

Abstract
The hallmark of enzymes from secondary metabolic pathways is the pairing of powerful reactivity with exquisite site selectivity. The application of these biocatalytic tools in organic synthesis, however, remains under-utilized due to limitations in substrate scope and scalability. Here we report the reactivity of a monooxygenase (PikC) from the pikromycin pathway is modified through computationally-guided protein and substrate engineering, and applied to the oxidation of unactivated methylene C-H bonds. Molecular dynamics and quantum mechanical calculations were employed to develop a predictive model for substrate scope, site selectivity, and stereoselectivity of PikC mediated C-H oxidation. A suite of menthol derivatives was screened computationally and evaluated through in vitro reactions where each substrate adhered to the predicted models for selectivity and conversion to product. This platform was also expanded beyond menthol-based substrates to the selective hydroxylation of a variety of substrate cores ranging from cyclic to fused bicyclic and bridged bicyclic compounds.
Introduction
Direct C–H functionalization has the potential to streamline existing synthetic routes and also to provide access to novel, high-value compounds. Research in this area has grown exponentially over the last two decades, however, the challenge of selectively targeting any given C–H bond for functionalization within a complex molecule has yet to be mastered1. Strategies for targeting the electronically weakest C–H bond or the most sterically accessible C–H bond have proven fruitful. Further, a plethora of directing groups has been developed to override the innate steric and electronic biases within a molecule to affect a reaction proximal to the directing group. Despite these efforts, certain types of C–H bonds remain difficult to target or entirely inaccessible, such as methylene C–H bonds remote from directing groups. While some successes have been realized in site selective methylene oxidations using small molecule catalysts2–4, the selective oxidation of a single C–H bond among several electronically and sterically similar methylene units remains one of the most challenging tasks in the field of C–H functionalizations. Enzymes have the potential to offer orthogonal reactivity compared to small molecule catalysts. In the C–H functionalization arena, cytochrome P450 biocatalysts have proven to be effective for selective oxidation of unactivated methylene C–H bonds5–9. However, the development of these biocatalysts often requires extensive protein engineering to achieve selectivity on a given substrate or to change the substrate scope of the enzyme10–13. Herein, we demonstrate a method for addressing this challenge that relies on a directing group distal to the site of functionalization and overrides steric or electronic effects inherent to the substrate with the ultimate goal of achieving a P450/directing group platform that can be broadly applied without requiring extensive protein engineering for each new substrate. While such a synthetic strategy using small molecule catalysts originates from the pioneering early studies by Breslow14,15 and has been applied in recent efforts to achieve meta-functionalizations of arenes16,17, the architectural complexity and unique anchoring mechanism of biological catalysts provides what is perhaps the ideal platform for engineering directed reactions to functionalize remote and unactivated structural elements18,19,20.
Menthol (1) was chosen as a model substrate based on its six-membered ring core, the variety of C–H bonds it contains (up to 18 potentially reactive positions, including primary, secondary and tertiary, see Fig. 1) and the depth of information available on its use in C–H hydroxylation studies. Several groups have demonstrated the ability to hydroxylate the weaker methine C–H bonds at C5 and C7 positions of menthol using Fe, Cr or Mn chemocatalysts21–25 or biotransformations (see 2 and 3, respectively). The stronger but sterically less hindered methyl C–H bonds have also been successfully targeted for C–H oxygenation (see 11 and 12) through group transfer reactions26,27 or biotransformation28. Selective functionalization of any of the six methylene C–H bonds of menthol has remained an exceptional chemical and biochemical challenge. Biotransformation studies have established the feasibility of a biocatalytic methylene oxidation of menthol to afford mixtures of six or more regioisomeric products29. However, to our knowledge, the only example of site-selective methylene hydroxylation of menthol involved a directed evolution approach in which Fasan and coworkers achieved a site and stereoselective hydroxylation at C6 (see 5) with a cytochrome P450 BM3 variant that was identified using a screening-based fingerprinting approach8.
Figure 1.

Selected oxidation products of (−)-menthol and calculated bond dissociation energies (BDEs) for the corresponding C–H bonds in kcal mol−1. The calculated BDEs display a general trend in C–H bond strength, tertiary < secondary < primary.
Our strategy was motivated by the selective oxygenation of unactivated C–H bonds catalyzed by the cytochrome P450 monooxygenase PikC from the pikromycin natural product biosynthetic pathway30, and exploits the native substrate anchoring mechanism utilized by this enzyme. PikC hydroxylates two types of endogenous substrates in Streptomyces venezuelae ATCC 15439. They include the 12- and 14-membered macrolides YC-17 (13) and narbomycin31 to produce methymycin (14), neomethymycin (15), novamethymycin32, pikromycin and neopikromycin. This level of substrate promiscuity is rare in secondary metabolic pathways, but is explained by the mechanism in which natural substrates bind within the PikC active site. Co-crystal structures of PikC with YC-17 (13) or narbomycin revealed salt bridge interactions between the protonated dimethylamino group of the substrate desosamine sugar, and an exposed carboxylate moiety within the active site (Fig. 2A, E94 for YC-17 and E85 for narbomycin)33,34. To assess the ability of salt bridge-mediated anchoring of unnatural substrates in the PikC active site, several desosamine-containing substrates were synthesized and tested for oxidation with PikC (Fig. 2B)35. Macrocyclic substrates were hydroxylated to afford multiple products (16); however, no reaction was observed with smaller cores such as desosaminyl cyclohexanol 17. We reasoned that the six-membered ring in 17 was too small to span the distance between the anchoring carboxylate residue and the PikC iron-oxo species where the reaction occurs. We previously demonstrated that the desosamine in the natural substrate YC-17 might be effectively replaced with synthetic alternatives (18, Fig. 2B)36. Thus, we envisioned that employing synthetic anchoring groups as well as introducing additional mutations in the biocatalyst could improve the activity of PikC with unnatural substrates.
Figure 2.


Substrate anchoring mechanism employed by P450 PikC. (a) Co-crystal structure of natural substrate, YC-17, and PikC (PDB ID 2CD8)33 depicting the anchoring salt bridge between E94 and the dimethylamino group of YC-17. (b) Evolution of the PikC substrate-engineering approach with the natural anchoring group, desosamine and dimethylamine containing synthetic anchoring groups.
Access to high-resolution co-crystal structures of PikC and its natural macrolide substrates has been instrumental in identifying the substrate-enzyme salt bridge at the center of our substrate-engineering strategy. Moreover, ligand-free x-ray structures (PDB ID 2BVJ)33 illustrate the differences in protein conformations attributed to a transition between the catalytically competent “closed” conformation and the “open” conformation, a prerequisite of the substrate binding and product release. However, these static conformation snapshots do not inform the dynamic nature of the protein-small molecule interactions. This has limited our view of the enzyme active site and the dynamic signature of the protein-ligand complex ultimately responsible for the unique site and stereoselectivity characteristics of the PikC biocatalyst. In order to address these experimental limitations, we turned to molecular dynamics (MD) simulations; this powerful computational tool that has proven to be useful in determining the structural origins of enhanced activity in evolved enzymes37. We conceptualized a suite of (−)- and (+)-menthol derivatives bearing synthetic anchoring groups including linear alkyl chains of various lengths (21–25) as well as benzylic amines (26–28). The binding efficiency of selected substrates (21, 25, and 26) to wild-type PikC and PikCD50N mutant was assessed by 0.5 µs MD simulations (see Online Methods).
Results and Discussion
Molecular dynamic simulations
MD simulations revealed a high dependence of the nature and persistence of the salt bridge contacts between the substrate and PikCwt on the structure of the anchoring group and the chirality of menthol (Supplementary Fig. 3). The (−)-menthol moiety was consistently retained in the hydrophobic pocket of the active site close to the heme cofactor. However, in the case of (−)-21, the two-carbon anchor was too short to establish strong interactions with residues E94 and E85 typically mediating catalytically productive substrate binding33,34. Instead, detrimental salt bridge interactions with the neighboring E246 located in the I-helix, were established with the 58% frequency throughout the simulation (Fig. 3), unproductively positioning the menthol core with less reactive primary and tertiary C–H bonds oriented towards the iron-oxo site. The lack of contacts with E85 and E94 distorted the tertiary structure of the protein. As a result, the active site channel adopted an open conformation similar to that observed crystallographically in the apo state of PikC33. These mechanisms of inactivation revealed through MD simulations, provided structural information to guide the optimization of catalysis through both substrate and protein engineering. The key objective was to maximize anchoring group-mediated substrate binding and maintain a closed, catalytically productive protein conformation.
Figure 3.

The persistence of the potential salt bridges (>4 Å) between a given substrate anchoring group and different PikC residues (percentages measured from MD simulations) are shown; (+)/(−) distinction refers to menthol absolute configuration.
MD revealed that less distortion of the PikC structure was observed with substrate bearing the longest linear anchoring group (see (−)-25). A new polar contact with E48, which had not been observed in the PikC complex bound to its natural macrolide substrates appeared to be relevant for the engineered substrate binding. The anchoring group in (−)-25 was sufficiently long to allow polar contacts between the dimethylamino group and binding residues located at the entrance of the active site (41%, 4% and 33% frequencies with E94, E85 and E48, respectively, Fig. 3) and avoid interacting with E246. Analogously, with the more rigid substrate (−)-26, the structural integrity of the protein and substrate binding are further improved as a persistent, short salt bridge is established with E94 (79% frequency, Fig. 3). Similar behavior was observed for the PikCD50N mutant, which has demonstrated an improved catalytic efficiency compared to PikCwt (Supplementary Fig. 4). Thus, we predicted that these longer anchoring groups, (−)-25 and (−)-26, should lead to improved enzymatic reactivities.
In contrast, the corresponding antipodal set of compounds showed inferior binding in MD simulations (Supplementary Figs. 5 and 6). Notably, the active site of PikCwt exhibited a higher shape complementarity towards (−)-menthol than (+)-menthol. For example, (+)-(25) completely abandoned the heme-containing hydrophobic pocket after ~100 ns, and (+)-21 unproductively oscillates in and out of the hydrophobic pocket (average FeO⋯C4 distance ~7 Å). MD revealed that the menthol core is held proximal to the heme group only in (+)-26, albeit with relatively weak binding contacts to E94, E85 or E48 (> 5 Å).
Enzymatic reactions
In vitro reactions of each substrate with PikC validated MD predictions (Table 1). Each of the menthol compounds was tested as a substrate for PikCwt-RhFRED and PikCD50N-RhFRED38 in the presence of the standard NADPH recycling system39. As observed with desosaminyl cyclohexanol (17), the desosamine derivative of (−)-menthol was not hydroxylated by PikCwt-RhFRED or PikCD50N-RhFRED. Similarly, short synthetic anchoring groups as those in (−)-21, (−)-22, and (−)-23 showed low total turnover numbers (TTN = # mol product/# mol enzyme) with PikCD50N-RhFRED and no product was detected for the reactions of these compounds with the wild type enzyme. However, increasing the length of the anchoring group by one or two additional methylene units improved catalysis with PikCD50N-RhFRED, as shown by TTN of 24 and 43 for (−)-24 and (−)-25, respectively (Table 1). Moreover, analysis of the benzylic amine substrates (−)-26, (−)-27, and (−)-28 demonstrated the superior nature of the para derivative over the meta and ortho analogs (TTN 96, 41 and 10, respectively). Only (−)-25 and (−)-26 were converted to hydroxylated products by PikC-RhFRED38, albeit in low conversion (TTN = 11 and 16, respectively).
Table 1.
Total turnover numbers (TTN, mol product/mol enzyme) for the hydroxylation of (−)- and (+)-menthol derived substrates using mutant forms of PikC.
 | ||||||
|---|---|---|---|---|---|---|
| PikCwt-RF | PikCD50N-RF | PikCD50ND176QE246A-RF | ||||
| substrate | (−) | (+) | (−) | (+) | (−) | (+) |
| 21 | 0 | 0 | 2.0 | 0 | 95 | 26 |
| 22 | 0 | 0 | 2.3 | 0 | 89 | 20 |
| 23 | 0 | 0 | 7.8 | 0 | 182 | 38 |
| 24 | 0 | 9.6 | 22 | 19 | 179 | 90 |
| 25 | 11 | 14 | 43 | 27 | 186 | 129 |
| 26 | 16 | 0 | 96 | 28 | 137 | 72 |
| 27 | 0 | 0 | 41 | 13 | 153 | 47 |
| 28 | 0 | 0 | 10 | 0 | 170 | 26 |
As predicted by MD simulations, PikC reactions with the (+)-menthol substrate panel demonstrated lower catalytic efficiency and selectivity compared to the (−)-menthol series. For example, TTN of 2–10 were measured for (−)-21, (−)-22, (−)-23 and (−)-28, while no product formation was observed for the (+)-menthol substrates with the same anchoring groups. Similarly, lower turnover numbers were observed for substrates (+)-24–27 ranging from 19 to 28 (Table 1).
MD-Guided Protein Engineering
Although the improved binding of (−)-25 and (−)-26 positioned the menthol core close to the reactive Fe-oxo group, fluctuations between open and closed conformations of the active site were still observed by MD. We hypothesized that the protein can adopt the open conformation when that anchoring moiety of the substrate does not persistently bind E85. Further, we anticipated that dynamic binding of the anchoring group to the triad of binding residues (E48, E85 and E94), not contacts with one or two residues as observed crystallographically would shift the open/closed equilibrium in favor of the closed, active conformation. The E246 and D176 positions were readily identified as targets for mutagenesis to promote catalysis.
Thus, we engineered the triple mutant monooxygenase PikCD50ND176QE246A-RhFRED, lacking carboxylate groups at both D176 and E246, to eliminate the potential for substrate binding at these residues. As predicted, a significant increase in TTN was observed for all substrates tested with this PikC triple mutant variant (Table 1). For example, the TTN for (−)-28 increased from 10 to 170, and substrates such as (−)-23, which showed no detectable product formation with PikCD50N-RhFRED had a TTN of 38 with the PikCD50ND176QE246A-RhFRED biocatalyst. MD simulations of PikCD50ND176QE246A-RhFRED alone and with substrate show a higher frequency of the closed conformation compared to PikCwt-RhFRED and PikCD50N-RhFRED (Fig. 4d–f and Supplementary Figs. 7–9), which supports our hypothesis that these mutations shift the equilibrium in favor of the catalytically active closed conformation. Similar impact of enzyme evolution on the active site configuration has been proposed in other engineered proteins37. Through these mutations, the dependence of the enzyme reactivity on an induced-fit mechanism, proposed to operate for the larger and more polar macrolactones, is alleviated by stabilizing the tertiary structure of the protein in a conformation that facilitates the binding in a productive orientation of the smaller and less polar unnatural ligands.
Figure 4. Substrate binding and tertiary structure of PikC variants observed in MD simulations.

(a) Improper binding of substrate (−)-21 to PikCwt showing detrimental interactions with E246, absence of contacts with E94, E85 or E48, and disruption of the hydrogen bond network in the R172/E146/W150 motif. (b and c) PikCwt in open conformation showing an open active site with widely spread binding residues (E94, E85 and E48). (d) Adequate binding of substrate (−)-26 to PikCD50ND176QE246A showing interactions with the all three binding residues simultaneously and the undistorted R172/E146/W150 motif. (e and f) PikCD50ND176QE246A in closed conformation showing a narrow active site channel and a tight arrangement of the binding residues.
Site Selectivity
Once the catalytic efficiency of PikC had been enhanced, we turned our attention to the selectivity of the hydroxylation reaction. Thus, in addition to driving the substrate and protein-engineering on the PikC platform, we investigated the site and stereoselectivity of this enzymatic hydroxylation reaction using a combination of QM and MD data. Density functional theory (DFT) transition state calculations of a model of Compound I reacting with (−)-menthol predicted that the methylene C–H bonds at the C4 position of menthol would be functionalized (Online Methods and Supplementary Table 1). Interestingly, the previously reported positions for site-selective C–H oxidation, including the sterically unhindered methyl C–H bonds and electronically activated methine C–H bonds, all involve higher activation barriers. Using this model system, the predicted selectivity between the axial and equatorial C4–H bonds is subtle, and abstraction of the axial hydrogen is favored by only 0.7 kcal mol−1 (3:1 kinetic ratio at 25 °C). We anticipated this selectivity to be modulated by the chiral environment of the enzyme, such that the C–H bond in closer proximity and more properly orientated towards the heme iron-oxo group in the preferred binding pose of the substrate in the active site, will be preferentially oxidized. Considering the transition structure for C4–Heq abstraction (Fig. 4A) as a reference, we screened all the binding poses sampled for each substrate bound to PikCD50N in the MD trajectories (compound 25 shown in Fig. 5B in order to detect the most favorably arranged C–H bond for oxidation. The hydrophobic active site of PikC consistently exposes the equatorial C4–Heq bond of (−)-menthol in a more appropriate orientation towards the iron-oxo group, irrespective of the anchoring group used and overriding the preference for C4–Hax predicted with the model non-enzymatic catalyst (Fig. 5C and Supplementary Fig. 10). In contrast, PikC was calculated to be unable to recognize any particular binding mode of the (+)-menthol substrates, leading to continuous reallocation of the cyclohexane ring inside the active site, and equally exposing H4eq and H4ax to the iron-oxo center (Fig. 5D and Supplementary Fig. 11). Simulations of the most active triple mutant PikCD50ND176QE246A gave analogous results (Supplementary Figs. 12 and 13). The combined QM and MD results predicted a high regioselectivity for both enantiomers of menthol at the C4 position, and a high stereoselectivity towards the equatorial C–H bond4 for the (−)-enantiomer.
Figure 5. Stereoselectivity in the C4–H abstraction catalyzed by PikC.

(a) Quantum mechanically (QM) optimized transition for hydrogen abstraction of H4eq in (−)-menthol (see Supplementary Fig. 2). (b) Representative snapshot of a MD trajectory of PikCD50N bound to 25 showing the substrate in close proximity to the catalytic heme iron-oxo species (Compound I). (c) and (d) Orientation of H4eq (in green) and H4ax (in orange) of (−)-menthol (c) and (+)-menthol (d) relative to the iron-heme oxo group in PikCD50N, derived from 0.5 µs MD trajectories. Each point represents a simulation snapshot. Deviations of the distances (x axis) and angles (y axis) from the quantum mechanically optimized transition structure (in blue) with a hypothetical model heme catalyst are shown. The greater separation between the two sets of points (H4eq and H4ax) in (−)-menthol is interpreted as a potential higher diastereoselectiviy in the associated transition states, and thus the hydroxylation reaction.
Experimentally, one major regioisomer was consistently formed from the (−)-menthol substrates, whereas at least five isomeric products were detected upon reaction of (+)-24–27 with PikCD50N-RhFRED (see Supplementary Information). To determine the structure of the major hydroxylation product, preparative-scale reactions were performed with (−)-25 and (−)-26, the two substrates with the highest TTN. Following enzymatic hydroxylation, the anchoring group was removed quantitatively (LiAlH4 reduction) for subsequent analysis. Diol 8 (Fig. 1), the result of H4eq oxidation, was obtained as the major product for both (−)-25 and (−)-26, confirming the prediction made through the QM and MD studies.
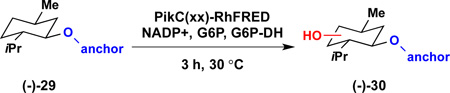
To evaluate the transferability of this approach to a structurally diverse panel of substrates, we tested the ability of the engineered P450, PikC(D50ND176QE246A)-RhFRED, to oxidize non-menthol small molecule cores bearing synthetic anchoring groups. As illustrated in in Fig. 6, six-membered ring substrates derived from (+)-α-terpineol and (−)-isopulegol are hydroxylated at C–H bonds remote from the anchoring group to afford 31 and 32, respectively. More sterically demanding fused bicyclic and bridged bicyclic substrates were also hydroxylated with high site and stereoselectivity to deliver alcohols 33–35. Notably, the relationship between the C–H bond oxidized in each case and the attachment point of the anchoring group is conserved, with C–H bonds remote to the anchoring group favored for hydroxylation. This directing effect can override electronic effects as more activated C–H bonds are untouched, for example a methylene C–H bond is hydroxylated over allylic C–H bonds to give 32. Further, the products generated consistently possess an anti relationship between the newly introduced hydroxyl group and the anchoring group as was predicted computationally and observed in the (−)-menthol model system.
Figure 6.
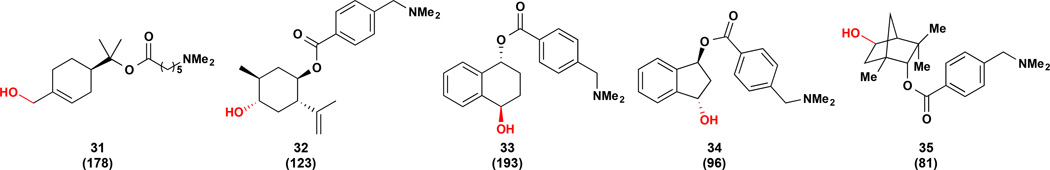
Site selective oxidation with PikCD50ND176QE246A-RhFRED. C–O bonds highlighted in red = hydroxyl group introduced in the P450 reaction. Numbers given in parentheses are TTN values determined in triplicate. Reaction conditions: 1 mM substrate, 5 µM enzyme, 1 mM NADP+, 5 mM glucose-6-phosphate, 1 U/mL glucose-6-phosphate dehydrogenase.
Conclusions
This work provides the foundation for a new method to site and stereoselectively oxygenate unactivated methylene C–H bonds that has eluded previous efforts using chemical catalysts with a substrate scope that exceeds what is typical of biocatalytic methods. Computation-driven substrate and protein engineering enabled the expansion of the substrate scope of PikC P450 monooxygenase from 12- and 14-membered macrolides to a six-membered ring scaffold. The efficiency of PikC oxidation of a given substrate as well as the selectivity was iteratively evaluated through QM and MD calculations with excellent correlation to experimental data. The optimized protocol devised through this initial study is described in Supplementary Fig. 14. This tool is currently being applied for the development of new anchoring groups to target specific classes of substrates, the prediction and improvement of enzyme selectivity, and the rational design of additional monooxygenases to generate superior C–H functionalization biocatalysts.
Online Methods
QM calculations
Full geometry optimizations, transition structure (TS) searches and single-point computations were carried out with the Gaussian 09 package (Frisch, M. J. et al., Gaussian, Inc., 2009). All geometry optimizations were carried out with the unrestricted version of the hybrid B3LYP functional,40 which has been widely used in the iron heme literature41,42. The relativistic LANL2DZ pseudopotential43, and associated basis-set was used for Fe. For, S, O, N, C, and H, the double-zeta basis-set 6–31G(d) was employed for obtaining the geometries, and the larger 6–311+G(d,p) basis-set was used to calculate single-point energies. Additional single-point energy calculations using functionals able to account for dispersion forces such as B3LYP-D3 with Becke-Johnson damping44 and M0645 in conjunction with the larger 6–311+G(2d,p) basis-set, and the SDD pseudopotential46 were performed (Supplementary Table 1). Thermal and entropic corrections to energy were calculated from vibrational frequencies. The nature of the stationary points was determined in each case according to the appropriate number of negative eigenvalues of the Hessian matrix from the frequency calculations. Scaled frequencies were not considered. In some problematic systems, the SCF convergence criterion was lowered (scfcon=6) to locate the transition states. Different spin states for Fe(III) and multiple conformers of the isopropyl and ester groups in the menthol derivatives were considered (see Supplementary Figs. 1 and 2). Mass-weighted intrinsic reaction coordinate calculations were carried out for a representative transition structure using the Gonzalez and Schlegel scheme47,50 in order to ensure that they indeed connected the appropriate reactants and products.
For the calculation of bond dissociation energies (BDE), single-point energy calculations using the correlated ab initio SCS-MP2 method49 in combination with the cc-pVTZ basis set50, were performed on the B3LYP/6–31G(d) optimized geometries. BDEs were defined as the difference in zero-point energies between menthol and the sum of the isolated radicals generated upon homolytic C–H cleavage (Supplementary Table 2).
Cartesian coordinates, electronic energies, entropies, enthalpies, Gibbs free energies, and lowest frequencies of the optimized structures are available in Supplementary Table 1).
MD simulations
The heme iron(IV)–oxo complex involved in the cytochrome-catalyzed oxidative hydroxylation cycle (Compound I) was used to model the active form of the PikC cofactor. Simulations were performed using the GPU code (pmemd)51 of the AMBER 12 package (Case, D. A. et al., UCSF, 2012). The Amber-compatible parameters developed by Cheatham III et al.52 were used for Compound I and its axial Cys ligand. Parameters for menthol substrates were generated within the antechamber module using the general AMBER force field (gaff)53, with partial charges set to fit the electrostatic potential generated at the HF/6–31G(d) level by the RESP model54. The charges were calculated according to the Merz-Singh-Kollman scheme55,56 using the Gaussian 09 package (Frisch, M. J. et al., Gaussian, Inc., 2009). Each protein was immersed in a pre-equilibrated truncated cuboid box with a 10 Å buffer of TIP3P57 water molecules using the leap module, resulting in the addition of around 15,000 solvent molecules. The systems were neutralized by addition of explicit counter ions (Na+ and Cl−). All subsequent calculations were done using the widely tested Stony Brook modification of the Amber 99 force field (ff99sb)58. A two-stage geometry optimization approach was performed. The first stage minimizes the positions of solvent molecules and ions imposing positional restraints on solute by a harmonic potential with a force constant of 500 kcal mol−1 Å−2, and the second stage was a minimization of all the atoms in the simulation cell except those involved in the harmonic distance restraint. The systems were gently heated using six 50 ps steps, incrementing the temperature 50 K each step (0–300 K) under constant-volume and periodic-boundary conditions. Water molecules were treated with the SHAKE algorithm such that the angle between the hydrogen atoms was kept fixed. Long-range electrostatic effects were modeled using the particle-mesh-Ewald method59. An 8 Å cutoff was applied to Lennard-Jones and electrostatic interactions. Harmonic restraints of 30 kcal/mol are applied to the solute, and the Andersen equilibration scheme is used to control and equalize the temperature. The time step was kept at 1 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Each system was then equilibrated for 2 ns with a 2 fs timestep at a constant volume. Production trajectories were then run for additional 500 ns under the same simulation conditions.
PikCxx-RhFRED analytical-scale enzymatic reactions
The standard assay contained 5 µM PikCxx-RhFRED, 1 mM substrate, 1 mM NADP+, 0.05 units of glucose-6-phosphate dehydrogenase, and 5 mM glucose-6-phosphate for NADPH regeneration in of reaction buffer (50 mM NaH2PO4, pH 7.3, 1 mM EDTA, 0.2 mM dithoerythritol, and 10% glycerol) total volume 50 µL. The reaction was carried out at 30 °C for 3 h and quenched by addition of 150 µL MeOH. The resulting mixture was briefly vortexed and centrifuged at 10,000 × g for 10 min. The subsequent liquid chromatography mass spectrometry (LC-MS) analysis was performed on an Agilent Q-TOF HPLC-MS (Department of Chemistry, University of Michigan) equipped with an high resolution electrospray mass spectrometry (ESI-MS) source and a Beckmann Coulter reverse-phase HPLC system using an Waters XBridge C18 3.5 µm, 2.1×150 mm under the following conditions: mobile phase (A = deionized water + 0.1% formic acid, B = acetonitrile + 0.1% formic acid), 10% to 100% B over 15 min, 100% B for 4 min; flow rate, 0.2 mL/min. Reactions were scanned for [M+16] (monohydoxylation) and [M+32] (dihydroxylation). The percent conversion was determined as outlined in Li et al35. Briefly, the percent conversion was calculated with AUCtotal products/(AUCtotal products + AUCunreacted substrate) by assuming ionization efficiency of substrate and hydroxylated products are the same, because the ionization site of this series of compounds is presumed to be the dimethylamino group.
Supplementary Material
Acknowledgments
This work was supported by the NSF under the CCI Center for Selective C−H Functionalization, CHE-1205646; NIH R01 grants GM078553 (D.H.S, J.M. and L.M.P.) and GM075962 (to K.N.H). The Extreme Science and Engineering Discovery Environment (XSEDE) is funded through the NSF (OCI-1053575) and the UCLA Institute of Digital Research and Education (IDRE). Additionally, the Life Sciences Research Foundation for a postdoctoral fellowship (A.R.H.N.) and the University of Michigan Chemistry-Biology Interface (CBI) training program (GM008597) (S.N.). Jessica Stachowski is thanked for helpful discussions.
Footnotes
Author contributions
A.R.H.N. and G.J.-O. contributed equally to this work. A.R.H.N., G.J.-O., P.L., S.N., J.M., K.N.H. and D.H.S. conceived, designed and supervised the project. G.J.-O., P.L., R.O.R., Y.-F.Y, L.R.F., and Z.L. performed the computational experiments. A.R.H.N, S.N., W.Z. and M.M.G. synthesized substrates and characterized products. A.R.H.N conducted the biochemical experiments. A.R.H.N, G.J.-O., P.L. L.M.P, J.M., K.N.H and D.H.S wrote and edited the manuscript.
Supplementary information is available in the online version of the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Newhouse T, Baran PS. If C-H bonds could talk: selective C-H bond oxidation. Angew. Chem. Int. Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gormisky PE, White MC. Catalyst-controlled aliphatic C-H oxidations with a predictive model for site-selectivity. J.. Am. Chem. Soc. 2013;135:14052–14055. doi: 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- 3.Chen MS, White MC. Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science. 2010;327:566–571. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- 4.Chen K, Eschenmoser A, Baran PS. Strain release in C-H bond activation. Angew. Chem. Int. Ed. 2009;48:9705–9708. doi: 10.1002/anie.200904474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roiban GD, Agudo R, Reetz MT. Cytochrome P450 catalyzed oxidative hydroxylation of achiral organic compounds with simultaneous creation of two chirality centers in a single C-H activation step. Angew. Chem. Int. Ed. 2014;53:8659–8663. doi: 10.1002/anie.201310892. [DOI] [PubMed] [Google Scholar]
- 6.Kille S, Zilly FE, Acevedo JP, Reetz MT. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 2011;3:738–743. doi: 10.1038/nchem.1113. [DOI] [PubMed] [Google Scholar]
- 7.Zhang KD, Shafer BM, Demars MD, Stern HA, Fasan R. Controlled oxidation of remote sp(3) C-H bonds in artemisinin via P450 catalysts with fine-tuned regio- and stereoselectivity. J. Am. Chem. Soc. 2012;134:18695–18704. doi: 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang KD, El Damaty S, Fasan R. P450 Fingerprinting method for rapid discovery of terpene hydroxylating P450 catalysts with diversified regioselectivity. J. Am. Chem. Soc. 2011;133:3242–3245. doi: 10.1021/ja109590h. [DOI] [PubMed] [Google Scholar]
- 9.Lewis JC, Coelho PS, Arnold FH. Enzymatic functionalization of carbon-hydrogen bonds. Chem. Soc. Rev. 2011;40:2003–2021. doi: 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fasan R. Tuning P450 Enzymes as oxidation catalysts. ACS Catal. 2012;2:647–666. [Google Scholar]
- 11.Agudo R, Roiban GD, Reetz MT. Achieving regio- and enantioselectivity of P450-catalyzed oxidative C–H activation of small functionalized molecules by structure-guided directed evolution. Chem Bio Chem. 2012;13:1465–1473. doi: 10.1002/cbic.201200244. [DOI] [PubMed] [Google Scholar]
- 12.Acevedo-Rocha CG, Hoebenreich S, Reetz MT. Iterative saturation mutagenesis: A powerful approach to engineer proteins by systematically simulating darwinian evolution in directed evolution library creation: Methods and protocols. In: Gillam EMJ, Copp JN, Ackerley DF, editors. Methods in Molecular Biology. 2nd Edition. Vol. 1179. Humana Press Inc; 2014. pp. 103–128. [DOI] [PubMed] [Google Scholar]
- 13.Jung ST, Lauchli R, Arnold FH. Cytochrome P450: Taming a wild type enzyme. Curr. Opin. Biotechnol. 2011;22:809–817. doi: 10.1016/j.copbio.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, Gabriele B, Belvedere S, Huang Y, Breslow R. Catalytic oxidations of steroid substrates by artificial cytochrome P-450 enzymes. J. Org. Chem. 2002;67:5057–5067. doi: 10.1021/jo020174u. [DOI] [PubMed] [Google Scholar]
- 15.Breslow R, Baldwin S, Flechtne T, Kalicky P, Liu S, Washburn W. Remote oxidation of steroids by photolysis of attached benzophenone groups. J. Am. Chem. Soc. 1973;95:3251–3262. doi: 10.1021/ja00791a031. [DOI] [PubMed] [Google Scholar]
- 16.Leow D, Li G, Mei TS, Yu JQ. Activation of remote meta-C-H bonds assisted by an end-on template. Nature. 2012;486:518–522. doi: 10.1038/nature11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang RY, Li G, Yu JQ. Conformation-induced remote meta-C-H activation of amines. Nature. 2014;507:215–220. doi: 10.1038/nature12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larsen AT, May EM, Auclair K. Predictable stereoselective and chemoselective hydroxylations and epoxidations with P450 3A4. J. Am. Chem. Soc. 2011;133:7853–7858. doi: 10.1021/ja200551y. [DOI] [PubMed] [Google Scholar]
- 19.Menard A, Fabra C, Huang Y, Auclair K. Type II ligands as chemical auxiliaries to favor enzymatic transformations by P450 2E1. Chem Bio Chem. 2012;13:2527–2536. doi: 10.1002/cbic.201200524. [DOI] [PubMed] [Google Scholar]
- 20.Polic V, Auclair K. Controlling Substrate specificity and product regio- and stereo-selectivities of P450 enzymes without mutagenesis. Bioorg. Med. Chem. 2014;22:5547–5554. doi: 10.1016/j.bmc.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vermeulen NA, Chen MS, White MC. The Fe(PDP)-catalyzed aliphatic C-H oxidation: A slow addition protocol. Tetrahedron. 2009;65:3078–3084. [Google Scholar]
- 22.Olivo G, Lanzalunga O, Mandolini L, Di Stefano S. Substituent effects on the catalytic activity of bipyrrolidine-based iron complexes. J. Org. Chem. 2013;78:11508–11512. doi: 10.1021/jo4020744. [DOI] [PubMed] [Google Scholar]
- 23.Yazerski VA, Spannring P, Gatineau D, Woerde CHM, Wieclawska SM, Lutz M, Kleijn H, Gebbink R. Making Fe(BPBP)-catalyzed C-H and C=C oxidations more affordable. Org. Biomol. Chem. 2014;12:2062–2070. doi: 10.1039/c3ob42249f. [DOI] [PubMed] [Google Scholar]
- 24.Lee S, Fuchs PL. An efficient C-H oxidation protocol for alpha-hydroxylation of cyclic steroidal ethers. Org. Lett. 2004;6:1437–1440. doi: 10.1021/ol049712a. [DOI] [PubMed] [Google Scholar]
- 25.Ottenbacher RV, Samsonenko DG, Talsi EP, Bryliakov KP. Highly efficient, regioselective, and stereospecific oxidation of aliphatic C-H groups with H2O2, catalyzed by aminopyridine manganese complexes. Org. Lett. 2012;14:4310–4313. doi: 10.1021/ol3015122. [DOI] [PubMed] [Google Scholar]
- 26.Chow YL, Hayasaka T, Tam JNS. Photoreaction of nitroso compounds in solution. 13. Photo-oxidation of alkyl nitrites. Can. J. Chem. 1970;48:508–511. [Google Scholar]
- 27.Petrovic G, Saicic RN, Cekovic Z. Regioselective free radical phenylsulfenation of a non-activated delta-carbon atom by the photolysis of alkyl benzenesulfenate. Tetrahedron. 2003;59:187–196. [Google Scholar]
- 28.Miyazawa M, Kumagae S, Kameoka H. Biotransformation of (+)- and (−)-menthol by the larvae of common cutworm (Spodoptera Litura) J. Agric. Food Chem. 1999;47:3938–3940. doi: 10.1021/jf980924u. [DOI] [PubMed] [Google Scholar]
- 29.Atta ur R, Yaqoob M, Farooq A, Anjum S, Asif F, Choudhary MI. Fungal transformation of (1R,2S,5R)-(−)-menthol by cephalosporium aphidicola. J. Nat. Prod. 1998;61:1340–1342. doi: 10.1021/np980057n. [DOI] [PubMed] [Google Scholar]
- 30.Xue YQ, Zhao LS, Liu HW, Sherman DH. A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: Architecture of metabolic diversity. Proc. Natl. Acad. Sci. U. S. A. 1998;95:12111–12116. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue YQ, Wilson D, Zhao LS, Liu HW, Sherman DH. Hydroxylation of macrolactones YC-17 and narbomycin is mediated by the PikC-encoded cytochrome P450 in Streptomyces venezuelae. Chem. Biol. 1998;5:661–667. doi: 10.1016/s1074-5521(98)90293-9. [DOI] [PubMed] [Google Scholar]
- 32.Zhang QB, Sherman DH. Isolation and structure determination of novamethymycin, a new bioactive metabolite of the methymycin biosynthetic pathway in Streptomyces venezuelae. J. Nat. Prod. 2001;64:1447–1450. doi: 10.1021/np010146r. [DOI] [PubMed] [Google Scholar]
- 33.Sherman DH, Li SY, Yermalitskaya LV, Kim YC, Smith JA, Waterman MR, Podust LM. The structural basis for substrate anchoring, active site selectivity, and product formation by P450 PikC from Streptomyces venezuelae. J. Biol. Chem. 2006;281:26289–26297. doi: 10.1074/jbc.M605478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li SY, Ouellet H, Sherman DH, Podust LM. Analysis of transient and catalytic desosamine-binding pockets in cytochrome P-450 PikC from Streptomyces venezuelae. J. Biol. Chem. 2009;284:5723–5730. doi: 10.1074/jbc.M807592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li SY, Chaulagain MR, Knauff AR, Podust LM, Montgomery J, Sherman DH. Selective oxidation of carbolide C-H bonds by an engineered macrolide P450 mono-oxygenase. Proc. Natl. Acad. Sci. U. S. A. 2009;106:18463–18468. doi: 10.1073/pnas.0907203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Negretti S, Narayan ARH, Chiou KC, Kells PM, Stachowski JL, Hansen DA, Podust LM, Montgomery J, Sherman DH. Directing group-controlled regioselectivity in an enzymatic C–H bond oxygenation. J. Am. Chem. Soc. 2014;136:4901–4904. doi: 10.1021/ja5016052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiménez-Osés G, Osuna S, Gao X, Sawaya MR, Gilson L, Collier SJ, Huisman GW, Yeates TO, Tang Y, Houk KN. The role of distant mutations and allosteric regulation on LovD active site dynamics. Nat. Chem. Biol. 2014;10:431–436. doi: 10.1038/nchembio.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li SY, Podust LM, Sherman DH. Engineering and analysis of a self-sufficient biosynthetic cytochrome P450 PikC fused to the RhFRED reductase domain. J. Am. Chem. Soc. 2007;129:12940–12941. doi: 10.1021/ja075842d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong C-H, Whitesides GM. Enzyme-catalyzed organic synthesis: NAD(P)H cofactor regeneration by using glucose-6-phosphate and glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides. J. Am. Chem. Soc. 1981;103:4890–4899. [Google Scholar]
- 40.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 41.Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. P450 enzymes: Their structure, reactivity, and selectivity—Modeled by QM/MM calculations. Chem. Rev. 2009;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 42.Rydberg P, Sigfridsson E, Ryde U. On the role of the axial ligand in heme proteins: A theoretical study. J. Biol. Inorg. Chem. 2004;9:203–223. doi: 10.1007/s00775-003-0515-y. [DOI] [PubMed] [Google Scholar]
- 43.Hay PJ, Wadt WR. Ab Initio Effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985;82:270–283. [Google Scholar]
- 44.Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011;32:1456–1465. doi: 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Truhlar D. The M06 Suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 46.Dolg M, Wedig U, Stoll H, Preuss H. Energy-adjusted abinitio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987;86:866–872. [Google Scholar]
- 47.Gonzalez C, Schlegel HB. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990;94:5523–5527. [Google Scholar]
- 48.Gonzalez C, Schlegel HB. An Improved algorithm for reaction path following. J. Chem. Phys. 1989;90:2154–2161. [Google Scholar]
- 49.Gerenkamp M, Grimme S. Spin-component scaled second-order Moller-Plesset perturbation theory for the calculation of molecular geometries and harmonic vibrational frequencies. Chem. Phys. Lett. 2004;392:229–235. [Google Scholar]
- 50.Dunning TH. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989;90:1007–1023. [Google Scholar]
- 51.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on Gpus. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 52.Shahrokh K, Orendt A, Yost GS, Cheatham TE. Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012;33:119–133. doi: 10.1002/jcc.21922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general Amber Force Field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 54.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A Well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 55.Besler BH, Merz KM, Kollman PA. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990;11:431–439. [Google Scholar]
- 56.Singh UC, Kollman PA. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984;5:129–145. [Google Scholar]
- 57.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 58.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 59.Darden T, York D, Pedersen L. Particle mesh ewald: An N-log(N) method for ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.