

Abstract
Farm animals are a potential reservoir for human Clostridium difficile infection (CDI), particularly PCR ribotype 078 which is frequently found in animals and humans. Here, whole genome single-nucleotide polymorphism (SNP) analysis was used to study the evolutionary relatedness of C. difficile 078 isolated from humans and animals on Dutch pig farms. All sequenced genomes were surveyed for potential antimicrobial resistance determinants and linked to an antimicrobial resistance phenotype. We sequenced the whole genome of 65 C. difficile 078 isolates collected between 2002 and 2011 from pigs (n = 19), asymptomatic farmers (n = 15) and hospitalised patients (n = 31) in the Netherlands. The collection included 12 pairs of human and pig isolates from 2011 collected at 12 different pig farms. A mutation rate of 1.1 SNPs per genome per year was determined for C. difficile 078. Importantly, we demonstrate that farmers and pigs were colonised with identical (no SNP differences) and nearly identical (less than two SNP differences) C. difficile clones. Identical tetracycline and streptomycin resistance determinants were present in human and animal C. difficile 078 isolates. Our observation that farmers and pigs share identical C. difficile strains suggests transmission between these populations, although we cannot exclude the possibility of transmission from a common environmental source.
Introduction
In the past decade Clostridium difficile has emerged rapidly to become the most common cause of antibiotic-associated diarrhoea in healthcare facilities worldwide. Antibiotic treatment, advanced age and hospitalisation are the major risk factors for developing C. difficile infection (CDI) leading to diarrhoea, pseudomembranous colitis or death [1,2]. CDI is increasingly recognised in the community setting [3-6] where exposure to antibiotics is an important risk factor [5], while the use of proton pump inhibitors [4], outpatient healthcare exposure [7], obesity and inflammatory bowel disease (IBD) [8] are potential risk factors. C. difficile virulence is primarily mediated by two potent enterotoxins, TcdA and TcdB, which are encoded in a pathogenicity locus (PaLoc) [9-11]. The binary toxin may contribute to the virulence of C. difficile as well [12], but its role in CDI is still under debate [13-16]. C. difficile produces highly resistant and infectious spores, which can survive in the environment for a long time and facilitate environmental transmission within the healthcare setting [17].
Symptomatic individuals are an important source of C. difficile transmission in a hospital setting, and patient isolation and antibiotic stewardship have been proven to be effective infection control measures [18,19]. The role of asymptomatic carriers as donors of transmission may also be significant [20-23], and diverse novel subtypes are continuously introduced in the healthcare system, highlighting a link to a large and diverse community reservoir [24]. Interestingly, C. difficile PCR ribotype 078, which is commonly found in the healthcare system of various European countries [25], is more often associated with community-acquired CDI [26]. Notably, this variant is the most common type found in pigs [27-30] and other farm animals [31-33].
Several studies have reported an overlap between C. difficile genotypes isolated from humans and animals [27,34-38] using conventional typing methods such as PCR ribotyping, multilocus sequence typing (MLST), and multilocus variable-number tandem repeat analysis (MLVA). However, these methods do not have the discriminatory power to distinguish between closely related strains as is required for transmission tracking. In this study, we used whole genome sequencing and phylogenetic analysis to track single clones in human and animal populations to demonstrate potential interspecies transmission.
Methods
Collection of Clostridium difficile isolates
In total, the genomes of 65 isolates designated PCR ribotype 078 were sequenced and analysed. Of these 65 isolates, 34 were derived from healthy humans (n=15) and pigs (n=19) on 19 Dutch pig farms (farm isolates) and 31 from hospitalised patients in various Dutch hospitals. Of the farm isolates, 24 isolates were paired by farm (i.e. 12 pairs of human and pig isolates from 12 farms), whereas the remaining 10 (from three farmers and seven pigs) were not paired. The majority of the farm isolates were collected in 2011 by the Institute for Risk Assessment Sciences of the Utrecht University as part of another study [34]. Thirty-one randomly selected clinical isolates originating from various Dutch hospitals between 2002 and 2011 were obtained from the Dutch National C. difficile reference laboratory at Leiden University Medical Center. In addition, one PCR ribotype 066 strain was included; this strain was obtained from our Leeds-Leiden/European Centre for Disease Prevention and Control (ECDC) reference strain collection [39]. Details of all sequenced isolates are listed in Table 1, including the European Nucleotide Archive (ENA) sample accession numbers. Two isolates were sequenced in duplicate.
Table 1A. Clostridium difficile type 078 isolates used in this study, the Netherlands, 2002–11 (n=65).
| R_L#Ta | Year | City | RT | Isolate | Source | Related isolates | Association | ENA IDb |
|---|---|---|---|---|---|---|---|---|
| 8080_2#24 | 2006 | Leiden | 078 | 6072310 | Clinic | Non-outbreak | Healthcare | ERS138026 |
| 8080_2#25 | 2006 | Nijmegen | 078 | 6086336 | Clinic | Non-outbreak | Healthcare | ERS138027 |
| 8080_2#26 | 2007 | Leiden | 078 | 7001233 | Clinic | Non-outbreak | Healthcare | ERS138028 |
| 8080_2#27 | 2007 | Groningen | 078 | 7004578 | Clinic | Non-outbreak | Unknown | ERS138029 |
| 8080_2#28 | 2007 | Utrecht | 078 | 7005405 | Clinic | Non-outbreak | Unknown | ERS138030 |
| 8080_2#29 | 2007 | Zwolle | 078 | 7021455 | Clinic | Non-outbreak | Healthcare | ERS138031 |
| 8080_2#30 | 2007 | Zwolle | 078 | 7044912 | Clinic | Non-outbreak | Community | ERS138032 |
| 8080_2#31 | 2007 | Zwolle | 078 | 7066827 | Clinic | Non-outbreak | Community | ERS138033 |
| 8080_2#32 | 2007 | Zwolle | 078 | 7071308 | Clinic | Non-outbreak | Healthcare | ERS138034 |
| 8080_2#33 | 2007 | Zwolle | 078 | 7086074 | Clinic | Non-outbreak | Healthcare | ERS138035 |
| 8080_2#34 | 2007 | Leiden | 078 | 7091952 | Clinic | Non-outbreak | Healthcare | ERS138036 |
| 8080_2#35 | 2008 | Leiden | 078 | 8011061 | Clinic | Non-outbreak | Healthcare | ERS138037 |
| 8080_2#36 | 2008 | Utrecht | 078 | 8013820 | Clinic | Non-outbreak | Healthcare | ERS138038 |
| 8080_2#37 | 2008 | Leiden | 078 | 8051728 | Clinic | Non-outbreak | Healthcare | ERS138039 |
| 8080_2#38 | 2008 | Leiden | 078 | 8055344 | Clinic | Non-outbreak | Healthcare | ERS138040 |
| 11250_1#22 | 2008 | Arnhem | 078 | 8056692 | Clinic | Non-outbreak | Unknown | ERS362924 |
| 8080_2#40 | 2008 | Breda | 078 | 8091554 | Clinic | Non-outbreak | Healthcare | ERS138042 |
| 8080_2#41 | 2009 | Harderwijk | 078 | 9012668 | Clinic | Non-outbreak | Healthcare | ERS138043 |
| 8080_2#42 | 2009 | Goes | 078 | 9019497 | Clinic | Non-outbreak | Unknown | ERS138044 |
| 8080_2#43 | 2009 | Hoorn | 078 | 9077637 | Clinic | Non-outbreak | Healthcare | ERS138045 |
| 8080_2#44 | 2010 | Roermond | 078 | 10005075 | Clinic | Non-outbreak | Healthcare | ERS138046 |
| 8080_2#45 | 2010 | Rotterdam | 078 | 10015222 | Clinic | Non-outbreak | Healthcare | ERS138047 |
| 8080_2#46 | 2010 | Velp | 078 | 10080193 | Clinic | Non-outbreak | Healthcare | ERS138048 |
| 8080_2#47 | 2011 | Zeeland | 078 | 11012929 | Clinic | Non-outbreak | Healthcare | ERS138049 |
| 8080_2#49 | 2009 | Zwolle | 078 | 1103 | Clinic | Non-outbreak | Unknown | ERS138051 |
| 8080_2#58 | NI | Leeds | 066 | 066 (root)c | Clinic | Reference collection | Unknown | ERS138052 |
| 8080_2#61 | 2002 | Rotterdam | 078 | 126065 | Clinic | Non-outbreak | Unknown | ERS138053 |
| 8080_2#62 | 2002 | Leiden | 078 | 126819 | Clinic | Non-outbreak | Healthcare | ERS138054 |
| 8080_2#63 | 2002 | Leiden | 078 | 126938 | Clinic | Non-outbreak | Healthcare | ERS138055 |
| 8080_2#64 | 2002 | Leiden | 078 | 129820 | Clinic | Non-outbreak | Healthcare | ERS138056 |
| 8080_2#71 | 2010 | Leiden | 078 | 53737 | Clinic | Non-outbreak | Healthcare | ERS138060 |
| 8080_2#72 | 2007 | Haarlem | 078 | 47337 | Clinic | Non-outbreak | Healthcare | ERS138063 |
| 8080_2#50 | 2009 | Gastel | 078 | P29 | Pig | Un-paired | Farm | ERS138064 |
| 8080_2#51 | 2009 | NI | 078 | P60 | Pig | Un-paired | Farm | ERS138065 |
| 8080_2#52 | 2009 | Flevoland | 078 | P27 | Pig | Un-paired | Farm | ERS138066 |
| 8080_2#53 | 2009 | NI | 078 | P70 | Pig | Un-paired | Farm | ERS138069 |
| 8080_2#54 | 2009 | Tolakker | 078 | P52 | Pig | Un-paired | Farm | ERS138070 |
| 8080_2#67 | 2011 | Aarle-Rixtel | 078 | H205d | Farmer | Pair 1 | Farm | ERS138073 |
| 8080_2#68 | 2011 | Aarle-Rixtel | 078 | B37_2e | Pig | Pair 1 | Farm | ERS138074 |
| 9221_6#55 | 2011 | NI | 078 | H102 | Farmer | Pair 12 | Farm | ERS199786 |
| 9221_6#56 | 2011 | Raamsdonksveer | 078 | B31_3 | Pig | Pair 9 | Farm | ERS199787 |
| 9221_6#57 | 2011 | Heino | 078 | B17_3 | Pig | Pair 4 | Farm | ERS199788 |
| 9221_6#58 | 2011 | Ulft | 078 | H121 | Farmer | Pair 11 | Farm | ERS199789 |
| 9221_6#59 | 2011 | Rijen | 078 | B27_7 | Pig | Pair 10 | Farm | ERS199790 |
| 9221_6#60 | 2011 | Baarle-Nassau | 078 | H230 | Farmer | Pair 2 | Farm | ERS199791 |
| 9221_6#61 | 2011 | Oirschot | 078 | H189 | Farmer | Pair 8 | Farm | ERS199792 |
| 9221_6#62 | 2011 | Lierop | 078 | B23_6 | Pig | Pair 6 | Farm | ERS199793 |
NI: not identified; RT: ribotyope.
R_L#T, run, lane and tag number.
European Nucleotide Archive sample submission number.
Included as root sequence.
Sequenced in duplicate.
Sequenced in duplicate.
Table 1B. Clostridium difficile type 078 isolates used in this study, the Netherlands, 2002–11 (n=65).
| R_L#Ta | Year | City | RT | Isolate | Source | Related isolates | Association | ENA IDb |
|---|---|---|---|---|---|---|---|---|
| 9221_6#63 | 2011 | Aarle-Rixtel | 078 | H205d | Farmer | Duplicate | Farm | ERS199794 |
| 9221_6#64 | 2011 | Hardenberg | 078 | B15_1 | Pig | Pair 3 | Farm | ERS199795 |
| 9221_6#65 | 2011 | Oirschot | 078 | H21 | Farmer | Un-paired | Farm | ERS199796 |
| 9221_6#66 | 2011 | Oirschot | 078 | B30_5 | Pig | Pair 8 | Farm | ERS199797 |
| 9221_6#67 | 2011 | Rijen | 078 | H122 | Farmer | Pair 10 | Farm | ERS199798 |
| 9221_6#68 | 2011 | Hardenberg | 078 | H95 | Farmer | Pair 3 | Farm | ERS199799 |
| 9221_6#69 | 2011 | Raamsdonk | 078 | B28_1 | Pig | Un-paired | Farm | ERS199800 |
| 9221_6#70 | 2011 | Bakel | 078 | H214 | Farmer | Un-paired | Farm | ERS199801 |
| 9221_6#71 | 2011 | Raamsdonksveer | 078 | H158 | Farmer | Pair 9 | Farm | ERS199802 |
| 9221_6#72 | 2011 | Heino | 078 | H88 | Farmer | Pair 4 | Farm | ERS199803 |
| 9221_6#73 | 2011 | Lemele | 078 | H111 | Farmer | Un-paired | Farm | ERS199804 |
| 9221_6#74 | 2011 | Baarle-Nassau | 078 | B39_4 | Pig | Pair 2 | Farm | ERS199805 |
| 9221_6#75 | 2011 | Moergestel | 078 | B4_2 | Pig | Pair 7 | Farm | ERS199806 |
| 9221_6#76 | 2011 | NI | 078 | B20_1 | Pig | Pair 12 | Farm | ERS199807 |
| 9221_6#77 | 2011 | Moergestel | 078 | H16 | Farmer | Pair 7 | Farm | ERS199808 |
| 9221_6#78 | 2011 | Aarle-Rixtel | 078 | B37_3e | Pig | Duplicate | Farm | ERS199809 |
| 9221_6#79 | 2011 | Ermelo | 078 | B1_5 | Pig | Un-paired | Farm | ERS199810 |
| 9221_6#80 | 2011 | Lierop | 078 | H170 | Farmer | Pair 6 | Farm | ERS199811 |
| 9221_6#81 | 2011 | Ulft | 078 | B22_6 | Pig | Pair 11 | Farm | ERS199812 |
| 9221_6#82 | 2011 | Houten | 078 | B29_10 | Pig | Pair 5 | Farm | ERS199813 |
| 9221_6#83 | 2011 | Houten | 078 | H141 | Farmer | Pair 5 | Farm | ERS199814 |
NI: not identified; RT: ribotyope.
R_L#T, run, lane and tag number.
European Nucleotide Archive sample submission number.
Included as root sequence.
Sequenced in duplicate.
Sequenced in duplicate.
Bacterial culture and genomic DNA preparation
C. difficile was cultured on blood agar plates (BioMérieux, the Netherlands), inoculated into liquid medium (brain–heart infusion (BHI) broth supplemented with yeast extract and cysteine) and grown over night (ca 16 hours) anaerobically at 37 °C. Cells were pelleted, washed with phosphate-buffered saline (PBS), and genomic DNA preparation was performed using a phenol–chloroform extraction as previously described [40].
Whole genome sequencing
Paired-end multiplex libraries were created as previously described [41]. Sequencing was performed on an Illumina HiSeq 2000 platform, with a read length of 100 bp.
In silico MLST
The alleles for the seven housekeeping genes used for C. difficile MLST [42] (http://pubmlst.org/cdifficile/), adk, atpA, dxr, glyA, recA, sodA, and tpi, were analysed in silico to determine the sequence type (ST). All sequenced genomes were aligned with the CDM120 genome using the multiple sequence alignment editor Seaview [43], after which each individual MLST allele was analysed for sequence variation.
SNP calling and recombination detection
Illumina sequence data were mapped to the C. difficile 078 reference genome, M120, (European Molecular Biology Laboratory (EMBL) accession number: FN665653) as paired-end reads using SMALT software (http://smalt.sourceforge.net/), and SNPs were identified as previously described [41]. A potential confounder within the downstream phylogenetic analysis is the effect of homologous recombination, which has the potential to interfere with the phylogenetic signal within the dataset. To alleviate this problem we used the approach developed by Croucher et al. [40] to identify regions in the genome of each isolate where there was evidence of recombination. We then removed those sites from our alignments used in downstream analyses.
Phylogeny and detection of non-phylogenetic SNPs
Phylogenetic trees were constructed using RAxML [44] with a general time reversible (GTR) model with a gamma correction for among-site rate variation combined with 100 random bootstrap replicates (default). Finally, metadata (source, year of isolation, geographical location) was transferred to the reconstructed tree.
Mutation rate estimation
The mutation rate across the population was estimated using the Bayesian evolutionary analysis sampling trees (BEAST) software v1.7.5 [45]. BEAST operates by utilising an explicit model of evolution to compute the mutation rate on each branch of a phylogenetic tree. This enables the translation of evolutionary time into calendar units: days or years. In order to ensure that the dataset was converging consistently, three independent Markov chain Monte Carlo (MCMC) chains were run, each of 100,000,000 states. From these, we removed an initial 10% as a burn-in (10,000,000 states) for each chain and joined the chains using LogCombiner (part of the BEAST suite), taking a sample every 10,000 states.
Genome-wide scan for antimicrobial resistance determinants
De novo assembly was performed for each sequenced genome using the Velvet assembler [46]. The assembled contigs were then ordered against the reference genome M120 using ABACAS [47], which was required for downstream analysis using Artemis Comparison Tool (ACT) [48]. The ordered contigs were used to perform BLAST homology searches for transposons and antimicrobial resistance determinants. The results of this analysis and the discovery of novel potential transposons were visualised using ACT [48]. In addition, the presence of antimicrobial resistance determinants located on the identified transposons were confirmed using the ResFinder 2.1 server [49], with an 98% threshold for identity.
Antibiotic resistance
The minimum inhibitory concentration (MIC) for tetracycline was determined using E-test (BioMérieux, the Netherlands) on Brucella plates (Mediaproducts BV, the Netherlands) under anaerobic conditions at 37 °C. Streptomycin resistance was tested by disk diffusion method, using Sensi-Neotabs 500 μg disks (Rosco, Denmark). Results were interpreted using the tetracycline breakpoints provided by the Clinical and Laboratory Standards Institute (CLSI) [50] and streptomycin breakpoints from Corver et al. [51].
Results
Mutation rate of Clostridium difficile 078 from the Netherlands
We performed whole genome sequencing on 65 C. difficile 078 strains isolated between 2002 and 2011 from various sources (animal or human) and locations in the Netherlands (Figure 1A; Table 1). The human isolates (n = 46) were obtained either from hospitalised patients suffering from CDI (n = 31) or from asymptomatic colonised humans working on Dutch pig farms (n = 15). C. difficile 078 was also isolated from asymptomatic pigs (n = 19). In total, 12 pairs of pig/farmer isolates were included, collected at the same time from the same farms where the sampled farmers resided and worked.
Figure 1. Transmission events and phylogeny of Clostridium difficile 078, the Netherlands 2002–11 (n=65).

A. Distribution of Dutch hospitals and pig farms included in this study. Only pig farms with a known location were plotted. Blue dots represent the hospitals (n = 16) where isolates from hospitalised patients were obtained, red dots represent pig farms (n = 12) where isolates from farmers and pigs were obtained. Brown dots represent the pig farms where pigs and farmers had identical C. difficile isolates. The green arrow indicates a potential (long-range) transmission event between two farms.
B. Phylogenetic tree revealing likely transmission between pigs and humans. Shown is the reconstructed phylogenetic tree based on 774 core genome single-nucleotide polymorphisms (SNPs). Samples are colour-coded according to their source: pig (red), farmer (green) and clinical isolate (blue). Identical genotypes with an epidemiological link (i.e. same location/farm) are marked with brown boxes. Long-range transmission events (i.e. different locations) are marked with a green box. The tip labels are coded with the city name followed by two numbers that represent year of isolation (’08 ⩠ 2008). The CDM120 genome (purple) is used for the reference-based mapping, RT066 (purple) is used as an out-group to root the tree. The scale indicates the branch length that correspond to 10 SNP differences. The numbers for the internal nodes show the support from 100 non-parametric bootstraps of a maximum likelihood reconstruction (only bootstrap values > 50 are shown).
We initially compared the genotypes of the C. difficile 078 isolates with MLST, the traditional gold standard for epidemiological typing of bacterial pathogens. MLST analysis was done using the DNA sequences of seven housekeeping genes [42], which were extracted from the whole-genome dataset. The concatenated sequence length of the MLST loci (3,501 nt) represents ca 0.09% of the whole genome. Our results demonstrated that all of the C. difficile 078 isolates belonged to ST11, and did therefore not provide a degree of resolution that could be used to track and understand the spread of this organism (data not shown).
To increase the discriminatory power of the analysis, we mapped the whole genome data for each sequenced isolate to the C. difficile 078 reference genome M120 [52] and identified all SNPs. Using this approach we identified 3,927 SNPs within the non-repetitive genome (95.2% of the entire genome). Of these, 3,153 SNPs were identified as acquired through horizontal gene transfer or homologous recombination. These SNPs were removed as they disrupt the true phylogeny, leaving a clonal frame of 774 phylogenetically informative SNPs for further downstream analysis. Of these, 373 SNPs were found only in the C. difficile 066 isolate (ST11), a close relative of C. difficile 078 [39], which was used to root the phylogenetic tree. A population-specific mutation rate of C. difficile 078 was estimated, using the isolation dates of our sequenced samples for calibrating the time scale of the phylogenetic tree. Based on our collection, the mutation rate for the C. difficile 078 lineage was estimated to be 2.72 × 10−7 substitutions per site per year (95% confidence interval (CI): 1.43 × 10−7–3.99 × 10−7) which is equivalent to 1.1 SNP per genome per year (95% CI: 0.6–1.6) when multiplied by the number of sites present in the C. difficile 078 genome. This mutation rate is comparable to published estimates for C. difficile 027 [53] and genomes obtained from a selection of 24 distinct STs [54].
Identical genotypes in humans and farm animals
In order to study potential transmission of C. difficile 078 between farm animals and humans, we compared 12 pairs of farmer and pig strains by whole genome SNP typing (Table 2). Interestingly, three farmer/pig pairs, collected at three farms located in Heino, Aarle-Rixtel and Moergestel, shared identical genotypes, i.e. had no SNP differences (Table 2). In addition, two pairs collected at farms in Hardenberg and Houten were separated by only one SNP difference. In all probability, one SNP difference is indicative of a very recent potential transmission event (less than one year earlier). Consequently, using one SNP difference as a threshold for defining suspected transmission on farms, the number of potential transmission events between farmers and animals increased to five, representing five of the 12 sequenced farmer/pig pairs. Of the remaining seven paired samples, only two differed more than 10 SNPs, whereas five had three (n = 3), four (n = 1) or seven (n = 1) SNP differences. The paired animal and human samples with only three to four SNP differences could suggest that a potential transmission event occurred a few years before, and from that moment, the bacterium evolved separately inside different hosts. The paired isolates with more than 10 SNPs difference were genetically so diverse that direct transmission was ruled out.
Table 2. Single nucleotide polymorphism differences between paired farm isolates of Clostridium difficile 078, the Netherlands 2009–11 (n=24).
| Pair number | R_L#Ta | Location of farm | RT | Source | ID | SNP differences |
|---|---|---|---|---|---|---|
| 1 | 8080_268_6#78 | Aarle-Rixtel | 078 | Pig | B37_2 | 0 |
| 8080_2#67 | 078 | Farmer | H205 | |||
| 2 | 9221_6#74 | Baarle-Nassau | 078 | Pig | B39_4 | 3 |
| 9221_6#60 | 078 | Farmer | H230 | |||
| 3 | 9221_6#64 | Hardenberg | 078 | Pig | B15_1 | 1 |
| 9221_6#68 | 078 | Farmer | H95 | |||
| 4 | 9221_6#57 | Heino | 078 | Pig | B17_3 | 0 |
| 9221_6#72 | 078 | Farmer | H88 | |||
| 5 | 9221_6#82 | Houten | 078 | Pig | B29_10 | 1 |
| 9221_6#83 | 078 | Farmer | H141 | |||
| 6 | 9221_6#62 | Lierop | 078 | Pig | B23_6 | 4 |
| 9221_6#80 | 078 | Farmer | H170 | |||
| 7 | 9221_6#75 | Moergestel | 078 | Pig | B4_2 | 0 |
| 9221_6#77 | 078 | Farmer | H16 | |||
| 8 | 9221_6#66 | Oirschot | 078 | Pig | B30_5 | 10 |
| 9221_6#61 | 078 | Farmer | H189 | |||
| 9 | 9221_6#56 | Raamsdonksveer | 078 | Pig | B31_3 | 3 |
| 9221_6#71 | 078 | Farmer | H158 | |||
| 10 | 9221_6#59 | Rijen | 078 | Pig | B27_7 | 19 |
| 9221_6#67 | 078 | Farmer | H122 | |||
| 11 | 9221_6#81 | Ulft | 078 | Pig | B22_6 | 7 |
| 9221_6#58 | 078 | Farmer | H121 | |||
| 12 | 9221_6#76 | NI | 078 | Pig | B20_1 | 3 |
| 9221_6#55 | 078 | Farmer | H102 |
ID: sample identifier. RT: ribotyope.
R_L#T, run, lane and tag number.
Population structure of Clostridium difficile 078 in the Netherlands
To study the closely related paired farm isolates in a broader evolutionary context, we compared the 12 pairs with 41 additional C. difficile samples that were epidemiologically unrelated to the farm isolates and collected over a longer period of time. These 41 samples included 10 individual (i.e. unpaired) farm isolates (from three farmers and seven pigs) collected between 2009 and 2011, and 31 independent (i.e. non-outbreak) clinical isolates obtained from hospitalised patients suffering from CDI collected at various Dutch hospitals between 2002 and 2011. According to the definitions described by Kuijper et al. [55], the majority of these clinical isolates (n = 23) were defined as healthcare-associated cases, while two cases were defined as community-associated; for six clinical isolates the onset was unknown (Table 1).
A maximum likelihood phylogeny was generated using the 401 phylogenetic SNPs identified in the genomes of the 65 sequenced isolates (Figure 1B). In total, 61 distinct SNP genotypes were observed among the 65 C. difficile 078 isolates. Two isolates (Oirschot ’11 and Leiden ‘06) at the periphery of the phylogenetic tree differed by 49 SNPs, which gave an indication of the extent of variation present in the phylogeny. Interestingly, the inferred phylogeny of Dutch C. difficile 078 revealed a general lack of clustering related to strain source (i.e. swine, farmer or clinical), as demonstrated by the mingling of strain sources in the phylogenetic tree (Figure 1B). Isolates from the same source group did not form distinct clusters, while several distinct heterogeneous groups were observed that included isolates obtained from diverse sources. This was especially apparent in the cluster consisting of a clinical isolate (Breda ‘08), a pig isolate (R’donksv.’11) and two farmer isolates (Oirschot ’11 and R’donksv.’11) that were all collected in the same region (Noord Brabant) of the Netherlands (Figure 2). Interestingly, only four SNP differences separated the clinical isolate (Breda ’08) from the nearest farm isolate (R’donksv.’11). Given the three year window in which these isolates were collected and the estimated mutation rate of 1.1 SNP difference per genome per year (95% CI: 0.6–1.6), one would expect to observe two to four SNP differences (prediction interval: 1.8–4.8) between these isolates in case of transmission during this time. Therefore, the observed four SNP difference in this cluster suggests a possible transmission link between farm and clinical isolates.
Figure 2. Phylogenetic cluster showing relatedness of Clostridium difficile clinical, pig and farmer isolates, the Netherlands, 2008–11 (n=4).
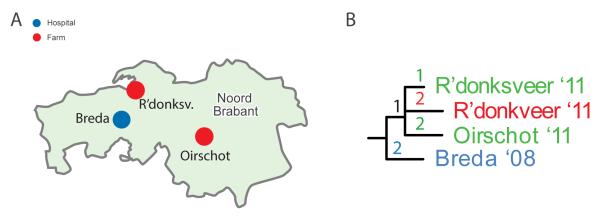
A. Geographical map showing the location of the isolates present in the phylogenetic cluster shown in panel B. Blue dot represents a hospital (Breda), red dots represent the two pig farms (R’donksv. and Oirschot).
B. Zoom-in on a phylogenetic cluster containing highly related isolates from different sources (swine, farmer and clinical isolates). The numbers on the tree branches represent the number of single-nucleotide polymorphisms differences in the cluster, the tip labels are coded with city (Breda) followed by two numbers that represent year of isolation (’08 ⩠ 2008).
The phylogenetic tree also demonstrated a general lack of geographic clustering (Figure 1B). This is particularly evident for the isolates from Leiden and Zwolle that were dispersed throughout the phylogeny. This observation suggested that related C. difficile 078 strains were widely distributed across the country and were frequently transmitted between locations. Interestingly, the analysis revealed two farmers with no obvious epidemiological link that were colonised with identical C. difficile 078 isolates (Figure 1B; green box). The farms were located at Lierop and Ulft (ca 100 km apart), emphasising the lack of geographic signal in these results.
Tetracycline and streptomycin resistance determinants are shared between Clostridium difficile 078 strains from humans and pig
C. difficile genomes carry a broad array of mobile genetic elements that are not included in our phylogenetic SNP analysis but often encode clinically relevant phenotypes such as antimicrobial resistance (AMR). We assembled and analysed the C. difficile 078 genomes to identify potential mobile elements containing AMR determinants and then mapped these onto the phylogenetic tree. We observed the presence of a mobile element with high homology (92.7%) to a previously described transposon Tn6190 [51] and a novel potential transposon that was designated as Tn6235 in this study (data not shown). These transposons potentially confer resistance to various antibiotics, including tetracycline, and each transposon grouped into distinct phylogenetic clusters (Figure 3).
Figure 3. Phylogeny of Clostridium difficle 078 isolates showing the presence of antimicrobial resistance determinants, the Netherlands, 2002–11 (n=65a).

NT = not phenotypically tested.
Circular representation of the C. difficile 078 phylogeny with coloured dots representing the distribution of antimicrobial resistance (AMR) determinants. The legend shows the identified transposons together with the AMR determinants (between brackets) located on the transposon. The coloured dotted lines represent the source of the respective isolates (swine, farmer and clinical isolate). The presence of Tn6190 (tetM) is associated with tetracycline resistance; 078 isolates phenotypically tested as tetracycline-resistant are indicated with orange tree branches, streptomycin-resistant isolates are indicated with purple tree branches, isolates resistant to both tetracycline and streptomycin are indicated with blue tree branches.
a Two isolates were sequenced in duplicate. Ne RT 066 sequence was included as root sequence. In total, 68 sequences are shown.
Mobile element Tn6190, harbouring tetracycline resistance determinant tetM (EMBL accession number: EU182585.1; 98.9% identity), was present in 24 of the sequenced genomes that were obtained from diverse hosts. The majority of these 24 genomes grouped together in a monophylogenetic cluster (Figure 3; orange dots). Tetracycline susceptibility testing confirmed that the presence of tetM correlated with tetracycline resistance (Figure 3 orange branches; Table 3). The novel mobile element Tn6235 was present in its full length (ca 40 kb; 100% homology) in 10 sequenced C. difficile 078 genomes that formed a distinct monophylogenetic cluster with strains from various sources (Figure 3; purple dots). Blast homology searches of this genomic region revealed an open reading frame with homology (100% identity) to a putative aminoglycoside 3′-phosphotransferase aphA1 (M26832) which may confer streptomycin resistance. Streptomycin susceptibility testing confirmed that all 10 isolates present in the monophylogenetic cluster were streptomycin-resistant (Figure 3 purple branches; Table 3).
Table 3. Results of Antimicrobial susceptibility testing.
| Isolate | Source | Streptomycin | Tn6235 | Tetracycline | Tn6190 |
|---|---|---|---|---|---|
| 6072310 | Clinic | Absent | Present | ||
| 6086336 | Clinic | Absent | Present | ||
| 7001233 | Clinic | NT | Absent | Absent | |
| 7004578 | Clinic | Absent | Present | ||
| 7005405 | Clinic | Present | Absent | ||
| 7021455 | Clinic | Absent | Present | ||
| 7044912 | Clinic | Present | Absent | ||
| 7066827 | Clinic | Absent | Present | ||
| 7071308 | Clinic | Absent | Absent | ||
| 7086074 | Clinic | Absent | Absent | ||
| 7091952 | Clinic | Absent | Present | ||
| 8011061 | Clinic | Absent | Absent | ||
| 8013820 | Clinic | Absent | Absent | ||
| 8051728 | Clinic | Absent | Present | ||
| 8055344 | Clinic | Present | Absent | ||
| 8056692 | Clinic | Absent | Absent | ||
| 8091554 | Clinic | Absent | Absent | ||
| 9012668 | Clinic | Absent | Absent | ||
| 9019497 | Clinic | Absent | Absent | ||
| 9077637 | Clinic | Absent | Present | ||
| 10005075 | Clinic | Absent | Present | ||
| 10015222 | Clinic | Absent | Present | ||
| 10080193 | Clinic | Absent | Present | ||
| 11012929 | Clinic | Absent | Absent | ||
| 1103 | Clinic | NT | Absent | NT | Present |
| P29 | Pig | NT | Absent | Absent | |
| P60 | Pig | NT | Absent | Present | |
| P27 | Pig | Absent | Absent | ||
| P70 | Pig | NT | Absent | Absent | |
| P52 | Pig | NT | Absent | Absent | |
| RT066 | Clinic | Absent | Present | ||
| 126065 | Clinic | NT | Absent | NT | Absent |
| 126819 | Clinic | NT | Absent | NT | Absent |
| 126938 | Clinic | NT | Absent | NT | Absent |
| 129280 | Clinic | Absent | Absent | ||
| H205 | Farmer | Present | Absent | ||
| B37.3 | Pig | Present | Absent | ||
| 53737 | Clinic | NT | Absent | NT | Present |
| 47337 | Clinic | Absent | Present | ||
| H102 | Farmer | Absent | Absent | ||
| B31.3 | Pig | Absent | Absent | ||
| B17.3 | Pig | Absent | Absent | ||
| H121 | Farmer | Absent | Present | ||
| B27.7 | Pig | Absent | Absent | ||
| H230 | Farmer | Absent | Present | ||
| H189 | Farmer | Absent | Absent | ||
| B23.6 | Pig | Absent | Present | ||
| H205 | Farmer | Present | Absent | ||
| B15.1 | Pig | Absent | Absent | ||
| H21 | Farmer | Absent | Absent | ||
| B30.5 | Pig | Absent | Absent | ||
| H122 | Farmer | Absent | Absent | ||
| H95 | Pig | Absent | Absent | ||
| B28.1 | Pig | Absent | Present | ||
| H214 | Farmer | Present | Absent | ||
| H158 | Farmer | Absent | Absent | ||
| H88 | Farmer | Absent | Absent | ||
| H111 | Farmer | Present | Absent | ||
| B39.4 | Pig | Absent | Present | ||
| B4.2 | Pig | Absent | Absent | ||
| B20.1 | Pig | Absent | Absent | ||
| H16 | Farmer | Absent | Absent | ||
| B37.3 | Pig | Present | Absent | ||
| B1.5 | Pig | Present | Absent | ||
| H170 | Farmer | Absent | Present | ||
| B22.6 | Pig | Absent | Present | ||
| B29.10 | Pig | Absent | Absent | ||
| H141 | Farmer | Absent | Absent |
NT: not available for testing; shown are the distribution of the mobile elements Tn6190 and Tn6235 among the 078 genomes.
Green: sensitive (S); orange: intermediate (I); red: resistant (R).
Minimum inhibitory concentration cut-off levels used:
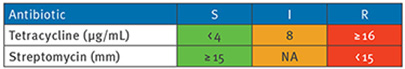
mm: zone diameter breakpoint in mm; NA: not applicable.
Discussion
We used whole genome sequencing and phylogenetic SNP analysis to study the overlap of C. difficile 078 genotypes in animals and humans. In three cases, Dutch farmers were colonised with identical C. difficile 078 clones as pigs kept on the same farms. We have also shown that the presence of clonal strains in pigs and farmers was common, as demonstrated by the number of farmer/pig pairs (five of 12) where clonality (defined as ≤ 1 SNP difference) was observed.
The clonal C. difficile 078 strains in farmers and farm animals that were identified indicate that interspecies transmission has occurred, although we cannot exclude the possibility that they shared a common (environmental) exposure source, e.g. acquisition of spores from a shared common environmental source. However, we believe that direct transmission is plausible. Firstly, the faecal–oral route is the main route of C. difficile transmission, and farmers have a high probability of exposure to pig faeces. Secondly, genomes with zero SNP differences were isolated from farmers and pigs. If acquisition of identical C. difficile strains in humans and animals was a result of transmission from a common source, then either it must have been a very recent environmental transmission event or it did not evolve inside either host after the exposure. Finally, the possibility of an intermediate host can be excluded for clonal cases because circulation via an intermediate host for a certain period is likely to result in SNP differences. If the cases of clonal C. difficile 078 strains in farmers and farm animals are indeed a result of direct interspecies transmission, it would be informative to know the direction of this transmission.
The faecal–oral route of acquisition makes it logical for the direction to be from pig to human. In addition, the high carriage rates of C. difficile among farmers [34,56] also suggest movement from pigs to farmers. This was further supported by the identical antibiotic resistance determinants (tetracycline and streptomycin) shared between animal and human strains, an observation that is in line with previous studies [35,57,58]. The independent insertion of Tn6235 or Tn6190 at the same locations in the C. difficile 078 genomes (data not shown) in combination with phylogenetic clustering of these isolates, suggest that Tn6235 and Tn6190 were introduced once in a progenitor genome that has since then spread in both human and animal hosts. Interestingly, tetracycline is not frequently used in the Dutch healthcare system, whereas it is still the preferred pharmacotherapeutic group for the veterinary industry in the Netherlands [59]. This suggests that tetracycline resistance could be arising in C. difficile isolates from pigs and passed on to the human population. Future, more systematic studies should provide more direct evidence for the direction of transmission.
In addition to the contribution of farm animals as a reservoir for human CDI, we want to emphasise that more than half (58%) of our sequenced farmer/pig pairs were not clonal. Two of the twelve pairs had a SNP difference above 10 SNPs. This suggests that exposure to multiple sources other than farm animals may be responsible for colonisation of the farmers and their pigs. C. difficile can be found almost anywhere in the environment (soil, water, and potentially food) making it hard to pinpoint which alternative reservoirs are significant contributors to the spread of C. difficile 078 in the community. Currently, several potential (environmental) vectors of transmission have been identified, including but not limited to birds, insects, pets and rodents such as rats and mice [60-64]. Our analysis also revealed two farmers who were not geographically linked but were colonised with identical C. difficile 078 isolates. These had been isolated ca 100 km apart from each other, which could suggest exposure to a common environmental source. Another possible explanation could be transport of pigs between the two involved farms that resulted in indirect transmission.
We further analysed the farm isolates in a broader context of clinical isolates with no obvious epidemiological links to the farms. Our analysis demonstrated that all sources, farmer, pig and clinical, were distributed throughout the entire phylogenetic tree and no single clusters per source were identified. These observations are in line with previous research on mixed human and animal C. difficile populations [35,65]. Both studies showed that animal isolates did not constitute a distinct lineage from human isolates. A possible explanation for this observation is that C. difficile 078 strains may have frequently been transmitted between sources, rather than persisting exclusively in one host. Consequently, the clonal strains in farmers and farm animals we identified may be part of a larger network that could have links with the healthcare system. The heterogeneous phylogenetic cluster with limited SNP diversity shown in Figure 2 is an example of potentially linked clinical and farm isolates. Additional patient data for the clinical isolate Breda ’08 showed that, although symptoms started five days before hospitalisation (suspected community onset), the patient was living in a long-term healthcare facility and therefore constituted a healthcare-associated case.
The strength of this study is that we applied for the first time the highly discriminatory method whole genome SNP typing to study the relatedness of C. difficile 078 isolates obtained from farmers and farm animals. A limitation of this study is the small number of clinical samples that were community-associated; such samples may have allowed us to demonstrate more links between farm animals, farmers and the wider community. In addition, the bacterial strain cohort was restricted to isolates obtained in one country, the Netherlands.
The recent trends in epidemiological data show that C. difficile 078 is an important type found in the Dutch healthcare system and its prevalence has remained stable between 2009 and 2013 (data not shown). Besides symptomatic patients, other sources play a major role in the spread of C. difficile within the healthcare system, for instance asymptomatic carriers visiting a healthcare facility [24]. Asymptomatic carriage can be common among hospitalised patients [20,21], although future large studies are needed to determine the precise scale of onward transmission by these carriers. The reservoirs from which these carriers in the community are colonised remain to be elucidated. Importantly, it is becoming clear now that the community reservoir for human CDI is much more diverse and larger than previously expected [24,66]. Here, we demonstrate that transmission from pigs to farmers is one of the potential routes by which C. difficile is entering the human population, and that these isolates also carry antimicrobial resistance determinants that might be a result of selection in response to antibiotic exposure in pigs.
Acknowledgements
We thank Miao He for her technical support during the initial data analysis. We thank the Wellcome Trust Sanger Institute core library and sequencing teams.
This study was supported by ZonMw grant 50-50800-98-079 the Netherlands Organization for Scientific Research (NWO), Wellcome Trust (grants 098051 and 086418) and Medical Research Council (TDL; grant 93614).
Footnotes
Conflict of interest
None declared.
References
- 1.Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med. 1994;330(4):257–62. doi: 10.1056/NEJM199401273300406. http://dx.doi.org/10.1056/NEJM199401273300406 PMID:8043060. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett JG, Gerding DN. Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis. 2008;46((s1) Suppl 1):S12–8. doi: 10.1086/521863. http://dx.doi.org/10.1086/521863 PMID:18177217. [DOI] [PubMed] [Google Scholar]
- 3.Dial S, Delaney JA, Barkun AN, Suissa S. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA. 2005;294(23):2989–95. doi: 10.1001/jama.294.23.2989. http://dx.doi.org/10.1001/jama.294.23.2989 PMID:16414946. [DOI] [PubMed] [Google Scholar]
- 4.Dial S, Delaney JA, Schneider V, Suissa S. Proton pump inhibitor use and risk of community-acquired Clostridium difficile-associated disease defined by prescription for oral vancomycin therapy. CMAJ. 2006;175(7):745–8. doi: 10.1503/cmaj.060284. http://dx.doi.org/10.1503/cmaj.060284 PMID:17001054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilcox MH, Mooney L, Bendall R, Settle CD, Fawley WN. A case-control study of community-associated Clostridium difficile infection. J Antimicrob Chemother. 2008;62(2):388–96. doi: 10.1093/jac/dkn163. http://dx.doi.org/10.1093/jac/dkn163 PMID:18434341. [DOI] [PubMed] [Google Scholar]
- 6.Khanna S, Pardi DS, Aronson SL, Kammer PP, Orenstein R, St Sauver JL, et al. The epidemiology of community-acquired Clostridium difficile infection: a population-based study. Am J Gastroenterol. 2012;107(1):89–95. doi: 10.1038/ajg.2011.398. http://dx.doi.org/10.1038/ajg.2011.398 PMID:22108454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chitnis AS, Holzbauer SM, Belflower RM, Winston LG, Bamberg WM, Lyons C, et al. Epidemiology of community-associated Clostridium difficile infection, 2009 through 2011. JAMA Intern Med. 2013;173(14):1359–67. doi: 10.1001/jamainternmed.2013.7056. http://dx.doi.org/10.1001/jamainternmed.2013.7056 PMID:23780507. [DOI] [PubMed] [Google Scholar]
- 8.Leung J, Burke B, Ford D, Garvin G, Korn C, Sulis C, et al. Possible association between obesity and Clostridium difficile infection. Emerg Infect Dis. 2013;19(11):1791–8. doi: 10.3201/eid1911.130618. PMID:24188730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18(2):247–63. doi: 10.1128/CMR.18.2.247-263.2005. http://dx.doi.org/10.1128/CMR.18.2.247-263.2005 PMID:15831824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458(7242):1176–9. doi: 10.1038/nature07822. http://dx.doi.org/10.1038/nature07822 PMID:19252482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature. 2010;467(7316):711–3. doi: 10.1038/nature09397. http://dx.doi.org/10.1038/nature09397 PMID:20844489. [DOI] [PubMed] [Google Scholar]
- 12.Schwan C, Stecher B, Tzivelekidis T, van Ham M, Rohde M, Hardt WD, et al. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog. 2009;5(10):e1000626. doi: 10.1371/journal.ppat.1000626. http://dx.doi.org/10.1371/journal.ppat.1000626 PMID:19834554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hensgens MP, Kuijper EJ. Clostridium difficile infection caused by binary toxin-positive strains. Emerg Infect Dis. 2013;19(9):1539–40. doi: 10.3201/eid1909.110814. http://dx.doi.org/10.3201/eid1909.110814 PMID:23977836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerding DN, Johnson S. Does infection with specific Clostridium difficile strains or clades influence clinical outcome? Clin Infect Dis. 2013;56(11):1601–3. doi: 10.1093/cid/cit133. http://dx.doi.org/10.1093/cid/cit133 PMID:23463642. [DOI] [PubMed] [Google Scholar]
- 15.Bacci S, Mølbak K, Kjeldsen MK, Olsen KE. Binary toxin and death after Clostridium difficile infection. Emerg Infect Dis. 2011;17(6):976–82. doi: 10.3201/eid1706.101483. http://dx.doi.org/10.3201/eid/1706.101483 PMID:21749757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walk ST, Micic D, Jain R, Lo ES, Trivedi I, Liu EW, et al. Clostridium difficile ribotype does not predict severe infection. Clin Infect Dis. 2012;55(12):1661–8. doi: 10.1093/cid/cis786. http://dx.doi.org/10.1093/cid/cis786 PMID:22972866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7(7):526–36. doi: 10.1038/nrmicro2164. http://dx.doi.org/10.1038/nrmicro2164 PMID:19528959. [DOI] [PubMed] [Google Scholar]
- 18.Wilcox MH, Shetty N, Fawley WN, Shemko M, Coen P, Birtles A, et al. Changing epidemiology of Clostridium difficile infection following the introduction of a national ribotyping-based surveillance scheme in England. Clin Infect Dis. 2012;55(8):1056–63. doi: 10.1093/cid/cis614. http://dx.doi.org/10.1093/cid/cis614 PMID:22784871. [DOI] [PubMed] [Google Scholar]
- 19.Price J, Cheek E, Lippett S, Cubbon M, Gerding DN, Sambol SP, et al. Impact of an intervention to control Clostridium difficile infection on hospital- and community-onset disease; an interrupted time series analysis. Clin Microbiol Infect. 2010;16(8):1297–302. doi: 10.1111/j.1469-0691.2009.03077.x. http://dx.doi.org/10.1111/j.1469-0691.2009.03077.x PMID:19832710. [DOI] [PubMed] [Google Scholar]
- 20.Alasmari F, Seiler SM, Hink T, Burnham CA, Dubberke ER. Prevalence and risk factors for asymptomatic Clostridium difficile carriage. Clin Infect Dis. 2014;59(2):216–22. doi: 10.1093/cid/ciu258. http://dx.doi.org/10.1093/cid/ciu258 PMID:24755858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eyre DW, Griffiths D, Vaughan A, Golubchik T, Acharya M, O’Connor L, et al. Asymptomatic Clostridium difficile colonisation and onward transmission. PLoS ONE. 2013;8(11):e78445. doi: 10.1371/journal.pone.0078445. http://dx.doi.org/10.1371/journal.pone.0078445 PMID:24265690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerrero DM, Becker JC, Eckstein EC, Kundrapu S, Deshpande A, Sethi AK, et al. Asymptomatic carriage of toxigenic Clostridium difficile by hospitalized patients. J Hosp Infect. 2013;85(2):155–8. doi: 10.1016/j.jhin.2013.07.002. http://dx.doi.org/10.1016/j.jhin.2013.07.002 PMID:23954113. [DOI] [PubMed] [Google Scholar]
- 23.Galdys AL, Nelson JS, Shutt KA, Schlackman JL, Pakstis DL, Pasculle AW, et al. Prevalence and duration of asymptomatic Clostridium difficile carriage among healthy subjects in Pittsburgh, Pennsylvania. J Clin Microbiol. 2014;52(7):2406–9. doi: 10.1128/JCM.00222-14. http://dx.doi.org/10.1128/JCM.00222-14 PMID:24759727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eyre DW, Cule ML, Wilson DJ, Griffiths D, Vaughan A, O’Connor L, et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med. 2013;369(13):1195–205. doi: 10.1056/NEJMoa1216064. http://dx.doi.org/10.1056/NEJMoa1216064 PMID:24066741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, et al. ECDIS Study Group Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377(9759):63–73. doi: 10.1016/S0140-6736(10)61266-4. http://dx.doi.org/10.1016/S0140-6736(10)61266-4 PMID:21084111. [DOI] [PubMed] [Google Scholar]
- 26.Goorhuis A, Bakker D, Corver J, Debast SB, Harmanus C, Notermans DW, et al. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis. 2008;47(9):1162–70. doi: 10.1086/592257. http://dx.doi.org/10.1086/592257 PMID:18808358. [DOI] [PubMed] [Google Scholar]
- 27.Debast SB, van Leengoed LA, Goorhuis A, Harmanus C, Kuijper EJ, Bergwerff AA. Clostridium difficile PCR ribotype 078 toxinotype V found in diarrhoeal pigs identical to isolates from affected humans. Environ Microbiol. 2009;11(2):505–11. doi: 10.1111/j.1462-2920.2008.01790.x. http://dx.doi.org/10.1111/j.1462-2920.2008.01790.x PMID:19196280. [DOI] [PubMed] [Google Scholar]
- 28.Squire MM, Carter GP, Mackin KE, Chakravorty A, Norén T, Elliott B, et al. Novel molecular type of Clostridium difficile in neonatal pigs, Western Australia. Emerg Infect Dis. 2013;19(5):790–2. doi: 10.3201/eid1905.121062. http://dx.doi.org/10.3201/eid1905.121062 PMID:23697508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Álvarez-Pérez S, Blanco JL, Peláez T, Astorga RJ, Harmanus C, Kuijper E, et al. High prevalence of the epidemic Clostridium difficile PCR ribotype 078 in Iberian free-range pigs. Res Vet Sci. 2013;95(2):358–61. doi: 10.1016/j.rvsc.2013.06.021. http://dx.doi.org/10.1016/j.rvsc.2013.06.021 PMID:23876331. [DOI] [PubMed] [Google Scholar]
- 30.Schneeberg A, Neubauer H, Schmoock G, Baier S, Harlizius J, Nienhoff H, et al. Clostridium difficile genotypes in piglet populations in Germany. J Clin Microbiol. 2013;51(11):3796–803. doi: 10.1128/JCM.01440-13. http://dx.doi.org/10.1128/JCM.01440-13 PMID:24025903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneeberg A, Neubauer H, Schmoock G, Grossmann E, Seyboldt C. Presence of Clostridium difficile PCR ribotype clusters related to 033, 078 and 045 in diarrhoeic calves in Germany. J Med Microbiol. 2013;62(Pt 8):1190–8. doi: 10.1099/jmm.0.056473-0. http://dx.doi.org/10.1099/jmm.0.056473-0 PMID:23639987. [DOI] [PubMed] [Google Scholar]
- 32.Keel K, Brazier JS, Post KW, Weese S, Songer JG. Prevalence of PCR ribotypes among Clostridium difficile isolates from pigs, calves, and other species. J Clin Microbiol. 2007;45(6):1963–4. doi: 10.1128/JCM.00224-07. http://dx.doi.org/10.1128/JCM.00224-07 PMID:17428945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hensgens MP, Keessen EC, Squire MM, Riley TV, Koene MG, de Boer E, et al. European Society of Clinical Microbiology and Infectious Diseases Study Group for Clostridium difficile (ESGCD) Clostridium difficile infection in the community: a zoonotic disease? Clin Microbiol Infect. 2012;18(7):635–45. doi: 10.1111/j.1469-0691.2012.03853.x. http://dx.doi.org/10.1111/j.1469-0691.2012.03853.x PMID:22536816. [DOI] [PubMed] [Google Scholar]
- 34.Keessen EC, Harmanus C, Dohmen W, Kuijper EJ, Lipman LJ. Clostridium difficile infection associated with pig farms. Emerg Infect Dis. 2013;19(6):1032–4. doi: 10.3201/eid1906.121645. http://dx.doi.org/10.3201/eid1906.121645 PMID:23735347</jrn>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bakker D, Corver J, Harmanus C, Goorhuis A, Keessen EC, Fawley WN, et al. Relatedness of human and animal Clostridium difficile PCR ribotype 078 isolates determined on the basis of multilocus variable-number tandem-repeat analysis and tetracycline resistance. J Clin Microbiol. 2010;48(10):3744–9. doi: 10.1128/JCM.01171-10. http://dx.doi.org/10.1128/JCM.01171-10PMID:20686080 PMID:20686080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koene MG, Mevius D, Wagenaar JA, Harmanus C, Hensgens MP, Meetsma AM, et al. Clostridium difficile in Dutch animals: their presence, characteristics and similarities with human isolates. Clin Microbiol Infect. 2012;18(8):778–84. doi: 10.1111/j.1469-0691.2011.03651.x. http://dx.doi.org/10.1111/j.1469-0691.2011.03651.x PMID:21919997. [DOI] [PubMed] [Google Scholar]
- 37.Keessen EC, Gaastra W, Lipman LJ. Clostridium difficile infection in humans and animals, differences and similarities. Vet Microbiol. 2011;153(3-4):205–17. doi: 10.1016/j.vetmic.2011.03.020. http://dx.doi.org/10.1016/j.vetmic.2011.03.020 PMID:21530110. [DOI] [PubMed] [Google Scholar]
- 38.Squire MM, Riley TV. Clostridium difficile infection in humans and piglets: a ‘One Health’ opportunity. Curr Top Microbiol Immunol. 2013;365:299–314. doi: 10.1007/82_2012_237. http://dx.doi.org/10.1007/82_2012_237 PMID:22695920. [DOI] [PubMed] [Google Scholar]
- 39.Knetsch CW, Terveer EM, Lauber C, Gorbalenya AE, Harmanus C, Kuijper EJ, et al. Comparative analysis of an expanded Clostridium difficile reference strain collection reveals genetic diversity and evolution through six lineages. Infect Genet Evol. 2012;12(7):1577–85. doi: 10.1016/j.meegid.2012.06.003. http://dx.doi.org/10.1016/j.meegid.2012.06.003 PMID:22705462. [DOI] [PubMed] [Google Scholar]
- 40.Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331(6016):430–4. doi: 10.1126/science.1198545. http://dx.doi.org/10.1126/science.1198545 PMID:21273480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327(5964):469–74. doi: 10.1126/science.1182395. http://dx.doi.org/10.1126/science.1182395 PMID:20093474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Griffiths D, Fawley W, Kachrimanidou M, Bowden R, Crook DW, Fung R, et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol. 2010;48(3):770–8. doi: 10.1128/JCM.01796-09. http://dx.doi.org/10.1128/JCM.01796-09 PMID:20042623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27(2):221–4. doi: 10.1093/molbev/msp259. http://dx.doi.org/10.1093/molbev/msp259 PMID:19854763. [DOI] [PubMed] [Google Scholar]
- 44.Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21(4):456–63. doi: 10.1093/bioinformatics/bti191. http://dx.doi.org/10.1093/bioinformatics/bti191 PMID:15608047. [DOI] [PubMed] [Google Scholar]
- 45.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7(1):214. doi: 10.1186/1471-2148-7-214. http://dx.doi.org/10.1186/1471-2148-7-214 PMID:17996036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–9. doi: 10.1101/gr.074492.107. http://dx.doi.org/10.1101/gr.074492.107 PMID:18349386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 2009;25(15):1968–9. doi: 10.1093/bioinformatics/btp347. http://dx.doi.org/10.1093/bioinformatics/btp347 PMID:19497936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21(16):3422–3. doi: 10.1093/bioinformatics/bti553. http://dx.doi.org/10.1093/bioinformatics/bti553 PMID:15976072. [DOI] [PubMed] [Google Scholar]
- 49.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–4. doi: 10.1093/jac/dks261. http://dx.doi.org/10.1093/jac/dks261 PMID:22782487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clinical and Laboratory Standards Institute (CLSI) Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria. Approved Standard-Seventh edition. CLSI; Wayne, Pennsylvania: 2007. CLSI document M11-A7. [ISBN 1-56238-626-3]. Available from: http://clsi.org. [Google Scholar]
- 51.Corver J, Bakker D, Brouwer MS, Harmanus C, Hensgens MP, Roberts AP, et al. Analysis of a Clostridium difficile PCR ribotype 078 100 kilobase island reveals the presence of a novel transposon, Tn6164. BMC Microbiol. 2012;12(1):130. doi: 10.1186/1471-2180-12-130. http://dx.doi.org/10.1186/1471-2180-12-130 PMID:22747711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci USA. 2010;107(16):7527–32. doi: 10.1073/pnas.0914322107. http://dx.doi.org/10.1073/pnas.0914322107 PMID:20368420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. 2013;45(1):109–13. doi: 10.1038/ng.2478. http://dx.doi.org/10.1038/ng.2478PMID: 23222960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Didelot X, Eyre DW, Cule M, Ip CL, Ansari MA, Griffiths D, et al. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol. 2012;13(12):R118. doi: 10.1186/gb-2012-13-12-r118. http://dx.doi.org/10.1186/gb-2012-13-12-r118 PMID:23259504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuijper EJ, Coignard B, Tüll P, ESCMID Study Group for Clostridium difficile, EU Member States, European Centre for Disease Prevention and Control Emergence of Clostridium difficile-associated disease in North America and Europe. Clin Microbiol Infect. 2006;12((s6) Suppl 6):2–18. doi: 10.1111/j.1469-0691.2006.01580.x. http://dx.doi.org/10.1111/j.1469-0691.2006.01580.x PMID:16965399. [DOI] [PubMed] [Google Scholar]
- 56.Loo VG, Bourgault AM, Poirier L, Lamothe F, Michaud S, Turgeon N, et al. Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med. 2011;365(18):1693–703. doi: 10.1056/NEJMoa1012413. http://dx.doi.org/10.1056/NEJMoa1012413 PMID:22047560. [DOI] [PubMed] [Google Scholar]
- 57.Keessen EC, Hensgens MP, Spigaglia P, Barbanti F, Sanders IM, Kuijper EJ, et al. Antimicrobial susceptibility profiles of human and piglet Clostridium difficile PCR-ribotype 078. Antimicrob Resist Infect Control. 2013;2(1):14. doi: 10.1186/2047-2994-2-14. http://dx.doi.org/10.1186/2047-2994-2-14 PMID:23566553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pirš T, Avberšek J, Zdovc I, Krt B, Andlovic A, Lejko-Zupanc T, et al. Antimicrobial susceptibility of animal and human isolates of Clostridium difficile by broth microdilution. J Med Microbiol. 2013;62(Pt 9):1478–85. doi: 10.1099/jmm.0.058875-0. http://dx.doi.org/10.1099/jmm.0.058875-0 PMID:23861298. [DOI] [PubMed] [Google Scholar]
- 59.Mevius D, Wit B, van Pelt W, van Geijlswijk IM, Bondt N, Puister LF, et al. Nethmap-MARAN 2013: Monitoring of Antimicrobial Resistance and Antibiotic Usage in Animals in the Netherlands in 2012. Central Veterinary Institute; Wageningen: 2013. Available from: http://www.swab.nl/swab/cms3.nsf/viewdoc/20BCD3983B5C390AC12575850031D33D. [Google Scholar]
- 60.Riley TV, Adams JE, O’Neill GL, Bowman RA. Gastrointestinal carriage of Clostridium difficile in cats and dogs attending veterinary clinics. Epidemiol Infect. 1991;107(3):659–65. doi: 10.1017/s0950268800049359. http://dx.doi.org/10.1017/S0950268800049359 PMID:1752313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weese JS, Finley R, Reid-Smith RR, Janecko N, Rousseau J. Evaluation of Clostridium difficile in dogs and the household environment. Epidemiol Infect. 2010;138(8):1100–4. doi: 10.1017/S0950268809991312. http://dx.doi.org/10.1017/S0950268809991312 PMID:19951453. [DOI] [PubMed] [Google Scholar]
- 62.Burt SA, Siemeling L, Kuijper EJ, Lipman LJ. Vermin on pig farms are vectors for Clostridium difficile PCR ribotypes 078 and 045. Vet Microbiol. 2012;160(1-2):256–8. doi: 10.1016/j.vetmic.2012.05.014. http://dx.doi.org/10.1016/j.vetmic.2012.05.014 PMID:22682200. [DOI] [PubMed] [Google Scholar]
- 63.Bandelj P, Trilar T, Blagus R, Ocepek M, Rousseau J, Weese JS, et al. Prevalence and molecular characterization of Clostridium difficile isolated from European Barn Swallows (Hirundo rustica) during migration. BMC Vet Res. 2014;10(1):40. doi: 10.1186/1746-6148-10-40. http://dx.doi.org/10.1186/1746-6148-10-40 PMID:24507706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Himsworth CG, Patrick DM, Mak S, Jardine CM, Tang P, Weese JS. Carriage of Clostridium difficile by wild urban Norway rats (Rattus norvegicus) and black rats (Rattus rattus) Appl Environ Microbiol. 2014;80(4):1299–305. doi: 10.1128/AEM.03609-13. http://dx.doi.org/10.1128/AEM.03609-13 PMID:24317079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lemee L, Dhalluin A, Pestel-Caron M, Lemeland JF, Pons JL. Multilocus sequence typing analysis of human and animal Clostridium difficile isolates of various toxigenic types. J Clin Microbiol. 2004;42(6):2609–17. doi: 10.1128/JCM.42.6.2609-2617.2004. http://dx.doi.org/10.1128/JCM.42.6.2609-2617.2004 PMID:15184441. [DOI] [PMC free article] [PubMed] [Google Scholar]