

Abstract
The α3β4 nicotinic acetylcholine receptor (nAChR) subtype is widely expressed in the peripheral and central nervous systems, including in airway sensory nerves. The nAChR subtype transduces the irritant effects of nicotine in tobacco smoke and, in certain brain areas, may be involved in nicotine addiction and/or withdrawal. Menthol, a widely used additive in cigarettes, is a potential analgesic and/or counterirritant at sensory nerves and may also influence nicotine’s actions in the brain. We examined menthol’s effects on recombinant human α3β4 nAChRs and native nAChRs in mouse sensory neurons. Menthol markedly decreased nAChR activity as assessed by Ca2+ imaging, 86Rb+ efflux, and voltage-clamp measurements. Coapplication of menthol with acetylcholine or nicotine increased desensitization, demonstrated by an increase in the rate and magnitude of the current decay and a reduction of the current integral. These effects increased with agonist concentration. Pretreatment with menthol followed by its washout did not affect agonist-induced desensitization, suggesting that menthol must be present during the application of agonist to augment desensitization. Notably, menthol acted in a voltage-independent manner and reduced the mean open time of single channels without affecting their conductance, arguing against a simple channel-blocking effect. Further, menthol slowed or prevented the recovery of nAChRs from desensitization, indicating that it probably stabilizes a desensitized state. Moreover, menthol at concentrations up to 1 mM did not compete for the orthosteric nAChR binding site labeled by [3H]epibatidine. Taken together, these data indicate that menthol promotes desensitization of α3β4 nAChRs by an allosteric action.
Introduction
Menthol is a monoterpene alcohol widely used in consumer products. Most notably, menthol is extensively used in cigarettes. More than 90% of tobacco products contain some amount of menthol; the majority, ∼75%, has only low levels of l-menthol as an additive (0.03%), and 25% contain higher levels (0.1–0.45%) and are designated mentholated cigarettes. Menthol may be added to tobacco to deliver a distinct oral sensation, as it imparts a characteristic cooling sensation via activation of transient receptor potential channel, subfamily M, member 8 (TRPM8) ion channels expressed in a population of thermosensory nerves (McKemy et al., 2002; Peier et al., 2002; Bautista et al., 2007). In turn, signaling downstream of TRPM8 can lead to analgesia through undefined mechanisms (Willis et al., 2011). In addition, menthol may produce analgesic/counterirritant effects through activation and desensitization of the nociceptive transient receptor potential channel, subfamily A, member 1 (TRPA1) channel (Macpherson et al., 2006; Karashima et al., 2007; Xiao et al., 2008) or by direct antagonism of the TRPA1 channel (Karashima et al., 2007; Xiao et al., 2008). Indeed, in the context of smoking, the analgesic effects of menthol may be desirable for the smoker. Cigarette smoke contains numerous noxious compounds that may be irritants to the airway. Further, nicotine itself can be noxious by activating nicotinic acetylcholine receptors (nAChRs) located on pulmonary sensory neurons; in fact, nicotine may be the primary mediator of airway irritation and cough evoked by cigarette smoke (Lee et al., 2007). Menthol may, therefore, reduce the harshness of cigarette smoke and nicotine, and thereby increase the tolerability and/or palatability of smoking. This might be particularly important to the person just beginning to smoke.
Interestingly, two recent studies revealed that menthol inhibits ACh and nicotine-stimulated currents in heterologously expressed α4β2 (Hans et al., 2012) and α7 (Ashoor et al., 2013) nAChRs, subtypes predominantly expressed in the brain, and produces a slow, time-dependent inhibition of ACh- and nicotine-evoked currents in cells from the trigeminal ganglia (Hans et al., 2012). In each case, menthol appeared to act allosterically. These data suggest that nAChRs may be additional pharmacological targets of menthol. However, the effects of menthol on α3β4 nAChRs, the major nicotinic subtype expressed in sensory nerves, and the potential mechanisms of menthol’s inhibition of nAChRs are unknown. Understanding the effects of menthol on α3β4 nAChRs in particular is relevant to nociceptive signaling in the airways arising from cigarette smoke and nicotine. In addition, α3β4 nAChRs are highly expressed in the habenula and intrapeduncular nuclei, brain regions implicated in reward processing and possibly addiction to and/or withdrawal from nicotine (Salas et al., 2004; McCallum et al., 2012). Here we show that menthol acts allosterically to inhibit the function of α3β4 nAChRs by increasing the rate and extent of agonist-induced desensitization. We propose that this mechanism may contribute to the analgesic actions of menthol in the bronchial airways in the presence of tobacco smoke, as well as to some of the effects of nicotine in the brain. Both of these actions may contribute to nicotine addiction.
Materials and Methods
All experimental procedures involving mice were approved by the Georgetown University Animal Care and Use Committee and conformed to National Institutes of Health guidelines.
(–)Menthol and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. [3H]epibatidine ([3H]EB) and 86RbCl were purchased from PerkinElmer (Boston, MA).
Cell Culture.
Human embryonic kidney cells stably expressing human α3β4 nAChRs were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 1% 100× minimum essential medium nonessential amino acids, and 1% penicillin/streptomycin (HyClone Laboratories/GE Healthcare Life Sciences, Logan, UT) at 37°C in a water-saturated atmosphere containing 5% CO2. Cell cultures were seeded in a culture flask (25 cm2; Sarstedt, Newton, NC) and subcultured twice a week. For Ca2+ imaging and electrophysiology, cells were plated on poly-d-lysine–coated coverslips and used for experiments within 1–3 days.
Nodose Ganglia Neurons.
For experiments with nodose ganglia neurons, adult C57BL/6J mice (25–30 g) were killed by CO2/decapitation and the nodose ganglia were dissected, digested with collagenase, and cultured in neurobasal medium plus 2% B-27 (Invitrogen/Life Technologies, Grand Island, NY), 0.1% l-glutamine and 1% penicillin/streptomycin on poly-d-lysine–coated glass coverslips at 37°C in 5% CO2. Neurons were used within 24–36 hours of culture.
Ca2+ Imaging.
Ca2+ imaging was performed using the dye Fluo4-AM (Invitrogen). The cells were loaded with 1 μM Fluo4-AM in a standard buffer containing 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1.2 mM CaCl2, 10 mM HEPES, 5 mM glucose, pH 7.3. The dye was excited at 480 ± 15 nm. Emitted fluorescence was filtered with a 535 ± 25-nm bandpass filter, captured by a SPOT RT digital camera (SPOT Imaging Solutions, Sterling Heights, MI) and read into a computer. Analysis was performed offline by using SimplePCI software (Compix Inc., University of New South Wales). Solutions were applied via a valve-controlled gravity-fed perfusion system with a 200-μm diameter outlet. The bathing solution contained 5 μM atropine to block muscarinic acetylcholine receptors.
[3H]Epibatidine Binding Competition Assays.
Binding competition assays were performed to determine menthol’s affinity for human α3β4 nAChRs. Cell membrane homogenates were prepared as described previously (Xiao and Kellar, 2004). Briefly, cells were washed, suspended in 50 mM Tris-HCl buffer, and homogenized with a Polytron homogenizer (Kinematica AG, Lucerne, Switzerland). The homogenate was centrifuged at 33,000g for 10 minutes at 4°C, the supernatant was discarded, and the pellet was then suspended in fresh buffer. This was repeated two more times before the final pellet was resuspended in 50 mM Tris-HCl buffer and used in subsequent assays. The membranes were incubated for 2 hours with ∼0.5 nM [3H]EB in the absence or presence of increasing concentrations of menthol or nicotine (for comparison). The membrane homogenates were filtered through Whatman GF/C filters treated with 0.5% polyethylenimine and then counted in a BeckmanCoulter scintillation counter (LS6500; Jersey City, NJ). The data were analyzed by nonlinear least-squares regression analysis in GraphPad Prism 5 (San Diego, CA).
Rubidium Efflux Assays.
Menthol’s effect on α3β4 nAChR function was initially examined by assessing 86Rb+ efflux through the receptor channel, as described previously (Xiao et al., 1998; Meyer et al., 2001). Cells were first loaded with 86RbCl by incubating them for ∼2 hours with 0.5 ml of media containing ∼100,000 dpm 86Rb+. To test menthol’s agonist activity, the cells were rinsed gently four times with 1 ml of buffer over 10 minutes, and then either buffer alone, buffer containing 100 μM nicotine, or 100 μM menthol was added for 2 minutes. To test for menthol’s antagonist activity, cells were incubated for 2 minutes with 100 μM nicotine in the absence or presence of increasing concentrations of menthol. In some experiments, menthol was added 10 minutes before and maintained during the 2-minute nicotine stimulation. In all cases, the background efflux was determined in the cells incubated in buffer alone, and maximal response was defined as the efflux elicited by 100 μM nicotine. The 86Rb+ efflux from the cells into the media was assessed using Cherenkov counting on a Beckman-Coulter LS6500 Scintillation Counter. After subtracting background efflux, stimulated efflux was calculated as the 86Rb+ in the media over the sum of the 86Rb+ in the media plus that in the cells. Results are expressed as the percent of efflux elicited by 100 μM nicotine, which elicits a maximal response (Meyer et al., 2001).
Electrophysiology.
Whole-cell and cell-attached voltage-clamp recordings were performed by using an EPC8 patch-clamp amplifier (HEKA Electronik, Bellmore, NY) that was controlled by the program Pulse (version 8.65; HEKA Electronik). Data were collected at 5 KHz and low-pass filtered at 3 KHz. Single-channel data were analyzed by Channel2 software (M. Smith and P. W. Gage, Australian National University). The bath solution contained 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1.2 mM CaCl2, 10 mM Hepes, 5 mM glucose, pH 7.3. The pipette solution contained 140 mM CsCl, 10 mM HEPES, 10 mM EGTA, 2 mM Mg-ATP, pH 7.3. For cell-attached recordings the pipette solution was the same as the bath solution and included 10 μM ACh. Solutions were applied via a valve-controlled gravity-fed perfusion system with an outlet (200-μm diameter) positioned ∼50 μm from the cell of interest. The solution exchange time constant (τ) was ∼1 second.
To characterize desensitization, currents were evoked by ACh or nicotine in the absence or presence of menthol for 40 seconds and the time constant was measured by fitting the current decays with one or two exponential equations using Origin 8.0 software (OriginLab Corporation, Northampton, MA). The fraction of the current remaining at the end of the 40-second drug application was designated as “D” and was used to quantify the extent of desensitization. All values were normalized to those produced by ACh for each individual cell.
Results
Menthol Inhibits Human α3β4 nAChR Activity in a Noncompetitive Manner.
Sensory and autonomic ganglia neurons predominantly express the α3β4 subtype of nAChR. To examine potential effects of menthol at these ion channels we performed Ca2+ imaging and 86Rb+ efflux measurements in HEK293 cells stably expressing human α3β4 nAChRs. As shown in Fig. 1, A and B, successive applications of 30 μM ACh (with an interstimulus interval of 5 minutes) evoked Ca2+ transients of similar magnitude (mean ΔF/Fo application 1 = 3.49 ± 0.18 versus application 2 = 3.45 ± 0.20, n = 39 cells). However, coapplication of menthol (100 μM) inhibited the ACh-induced peak Ca2+ signal by 47% (ΔF/Fo = 3.30 ± 0.16 versus 1.90 ± 0.18, n = 37 cells) (Fig. 1, A and B).
Fig. 1.

Effects of menthol on human α3β4 nAChR-mediated [Ca2+] influx and nicotine-stimulated 86Rb+ efflux. (A) Representative Fluo-4 fluorescence images of α3β4-expressing HEK293 cells captured during control, repeated application of 30 μM ACh (upper images), or application of 30 μM ACh alone and ACh plus 100 μM menthol (lower images). (B, upper trace) Mean Fluo-4 fluorescence during repeated application of ACh (n = 39 cells) or (B, lower trace) ACh and ACh plus menthol (n = 37 cells). The interstimulus interval was 5 minutes. (C) Inhibition of nicotine-stimulated 86Rb+ efflux in the presence of menthol or (D) 10-minute preincubation with menthol; n = 3 independent assays.
We next examined menthol’s effect on nAChR channel function by measuring nicotine-stimulated 86Rb+ efflux in these cells. Menthol coapplied with 100 μM nicotine inhibited nicotine-stimulated 86Rb+ efflux in a concentration-dependent manner, with an IC50 of 100 ± 8 μM (Fig. 1C). Preincubation with menthol for 10 minutes before addition of nicotine decreased menthol’s IC50 only slightly, to 69 ± 8 μM (Fig. 1D).
To determine if menthol acted at the agonist binding site of α3β4 nAChRs, we examined its competition for nAChR binding sites labeled by [3H]EB in cell membrane homogenates, and compared it to nicotine. As shown in Fig. 2, in contrast to nicotine, which competed effectively for binding with a Ki of ∼250 nM, menthol at concentrations up to 1 mM was completely ineffective.
Fig. 2.

Menthol does not compete for the nAChR agonist binding site labeled by [3H]epibatidine. Membranes from human embryonic kidney cells expressing α3β4 nAChRs were incubated for 2 hours with ∼0.5 nM [3H]EB in the absence or presence of increasing concentrations of menthol or nicotine. The membranes were then filtered and counted. Data were analyzed by nonlinear least-squares regression analysis. The Ki of nicotine in these studies was 256 nM. Menthol at concentrations up to 1 mM did not compete for these receptors. Data shown are the mean ± S.E.M. of five independent assays.
Menthol Enhances the Apparent Desensitization of Human α3β4 nAChR Activity.
To further explore how menthol inhibits the function of α3β4 nAChRs we performed whole-cell voltage-clamp recordings. Application of 30 or 100 μM ACh to cells expressing α3β4 nAChRs evoked inward currents with rapid onset and minimal desensitization (Fig. 3, A and B). However, coapplication of menthol (100 μM) with ACh markedly increased the rate and magnitude of desensitization without appreciably affecting the peak response to ACh (Fig. 3, A and B). These effects were nearly completely reversed 60 seconds after removal of menthol (Fig. 3A, right trace).
Fig. 3.

Menthol enhances the decay of ACh-evoked currents. Representative inward currents in HEK293 cells expressing α3β4 nAChRs in response to (A) 30 μM ACh and (B) 100 μM, with or without menthol (100 μM) and after 60-second washout (A). The holding potential was –50 mV. (C) Menthol (100 μM) alone evokes no current. (D) A 5-minute treatment with menthol (100 μM) alone does not affect the subsequent current evoked by ACh (30 μM). (E) Summary of the peak current evoked by ACh and amount of current decay following 5-minute pretreatment with either control or menthol (n = 3). D, desensitization.
Menthol could potentially act as an agonist or partial agonist to increase receptor desensitization; however, consistent with its lack of binding to the receptor, we found that application of menthol alone (1–1000 μM) did not elicit inward currents (Fig. 3C). Further, a 5-minute treatment with 100 μM menthol alone followed by washout did not affect a subsequent ACh-evoked response (Fig. 3D). Neither the peak ACh-evoked current nor the level of desensitization was affected by menthol pretreatment (Fig. 3, D and E). These data indicate that menthol alone produces little to no desensitization, and that its effects thus require the presence of ACh.
Inhibitory Effects of Menthol Are Dependent on the Concentration of ACh.
We next tested the effect of menthol (300 μM) on inward currents evoked by different concentrations of ACh ranging from 3–300 μM. We analyzed these currents to determine the level of desensitization (D) and the time constant (τ) or weighted time constant (τw) for current decay obtained by the best fits to single- or double-exponential functions. As shown in Fig. 4A, menthol had no measurable effect on currents evoked by a low concentration of ACh (3 μM), which exhibited a very slow rate of desensitization (τ ∼50 seconds; Fig. 4B) and a low level of desensitization (∼25%; Fig. 4C) with or without menthol. However, at ACh concentrations of 30 and 300 μM, menthol markedly increased the rate and extent of desensitization (Fig. 4A). Thus, as shown in Fig. 4, B and C, at 30 μM ACh, menthol decreased the τw from 35 seconds to 2.4 seconds and increased D from 35 to 91%; and at 300 μM ACh, menthol decreased the τw from 21 to 0.9 seconds and increased D from 74 to 94%. At 300 μM ACh, the τw approaches the time to activation of ∼1 second (limited by the solution exchange time). Consequently, at these higher concentrations of ACh, the peak currents evoked in the presence of menthol were very much decreased (Fig. 4A). These results demonstrate the importance of the ACh concentration in the actions of menthol at α3β4 nAChRs. Moreover, the fact that menthol had little effect on desensitization parameters at the lowest ACh concentration used here, but a large effect as the ACh concentrations were increased and open-channel probability increased, indicates that menthol acts in a state-dependent manner; that is, menthol acts preferentially on the open or desensitized state of the channel.
Fig. 4.

Menthol modulation of α3β4 nAChRs is dependent on the concentration of ACh. (A) Representative whole-cell inward currents in response to different concentrations of ACh in the absence (left) and in the presence (right) of 300 μM menthol in α3β4 nAChR-expressing cells. The current decay during desensitization was best fit to a one- or two-exponential function, yielding the indicated time constants. (B) Mean weighted average time constants for decay and (C) percent of desensitization (D) of the ACh-mediated current obtained in the absence of menthol (open circle) and in the presence of 300 μM of menthol (filled circle). Data are mean ± S.E.M., n = 3–5.
Menthol Inhibits in a Voltage-Independent Manner and Without Affecting Single-Channel Conductance.
The observation that the effects of menthol depend on ACh concentration raised the possibility of an open-channel block mechanism. To explore this hypothesis we tested for voltage-dependent effects of menthol. Figure 5A shows the current-voltage relationship (elicited by 200-millisecond voltage ramps) during the peak response to ACh, and 20 seconds after addition of menthol (100 μM). Both traces exhibit characteristic inward rectification that is relieved at high positive membrane potentials. The inset reveals the fractional current (menthol/control) at different voltages and shows that menthol likewise reduced the current by ∼50% at all potentials. Therefore, menthol inhibits nAChRs in a voltage-independent manner. In addition, these data show that menthol acts independently of the direction of net current flow, which is inward and outward, respectively, at negative and positive potentials. We next tested for use-dependent effects that are characteristic of many open-channel blockers. Figure 5, B and C, shows the response of repeated, 5-second applications of ACh with or without continued presence of 200 μM menthol. The relatively brief application duration was chosen to minimize desensitization. In both cases the peak responses exhibited a marginal decrease with successive ACh applications (Fig. 5C) but there was no difference between control and menthol treatments. Thus, menthol does not produce a rapid, use-dependent block, although we cannot exclude the possibility that menthol binds very slowly to the pore (>>5 seconds) and therefore failed to significantly inhibit current during brief applications of ACh used here. Finally, we tested the effects of menthol on single-channel activity. Figure 5D shows representative current traces from a cell-attached recording before and after application of 200 μM menthol. Under control conditions channel activity consisted of bursts of openings separated by long and variable closed times. Menthol markedly reduced the mean open time from 32.4 ± 6.4 milliseconds to 5.7 ± 1.1 milliseconds (Fig. 5, D and F) without affecting the single-channel conductance (Fig. 5E; slope conductance 30.9 versus 30.3 pS). Taken together, these data suggest that menthol inhibits α3β4 nAChRs in an allosteric manner by altering gating rather than simply blocking the channel pore.
Fig. 5.

Menthol inhibits α3β4 nAChRs in a voltage- and use-independent manner and without affecting single-channel conductance. (A) Current voltage relationship for peak response to ACh (30 μM) and after 20-second application of menthol (100 μM). The background current in the absence of ACh is subtracted. The inset shows the fraction of the menthol versus control current at indicated potentials. (B) Responses to repeated, 5-second stimulation with ACh (30 μM) under control conditions (upper trace) or in the presence of 200 μM menthol (lower trace). Scale bars, 1 nA and 20 seconds. (C) Mean peak responses to ACh from experiments depicted in (B) (n = 3). (D) Representative single-channel currents from a cell-attached patch (VM, –110 mV, 10 μM ACh in pipette) under control conditions (upper trace) and in the presence of 200 μM menthol. (E) All-points histograms constructed from 2 seconds of continuous data. The smooth lines represent best-fits to Gaussian functions yielding similar conductances (G) of 30.9 and 30.3 pS respectively. (F) Mean open time measured from data in (E) (measured from >50 events).
Increasing Menthol Concentration Enhances the Desensitization of the ACh-Induced Currents.
We next examined the concentration dependent effects of menthol on currents evoked by 30 μM ACh. Figure 6A shows that both the rate and extent of desensitization increases with increasing menthol concentration. In assessing the desensitization parameters, τw and D, values were normalized to data obtained from the same cell in menthol-free conditions. Menthol increased the rate of desensitization in a concentration dependent manner and at 300 μM almost completely desensitized currents within 1 second of ACh/menthol application (Fig. 6, A and B). The extent of desensitization, D, also increased with increasing menthol concentration, and at the end of the 40-second application of ACh in the presence of 300 μM menthol, the extent of desensitization was more than twice as great as in the absence of menthol (Fig. 6C). Figure 6D shows the current integral (the total current passed during the 40-second application of agonist) as a function of menthol concentration. Half-maximal inhibition (obtained by the best fit to a Hill equation) occurred at 43 μM, similar to the values obtained by the 86Rb+ efflux measurements (see Fig. 1, C and D). Notably, these values are also similar to the EC50 for menthol activity at TRPM8 receptors of ∼55–80 μM at room temperature (McKemy et al., 2002; Premkumar et al., 2005) and ∼30 μM for activation of human TRPA1 (Xiao et al., 2008). Thus, the concentration of menthol needed to drive its sensory perception (cooling sensation and pungency) in airway C fibers is also sufficient to decrease channel activity through α3β4 nAChRs by promoting desensitization.
Fig. 6.

Concentration-dependent effects of menthol. (A) Representative currents activated by ACh (30 μM) and ACh plus menthol (30 and 300 μM); (B) Bar graph of time constant; (C) the fraction of desensitized (D) receptors; and (D) the current integrals in response to the concentration of menthol from 10 to 300 μM, which was coapplied with 30 μM ACh. Data are the normalized means ± S.E.M. of three to six experiments.
Menthol Enhances Desensitization of Currents Evoked by Nicotine.
To corroborate and further explore the effects of menthol on nAChRs, we determined if these effects extended to nicotine, the addictive component of tobacco. As with its effect on the ACh-induced currents, we observed that menthol enhanced the apparent desensitization of the nicotine-induced currents by reducing the time constant and increasing current decay during a 40-second concomitant application of nicotine and 100 μM menthol. Figure 7, A–D, illustrate the currents induced by nicotine alone at concentrations of 1–100 μM (left column); the combination of nicotine and 100 μM menthol (middle); and a second application of nicotine alone after a 5-minute washout of the menthol (right). At a low concentration of nicotine (1 μM), menthol had no measurable effect on the currents. At concentrations of 3 and 10 μM nicotine, the decay of current during the 40-second nicotine exposure was fit best to a single exponential with time constants of 18.1 and 15.9 seconds, respectively, and the current desensitized by 26 and 22%. In the presence of menthol, the time constants of the current decay were decreased to 9.5 and 10.8 seconds, respectively, and there was a nearly 2-fold increase in the extent of desensitization at both concentrations of nicotine. After the 5-minute washout, the time constant and the degree of desensitization to 3 μM nicotine nearly fully recovered to the initial control levels. The time constant to 10 μM nicotine did not fully recover, but the degree of desensitization did. At a nicotine concentration of 100 μM, the decay current during the 40-second nicotine exposure was fit best to a single exponential with a time constant of 9.4 seconds, and the current desensitized by 60%. In the presence of menthol, the decay current was best fit to two exponentials with time constants of 5.1 seconds and 1.9 seconds, which yielded a weighted time constant, τw, of 3.2 seconds; the current in the presence of menthol desensitized by 78%. After the 5-minute washout, the decay constant again fit best to a single exponential but had only partially recovered to the original control value. Likewise, the degree of desensitization had not recovered completely. Taken together, these findings indicate menthol enhances the apparent desensitization of human α3β4 nAChRs to nicotine.
Fig. 7.

Menthol enhances the desensitization of the currents evoked by nicotine. Representative inward currents induced by (A) 1 μM, (B) 3 μM, (C) 10 μM, and (D) 100 μM nicotine in the absence of menthol (left), presence of 100 μM menthol (middle), and 5 minutes after washout of the menthol (right). Time constant (τ) and the extent of desensitization (D) were used to characterize the desensitization.
Menthol Traps α3β4 nAChRs in the Desensitized State.
We examined next whether continued presence of menthol would hinder the receptor’s recovery from desensitization. We stimulated receptors with 30 μM ACh in the presence of menthol to promote desensitization, and subsequently measured the time course for recovery (over 1–5 minutes) in either the presence or absence of 100 μM menthol. Figure 8 shows that in the absence of menthol the α3β4 receptors recovered rapidly from desensitization with ACh (Fig. 8A), with 83 ± 5% (n = 3) recovery after 1 minute and 97 ± 2% (n = 5) recovery after 5 minutes (Fig. 8B). In contrast, in the presence of menthol, recovery from desensitization was greatly attenuated (Fig. 8A), with only 20 ± 3% (n = 3), 26 ± 0.6 (n = 3), and 33 ± 5% (n = 5) recovery after 1, 3, and 5 minutes, respectively (Fig. 8B). As shown in Fig. 3D, treating nondesensitized α3β4 receptors with menthol had minimal effect on subsequent recovery, suggesting that menthol acts mainly on the desensitized state. To further discriminate actions of menthol at open or desensitized states, we tested effects of menthol on α3β4 nAChRs that were already fully desensitized by a high concentration (300 μM) of ACh. Figure 8C shows that menthol (100 μM) almost fully prevented recovery of these receptors from desensitization; however, following removal of menthol, channel activity was nearly fully restored (n = 3). Taken together, these data suggest that menthol traps α3β4 nAChRs preferentially in their desensitized state(s), thereby markedly slowing their recovery from desensitization. Thus, menthol may both augment initial agonist-induced desensitization and prolong it.
Fig. 8.

Menthol hinders the recovery of α3β4 nAChRs from desensitization. (A) Representative inward currents showing desensitization induced by coapplied 30 μM ACh and 100 μM menthol and the subsequent response to ACh following a 5-minute wash in either control bath solution (upper traces) or 100 μM menthol (lower traces). (B) Time course for recovery following the treatment described in (A), control (open circles), and menthol (closed circles). Data are the means ± S.E.M. of three to five experiments. (C) Menthol (100 μM) prevents recovery of α3β4 nAChRs following desensitization with 300 μM Ach, but receptors almost fully recover after 1 minute washout with control solution (n = 3).
Menthol Inhibits ACh-Evoked Currents in Nodose Ganglia Neurons.
To test whether menthol likewise affects native α3β4 nAChRs, we examined responses in cultured nodose ganglia neurons. These neurons send vagal projections to the lung and predominantly express the α3β4 nAChR subtype (Mao et al., 2006). Figure 9 shows that ACh evoked a slowly desensitizing current in a voltage-clamped sensory neuron. Coapplication of menthol markedly reduced the peak current and increased the extent of desensitization from ∼50 to ∼90%. The current responses to ACh nearly fully recovered following washout of the menthol. These responses in nodose neurons are therefore consistent with menthol increasing the speed and magnitude of desensitization and mirror the responses observed with recombinant α3β4 nAChRs.
Fig. 9.
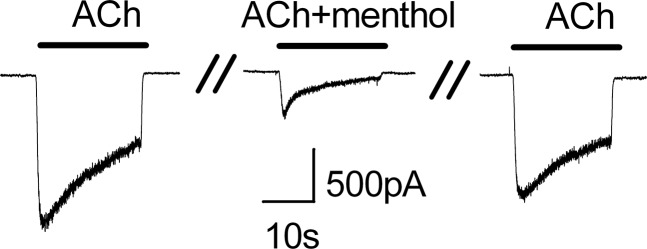
Menthol inhibits ACh-evoked currents in nodose ganglia neurons. Representative current traces in a voltage-clamped neuron evoked by ACh (30 μM) and ACh plus menthol (100 μM). Note that menthol increases the speed and extent of desensitization. The cell was washed for 60 seconds between ACh applications.
Discussion
The studies presented here demonstrate that menthol attenuates signaling through human α3β4 nAChRs. This was shown with three different kinds of measurements: ACh-stimulated Ca2+ signaling, nicotine-stimulated 86Rb+ efflux, and both ACh- and nicotine-stimulated currents measured by whole-cell and single-channel voltage-clamp recordings. Our data also demonstrate that menthol does not bind to the orthosteric site, indicating that its actions are via an allosteric mechanism. Importantly, these effects of menthol at nAChRs occurred at pharmacologically relevant concentrations similar to those required to activate TRPM8 and TRPA1 receptors.
Previous studies also found that menthol attenuated nAChR functions. Hans et al. (2012) found that menthol at concentrations similar to those used here decreased nicotine-stimulated currents through unidentified nAChRs in trigeminal ganglia cells and reduced single-channel currents in cells heterologously expressing α4β2 nAChRs. Their data also suggested that menthol acted as a negative allosteric modulator. Ashoor et al. (2013) found that menthol attenuated function of α7 nAChRs expressed in Xenopus oocytes but did not compete for α7 receptor binding sites, again suggesting a negative allosteric effect.
Our studies extend these findings to human α3β4 nAChRs and, importantly, identify augmented desensitization as the mechanism by which menthol attenuates this receptor’s function. This conclusion is supported by the following observations: First, addition of menthol to cells for 5 minutes and its removal immediately before addition of agonist did not alter the response of α3β4 nAChRs to ACh, indicating that menthol did not produce a long-lasting effect in the absence of an agonist. Second, the effects of menthol on channel function are minimal or absent in the presence of low concentrations of agonist but become prominent as the agonist concentration is increased to the level where desensitization begins to occur. This indicates that menthol acts preferentially on the open state or desensitized state(s) of the channel. Third, the effects of menthol are independent of the membrane voltage and the net current direction, two parameters that can affect the actions of open-channel blockers. Furthermore, menthol did not produce a rapid, use-dependent inhibition nor reduce the single-channel conductance characteristic of open-channel blockade. Rather, menthol reduced the open probability and mean open time of single α3β4 nAChR channels consistent with alterations in channel gating. Interestingly, Hans et al. (2012) reported similar effects of menthol on single-channel properties of α4β2 nAChRs. Fourth, menthol markedly delayed recovery from desensitization; in particular, menthol prevented the recovery of receptors already fully desensitized by a high concentration of ACh. This suggests that menthol binds and traps the receptor in a desensitized conformation(s). Importantly, these data argue strongly against the possibility that menthol acts as a slow, open-channel blocker. Instead, menthol appears to speed and magnify nAChR desensitization and stabilize a desensitized conformation. Thus, we propose that menthol augments and facilitates the normally weak desensitizing effect of ACh and low concentrations of nicotine, with the result that menthol, acting via an allosteric site, markedly decreases the current carried by the receptor.
Allosteric modulators, both positive and negative, of nAChRs have been studied previously (for review, see Pandya and Yakel, 2011; Williams et al., 2011). These modulators include metal ions (Vernino et al., 1992; Zwart et al., 1995; Hsiao et al., 2001), steroid hormones (Valera et al., 1992; Ke and Lukas, 1996; Paradiso et al., 2000; Curtis et al., 2002), and small synthetic ligands (Bertrand and Gopalakrishnan, 2007; Moaddel et al., 2007; Henderson et al., 2010). In fact, menthol has recently been reported to allosterically inhibit currents mediated by α4β2 (Hans et al., 2012) and α7 (Ashoor et al., 2013) nAChRs. However, to our knowledge menthol is the first example of a drug demonstrated to act allosterically to augment desensitization of a neuronal nAChR without activating or even binding to the receptor’s orthosteric site.
nAChRs are desensitized immediately after or even during their activation by high concentrations of ACh or by nicotine and other nicotinic agonists and partial agonists. Moreover, this desensitization usually lasts much longer than the brief agonist-induced activation (Katz and Thesleff, 1957; Sharp and Beyer, 1986; Hulihan-Giblin et al., 1990). We do not yet know whether menthol’s effect of augmenting desensitization extends to other nAChR subtypes, but interestingly, the α3β4 nAChR subtype is one of the slowest to desensitize and fastest to resensitize (Cachelin and Jaggi, 1991; Fenster et al., 1997; Quick and Lester, 2002); thus, the effect of menthol-augmented desensitization at this receptor may be especially important.
The suppression of α3β4 nAChR activity by menthol has potentially important implications for its analgesic effects in sensory nerves and in airways. Rat trigeminal ganglia neurons innervating the mouth and throat express predominantly α3β4 nAChRs (Flores et al., 1996), as do nodose ganglia neurons innervating the airways (Mao et al., 2006). Bronchial epithelial cells express this subtype, as well as other nAChRs (Maus et al., 1998; Wang et al., 2001). Notably, compared with other subtypes, the α3β4 nAChR is more resistant to agonist-induced desensitization (Olale et al., 1997); thus, menthol may augment the desensitization effects of nicotine at these receptors. Indeed, we found that menthol markedly inhibited ACh-evoked currents in nodose sensory neurons apparently by increasing the speed and magnitude of desensitization. The consequences of this effect are not known with certainty, but one possibility is that it could offset the irritant effects of nicotine in the airways, allowing cigarette smoke to be inhaled deeper into the lungs and held there for a longer time. Thus, the known analgesic effects of menthol, acting via sensory nerve TRPM8 (Willis et al., 2011) and TRPA1 channels, may be augmented by enhanced desensitization of sensory nerve nAChRs.
Interestingly, desensitization of brain α3β4 nAChRs may also be an important component of nicotine addiction. For example, mice null for the β4 nAChR subunit display fewer signs of withdrawal from nicotine (Salas et al., 2004). Accordingly, by augmenting desensitization of brain α3β4 nAChRs, menthol may likewise delay or blunt signs and symptoms of nicotine withdrawal. Additionally, one hypothesis supporting an underlying mechanism of nicotine addiction predicts that the drive to smoke a cigarette is prompted by a cyclical need to desensitize overactive brain nAChRs, some of which are upregulated by chronic administration of nicotine (Dani and Heinemann, 1996; Hussmann et al., 2012). In both of these cases, menthol’s effect of increasing desensitization of the nAChRs that underlies addiction and/or withdrawal may actually result in less nicotine being needed to desensitize the overactive receptors and to blunt withdrawal effects. If menthol were to modulate nAChRs in the central nervous system, then sufficient levels of menthol must accumulate in the brain along with nicotine. Although precise concentrations of menthol in the brains of smokers are unknown, the results of an animal study show that menthol can readily penetrate the central nervous system. For example, Pan et al. (2012) showed that menthol can reach high levels in the brains of mice within 5 minutes of a bolus intraperitoneal injection and was still measurable 60 minutes after injection.
In conclusion, we have shown that menthol in the presence of ACh or nicotine acts allosterically to augment desensitization of α3β4 nAChRs, resulting in decreased agonist-induced intracellular Ca2+ and currents. In addition, menthol appears to prolong the time that the receptor resides in a desensitized state.
Acknowledgments
The authors thank Phan Thieu for assistance with cell culture.
Abbreviations
- [3H]EB
[3H]epibatidine
- nAChR
nicotinic acetylcholine receptor
Author Contributions
Participated in research design: Ton, Kellar, Ahern.
Conducted experiments: Ton, Smart, Aguilar, Olson, Ahern.
Performed data analysis: Ton, Kellar, Ahern.
Wrote or contributed to the writing of the manuscript: Ton, Aguilar, Kellar, Ahern.
Footnotes
This study was supported by the National Institutes of Health National Institute on Drug Abuse [Grant R01-DA012976] and National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant U01-DK101040].
References
- Ashoor A, Nordman JC, Veltri D, Yang K-HS, Al Kury L, Shuba Y, Mahgoub M, Howarth FC, Sadek B, Shehu A, et al. (2013) Menthol binding and inhibition of α7-nicotinic acetylcholine receptors. PLoS ONE 8:e67674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, Jordt S-E, Julius D. (2007) The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 448:204–208. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. (2007) Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol 74:1155–1163. [DOI] [PubMed] [Google Scholar]
- Cachelin AB, Jaggi R. (1991) Beta subunits determine the time course of desensitization in rat alpha 3 neuronal nicotinic acetylcholine receptors. Pflugers Arch 419:579–582. [DOI] [PubMed] [Google Scholar]
- Curtis L, Buisson B, Bertrand S, Bertrand D. (2002) Potentiation of human alpha4beta2 neuronal nicotinic acetylcholine receptor by estradiol. Mol Pharmacol 61:127–135. [DOI] [PubMed] [Google Scholar]
- Dani JA, Heinemann S. (1996) Molecular and cellular aspects of nicotine abuse. Neuron 16:905–908. [DOI] [PubMed] [Google Scholar]
- Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. (1997) Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci 17:5747–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CM, DeCamp RM, Kilo S, Rogers SW, Hargreaves KM. (1996) Neuronal nicotinic receptor expression in sensory neurons of the rat trigeminal ganglion: demonstration of alpha3beta4, a novel subtype in the mammalian nervous system. J Neurosci 16:7892–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans M, Wilhelm M, Swandulla D. (2012) Menthol suppresses nicotinic acetylcholine receptor functioning in sensory neurons via allosteric modulation. Chem Senses 37:463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BJ, Pavlovicz RE, Allen JD, González-Cestari TF, Orac CM, Bonnell AB, Zhu MX, Boyd RT, Li C, Bergmeier SC, et al. (2010) Negative allosteric modulators that target human alpha4beta2 neuronal nicotinic receptors. J Pharmacol Exp Ther 334:761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao B, Dweck D, Luetje CW. (2001) Subunit-dependent modulation of neuronal nicotinic receptors by zinc. J Neurosci 21:1848–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulihan-Giblin BA, Lumpkin MD, Kellar KJ. (1990) Acute effects of nicotine on prolactin release in the rat: agonist and antagonist effects of a single injection of nicotine. J Pharmacol Exp Ther 252:15–20. [PubMed] [Google Scholar]
- Hussmann GP, Turner JR, Lomazzo E, Venkatesh R, Cousins V, Xiao Y, Yasuda RP, Wolfe BB, Perry DC, Rezvani AH, et al. (2012) Chronic sazetidine-A at behaviorally active doses does not increase nicotinic cholinergic receptors in rodent brain. J Pharmacol Exp Ther 343:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karashima Y, Damann N, Prenen J, Talavera K, Segal A, Voets T, Nilius B. (2007) Bimodal action of menthol on the transient receptor potential channel TRPA1. J Neurosci 27:9874–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Thesleff S. (1957) A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol 138:63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke L, Lukas RJ. (1996) Effects of steroid exposure on ligand binding and functional activities of diverse nicotinic acetylcholine receptor subtypes. J Neurochem 67:1100–1112. [DOI] [PubMed] [Google Scholar]
- Lee L-Y, Burki NK, Gerhardstein DC, Gu Q, Kou YR, Xu J. (2007) Airway irritation and cough evoked by inhaled cigarette smoke: role of neuronal nicotinic acetylcholine receptors. Pulm Pharmacol Ther 20:355–364. [DOI] [PubMed] [Google Scholar]
- Macpherson LJ, Hwang SW, Miyamoto T, Dubin AE, Patapoutian A, Story GM. (2006) More than cool: promiscuous relationships of menthol and other sensory compounds. Mol Cell Neurosci 32:335–343. [DOI] [PubMed] [Google Scholar]
- Mao D, Yasuda RP, Fan H, Wolfe BB, Kellar KJ. (2006) Heterogeneity of nicotinic cholinergic receptors in rat superior cervical and nodose Ganglia. Mol Pharmacol 70:1693–1699. [DOI] [PubMed] [Google Scholar]
- Maus AD, Pereira EF, Karachunski PI, Horton RM, Navaneetham D, Macklin K, Cortes WS, Albuquerque EX, Conti-Fine BM. (1998) Human and rodent bronchial epithelial cells express functional nicotinic acetylcholine receptors. Mol Pharmacol 54:779–788. [DOI] [PubMed] [Google Scholar]
- McCallum SE, Cowe MA, Lewis SW, Glick SD. (2012) α3β4 nicotinic acetylcholine receptors in the medial habenula modulate the mesolimbic dopaminergic response to acute nicotine in vivo. Neuropharmacology 63:434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKemy DD, Neuhausser WM, Julius D. (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416:52–58. [DOI] [PubMed] [Google Scholar]
- Meyer EL, Xiao Y, Kellar KJ. (2001) Agonist regulation of rat alpha 3 beta 4 nicotinic acetylcholine receptors stably expressed in human embryonic kidney 293 cells. Mol Pharmacol 60:568–576. [PubMed] [Google Scholar]
- Moaddel R, Jozwiak K, Wainer IW. (2007) Allosteric modifiers of neuronal nicotinic acetylcholine receptors: new methods, new opportunities. Med Res Rev 27:723–753. [DOI] [PubMed] [Google Scholar]
- Olale F, Gerzanich V, Kuryatov A, Wang F, Lindstrom J. (1997) Chronic nicotine exposure differentially affects the function of human alpha3, alpha4, and alpha7 neuronal nicotinic receptor subtypes. J Pharmacol Exp Ther 283:675–683. [PubMed] [Google Scholar]
- Pan R, Tian Y, Gao R, Li H, Zhao X, Barrett JE, Hu H. (2012) Central mechanisms of menthol-induced analgesia. J Pharmacol Exp Ther 343:661–672. [DOI] [PubMed] [Google Scholar]
- Pandya A, Yakel JL. (2011) Allosteric modulators of the α4β2 subtype of neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 82:952–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso K, Sabey K, Evers AS, Zorumski CF, Covey DF, Steinbach JH. (2000) Steroid inhibition of rat neuronal nicotinic alpha4beta2 receptors expressed in HEK 293 cells. Mol Pharmacol 58:341–351. [DOI] [PubMed] [Google Scholar]
- Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, et al. (2002) A TRP channel that senses cold stimuli and menthol. Cell 108:705–715. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Raisinghani M, Pingle SC, Long C, Pimentel F. (2005) Downregulation of transient receptor potential melastatin 8 by protein kinase C-mediated dephosphorylation. J Neurosci 25:11322–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RAJ. (2002) Desensitization of neuronal nicotinic receptors. J Neurobiol 53:457–478. [DOI] [PubMed] [Google Scholar]
- Salas R, Pieri F, De Biasi M. (2004) Decreased signs of nicotine withdrawal in mice null for the beta4 nicotinic acetylcholine receptor subunit. J Neurosci 24:10035–10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp BM, Beyer HS. (1986) Rapid desensitization of the acute stimulatory effects of nicotine on rat plasma adrenocorticotropin and prolactin. J Pharmacol Exp Ther 238:486–491. [PubMed] [Google Scholar]
- Valera S, Ballivet M, Bertrand D. (1992) Progesterone modulates a neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci USA 89:9949–9953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernino S, Amador M, Luetje CW, Patrick J, Dani JA. (1992) Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron 8:127–134. [DOI] [PubMed] [Google Scholar]
- Wang Y, Pereira EF, Maus AD, Ostlie NS, Navaneetham D, Lei S, Albuquerque EX, Conti-Fine BM. (2001) Human bronchial epithelial and endothelial cells express alpha7 nicotinic acetylcholine receptors. Mol Pharmacol 60:1201–1209. [DOI] [PubMed] [Google Scholar]
- Williams DK, Wang J, Papke RL. (2011) Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem Pharmacol 82:915–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis DN, Liu B, Ha MA, Jordt S-E, Morris JB. (2011) Menthol attenuates respiratory irritation responses to multiple cigarette smoke irritants. FASEB J 25:4434–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Dubin AE, Bursulaya B, Viswanath V, Jegla TJ, Patapoutian A. (2008) Identification of transmembrane domain 5 as a critical molecular determinant of menthol sensitivity in mammalian TRPA1 channels. J Neurosci 28:9640–9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Kellar KJ. (2004) The comparative pharmacology and up-regulation of rat neuronal nicotinic receptor subtype binding sites stably expressed in transfected mammalian cells. J Pharmacol Exp Ther 310:98–107. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Meyer EL, Thompson JM, Surin A, Wroblewski J, Kellar KJ. (1998) Rat alpha3/beta4 subtype of neuronal nicotinic acetylcholine receptor stably expressed in a transfected cell line: pharmacology of ligand binding and function. Mol Pharmacol 54:322–333. [DOI] [PubMed] [Google Scholar]
- Zwart R, Van Kleef RG, Milikan JM, Oortgiesen M, Vijverberg HP. (1995) Potentiation and inhibition of subtypes of neuronal nicotinic acetylcholine receptors by Pb2+. Eur J Pharmacol 291:399–406. [DOI] [PubMed] [Google Scholar]