

Summary
Taxol and other antimitotic agents are frontline chemotherapy agents but the mechanisms responsible for patient benefit remain unclear. Following a genome-wide siRNA screen, we identified the oncogenic transcription factor Myc as a taxol sensitizer. Using time-lapse imaging to correlate mitotic behavior with cell fate, we show that Myc sensitizes cells to mitotic blockers and agents that accelerate mitotic progression. Myc achieves this by upregulating a cluster of redundant pro-apoptotic BH3-only proteins and suppressing pro-survival Bcl-xL. Gene expression analysis of breast cancers indicates that taxane responses correlate positively with Myc and negatively with Bcl-xL. Accordingly, pharmacological inhibition of Bcl-xL restores apoptosis in Myc-deficient cells. These results open up opportunities for biomarkers and combination therapies that could enhance traditional and second-generation antimitotic agents.
Graphical Abstract
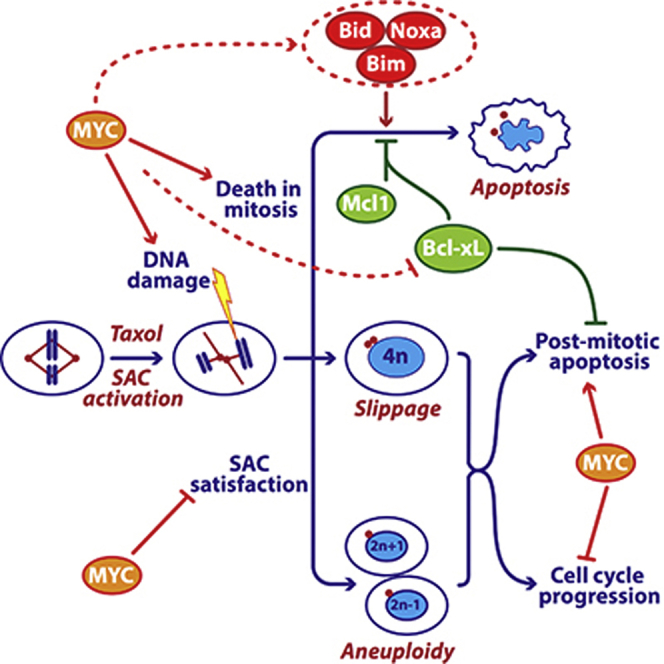
Highlights
-
•
Genome-wide screen shows that Myc and Egr1 promote apoptosis during mitotic arrest
-
•
Myc upregulates BH3-only proteins and downregulates Bcl-xL
-
•
Myc sensitizes lung, breast, ovarian, and colon cancer cells to antimitotic drugs
-
•
Pharmacological inhibition of Bcl-xL restores apoptosis in Myc-low cells
Topham et al. show that Myc sensitizes cancer cells to mitotic blockers and agents that accelerate mitotic progression by regulating the expression of an apoptotic network. Taxane responses in human breast cancers correlate positively with the Myc level and negatively with the Bcl-xL level.
Significance
Antimitotic agents such as the taxanes are used widely to treat various cancers. To address limitations with these agents, a new generation of inhibitors that disrupt mitosis without affecting microtubule dynamics is being evaluated, including drugs targeting mitotic kinesins and mitotic kinases. However, we still have limited understanding of the mechanisms that dictate cell fate in response to mitotic disruption. Here we show that Myc drives expression of an apoptotic network that sensitizes breast, ovarian, lung, and colon cancer cells to drugs that both activate and override the spindle assembly checkpoint. Moreover, we show that Myc promotes both p53-independent death in mitosis and p53-dependent post-mitotic responses. Our results raise opportunities to explore biomarkers and combination therapies aimed at enhancing antimitotic efficacy.
Introduction
Antimitotic drugs are frontline treatments for breast, ovarian, and lung cancer, as well as various hematological malignancies (Dumontet and Jordan, 2010). These drugs bind tubulin and inhibit microtubule dynamics, and although many cancers initially respond well, some are intrinsically resistant and others acquire resistance (Murray et al., 2012). Predicting which cancers will respond is hampered by our limited understanding of the molecular mechanisms responsible for patient benefit (Gascoigne and Taylor, 2009; Weaver, 2014). At high concentrations, antimitotic drugs disrupt spindle assembly, leading to mitotic arrest by persistent activation of the spindle assembly checkpoint (SAC) (Lara-Gonzalez et al., 2012). SAC activation blocks the anaphase promoting complex/cyclosome (APC/C), thereby preventing ubiquitination and degradation of cyclin B1, in turn maintaining the mitotic state. Following prolonged arrest, cells either die in mitosis or undergo “slippage,” returning to interphase without completing cell division (Brito and Rieder, 2006). Following slippage, p53-dependent post-mitotic responses then induce cell cycle arrest, senescence, or apoptosis (Rieder and Maiato, 2004). At lower taxol concentrations, the SAC becomes satisfied, allowing cells to progress through mitosis, albeit with spindle abnormalities and chromosome segregation errors (Zasadil et al., 2014). Bypassing both death in mitosis (DiM) and post-mitotic responses can fuel chromosome instability and taxane resistance (A’Hern et al., 2013).
The competing-networks model helps explain whether a cell either dies in mitosis or undergoes slippage (Gascoigne and Taylor, 2008). According to this model, two independent networks dictate mitotic cell fate, one slowly generating a death signal, the other slowly degrading cyclin B1, leading to slippage. During a prolonged arrest, these networks work in opposite directions: while cell death signals become stronger, cyclin B1 levels slowly fall due to incomplete penetrance of SAC-mediated APC/C inhibition (Brito and Rieder, 2006). Both networks have thresholds and the fate of the cell is dictated by which threshold is breached first. Whereas our understanding of the mechanisms regulating cyclin B1 degradation is well advanced, less is known about death in mitosis. It involves the intrinsic apoptosis pathway; however, how this is regulated during mitosis is unclear (Topham and Taylor, 2013). The nature of the apoptotic trigger is also unclear, but DNA damage seems a likely candidate, with one source being partial activation of caspase-activated DNase (CAD), caused by cytochrome c leakage from mitochondria (Orth et al., 2012). A second source is telomere deprotection, driven by the mitotic kinase Aurora B (Hayashi et al., 2012). In light of our limited understanding regarding the mechanisms responsible for apoptosis during a mitotic arrest, we adopted an unbiased approach and screened a genome-wide library for siRNAs that suppress taxol-induced cell death. To define how genes identified in the screen modulate antimitotic responses, we then used single-cell time-lapse imaging to directly correlate mitotic behavior with subsequent cell fate.
Results
A Genome-wide Screen for Regulators of Mitotic Cell Fate
The competing-networks model predicts that suppressing death signals during mitotic arrest provides more time for cyclin B1 degradation, thereby shifting cell fate from death to slippage (Figure 1A). To test this, we screened an siRNA library to identify genes required for DiM. Because slippage results in cell survival, we based the screen on a viability assay (Figure S1A). To maximize the assay’s dynamic range, we treated RKO cells, which predominantly undergo DiM (Gascoigne and Taylor, 2008), with a saturating concentration of taxol to ensure maximal mitotic blockage and apoptotic response. We also synchronized the cells to maximize cell death by 48 hr (Figure 1B). The primary screen identified 325 hits (Figure S1B). To filter out off-target hits, we performed a secondary screen using a pool of four different siRNAs, yielding 100 hits. Because taxol-induced death requires mitotic entry and robust spindle checkpoint activation, we predicted that in addition to DiM genes, the screen would also uncover genes required for cell cycle progression and SAC function. Indeed, we identified all the known SAC components, several kinetochore proteins required for SAC function and the entire chromosomal passenger complex, plus several genes required for mitotic entry (Figure 1C). To distinguish cell cycle and SAC genes from potential DiM genes, we performed a tertiary screen measuring mitotic index at 24 hr (Figure 1B) and plotted it against viability at 48 hr (Figure 1C). To hone in on potential DiM genes, we focused on hits with a high mitotic index at 24 hr and a substantial viability score at 48 hr (Figure 1C). Time-lapse microscopy showed that siRNA pools targeting KCNK1, ZNF791, SNTA1, and MYC shifted cell fate from death to slippage (Figure S1C). Importantly, mitotic exit was not accelerated, indicating inhibition of apoptosis rather than SAC override.
Figure 1.

A Genome-wide siRNA Screen for Regulators of Mitotic Cell Fate
(A) Rationale for the screen based on the competing-networks model.
(B) Timeline of siRNA transfection procedure.
(C) Scatterplot of mitotic index at 24 hr against viability at 48 hr.
Myc Is a Regulator of Cell Fate following Prolonged Mitotic Arrest
Of the four hits, we first focused on MYC, which encodes the bHLH-Zip transcription factor c-Myc (hereafter Myc). Myc, a potent oncogene deregulated in many cancers, regulates a multitude of genes via both transcriptional amplification and co-factor-dependent activation/repression (Conacci-Sorrell et al., 2014; Eilers and Eisenman, 2008; Hann, 2014; Wolf et al., 2015). Myc thus drives numerous biological pathways including proliferation, biogenesis, and metabolism which, when deregulated, promote transformation and tumorigenesis. Because Myc can also drive apoptosis, primarily via the ARF-MDM2-p53 pathway (Lowe et al., 2004; McMahon, 2014), we considered it an attractive candidate for a DiM gene. To validate Myc as a bona fide on-target hit, we deconvolved the siRNA pools, identifying four distinct siRNAs that repressed Myc and inhibited DiM (Figures S1D and S2A). When combined, these four siRNAs reduced Myc protein levels by 90% and shifted cell fate in favor of slippage (Figures 2A and 2B). In nine control experiments, quantitating 100 cells per population, 82% of cells underwent DiM, while 18% slipped (Figure 2C). In five Myc RNAi populations, 45% of cells died, while 55% slipped. Moreover, titrating the siRNAs revealed a correlation between Myc protein levels and cell fate (Figure 2D). In addition, an RNAi-resistant Myc transgene reverted the fate profile back toward DiM (Figure S2B). To further validate Myc, we turned to non-RNAi modalities, in particular the small molecules DMSO and JQ1 (Figure S2C). DMSO, which blocks transcriptional elongation of MYC (Eick and Bornkamm, 1986), efficiently suppressed Myc in RKO cells (Figure S2C) and reduced DiM from 92% to 58% (Figure S2D). JQ1 displaces the Brd4 transcriptional elongation factor from the MYC promoter (Filippakopoulos et al., 2010; McKeown and Bradner, 2014) and accordingly JQ1 inhibited Myc expression in RKO cells (Figure S2C). This was accompanied by a substantial effect on proliferation (Figure 2E). However, of the cells that did enter mitosis, only 56% were killed by taxol, demonstrating a shift in favor of slippage (Figure 2E). Significantly, a Myc cDNA resisted the DMSO and JQ1 effects and restored DiM (Figure S2D). To determine whether Myc’s role in DiM depends on its ability to modulate gene expression, we turned to Omomyc, a mutant bHLH-Zip domain that sequesters Myc in complexes unable to bind to E-boxes (Soucek et al., 2002). Inducing Omomyc in RKO cells inhibited DiM (Figure S2B), indicating that Myc most likely promotes DiM via its canonical role as a transcription factor. Interestingly Myc V394D, which cannot bind the Miz1 transcriptional repressor (Wiese et al., 2013), rescued Myc RNAi (Figure S2B), suggesting that Myc promotes DiM largely via transcriptional activation. Taking together the RNAi data, the DMSO, JQ1, and Omomyc experiments, we conclude that Myc is a key determinant of cell fate following prolonged mitotic arrest.
Figure 2.

Myc Is a Regulator of Mitotic Cell Fate
(A) Immunoblots showing Myc inhibition.
(B) Fate profiles of RKO cells exposed to 0.1 μM taxol following Myc RNAi.
(C) Cell fate in nine control and five Myc RNAi populations.
(D) Correlation between Myc protein levels and cell fate.
(E) Fate profiles of RKO cells exposed to 0.1 μM taxol and 0.5 μM JQ1.
(F) Time spent arrested in mitosis; entire population (gray), cells that die (red), and cells that slip (blue).
(G) Time arrested in mitosis with lines connecting cells from the same population. ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
See also Figure S2.
Using Myc to Test the Competing-Networks Model
The competing-networks model predicts that suppressing mitotic death provides more time for cyclin B1 degradation, thus shifting the balance toward slippage. A corollary is that the average time spent in mitosis should increase (Figure 1A). Consistently, whereas controls spent 17.1 hr arrested in mitosis, Myc-deficient cells spent 21.3 hr (Figure 2F) arrested in mitosis. Moreover, when we compared the cells that died, controls took 16.0 hr, whereas Myc-deficient cells took 20.4 hr; thus, even if a cell did not escape death, inhibiting Myc delayed its onset. Slippage typically took longer than DiM (Figure 2G) and the time from mitotic entry to slippage was not significantly affected by Myc RNAi (Figure 2F), consistent with the notion that the two competing networks are independent, and that Myc influences the death pathway but not the slippage pathway.
Myc Inhibition Deregulates an Apoptosis Module
To define how Myc promotes DiM, we interrogated mitosis and apoptosis gene expression modules using Nanostring technology. With the exception of Cenp-T, all the mitosis genes were suppressed following Myc RNAi (Figure 3A), reflecting Myc’s role as a transcriptional amplifier and/or cell cycle driver. Of the three notably repressed genes, Survivin and Mad2 promote chromosome alignment and SAC function. Consistently, in the absence of taxol, whereas overall mitotic timing was normal in Myc RNAi cells, chromosome alignment was delayed slightly and anaphase onset slightly accelerated (Figure S3A). Nevertheless, despite these subtle effects on an unperturbed mitosis, Figure 2 clearly demonstrates that Myc-deficient cells mount a robust SAC response in 100 nM taxol, suggesting that mitotic deregulation is unlikely to account for the shift in cell fate. We therefore turned to the apoptosis module, which included 12 upregulated and six downregulated genes (Figure 3A). Because Myc RNAi promotes survival, the dominant effectors are likely to be upregulated pro-survival genes and/or downregulated pro-death genes. Of the upregulated genes, Bcl-xL is a well-established pro-survival factor, while three of the downregulated genes, namely Bid, Bim, and Noxa, encode BH3-only pro-apoptotic proteins (Figure 3A). Because these are Myc effectors in other contexts (McMahon, 2014), we analyzed them in more detail.
Figure 3.

A Cluster of Redundant BH3-Only Proteins Promote Death in Mitosis
(A) Gene expression changes following Myc RNAi; y axis shows the fold change and circle sizes reflect the number of transcripts detected. Horizontal lines represent mean ± 1 SD.
(B–E) Fate profiles of RKO cells exposed to 0.1 μM taxol following tet-induced overexpression of Bcl-xL (B); RNAi-mediated co-repression of Myc and Bcl-xL (C); RNAi-mediated co-repression of Bim, Bid, and Noxa (D); and tet-induced overexpression of Bim following Myc RNAi (E).
BH3-Only Pro-apoptotic Proteins Are Redundant Effectors of Myc
Consistent with Myc’s known ability to repress Bcl-xL (Eischen et al., 2001), Myc RNAi elevated Bcl-xL protein levels in RKO cells (Figure S3B). Ectopic overexpression of Bcl-xL suppressed both DiM and post-mitotic apoptosis (Figures 3B and S3C), supporting the notion that Bcl-xL is a potent mitotic survival factor (Bah et al., 2014; Minn et al., 1996; Upreti et al., 2008). However, ectopic Bcl-xL enhanced survival more potently than Myc RNAi, suggesting that other consequences of Myc inhibition attenuate the pro-survival effect of increased Bcl-xL (Eichhorn et al., 2014). Indeed, whereas Mcl1 transcripts fell only marginally upon Myc RNAi, Mcl1 protein levels fell substantially (Figures S3B and S3E), possibly due to deregulation of factors involved in Mcl1 turnover. However, in taxol-arrested cells, this residual Mcl1 appeared to resist mitotic degradation (Figure S3E). Nevertheless, despite these complexities, we reasoned that Bcl-xL upregulation alone is unlikely to explain the Myc RNAi phenotype, and therefore we turned our attention to the downregulated pro-death genes.
The downregulated BH3-only proteins (Figure 3A), namely Bid, Bim, and Noxa, are known to be upregulated by Myc, either directly or via the ARF-MDM2-p53 pathway (McMahon, 2014). If Bid, Bim, and Noxa are important Myc DiM effectors, then their inhibition should mimic Myc RNAi. However, because they did not manifest in the screen they are unlikely to be essential for DiM. Indeed, repression of each in isolation or in pairs had little effect on mitotic fate (Figure S3G). In contrast, co-repression of Bim, Bid, and Noxa tipped the balance in favor of slippage (Figure 3D), consistent with them being redundant downstream effectors of Myc. A corollary is that overexpression of any one should revert the Myc RNAi phenotype. Indeed, transgenic Bim restored DiM in Myc RNAi cells (Figures 3E and S3H). Consistent with the competing-networks concept, Bim/Bid/Noxa RNAi extended mitotic timing, whereas induction of Bim accelerated the onset of DiM (Figure S3I). We conclude therefore that Bim, Bid, and Noxa are redundant Myc effectors required for DiM.
MYC Sensitizes Various Cancer Lines to Antimitotic Drugs
To test the role of Myc in a wider context, we inhibited Myc in 12 cell lines derived from colon, lung, breast, cervical, and ovarian cancers (Figure S4A), then exposed them to a panel of antimitotic drugs including agents targeting Eg5/KSP, Plk1, Cenp-E, Aurora A, Aurora B, and Mps1. To monitor apoptosis, we used time-lapse imaging to measure caspase-3/7 activity. The effects of inhibiting Myc were strikingly consistent, significantly attenuating apoptosis in nine lines (Figure 4A). Interestingly, Myc inhibition had little effect in three lines, namely DLD-1, H1703, and Caov-3. The competing-networks model may explain these exceptions. DLD-1 cells slip very quickly (Gascoigne and Taylor, 2008); therefore, despite inhibiting DiM, slippage would be expected to continue such that Myc RNAi has little effect. Conversely, H1703 cells die very quickly and rarely slip, suggesting that despite delaying DiM, slippage may not be fast enough to permit exit. Nevertheless, Myc promotes apoptosis in a variety of cancer lines exposed to various antimitotic agents.
Figure 4.

Myc Promotes Post-mitotic Death
(A) Apoptosis induction in cell lines indicated exposed to various antimitotic agents following Myc RNAi.
(B) Graph quantitating death following slippage in the presence of 0.1 μM taxol.
(C) Fate profiles of wild-type and p53-deficient HCT116 cells following Myc RNAi then exposed to the Mps1 inhibitor AZ3146 (2 μM). Numbers indicate percentage of cells that undergo one division (white), multiple divisions (black), post-mitotic death (green), and DiM (red).
(D) Fate profiles of RKO cells exposed to 10 nM taxol in combination with 100 nM WEHI-539. In (C), 0 hr is when imaging started.∗p < 0.05, ∗∗p < 0.01.
MYC Promotes Apoptosis following Slippage
In contrast to taxol, drugs targeting Aurora B and Mps1 drive cells through an aberrant mitosis (Keen and Taylor, 2009), suggesting that Myc also promotes apoptosis following slippage. Indeed, following exit from a prolonged taxol arrest, Myc RNAi reduced cell death from 60% to 25% (Figure 4B). Moreover, in response to an Mps1 inhibitor, Myc RNAi reduced post-mitotic apoptosis from 40% to 18% (Figure 4C) and enhanced colony formation (Figure S4B). Canonical Myc-driven apoptosis involves the ARF-MDM2-p53 pathway; however, because p53 is disengaged during mitosis, Myc-dependent DiM is likely p53-independent. Indeed, Myc RNAi suppressed apoptosis in p53-deficient HCT116 cells treated with mitotic blockers (Figure 4A). Consistent with p53 restraining further cell cycle progression following an aberrant mitosis (Thompson and Compton, 2010), p53 deletion increased the number of HCT116 cells entering a second mitosis from 32% to 68% (Figure 4C). However, apoptosis was only slightly affected by p53 loss, 30% versus 40% in controls, indicating that post-mitotic apoptosis is largely p53-independent. Interestingly, whereas Myc RNAi only had a marginal effect on post-mitotic apoptosis in p53-deficient cells, it increased the number of p53-proficient cells entering a second mitosis from 32% to 58% (Figure 4C). Thus, following an aberrant mitosis, Myc not only enhances post-mitotic apoptosis but also suppresses cell cycle progression, possibly via the ARF-MDM2-p53 pathway.
Myc Enhances Survival in Low-Dose Taxol
In breast cancers, taxol does not accumulate to concentrations high enough to induce prolonged mitotic arrest; rather cells progress through mitosis, albeit with chromosome segregation errors (Zasadil et al., 2014). Because Myc promotes post-mitotic death, we reasoned that Myc would also influence low-dose taxol responses. To test this, we reduced the taxol concentration to 10 nM (Figure S4C), a concentration in cell culture medium that results in intracellular concentrations similar to those measured in breast cancer (Zasadil et al., 2014). In 10 nM taxol, most RKO cells died in mitosis but 31% divided, indicating that the taxol concentration was “on the edge” (Figure 4D). Of those that divided, 12.5% died in the next interphase. Strikingly, Myc RNAi cells spent considerably less time in mitosis then divided, indicating that the SAC became satisfied (Figure 4D). Consistently, Myc RNAi slightly accelerated anaphase onset during an unperturbed mitosis (Figure S3A). Following division in 10 nM taxol, Myc RNAi cells survived, at least for the duration of the experiment. These divisions are unlikely to be normal; indeed, when we added 100 nM WEHI-539, a selective Bcl-xL inhibitor (Lessene et al., 2013), all the cells that divided subsequently died (Figures 4D and S4C). Thus, inhibiting Myc enhances survival in low-dose taxol but this can be ameliorated by inhibition of Bcl-xL.
Tumor Cells Overexpressing MYC Are Sensitive to Antimitotic Agents
Because inhibiting Myc suppresses apoptosis in response to antimitotic agents, we asked whether elevating Myc expression had the opposite effect. Indeed, tet-induction of a Myc transgene in RKO cells accelerated DiM by 2.3 hr and reduced slippage, albeit modestly (Figure 5A). Moreover, of the cells that slipped, overexpressing Myc increased post-mitotic death from 46% to 78%. Consistently, overexpressing Myc in Rat1a cells enhances colcemid-induced apoptosis (Li and Dang, 1999). To examine Myc overexpression in a wider context, we interrogated the Genomics of Drug Sensitivity in Cancer database (Garnett et al., 2012), which describes 665 cell lines, 47 of which overexpress Myc, in response to 141 drugs, 11 of which target microtubules or mitotic regulators. The mean half-maximal inhibitory concentration (IC50) effect for the 11 antimitotic drugs was 0.47 compared to 0.83 for the other 130 drugs (Figures 5B and S5A), confirming that tumor cells overexpressing Myc are more sensitive to antimitotic agents compared to drugs in general.
Figure 5.

Overexpression of Myc Sensitizes Cancer Cells to Antimitotic Agents
(A) Fate profiles and box-and-whisker plot showing time to DiM in RKO cells exposed to 0.1 μM taxol following tet-induced overexpression of Myc.
(B) Gene IC50 effects for MYC comparing antimitotic agents with other drugs.
(C) Heatmaps showing gene expression profiles of 22 breast tumors (six non-responders and 16 complete/near-complete responders) treated with capecitabine and docetaxel.
(D) Box-and-whisker plots showing Myc and Bcl-xL expression levels in non-responsive (N) and responsive tumors (C).
(E) Bar graphs showing correlations between MYC and the SAC, cell cycle, and apoptosis genes. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To determine whether the Myc overexpression effect extended to patient chemotherapy responses, we interrogated microarray datasets from XeNA, a clinical trial examining response rates in women with operable, early stage breast cancer receiving neoadjuvant capecitabine plus the antimitotic agent docetaxel (Glück et al., 2012). Tumors from patients showing complete or near-complete responses tended to have elevated Myc (Figures 5C and 5D). Next, we analyzed the SAC and cell cycle genes identified by the siRNA screen (Figure 1C), and the Myc regulated genes identified by our Nanostring analysis (Figure 3A). Although there was no obvious overall correlation between Myc and several housekeeping genes, Myc correlated positively with the SAC, cell cycle, and apoptosis genes (Figures 5C and 5E). Moreover, the SAC, cell cycle, and Myc clusters were elevated in the responsive tumors (Figure S5B). The elevation was not simply due to a global increase in gene expression because Bcl-xL displayed a negative correlation (Figures 5C–5E), consistent with Myc-induced suppression (Figure 3A). Moreover, the correlation between Myc and cell cycle/SAC genes was not simply due to increased proliferation, because Her2-positive tumors did not show a similar pattern (Figures 5E and S5C). These results suggest that a positive response to antimitotic chemotherapy requires entry into mitosis, a robust SAC response, and the ability to undergo Myc-dependent apoptosis.
Myc Is Required for Taxol-Induced Apoptosis in Mouse Intestinal Crypts
The correlation between Myc expression and chemotherapy responses is provocative. However, Her2-negative breast cancers include various tumor subtypes and XeNA used multiple chemotherapy agents. We therefore turned to a genetically constrained model system that allows single agent exposure to validate the role of Myc in the context of an intact tissue. Mice harboring a conditional MYC allele provided such a system (Phesse et al., 2014). AhCre+ MYCfl/fl mice were injected with β-napthoflavone to delete MYC in the small intestine. Four days later, taxol was administered to induce mitotic arrest and then apoptosis was measured with caspase 3 staining (Radulescu et al., 2010). In Myc-deficient intestines, we observed 0.2 apoptotic cells per intestinal crypt compared to 1.2 in Myc-proficient controls (Figure 6). We conclude therefore that Myc is a determinant of mitotic cell fate in the mouse intestine.
Figure 6.
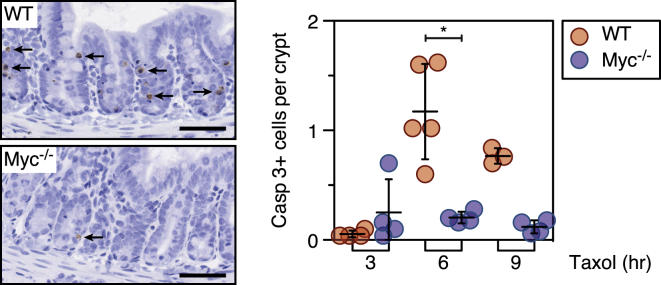
MYC-Deficient Crypts Are Resistant to Taxol-Induced Apoptosis
Immunohistochemical staining and quantitation of cleaved caspase 3 in intestinal sections from wild-type and MYC mutant mice following 3, 6, and 9 hr exposure to taxol. Bar represents 50 μm. ∗p < 0.05.
Interrogating Kcnk1, Snta1, and Znf791
The transcript profiling and functional experiments indicate that Myc enhances DiM by suppressing Bcl-xL and upregulating BH3-only proteins (Figure 3). However, a defining feature of Myc is its ability to modulate numerous genes thereby influencing various biological processes, including biosynthesis and metabolism pathways (Conacci-Sorrell et al., 2014; Eilers and Eisenman, 2008). Consequently, Myc targets not included in the Nanostring analysis could contribute to the phenotype. Moreover, the screen identified KCNK1, ZNF791 and SNTA1 (Figure S1C), but it is not immediately obvious how they might modulate apoptosis. To address these issues, we deconvolved the Kcnk1, Znf791, and Snta1 siRNA pools. In each case, only a single siRNA sequence enhanced viability, suggesting that they were “off-target” hits (Figure S1D). When transfected in isolation, the active Znf791 and Snta1 siRNAs accelerated mitotic exit rather than delaying DiM (Figure S1E). In contrast, the active Kcnk1 siRNA induced a Myc-like phenotype, suppressing DiM without accelerating mitotic exit. Therefore, to identify the target of this siRNA, and to interrogate Myc target genes not included in the Nanostring analysis, we turned to global gene expression profiling.
Egr1 Promotes Death in Mitosis
RKO cells were transfected with Myc, Kcnk1, and Snta1 siRNAs and then cDNA libraries were sequenced using Illumina HiSeq technology. Myc RNAi induced numerous changes, with 955 downregulated genes and 1,214 upregulated genes (Figure 7A). The effect on Myc itself was relatively modest, possibly reflecting negative auto-regulation (Conacci-Sorrell et al., 2014). Gene ontology analysis highlighted ribosome biogenesis, metabolism, gene expression, cell cycle, and apoptosis pathways (Figure S6C), consistent with known Myc functions. The Kcnk1 siRNA affected 424 genes, with KCNK1 itself one of the most repressed (Figure 7A). Whereas gene ontology analysis also highlighted metabolism and biosynthesis pathways, the p values and fold enrichment scores were substantially lower (Figure S6C), indicating that DiM can be suppressed without major effects on metabolism and biosynthesis pathways.
Figure 7.

Egr1 Is a Regulator of Mitotic Cell Fate
(A) Volcano plots showing gene expression changes following Myc and Kcnk1 RNAi.
(B) Venn diagram and scatterplot showing common downregulated genes.
(C) Fate profiles of RKO cells exposed to 0.1 μM taxol following Egr1 RNAi and immunoblots showing reduced Egr1 following Myc RNAi.
To understand how the active Kcnk1 siRNA suppresses DiM, we focused on the 58 downregulated genes in common with Myc (Figure 7B). Only two were repressed more than 2-fold in both conditions, namely SNORD102 and EGR1. Of these, Egr1, a zinc finger transcription factor, stands out as it is an established Myc target required for Myc-dependent, p53-independent apoptosis, and it cooperates with Myc to upregulate Bim and Noxa (Boone et al., 2011; Wirth et al., 2014). We reasoned therefore that the Kcnk1 siRNA might suppress DiM via inhibition of Egr1. Consistently, transcript profiling indicated that Bim, Bid, and Noxa were reduced following Kcnk1 siRNA (not shown). To test directly whether Egr1 promotes DiM, we transfected RKO cells with siRNAs specifically targeting Egr1. Strikingly, this shifted cell fate from DiM to slippage in a manner comparable to Myc siRNA (Figure 7C). Thus, these observations identify EGR1 as a “DiM” gene and suggest that KCNK1 manifested in the screen because of off-target activity toward Egr1.
Myc Modulates DNA Damage Accumulation in Mitosis
Although Myc and Egr1 appear to set the stage for DiM, what actually triggers apoptosis during a prolonged mitotic arrest is unclear. During the course of this work, we made two observations suggesting that Myc may modulate two recently identified mechanisms (Hayashi et al., 2012; Orth et al., 2012). First, we noted that ICAD, the inhibitor of CAD, was markedly reduced by Myc RNAi (Figure 3A). This was intriguing in light of the demonstration that CAD-dependent DNA damage incurred during mitosis activates p53 following slippage (Orth et al., 2012). In addition to being an inhibitor of CAD, ICAD is also a chaperone essential for CAD function (Nagase et al., 2003), and accordingly, inhibition of both ICAD and Myc reduced CAD (Figure S7A). Moreover, ICAD RNAi suppressed DiM (Figure 8A), suggesting that by stabilizing CAD, Myc promotes accumulation of DNA damage during mitosis thereby accelerating DiM. Consistently, γ-H2AX accumulation was less prevalent in taxol-treated Myc RNAi cells (Figure S7B).
Figure 8.

Inhibition of ICAD Enhances Slippage
(A) Fate profiles of RKO cells exposed to 0.1 μM taxol following ICAD RNAi.
(B) Fate profiles of RKO cells in the absence of taxol following RNAi-mediated co-repression Bcl-xL/Mcl1 plus tet-induced overexpression of TRF2. In (B), 0 hr represents when imaging started.
(C) Mechanistic model, see text for details.
See also Figure S7.
We were also intrigued by the very rapid DiM in cells lacking Bcl-xL and Mcl1 (Figure S3F). In addition, we noticed that in the absence of taxol, Bcl-xL/Mcl1-deficient cells often died upon mitotic entry (Figure S7C). However, it seems unlikely that ICAD/CAD-dependent damage accumulates fast enough to trigger apoptosis during an unperturbed mitosis. Indeed, ICAD RNAi had little protective effect in cells co-depleted for Bcl-xL and Mcl1 (not shown). In contrast, telomere deprotection might cause a burst of DNA damage upon mitotic entry (Hayashi et al., 2012). Indeed, inhibiting Aurora B in Bcl-xL/Mcl1-deficient cells reduced DiM from 69% to 34% (Figure S7C) and suppressing telomere deprotection by overexpressing TRF2 also had a protective effect (Figures 8B and S7D). Inhibiting Myc in Bcl-xL/Mcl1-deficient cells had an even more penetrant effect, reducing DiM in the absence of taxol from 69% to 10% (Figure S7C). Although this could simply reflect Myc’s role setting the balance between pro-survival and pro-death factors, these observations raise the possibility that Myc may also modulate the DNA damage-inducing pathways that trigger apoptosis during a prolonged mitotic arrest.
Discussion
The success of the siRNA screen was predicated on the existence of genes essential for DiM. Consistent with the SAC being indirectly required for DiM (Taylor and McKeon, 1997), we identified all the known SAC components. Indeed, SAC genes frequently manifest in antimitotic RNAi screens, yet apoptotic regulators rarely do (Díaz-Martínez et al., 2014). This suggests that the two networks governing mitotic fate are rather different: while the SAC consists of essential genes, the DiM network involves redundant sub-networks. Myc drives expression of the apoptotic network required for DiM, providing a simple explanation for why it manifested in the screen. The different architectures of the two networks may reflect evolutionary origins and/or buffering capacities. The SAC, which is conserved from yeast to man, is an “all-or-nothing” mechanism that responds to a single input, unattached kinetochores, and is not buffered by transcription (Lara-Gonzalez et al., 2012). In contrast, apoptosis, a metazoan characteristic, responds to multiple inputs and can be “fine-tuned” by transcriptional buffering depending on developmental context and homeostatic pressures (Barkett and Gilmore, 1999). The differing architectures also support the notion that they are largely independent (Gascoigne and Taylor, 2008; Huang et al., 2010).
In addition to driving proliferation, Myc overexpression drives apoptosis via ARF-MDM2-p53 (McMahon, 2014). Because p53 is disengaged during mitosis, it is not clear how this mechanism could contribute to DiM, and indeed p53 is not required for Myc-dependent mitotic death. Moreover, Myc can upregulate Bim, Bid, and Noxa independently of p53 (Campone et al., 2011; Egle et al., 2004; Eischen et al., 2001; Hemann et al., 2005; Iaccarino et al., 2003; Muthalagu et al., 2014; Nikiforov et al., 2007). Recent evidence shows that Myc drives p53-independent apoptosis by cooperating with Egr1, itself a Myc target (Boone et al., 2011). Myc promotes EGR1 expression via a non-canonical mechanism involving ARF and in turn, Myc and Egr1 are co-recruited to the promoters of BIM and NOXA (Boone et al., 2011; Wirth et al., 2014). It seems likely, therefore, that in interphase, Myc and Egr1 upregulate a cluster of redundant pro-apoptotic BH3-only proteins and suppress Bcl-xL, establishing the apoptotic network which can later induce DiM without the need for p53 engagement and de novo gene expression. Upon entry into mitosis, the apoptotic network is balanced so that a pro-survival environment is maintained (Figure 8C). However, in the presence of mitotic blockers, the balance slowly tips in favor of the pro-apoptotic BH3-only proteins, eventually triggering cell death. Several processes help tip the balance, including accumulation of DNA damage due to partial CAD activation and telomere deprotection (Hayashi et al., 2012; Orth et al., 2012). Also, slow degradation of Mcl1, possibly due to incomplete APC/C inhibition (Harley et al., 2010), weakens pro-survival function. When Myc is inhibited, the initial balance is more heavily weighted toward pro-survival, mitotic death is thus delayed providing more time for CyclinB1 degradation and slippage. Myc inhibition may also suppress DNA damage accumulation, weakening the apoptotic trigger. When Bcl-xL and Mcl1 are co-inhibited, the balance is so heavily weighted toward pro-death that cells cannot survive a short mitotic arrest, and even an unperturbed mitosis can induce apoptosis.
Myc is also required for efficient apoptosis in response to drugs that drive cells through an aberrant mitosis. Whether this is because these cells inherit an apoptotic balance tipped in favor of pro-survival or cannot initiate a robust post-mitotic response remains to be seen. Consistent with the latter, Myc also promotes cell cycle restraint following SAC override. One possibility to account for this is the ARF-MDM2-p53 pathway; by inducing ARF and thus suppressing MDM2, Myc may sensitize the p53-dependent mechanism that detects DNA damage incurred when chromosomes missegregate (Janssen et al., 2011). This may explain why ARF-deficient mouse embryonic fibroblasts tolerate aneuploidies (Silk et al., 2013). Alternatively, following chromosome missegregation induced by SAC override, Myc’s ability to drive global gene expression might elevate the proteotoxic burden that arises in aneuploid daughter cells, thus enhancing cell cycle suppression (Tang and Amon, 2013). Interestingly, several mitotic regulators are synthetic lethal with Myc overexpression, including Cdk1, Survivin, Aurora B, and the SUMO-activating enzyme SAE-2 (den Hollander et al., 2010; Goga et al., 2007; Kessler et al., 2012; Yang et al., 2010). Whether this is due to deregulation of mitosis per se as opposed to deregulation of cellular responses to mitotic abnormalities is unclear. Consistent with the former, SAE-2 modulates a spindle assembly gene expression program (Kessler et al., 2012). Consistent with the latter, our observations show that Myc enhances both DiM and post-mitotic responses.
Antimitotic agents continue to be important frontline drugs, emphasized by the impressive effect of combining taxanes with targeted therapies in the treatment of breast cancer (Slamon et al., 2001). Whether taxanes inhibit tumor growth via antimitotic or other tubulin-dependent mechanisms remains unclear (Komlodi-Pasztor et al., 2012; Mitchison, 2012). Consistent with Myc enhancing antimitotic apoptosis, ovarian cancers treated with taxol and carboplatin responded better if Myc was more highly expressed (Iba et al., 2004). Consistently, Her2-negative breast cancers that responded to docetaxel and capecitabine had higher Myc levels. The correlation between Myc and cell cycle/SAC genes is especially striking because Her2-positive tumors do not show a similar pattern. This suggests that docetaxel-capecitabine responses require cell cycle progression and a robust SAC response. In contrast, anti-tumor effects mediated by trastuzumab-docetaxel-capecitabine are more likely dominated by inhibition of Her2-dependent PI3K/Akt survival signaling (Berns et al., 2007), and therefore less dependent on mitotic entry and SAC activation. Taken together, these observations suggest that the Myc network may yield potential biomarkers. However, a recent study found that while triple-negative breast cancers exhibited elevated Myc expression, this did not predict responses to neoadjuvant chemotherapy (Horiuchi et al., 2012). Consistent with the mechanisms we describe here, this study did however observe that elevated Myc sensitized triple-negative cells to Cdk1 inhibition in a Bim-dependent, p53-independent manner. Thus, taking together our observations, the synthetic lethality relationships described above, and the provocative clinical observations, there is considerable merit in further exploring the links between the BH3-only/Bcl-xL pathway and mitotic regulators in the context of Myc-driven tumors. Interestingly, Myc inhibition had little effect on three cell lines we studied, suggesting that this avenue may provide insight into intrinsic resistance, while changes and/or heterogeneity in Myc expression may provide insight into acquired taxane resistance.
Myc suppresses Bcl-xL in various contexts (Eischen et al., 2001) and they inversely correlate in the breast cancer gene expression profiles we analyzed. Moreover, Bcl-xL overexpression potently blocks Myc-driven apoptosis (Pelengaris et al., 2002) and our observations reaffirm Bcl-xL as a potent mitotic survival factor. Although Mcl1 and Bcl-xL can partially compensate for each other during mitosis (Shi et al., 2011), degradation of Mcl1 during a mitotic arrest means that Bcl-xL becomes particularly critical following slippage. Because slippage is a clinically relevant phenotype (Zasadil et al., 2014), these observations make a compelling case for combining Bcl-xL inhibitors with antimitotic agents. Indeed, the Bcl2/Bcl-xL inhibitor navitoclax sensitizes ovarian cancer cell lines to taxol (Wong et al., 2012). Similar combination strategies may also help revive the prospects of targeted antimitotic agents that have thus far been disappointing in the clinic (Komlodi-Pasztor et al., 2012; Mitchison, 2012). Exploring Myc-dependent apoptotic pathways for predicative biomarkers may also facilitate better clinical evaluation of these agents. Finally, as a potent driver of tumorigenesis, Myc is itself an attractive anti-cancer target (McKeown and Bradner, 2014; Sodir and Evan, 2011). However, if superimposed on existing taxane chemotherapy regimens, targeting Myc may be counterproductive, weakening both the SAC and post-mitotic apoptosis, thereby fueling genomic instability. This should not detract from Myc as a target as long as mitigating strategies are also explored. Our observation that pharmacological inhibition of Bcl-xL potently restores apoptosis in Myc-deficient cells exposed to low-dose taxol further supports the case for exploring Bcl-xL inhibitors in the context of antimitotic agents.
Experimental Procedures
siRNA Library Screen
RKO cells were synchronized for 16 hr using 2 mM thymidine, released then seeded in 96-well plates (Greiner Bio-One) containing Opti-MEM media (LifeTechnologies), DharmaFECT 1 transfection reagent (Dharmacon), and siRNAs at a final concentration of 66 nM, after which 0.1 μM taxol and viability reagent (CellTiter 96 AQueousOne Solution Cell Proliferation Assay, Promega) were added after 24 and 68 hr, respectively, and the absorbance at 490 nm measured after 72 hr. For the tertiary screen, the mitotic index at 24 hr was determined using a BD Pathway (BD Biosciences). Cell lines and small molecule inhibitors are described in the Supplemental Experimental Procedures.
Functional Experiments
siRNAs and DharmaFECT 1 combined in Opti-MEM media were added to RKO cells plated at 10 × 104 cells/ml, yielding a final siRNA concentration of 66 nM. For siRNA sequences, see the Supplemental Experimental Procedures. Open reading frames described in the Supplemental Experimental Procedures were cloned into pcDNA5/FRT/TO based vectors and isogenic, tetracycline-inducible, stable cell lines generated by co-transfection with pOG44 (Invitrogen) into Flp-In T-REx RKO cells. Phase contrast imaging, cell proliferation, and apoptosis measurements were performed on an IncuCyte ZOOM (EssenBioScience) with CellPlayer Kinetic Caspase-3/7 Apoptosis Assay Kit (EssenBioSciences). Image sequences were analyzed manually and statistical analysis performed with GraphPad Prism. On fate profiles, 0 hr corresponds to mitotic entry unless stated otherwise in the legend. Immunoblotting was performed using antibodies described in the Supplemental Experimental Procedures.
Gene Expression Profiling
Cells transfected with siRNAs were synchronized, released for 5 hr, then RNA was prepared using Trizol (Life Technologies). One hundred nanograms of RNA was hybridized with custom nCounter Reporter and Capture probe sets (Nanostring Technologies) at 65°C overnight, unhybridized probes removed, complexes bound to the imaging surface, and images acquired using the nCounter Digital Analyzer. Transcript counts were normalized to housekeeping genes using nSolver Analysis Software. For global gene expression profiling, total RNA was processed using the Illumina TruSeq Stranded mRNA Sample Preparation Kit, then cDNA libraries sequenced on an Illumina HiSeq 2000 using single read, 50 cycle runs. Quality of sequencing reads was assessed using FastQC (Babraham Bioinformatics) and aligned to a reference genome (hg19, UCSC Genome Browser) using TopHat. Sequencing yielded on average 23.7 million unique reads per sample with a 60.7%–65.7% mapping rate. Cufflinks was used to generate transcript abundance as fragments per kilobase of transcript per million mapped reads (FPKM), and statistical analysis of FPKM values was calculated using R (Bioconductor).
Inactivation of Myc in the Mouse Intestine
Cre-mediated inactivation of MYC in the intestinal epithelium was induced via three intraperitoneal (i.p.) injections of 80 mg/kg β-naphthoflavone in 1 day. Four days later, 10 mg/kg taxol was administered via i.p. injection, tissue harvested after various time points, fixed in 4% formaldehyde, then stained for cleaved caspase 3 (R&D systems). All animal experiments were conducted under an appropriate animal project license approved by the UK home office and in accordance with the Animal Welfare and Experimental Ethics Committee at the University of Glasgow.
Statistical Methods
Statistical analysis was performed in GraphPad Prism 6 as follows: ANOVA plus Bonferroni (Figures 2C and 2F); linear regression (Figure 2D); correlation (Figures S1B and 5E); Paired t test (Figure 2G); Wilcoxon t test (Figure 4A); Mann-Whitney (Figures 4B, 5A, 5B, 5D, 6, S3A, S3I, and S5B); Kruskal-Wallis (Figure S1E). In figures, p values were ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Scatterplots show mean and SD. Unless stated otherwise in the figure legend, box-and-whisker plots show median, interquartile ranges, plus min to max range. Figure S1D, mean ± SD; Figure S4B, mean ± SEM.
Author Contributions
C.T. performed the RNAi screen, Nanostring analysis, and functional experiments. A.T., A.B., O.S., L.N., S.L., and C.S. contributed to the functional experiments. R.A.R., D.H., and O.J.S. contributed the mouse data. B.B. and D.J.P. synthesized the Cenp-E inhibitor. P.L., Y.S., and D.W.C. contributed the HiSeq analysis. S.S.T. conceived the study and wrote the manuscript. All authors read and commented on the manuscript.
Acknowledgments
This work was supported by the Genomic Technologies and Bioimaging Core Facilities in the Faculty of Life Sciences. We thank Stefan Knapp (University of Oxford) and Bert Vogelstein (Johns Hopkins) for reagents; and Andy Hayes, Leo Zeef, Dave Spiller, Andy Sharrocks, Mike White, Gino Poulin, Donald Ogilvie, Dean Jackson, and William Weiss for useful discussions. Funding was provided by Cancer Research UK, the Medical Research Council, and the Wellcome Trust. S.S.T. is supported by a Cancer Research UK Senior Fellowship.
Published: July 13, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2015.06.001.
Accession Numbers
The accession number for the global gene expression data reported in this paper is GEO: GSE68219.
Supplemental Information
References
- A’Hern R.P., Jamal-Hanjani M., Szász A.M., Johnston S.R., Reis-Filho J.S., Roylance R., Swanton C. Taxane benefit in breast cancer—a role for grade and chromosomal stability. Nat. Rev. Clin. Oncol. 2013;10:357–364. doi: 10.1038/nrclinonc.2013.67. [DOI] [PubMed] [Google Scholar]
- Bah N., Maillet L., Ryan J., Dubreil S., Gautier F., Letai A., Juin P., Barillé-Nion S. Bcl-xL controls a switch between cell death modes during mitotic arrest. Cell Death Dis. 2014;5:e1291. doi: 10.1038/cddis.2014.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkett M., Gilmore T.D. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- Berns K., Horlings H.M., Hennessy B.T., Madiredjo M., Hijmans E.M., Beelen K., Linn S.C., Gonzalez-Angulo A.M., Stemke-Hale K., Hauptmann M. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Boone D.N., Qi Y., Li Z., Hann S.R. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc. Natl. Acad. Sci. USA. 2011;108:632–637. doi: 10.1073/pnas.1008848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito D.A., Rieder C.L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 2006;16:1194–1200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campone M., Noël B., Couriaud C., Grau M., Guillemin Y., Gautier F., Gouraud W., Charbonnel C., Campion L., Jézéquel P. c-Myc dependent expression of pro-apoptotic Bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1. Mol. Cancer. 2011;10:110. doi: 10.1186/1476-4598-10-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M., McFerrin L., Eisenman R.N. An overview of MYC and its interactome. Cold Spring Harb Perspect Med. 2014;4:a014357. doi: 10.1101/cshperspect.a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander J., Rimpi S., Doherty J.R., Rudelius M., Buck A., Hoellein A., Kremer M., Graf N., Scheerer M., Hall M.A. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Martínez L.A., Karamysheva Z.N., Warrington R., Li B., Wei S., Xie X.J., Roth M.G., Yu H. Genome-wide siRNA screen reveals coupling between mitotic apoptosis and adaptation. EMBO J. 2014;33:1960–1976. doi: 10.15252/embj.201487826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumontet C., Jordan M.A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle A., Harris A.W., Bouillet P., Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorn J.M., Alford S.E., Sakurikar N., Chambers T.C. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp. Cell Res. 2014;322:415–424. doi: 10.1016/j.yexcr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eick D., Bornkamm G.W. Transcriptional arrest within the first exon is a fast control mechanism in c-myc gene expression. Nucleic Acids Res. 1986;14:8331–8346. doi: 10.1093/nar/14.21.8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M., Eisenman R.N. Myc’s broad reach. Genes Dev. 2008;22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen C.M., Woo D., Roussel M.F., Cleveland J.L. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol. 2001;21:5063–5070. doi: 10.1128/MCB.21.15.5063-5070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X., Soares J. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne K.E., Taylor S.S. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Gascoigne K.E., Taylor S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009;122:2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- Glück S., Ross J.S., Royce M., McKenna E.F., Jr., Perou C.M., Avisar E., Wu L. TP53 genomics predict higher clinical and pathologic tumor response in operable early-stage breast cancer treated with docetaxel-capecitabine ± trastuzumab. Breast Cancer Res. Treat. 2012;132:781–791. doi: 10.1007/s10549-011-1412-7. [DOI] [PubMed] [Google Scholar]
- Goga A., Yang D., Tward A.D., Morgan D.O., Bishop J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007;13:820–827. doi: 10.1038/nm1606. [DOI] [PubMed] [Google Scholar]
- Hann S.R. MYC cofactors: molecular switches controlling diverse biological outcomes. Cold Spring Harb Perspect Med. 2014;4:a014399. doi: 10.1101/cshperspect.a014399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley M.E., Allan L.A., Sanderson H.S., Clarke P.R. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–2420. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M.T., Cesare A.J., Fitzpatrick J.A.J., Lazzerini-Denchi E., Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol. 2012;19:387–394. doi: 10.1038/nsmb.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann M.T., Bric A., Teruya-Feldstein J., Herbst A., Nilsson J.A., Cordon-Cardo C., Cleveland J.L., Tansey W.P., Lowe S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D., Kusdra L., Huskey N.E., Chandriani S., Lenburg M.E., Gonzalez-Angulo A.M., Creasman K.J., Bazarov A.V., Smyth J.W., Davis S.E. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012;209:679–696. doi: 10.1084/jem.20111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.C., Mitchison T.J., Shi J. Stochastic competition between mechanistically independent slippage and death pathways determines cell fate during mitotic arrest. PLoS ONE. 2010;5:e15724. doi: 10.1371/journal.pone.0015724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino I., Hancock D., Evan G., Downward J. c-Myc induces cytochrome c release in Rat1 fibroblasts by increasing outer mitochondrial membrane permeability in a Bid-dependent manner. Cell Death Differ. 2003;10:599–608. doi: 10.1038/sj.cdd.4401211. [DOI] [PubMed] [Google Scholar]
- Iba T., Kigawa J., Kanamori Y., Itamochi H., Oishi T., Simada M., Uegaki K., Naniwa J., Terakawa N. Expression of the c-myc gene as a predictor of chemotherapy response and a prognostic factor in patients with ovarian cancer. Cancer Sci. 2004;95:418–423. doi: 10.1111/j.1349-7006.2004.tb03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A., van der Burg M., Szuhai K., Kops G.J., Medema R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- Keen N., Taylor S. Mitotic drivers—inhibitors of the Aurora B Kinase. Cancer Metastasis Rev. 2009;28:185–195. doi: 10.1007/s10555-009-9184-9. [DOI] [PubMed] [Google Scholar]
- Kessler J.D., Kahle K.T., Sun T., Meerbrey K.L., Schlabach M.R., Schmitt E.M., Skinner S.O., Xu Q., Li M.Z., Hartman Z.C. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E., Sackett D.L., Fojo A.T. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin. Cancer Res. 2012;18:51–63. doi: 10.1158/1078-0432.CCR-11-0999. [DOI] [PubMed] [Google Scholar]
- Lara-Gonzalez P., Westhorpe F.G., Taylor S.S. The spindle assembly checkpoint. Curr. Biol. 2012;22:R966–R980. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Lessene G., Czabotar P.E., Sleebs B.E., Zobel K., Lowes K.N., Adams J.M., Baell J.B., Colman P.M., Deshayes K., Fairbrother W.J. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013;9:390–397. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- Li Q., Dang C.V. c-Myc overexpression uncouples DNA replication from mitosis. Mol. Cell. Biol. 1999;19:5339–5351. doi: 10.1128/mcb.19.8.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe S.W., Cepero E., Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- McKeown M.R., Bradner J.E. Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med. 2014;4:a014266. doi: 10.1101/cshperspect.a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon S.B. MYC and the control of apoptosis. Cold Spring Harb Perspect Med. 2014;4:a014407. doi: 10.1101/cshperspect.a014407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn A.J., Boise L.H., Thompson C.B. Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage. Genes Dev. 1996;10:2621–2631. doi: 10.1101/gad.10.20.2621. [DOI] [PubMed] [Google Scholar]
- Mitchison T.J. The proliferation rate paradox in antimitotic chemotherapy. Mol. Biol. Cell. 2012;23:1–6. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray S., Briasoulis E., Linardou H., Bafaloukos D., Papadimitriou C. Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat. Rev. 2012;38:890–903. doi: 10.1016/j.ctrv.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Muthalagu N., Junttila M.R., Wiese K.E., Wolf E., Morton J., Bauer B., Evan G.I., Eilers M., Murphy D.J. BIM is the primary mediator of MYC-induced apoptosis in multiple solid tissues. Cell Rep. 2014;8:1347–1353. doi: 10.1016/j.celrep.2014.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H., Fukuyama H., Tanaka M., Kawane K., Nagata S. Mutually regulated expression of caspase-activated DNase and its inhibitor for apoptotic DNA fragmentation. Cell Death Differ. 2003;10:142–143. doi: 10.1038/sj.cdd.4401139. [DOI] [PubMed] [Google Scholar]
- Nikiforov M.A., Riblett M., Tang W.H., Gratchouck V., Zhuang D., Fernandez Y., Verhaegen M., Varambally S., Chinnaiyan A.M., Jakubowiak A.J., Soengas M.S. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc. Natl. Acad. Sci. USA. 2007;104:19488–19493. doi: 10.1073/pnas.0708380104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth J.D., Loewer A., Lahav G., Mitchison T.J. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell. 2012;23:567–576. doi: 10.1091/mbc.E11-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelengaris S., Khan M., Evan G.I. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109:321–334. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- Phesse T.J., Myant K.B., Cole A.M., Ridgway R.A., Pearson H., Muncan V., van den Brink G.R., Vousden K.H., Sears R., Vassilev L.T. Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ. 2014;21:956–966. doi: 10.1038/cdd.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulescu S., Ridgway R.A., Appleton P., Kroboth K., Patel S., Woodgett J., Taylor S., Nathke I.S., Sansom O.J. Defining the role of APC in the mitotic spindle checkpoint in vivo: APC-deficient cells are resistant to Taxol. Oncogene. 2010;29:6418–6427. doi: 10.1038/onc.2010.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder C.L., Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Shi J., Zhou Y., Huang H.C., Mitchison T.J. Navitoclax (ABT-263) accelerates apoptosis during drug-induced mitotic arrest by antagonizing Bcl-xL. Cancer Res. 2011;71:4518–4526. doi: 10.1158/0008-5472.CAN-10-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk A.D., Zasadil L.M., Holland A.J., Vitre B., Cleveland D.W., Weaver B.A. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. USA. 2013;110:E4134–E4141. doi: 10.1073/pnas.1317042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon D.J., Leyland-Jones B., Shak S., Fuchs H., Paton V., Bajamonde A., Fleming T., Eiermann W., Wolter J., Pegram M. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Sodir N.M., Evan G.I. Finding cancer’s weakest link. Oncotarget. 2011;2:1307–1313. doi: 10.18632/oncotarget.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L., Jucker R., Panacchia L., Ricordy R., Tatò F., Nasi S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002;62:3507–3510. [PubMed] [Google Scholar]
- Tang Y.C., Amon A. Gene copy-number alterations: a cost-benefit analysis. Cell. 2013;152:394–405. doi: 10.1016/j.cell.2012.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S.S., McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Thompson S.L., Compton D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010;188:369–381. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham C.H., Taylor S.S. Mitosis and apoptosis: how is the balance set? Curr. Opin. Cell Biol. 2013;25:780–785. doi: 10.1016/j.ceb.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Upreti M., Galitovskaya E.N., Chu R., Tackett A.J., Terrano D.T., Granell S., Chambers T.C. Identification of the major phosphorylation site in Bcl-xL induced by microtubule inhibitors and analysis of its functional significance. J. Biol. Chem. 2008;283:35517–35525. doi: 10.1074/jbc.M805019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell. 2014;25:2677–2681. doi: 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese K.E., Walz S., von Eyss B., Wolf E., Athineos D., Sansom O., Eilers M. The role of MIZ-1 in MYC-dependent tumorigenesis. Cold Spring Harb Perspect Med. 2013;3:a014290. doi: 10.1101/cshperspect.a014290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M., Stojanovic N., Christian J., Paul M.C., Stauber R.H., Schmid R.M., Häcker G., Krämer O.H., Saur D., Schneider G. MYC and EGR1 synergize to trigger tumor cell death by controlling NOXA and BIM transcription upon treatment with the proteasome inhibitor bortezomib. Nucleic Acids Res. 2014;42:10433–10447. doi: 10.1093/nar/gku763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E., Lin C.Y., Eilers M., Levens D.L. Taming of the beast: shaping Myc-dependent amplification. Trends Cell Biol. 2015;25:241–248. doi: 10.1016/j.tcb.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M., Tan N., Zha J., Peale F.V., Yue P., Fairbrother W.J., Belmont L.D. Navitoclax (ABT-263) reduces Bcl-x(L)-mediated chemoresistance in ovarian cancer models. Mol. Cancer Ther. 2012;11:1026–1035. doi: 10.1158/1535-7163.MCT-11-0693. [DOI] [PubMed] [Google Scholar]
- Yang D., Liu H., Goga A., Kim S., Yuneva M., Bishop J.M. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc. Natl. Acad. Sci. USA. 2010;107:13836–13841. doi: 10.1073/pnas.1008366107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadil L.M., Andersen K.A., Yeum D., Rocque G.B., Wilke L.G., Tevaarwerk A.J., Raines R.T., Burkard M.E., Weaver B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014;6:229–243. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.