P2YRs have more than one binding cleft in the upper part of the TM region. However, a structural explanation for the recognition of dinucleotides at P2Y1R is still lacking.
Abbreviations
- 2MeSADP
2-methylthioadenosine 5′-diphosphate
- Ap4A
P1-(5′-adenosinyl)-P4-(5′-adenosinyl)-tetraphosphate
- AZD1283
ethyl 6-(4-[(benzylsulfonyl)carbamoyl]piperidin-1-yl)-5-cyano-2-methylnicotinate
- BPTU
1-(2-[2-(tert-butyl)phenoxy]pyridin-3-yl)-3-[4-(trifluoromethoxy)phenyl]urea
- CD39
ecto-nucleoside triphosphate diphosphohydrolase
- EL
extracellular loop
- GPCR
G protein–coupled receptor
- MRS2500
(1′R,2′S,4′S,5′S)-4-(2-iodo-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane
- P2YR
P2Y receptor
- SAR
structure activity relationship
- TM
transmembrane helix
- UDPG
UDP glucose
- Up4A
P1-(5′-adenosinyl)-P4-(5′-uridinyl)-tetraphosphate
Authorship Contributions
Performed data analysis: Paoletta.
Wrote or contributed to the writing of the manuscript: Jacobson, Kiselev, Katritch, Wu, Gao, Zhao, Stevens.
Footnotes
This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant Z01-DK031126-08].
References
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, et al. (2006) International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright RA, Chang WC, Robert D, Ornstein DL, Cao W, Liu L, Redick ME, Young JI, De La Cruz EM, Braddock DT. (2012) NPP4 is a procoagulant enzyme on the surface of vascular endothelium. Blood 120:4432–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azran S, Förster D, Danino O, Nadel Y, Reiser G, Fischer B. (2013) Highly efficient biocompatible neuroprotectants with dual activity as antioxidants and P2Y receptor agonists. J Med Chem 56:4938–4952. [DOI] [PubMed] [Google Scholar]
- Balasubramanian R, Maruoka H, Jayasekara PS, Gao ZG, Jacobson KA. (2013) AMP-activated protein kinase as regulator of P2Y(6) receptor-induced insulin secretion in mouse pancreatic β-cells. Biochem Pharmacol 85:991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, Romero-Avila T, Schachter JB, Harden TK. (1996a) Identification of competitive antagonists of the P2Y1 receptor. Mol Pharmacol 50:1323–1329. [PubMed] [Google Scholar]
- Boyer JL, Siddiqi S, Fischer B, Romero-Avila T, Jacobson KA, Harden TK. (1996b) Identification of potent P2Y-purinoceptor agonists that are derivatives of adenosine 5′-monophosphate. Br J Pharmacol 118:1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinson AE, Harden TK. (2001) Differential regulation of the uridine nucleotide-activated P2Y4 and P2Y6 receptors. SER-333 and SER-334 in the carboxyl terminus are involved in agonist-dependent phosphorylation desensitization and internalization of the P2Y4 receptor. J Biol Chem 276:11939–11948. [DOI] [PubMed] [Google Scholar]
- Brunschweiger A, Müller CE. (2006) P2 receptors activated by uracil nucleotides—an update. Curr Med Chem 13:289–312. [DOI] [PubMed] [Google Scholar]
- Carter RL, Fricks IP, Barrett MO, Burianek LE, Zhou Y, Ko H, Das A, Jacobson KA, Lazarowski ER, Harden TK. (2009) Quantification of Gi-mediated inhibition of adenylyl cyclase activity reveals that UDP is a potent agonist of the human P2Y14 receptor. Mol Pharmacol 76:1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castany M, Jordi I, Catala J, Gual A, Morales M, Gasull X, Pintor J. (2011) Glaucoma patients present increased levels of diadenosine tetraphosphate, Ap(4)A, in the aqueous humour. Exp Eye Res 92:221–226. [DOI] [PubMed] [Google Scholar]
- Chambers JK, Macdonald LE, Sarau HM, Ames RS, Freeman K, Foley JJ, Zhu Y, McLaughlin MM, Murdock P, McMillan L, et al. (2000) A G protein-coupled receptor for UDP-glucose. J Biol Chem 275:10767–10771. [DOI] [PubMed] [Google Scholar]
- Chang H, Yanachkov IB, Dix EJ, Li YF, Barnard MR, Wright GE, Michelson AD, Frelinger AL., 3rd (2012) Modified diadenosine tetraphosphates with dual specificity for P2Y1 and P2Y12 are potent antagonists of ADP-induced platelet activation. J Thromb Haemost 10:2573–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Yanachkov IB, Dix EJ, Yanachkova M, Li Y, Barnard MR, Wright GE, Michelson AD, Frelinger AL., 3rd (2014) Antiplatelet activity, P2Y₁ and P2Y₁₂ inhibition, and metabolism in plasma of stereoisomers of diadenosine 5′,5′″-P¹, P⁴-dithio-P²,P³-chloromethylenetetraphosphate. PLoS One 9:e94780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Yanachkov IB, Michelson AD, Li Y, Barnard MR, Wright GE, Frelinger AL., 3rd (2010) Agonist and antagonist effects of diadenosine tetraphosphate, a platelet dense granule constituent, on platelet P2Y1, P2Y12 and P2X1 receptors. Thromb Res 125:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H, Turdi H, Herpin TF, Roberge JY, Liu Y, Schnur DM, Poss MA, Rehfuss R, Hua J, Wu Q, et al. (2013) Discovery of 2-(phenoxypyridine)-3-phenylureas as small molecule P2Y1 antagonists. J Med Chem 56:1704–1714. [DOI] [PubMed] [Google Scholar]
- Costanzi S, Mamedova L, Gao ZG, Jacobson KA. (2004) Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem 47:5393–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S, Tikhonova IG, Ohno M, Roh EJ, Joshi BV, Colson A-O, Houston D, Maddileti S, Harden TK, Jacobson KA. (2007) P2Y1 antagonists: combining receptor-based modeling and QSAR for a quantitative prediction of the biological activity based on consensus scoring. J Med Chem 50:3229–3241. [DOI] [PubMed] [Google Scholar]
- Cosyn L, Van Calenbergh S, Joshi BV, Ko H, Carter RL, Kendall Harden T, Jacobson KA. (2009) Synthesis and P2Y receptor activity of nucleoside 5′-phosphonate derivatives. Bioorg Med Chem Lett 19:3002–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Ko H, Burianek LE, Barrett MO, Harden TK, Jacobson KA. (2010) Human P2Y(14) receptor agonists: truncation of the hexose moiety of uridine-5′-diphosphoglucose and its replacement with alkyl and aryl groups. J Med Chem 53:471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delicado EG, Miras-Portugal MT, Carrasquero LM, León D, Pérez-Sen R, Gualix J. (2006) Dinucleoside polyphosphates and their interaction with other nucleotide signaling pathways. Pflugers Arch 452:563–572. [DOI] [PubMed] [Google Scholar]
- Djerada Z, Millart H. (2013) Intracellular NAADP increase induced by extracellular NAADP via the P2Y11-like receptor. Biochem Biophys Res Commun 436:199–203. [DOI] [PubMed] [Google Scholar]
- Deterding RR, Lavange LM, Engels JM, Mathews DW, Coquillette SJ, Brody AS, Millard SP, Ramsey BW, Cystic Fibrosis Therapeutics Development Network and the Inspire 08-103 Working Group (2007) Phase 2 randomized safety and efficacy trial of nebulized denufosol tetrasodium in cystic fibrosis. Am J Respir Crit Care Med 176:362–369. [DOI] [PubMed] [Google Scholar]
- Douglass JG, Patel RI, Yerxa BR, Shaver SR, Watson PS, Bednarski K, Plourde R, Redick CC, Brubaker K, Jones AC, et al. (2008) Lipophilic modifications to dinucleoside polyphosphates and nucleotides that confer antagonist properties at the platelet P2Y12 receptor. J Med Chem 51:1007–1025. [DOI] [PubMed] [Google Scholar]
- Durnin L, Hwang SJ, Kurahashi M, Drumm BT, Ward SM, Sasse KC, Sanders KM, Mutafova-Yambolieva VN. (2014) Uridine adenosine tetraphosphate is a novel neurogenic P2Y1 receptor activator in the gut. Proc Natl Acad Sci USA 111:15821–15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Tayeb A, Qi A, Nicholas RA, Müller CE. (2011) Structural modifications of UMP, UDP, and UTP leading to subtype-selective agonists for P2Y2, P2Y4, and P2Y6 receptors. J Med Chem 54:2878–2890. [DOI] [PubMed] [Google Scholar]
- Erb L, Garrad R, Wang Y, Quinn T, Turner JT, Weisman GA. (1995) Site-directed mutagenesis of P2U purinoceptors. Positively charged amino acids in transmembrane helices 6 and 7 affect agonist potency and specificity. J Biol Chem 270:4185–4188. [DOI] [PubMed] [Google Scholar]
- Ferri N, Corsini A, Bellosta S. (2013) Pharmacology of the new P2Y12 receptor inhibitors: insights on pharmacokinetic and pharmacodynamic properties. Drugs 73:1681–1709. [DOI] [PubMed] [Google Scholar]
- Gao ZG, Hechler B, Besada P, Gachet C, Jacobson KA. (2008) Caged agonist of P2Y1 and P2Y12 receptors for light-directed facilitation of platelet aggregation. Biochem Pharmacol 75:1341–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Aranguez A, Díez LM, Martín-Gil A, Gualix J, Miras-Portugal MT, Pintor J. (2012) Effect of diinosine polyphosphates on intraocular pressure in normotensive rabbits. Exp Eye Res 101:49–55. [DOI] [PubMed] [Google Scholar]
- Haas M, Ginsburg-Shmuel T, Fischer B, Reiser G. (2014) 5-OMe-uridine-5′-O-(α-boranodiphosphate), a novel nucleotide derivative highly active at the human P2Y(6) receptor protects against death-receptor mediated glial apoptosis. Neurosci Lett 578:80–84. [DOI] [PubMed] [Google Scholar]
- Herold CL, Qi AD, Harden TK, Nicholas RA. (2004) Agonist versus antagonist action of ATP at the P2Y4 receptor is determined by the second extracellular loop. J Biol Chem 279:11456–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuts DPHM, Weissenborn MJ, Olkhov RV, Shaw AM, Gummadova J, Levy C, Scrutton NS. (2012) Crystal structure of a soluble form of human CD73 with ecto-5′-nucleotidase activity. ChemBioChem 13:2384–2391. [DOI] [PubMed] [Google Scholar]
- Hillmann P, Ko GY, Spinrath A, Raulf A, von Kügelgen I, Wolff SC, Nicholas RA, Kostenis E, Höltje HD, Müller CE. (2009) Key determinants of nucleotide-activated G protein-coupled P2Y(2) receptor function revealed by chemical and pharmacological experiments, mutagenesis and homology modeling. J Med Chem 52:2762–2775. [DOI] [PubMed] [Google Scholar]
- Hoffmann K, Sixel U, Di Pasquale F, von Kügelgen I. (2008) Involvement of basic amino acid residues in transmembrane regions 6 and 7 in agonist and antagonist recognition of the human platelet P2Y(12)-receptor. Biochem Pharmacol 76:1201–1213. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Soltysiak K, West PL, Jacobson KA. (2004) Shift in purine/pyrimidine base recognition upon exchanging extracellular domains in P2Y 1/6 chimeric receptors. Biochem Pharmacol 68:2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston D, Costanzi S, Jacobson KA, Harden TK. (2008) Development of selective high affinity antagonists, agonists, and radioligands for the P2Y1 receptor. Comb Chem High Throughput Screen 11:410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatovica V, Megnis K, Lapins M, Schiöth HB, Klovins J. (2012) Identification and analysis of functionally important amino acids in human purinergic 12 receptor using a Saccharomyces cerevisiae expression system. FEBS J 279:180–191. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Boeynaems JM. (2010) P2Y nucleotide receptors: promise of therapeutic applications. Drug Discov Today 15:570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Jayasekara MPS, Costanzi S. (2012) Molecular structure of P2Y receptors: mutagenesis, modelling and chemical probes. WIREs Membr Transp Signal 1:815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski V, van der Giet M, Mischak H, Morgan M, Zidek W, Jankowski J. (2009) Dinucleoside polyphosphates: strong endogenous agonists of the purinergic system. Br J Pharmacol 157:1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen S, Perrakis A, Ulens C, Winkler C, Andries M, Joosten RP, Van Acker M, Luyten FP, Moolenaar WH, Bollen M. (2012) Structure of NPP1, an ectonucleotide pyrophosphatase/phosphodiesterase involved in tissue calcification. Structure 20:1948–1959. [DOI] [PubMed] [Google Scholar]
- Jayasekara PS, Barrett MO, Ball CB, Brown KA, Hammes E, Balasubramanian R, Harden TK, Jacobson KA. (2014) 4-Alkyloxyimino derivatives of uridine-5′-triphosphate: distal modification of potent agonists as a strategy for molecular probes of P2Y2, P2Y4, and P2Y6 receptors. J Med Chem 57:3874–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffenstein G, Hechler B, Cazenave JP, Gachet C. (2004) Adenine triphosphate nucleotides are antagonists at the P2Y receptor. J Thromb Haemost 2:1980–1988. [DOI] [PubMed] [Google Scholar]
- Kennedy C, Qi A-D, Herold CL, Harden TK, Nicholas RA. (2000) ATP, an agonist at the rat P2Y(4) receptor, is an antagonist at the human P2Y(4) receptor. Mol Pharmacol 57:926–931. [PubMed] [Google Scholar]
- Klein C, Grahnert A, Abdelrahman A, Müller CE, Hauschildt S. (2009) Extracellular NAD(+) induces a rise in [Ca(2+)](i) in activated human monocytes via engagement of P2Y(1) and P2Y(11) receptors. Cell Calcium 46:263–272. [DOI] [PubMed] [Google Scholar]
- Kim HS, Ohno M, Xu B, Kim HO, Choi Y, Ji XD, Maddileti S, Marquez VE, Harden TK, Jacobson KA. (2003) 2-Substitution of adenine nucleotide analogues containing a bicyclo[3.1.0]hexane ring system locked in a northern conformation: enhanced potency as P2Y1 receptor antagonists. J Med Chem 46:4974–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko H, Carter RL, Cosyn L, Petrelli R, de Castro S, Besada P, Zhou Y, Cappellacci L, Franchetti P, Grifantini M, et al. (2008) Synthesis and potency of novel uracil nucleotides and derivatives as P2Y2 and P2Y6 receptor agonists. Bioorg Med Chem 16:6319–6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko H, Das A, Carter RL, Fricks IP, Zhou Y, Ivanov AA, Melman A, Joshi BV, Kovác P, Hajduch J, et al. (2009) Molecular recognition in the P2Y(14) receptor: probing the structurally permissive terminal sugar moiety of uridine-5′-diphosphoglucose. Bioorg Med Chem 17:5298–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukulski F, Lévesque SA, Sévigny J. (2011) Impact of ectoenzymes on p2 and p1 receptor signaling. Adv Pharmacol 61:263–299. [DOI] [PubMed] [Google Scholar]
- Lau OCF, Samarawickrama C, Skalicky SE. (2014) P2Y2 receptor agonists for the treatment of dry eye disease: a review. Clin Ophthalmol 8:327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarowski ER, Boucher RC, Harden TK. (2003) Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol Pharmacol 64:785–795. [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Watt WC, Stutts MJ, Boucher RC, Harden TK. (1995) Pharmacological selectivity of the cloned human P2U-purinoceptor: potent activation by diadenosine tetraphosphate. Br J Pharmacol 116:1619–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Zhang L, Jin J, Ashby B, Kunapuli SP. (2010) Mutational analysis of residues important for ligand interaction with the human P2Y(12) receptor. Eur J Pharmacol 644:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruoka H, Barrett MO, Ko H, Tosh DK, Melman A, Burianek LE, Balasubramanian R, Berk B, Costanzi S, Harden TK, et al. (2010) Pyrimidine ribonucleotides with enhanced selectivity as P2Y(6) receptor agonists: novel 4-alkyloxyimino, (S)-methanocarba, and 5′-triphosphate γ-ester modifications. J Med Chem 53:4488–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruoka H, Jayasekara MPS, Barrett MO, Franklin DA, de Castro S, Kim N, Costanzi S, Harden TK, Jacobson KA. (2011) Pyrimidine nucleotides with 4-alkyloxyimino and terminal tetraphosphate δ-ester modifications as selective agonists of the P2Y(4) receptor. J Med Chem 54:4018–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miras-Portugal MT, Gualix J, Mateo J, Díaz-Hernández M, Gómez-Villafuertes R, Castro E, Pintor J. (1999) Diadenosine polyphosphates, extracellular function and catabolism. Prog Brain Res 120:397–409. [DOI] [PubMed] [Google Scholar]
- Monds RD, Newell PD, Wagner JC, Schwartzman JA, Lu W, Rabinowitz JD, O’Toole GA. (2010) Di-adenosine tetraphosphate (Ap4A) metabolism impacts biofilm formation by Pseudomonas fluorescens via modulation of c-di-GMP-dependent pathways. J Bacteriol 192:3011–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreschi I, Bruzzone S, Nicholas RA, Fruscione F, Sturla L, Benvenuto F, Usai C, Meis S, Kassack MU, Zocchi E, et al. (2006) Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem 281:31419–31429. [DOI] [PubMed] [Google Scholar]
- Moro S, Hoffmann C, Jacobson KA. (1999) Role of the extracellular loops of G protein-coupled receptors in ligand recognition: a molecular modeling study of the human P2Y1 receptor. Biochemistry 38:3498–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutafova-Yambolieva VN, Hwang SJ, Hao X, Chen H, Zhu MX, Wood JD, Ward SM, Sanders KM. (2007) Beta-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci USA 104:16359–16364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Jarvis MF. (2013) P2X receptors as drug targets. Mol Pharmacol 83:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmann P, Lecchi A, El-Tayeb A, Müller CE, Cattaneo M, Gachet C. (2013) The platelet P2Y(12) receptor under normal and pathological conditions. Assessment with the radiolabeled selective antagonist [(3)H]PSB-0413. Purinergic Signal 9:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K, Barnes A, Camacho J, Paterson C, Boughtflower R, Cousens D, Marshall F. (2001) Activity of diadenosine polyphosphates at P2Y receptors stably expressed in 1321N1 cells. Eur J Pharmacol 430:203–210. [DOI] [PubMed] [Google Scholar]
- Pintor J, Díaz-Hernández M, Gualix J, Gómez-Villafuertes R, Hernando F, Miras-Portugal MT. (2000) Diadenosine polyphosphate receptors. from rat and guinea-pig brain to human nervous system. Pharmacol Ther 87:103–115. [DOI] [PubMed] [Google Scholar]
- Rapaport E, Zamecnik PC. (1976) Presence of diadenosine 5′,5″’ -P1, P4-tetraphosphate (Ap4A) in mamalian cells in levels varying widely with proliferative activity of the tissue: a possible positive “pleiotypic activator”. Proc Natl Acad Sci USA 73:3984–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter H, Offers E, Brüggemann G, van der Giet M, Tepel M, Nordhoff E, Karas M, Spieker C, Witzel H, Zidek W. (1994) Diadenosine phosphates and the physiological control of blood pressure. Nature 367:186–188. [DOI] [PubMed] [Google Scholar]
- Schmidt P, Ritscher L, Dong EN, Hermsdorf T, Cöster M, Wittkopf D, Meiler J, Schöneberg T. (2013) Identification of determinants required for agonistic and inverse agonistic ligand properties at the ADP receptor P2Y12. Mol Pharmacol 83:256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patel RI, Redick CC, Jones AC, Picher M, et al. (2005) Structure-activity relationships of dinucleotides: potent and selective agonists of P2Y receptors. Purinergic Signal 1:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springthorpe B, Bailey A, Barton P, Birkinshaw TN, Bonnert RV, Brown RC, Chapman D, Dixon J, Guile SD, Humphries RG, et al. (2007) From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg Med Chem Lett 17:6013–6018. [DOI] [PubMed] [Google Scholar]
- Takasaki J, Kamohara M, Saito T, Matsumoto M, Matsumoto S, Ohishi T, Soga T, Matsushime H, Furuichi K. (2001) Molecular cloning of the platelet P2T(AC) ADP receptor: pharmacological comparison with another ADP receptor, the P2Y(1) receptor. Mol Pharmacol 60:432–439. [PubMed] [Google Scholar]
- Trujillo K, Paoletta S, Kiselev E, Jacobson KA. (2015) Molecular modeling of the human P2Y14 receptor: a template for structure-based design of selective agonist ligands. Bioorg Med Chem DOI: 10.1016/j.bmc.2015.03.042 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN Giezen JJJ, Nilsson L, Berntsson P, Wissing B-M, Giordanetto F, Tomlinson W, Greasley PJ. (2009) Ticagrelor binds to human P2Y(12) independently from ADP but antagonizes ADP-induced receptor signaling and platelet aggregation. J Thromb Haemost 7:1556–1565. [DOI] [PubMed] [Google Scholar]
- Webb TE, Simon J, Krishek BJ, Bateson AN, Smart TG, King BF, Burnstock G, Barnard EA. (1993) Cloning and functional expression of a brain G-protein-coupled ATP receptor. FEBS Lett 324:219–225. [DOI] [PubMed] [Google Scholar]
- Wiedon A, Tölle M, Bastine J, Schuchardt M, Huang T, Jankowski V, Jankowski J, Zidek W, van der Giet M. (2012) Uridine adenosine tetraphosphate (Up4A) is a strong inductor of smooth muscle cell migration via activation of the P2Y2 receptor and cross-communication to the PDGF receptor. Biochem Biophys Res Commun 417:1035–1040. [DOI] [PubMed] [Google Scholar]
- Yanachkov IB and Wright GE (2010) inventors, Glsynthesis Inc., assignee. Novel antithrombotic diadenosine tetraphosphates and related analogs. Patent WO 2010059215 A1. 2010 May 27.
- Yang W, Wang Y, Lai A, Qiao JX, Wang TC, Hua J, Price LA, Shen H, Chen XQ, Wong P, et al. (2014) Discovery of 4-aryl-7-hydroxyindoline-based P2Y1 antagonists as novel antiplatelet agents. J Med Chem 57:6150–6164. [DOI] [PubMed] [Google Scholar]
- Yelovitch S, Camden J, Weisman GA, Fischer B. (2012) Boranophosphate isoster controls P2Y-receptor subtype selectivity and metabolic stability of dinucleoside polyphosphate analogues. J Med Chem 55:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerxa BR, Sabater JR, Davis CW, Stutts MJ, Lang-Furr M, Picher M, Jones AC, Cowlen M, Dougherty R, Boyer J, et al. (2002) Pharmacology of INS37217 [P(1)-(uridine 5′)-P(4)- (2′-deoxycytidine 5′)tetraphosphate, tetrasodium salt], a next-generation P2Y(2) receptor agonist for the treatment of cystic fibrosis. J Pharmacol Exp Ther 302:871–880. [DOI] [PubMed] [Google Scholar]
- Zamecnik PC, Kim B, Gao MJ, Taylor G, Blackburn GM. (1992) Analogues of diadenosine 5′,5″’-P1,P4-tetraphosphate (Ap4A) as potential anti-platelet-aggregation agents. Proc Natl Acad Sci USA 89:2370–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Gao Z-G, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, et al. (2015) Two disparate ligand-binding sites in the human P2Y1 receptor. Nature DOI: 10.1038/nature14287 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FL, Luo L, Gustafson E, Palmer K, Qiao X, Fan X, Yang S, Laz TM, Bayne M, Monsma F., Jr (2002) P2Y(13): identification and characterization of a novel Galphai-coupled ADP receptor from human and mouse. J Pharmacol Exp Ther 301:705–713. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Müller CE, et al. (2014b) Agonist-bound structure of the human P2Y12 receptor. Nature 509:119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, et al. (2014a) Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 509:115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H, Zebisch M, Sträter N. (2012) Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8:437–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylberg J, Ecke D, Fischer B, Reiser G. (2007) Structure and ligand-binding site characteristics of the human P2Y11 nucleotide receptor deduced from computational modelling and mutational analysis. Biochem J 405:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]